DOI:
10.1039/C8RA06576D
(Paper)
RSC Adv., 2018,
8, 33059-33064
Facile and template-free solvothermal synthesis of mesoporous/macroporous metal–organic framework nanosheets
Received
5th August 2018
, Accepted 18th September 2018
First published on 24th September 2018
Abstract
A facile and template-free solvothermal method was developed as a bottom-up approach to synthesize mesoporous/macroporous MOF nanosheets in a simple and scalable way. It was found that starting coordination complexes of different copper(II)-ligand compounds mediated the controlled growth and morphology of MOF crystals. By controlling the size and shape of the MOF crystals, the possibility to adjust and tailor the structure and performances of the assemblies was demonstrated. This work provides a bottom-up approach to synthesize MOF films and nanosheets in a simple and scalable way, which may have potential in energy and biomedical applications.
Introduction
Metal–organic-frameworks (MOFs) are a class of compounds composed of metal ions or clusters within a network of organic ligand linkages. Similar to other porous materials, MOFs have attracted interest for use in gaseous or liquid adsorption, sensor materials,1 catalysis, and biomedical devices.2,3 Large pore volumes and surface areas in conjunction with the well-organized structures that MOFs display set them apart from other porous materials such as zeolites and activated carbons.4 Furthermore, MOFs have enormous flexibility in composition and structure, can be synthesized quickly at low cost, and can be modified to have distinct functionalities.5
Theoretically, MOF pore size and functionality can be controlled by modification of metal ions and organic linkers. However, the conventional MOF synthesis limits the applicability as agglomeration decreases porosity and complicates gas adsorption and separation.4,6 Recent work combined MOFs with other macroporous compounds to form materials with both micro- and macroporous features.7,8 Other methods avoid agglomeration and add porosity by arranging the MOFs into macroporous and/or mesoporous frameworks through packing into monoliths, dispersing in aerogels or arranging in films or membranes.2,3,9,10
MOF films and nanosheets, in particular, have been of interest for commercial applications, especially in molecular sieving,12,13 catalytic membranes and semiconductor materials14 due to their tendency to form mesopores and macropores.11 Other applications include liquid phase separations and chromatography as well as the storage, separation, or catalytic conversion of small molecules.9,15,16 The applications of MOF nanofilms have even extended to photonic devices.17 However, the control of nanosheet thickness, crystallinity, and growth direction in MOF synthesis remains a work in progress.6,18,19
Many different methods have been employed to create MOF films and nanosheets.17,20–22 Most methods require templating MOFs onto other substances, including surfactant templating,23,24 liquid phase epitaxy25 among others.2,12,26 Though templating methods produce thin layers and homogeneous coatings, direct synthesis of template-free MOF nanosheets or films could significantly reduce the cost and complexity of synthesis, but this is largely unavailable.
Efficient, high-yielding methods that can produce MOF nanosheets with stable mesoporous frameworks are rare.19 Synthesizing MOFs as freestanding and template-free nanosheets that retain morphological and structural integrity has been a challenge.6 To create films without templates, several groups have synthesized agglomerated nanosheets with organic solvents, followed by lengthy ultrasonication.14,19 Recently, Li et al. developed a top-down delamination method to synthesize crystalline MOF nanosheets from bulk crystals.27 Rodenas et al.6 and Wu et al.17 employed a diffusion-mediated modulation of MOF molecular assembly to form a small layer within glass tubes. Nonetheless, applications are limited by the scalability issue of their synthetic methods.
In this study, a facile and template-free solvothermal method has been developed to synthesize mesoporous/macroporous MOF nanosheets. In our method, cupric salts (i.e., Ac−, Cl−, NO3−, SO42−) and terephthalic acid (TPA) were dissolved in organic solvents containing dimethylformamide (DMF) and methanol (MeOH). The entire synthesis was performed in a Teflon-lined solvothermal autoclave reactor. The growth rate and morphology of the MOFs were controlled using starting coordination complexes of different copper(II)-ligands.
Experimental
Solvothermal synthesis of MOF
Cupric salts, terephthalic acid (TPA), dimethylformamide (DMF), methanol and ethanol were purchased from Sigma Aldrich (St. Louis, MO, USA). The MOF matrices were synthesized via a modified solvothermal method.28 Cupric salt (55 mg of copper(II) chloride, 76 mg of copper(II) nitrate, 74 mg of copper(II) acetate, or 65 mg of copper(II) sulfate) and 92 mg of terephthalic acid (TPA) were dissolved in organic solvents containing 10 mL dimethylformamide (DMF) and 10 mL methanol or a solvent system inside a Teflon lined autoclave reactor (Col-Int Tech, Columbia, SC, USA). The solution was sonicated for 5 minutes to dissolve all particles and incubated for 24 hours at room temperature. The MOF product was synthesized by heating the reactor at either 120 °C for 48 hours or 180 °C for 24 hours. The crystalline bluish solids were centrifuged (5000 × g for 10 minutes) and washed three times with pure ethanol. The product was dried and activated in a vacuum oven at 100 °C.
X-ray diffraction crystallography (XRD)
The MOF powder diffraction was characterized using a Bruker D8 Advance powder diffractometer (Bruker AXS Inc., Madison, WI, USA). The equipment was equipped with a CuKα sealed tube (wavelength of 1.541 78 Å), Ni β-filter, and position-sensitive LynxEye detector and operated in Bragg–Brentano mode (θ–θ geometry). Phase identification was performed using the international center for diffraction data (ICDD) powder diffraction database, and the data were further analyzed using Le Bail and Rietveld refinement on the supplier's TOPAS software (Bruker AXS Inc., Madison, WI, USA).
Electron microscopy
Scanning electron photomicrographs were captured using an S-4700 low-temperature field emission scanning electron microscope (LT-SEM) (Hitachi High Technologies America, Inc., Pleasanton, CA, USA) with a Quorum Cryo-Prep Chamber (Quorum Technologies, East Sussex, UK). All MOF samples were pretreated with a thin layer of Pt coating. All images were captured at 10 kV accelerating voltage, and 10 mm working distance with a 4 pi Analysis System (Hillsborough, NC, USA). For transmission electron microscopy (TEM), ethanol solutions containing MOF samples were directly applied onto 400 mesh carbon-coated copper grids and allowed to absorb for 30 minutes, excess solution was wicked off and grids were air dried. Grids were imaged at 80 kV with a Hitachi HT-7700 TEM (Hitachi High Technologies America, Inc., Dallas, TX, USA).
Characterization of MOF porosity and surface area
Nitrogen (N2) isotherms were measured on a Micromeritics TriStar II Plus (Micromeritics Instrument Co., Norcross, GA, USA) unit to full saturation, i.e., a relative pressure of approximately ∼1.0 at 77 K to enable BET surface area and TOPV analyses. In addition, a low pressure (P < 1 atm) ethylene isotherm was measured on a Micromeritics Accelerated Surface Area and Porosimetry System (ASAP, Norcross, GA, USA) unit at 25 °C. The samples were activated on a Smart VacPrep degas unit by degassing in stages with a series of ramp/soak steps under dynamic vacuum till the final temperature reached 150 °C with a vacuum level of < 10−4 torr. Measurements of N2 BET on standard materials suggest accuracy to within approximately 5% at surface area values of 10 m2 g−1 and approximately 10% at levels of ∼0.5 m2 g−1 when at least 50 m2 of material are available for testing within the sample cell. Reproducibility for any given sample is dependent on the ability to regenerate the sample to the same degree of activation without modifying the surface or the pore structure. The cumulative volume was calculated based on a BJH analysis which relates pore size to relative pressure. In this analysis, a Halsey correction was used to account for monolayer thickness coverage.
Results and discussion
Synthesis
Fig. 1 shows the MOF morphologies resulting from starting coordination complexes of different copper(II)-ligands, including Cu2+–Ac−, Cu2+–SO42−, Cu2+–Cl−, and Cu2+–NO3−. Fig. 2a highlights the TEM micrograph of the as-obtained MOF nanosheets mediated via the Cu2+–Cl− coordination complex. The free-standing nanosheets consisted of both individual films and macroporous agglomerated stacks. Fig. 2b shows an SEM micrograph of a cross-sectional view of the sponge-like MOF nanosheets in the agglomerated form. The thickness of an individual nanosheet was about 50 nm, and the width of the nanosheet was over 15 μm (Fig. 1a). Fig. 1e showed nanosheet structure at higher magnification power, and its selected area electron diffraction (SAED) pattern suggests that the nanosheet is polycrystalline. Similar structures were observed in the MOF synthesized via Cu2+–NO3− coordination complex (Fig. 1b), in which the MOFs had the form of porous and fragmented nanosheets. The high degree of crystallinity of both nanocubes and nanosheets produces clearly identifiable peaks in the X-ray powder diffraction (XRD) patterns.
 |
| | Fig. 1 TEM photomicrograph of MOF morphologies controlled via different copper(II)-ligand coordination complexes: (a) Cu2+–Cl−; (b) Cu2+–NO3−; (c) Cu2+–Ac−; (d) Cu2+–SO42−; (e) Cu2+–Cl− at higher magnification; (f) selected area electron diffraction (SAED) pattern of Cu2+–Cl− nanosheet (a and e). | |
 |
| | Fig. 2 (a) Micrograph of MOF nanosheet from transmission electron microscopy (TEM); (b) micrograph of MOF nanosheet from scanning electron microscopy (SEM). | |
XRD
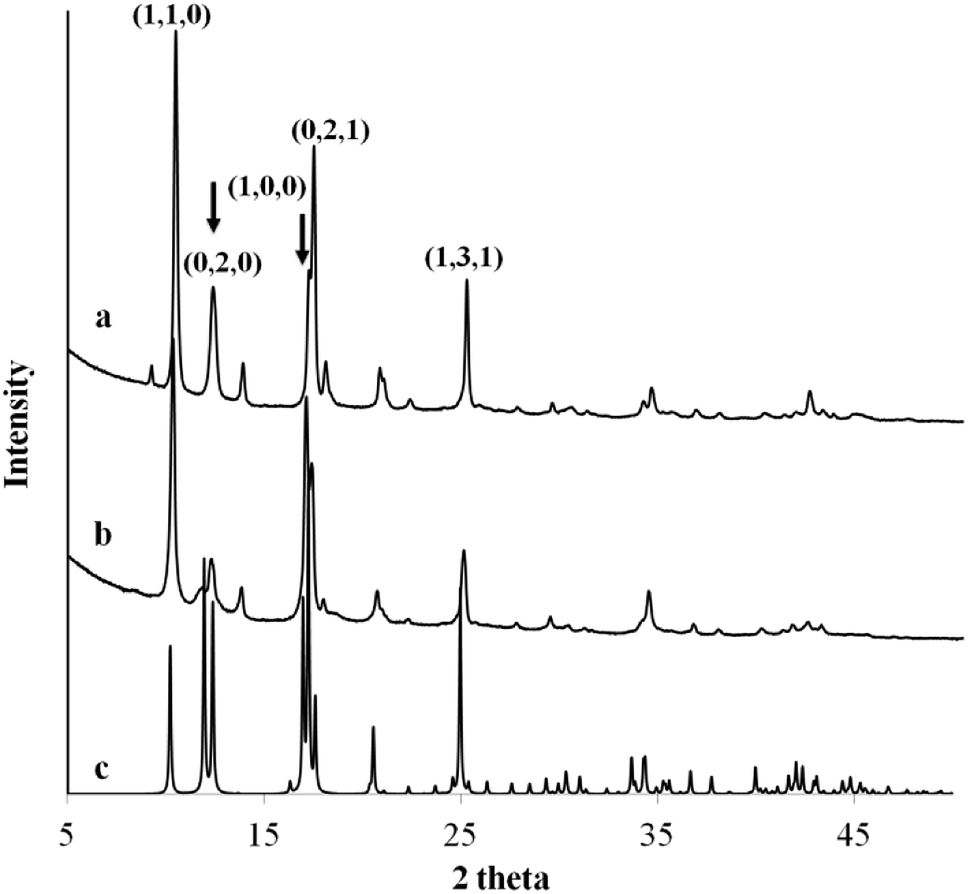
Fig. 3 compares the XRD powders diffraction pattern between the anisotropic MOF nanosheet prepared from Cu2+–Cl− coordination complexes and the MOF nanocubes formed via Cu2+–Ac− coordination complexes. The XRD analyses of the as-obtained MOFs with different morphologies revealed the same cupric-TPA-based MOF.29 However, changes in the original cupric-ligand coordination complex could help engineer the size and shape of the MOF nanocrystals by determining the packing and growth rate of the MOF crystals during the solvothermal synthesis. The copper-ligand coordination complexes have different initial solubilities, in the order of NO3− > SO42 > Cl− > Ac−. It was found that lower solubility with the Ksp below 1 × 10−6 could restrain the growth of MOF crystals in sizes and in 2-dimension, and therefore, nanosheet were only obtained from Cl− and Ac−. In this study, the differences in crystal packing and gas storage capacity were investigated between two representative samples, which are MOF nanocubes (Cu2+–Ac−) and MOF nanosheets (Cu2+–Cl−).
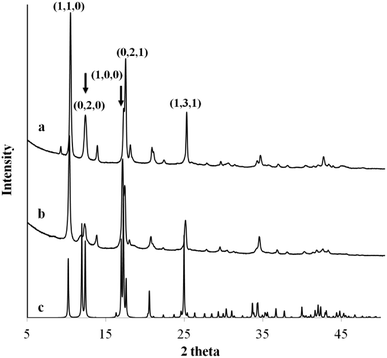 |
| | Fig. 3 The XRD powder diffraction pattern of MOF (a) nanocubes formed via Cu2+–Ac− coordination complex, (b) nanosheet formed via Cu2+–Cl− coordination complex, and (c) the identified single crystal diffraction pattern from ICDD database. | |
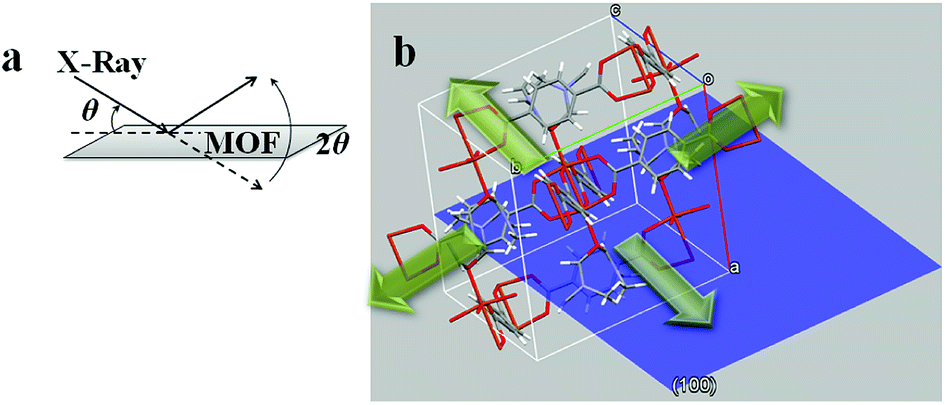
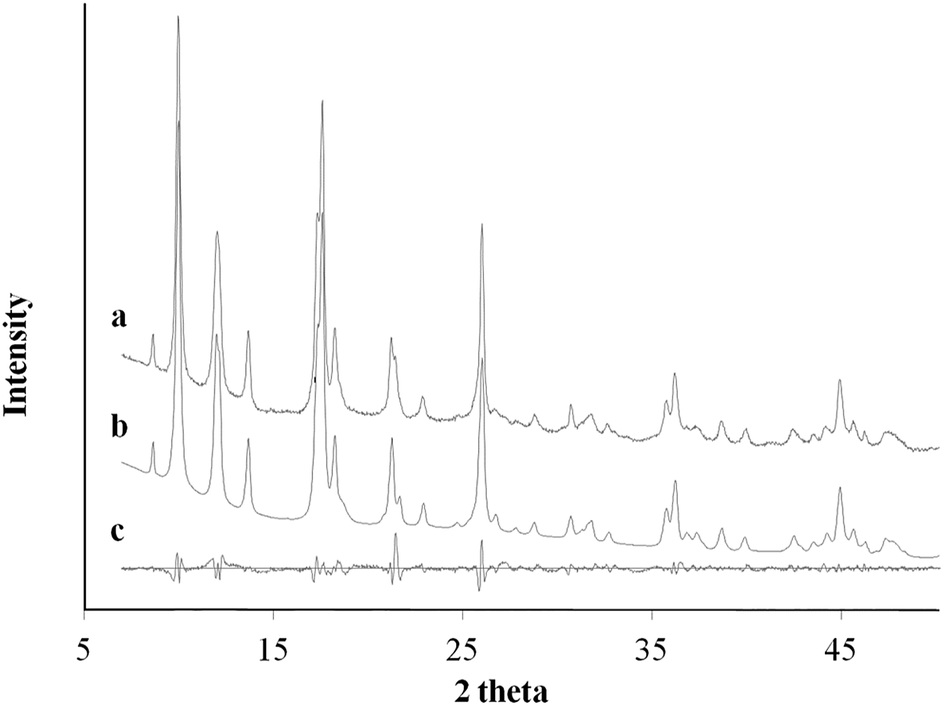
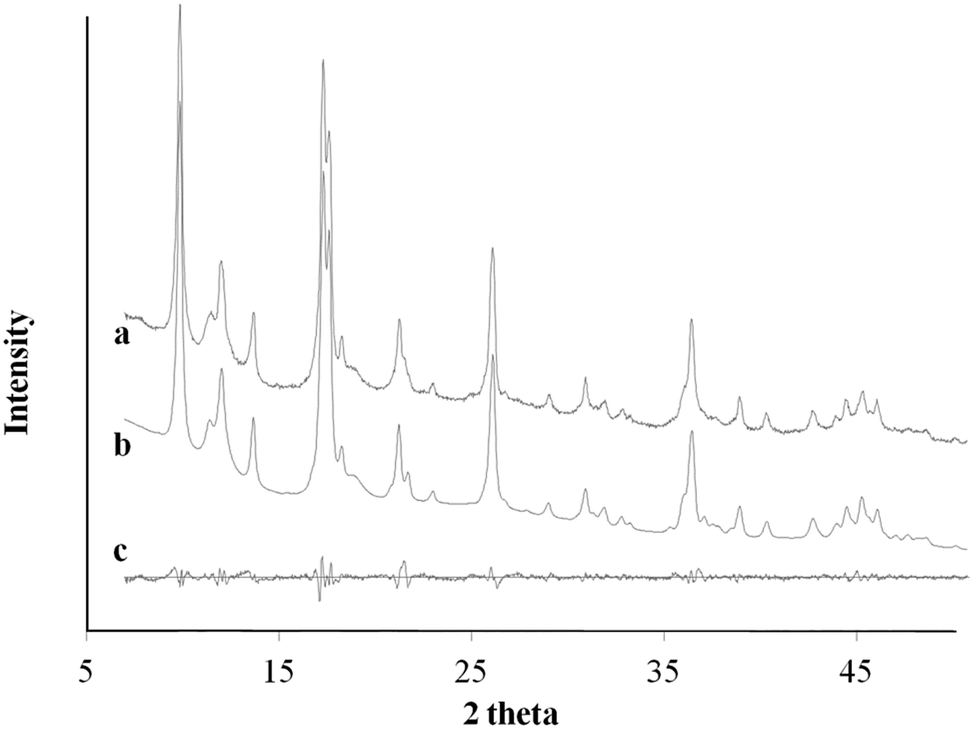
Ac− is a weaker ligand to Cu2+ and the organic ligand has better solubility in the organic solvent base of DMF and MeOH comparing to the Cl− ligand. The differences in ligand affinity and solubility could have led to a slower reaction rate and the anisotropy in the crystal growth kinetics.30 The MOF nanosheet revealed the significant change in the ratio of peak intensity (Fig. 3). The increase in peak intensity at 2θ = 16.380° was assigned to (h,k,l = 2,0,0) crystallographic plane, and the drop in peak intensity at 2θ = 12.165° was assigned to (h,k,l = 0,2,0) crystallographic plane. The significant change in (020 and 200) peak intensity (Fig. 3b) was a result of out-of-plane XRD (Fig. 4a). The nanosheet pattern was further fitted using Le Bail refinement fitting for preferred orientation (PO) tendency analysis (Fig. 5 and 6). The results suggest that the stronger Cu2+–Cl− coordination complex could either induce preferred covalent bond formation in the (100) plane along the b, c axes or inhibit the crystallization process along the a axis (Fig. 4b), in order to form the anisotropic MOF nanosheet. Nonetheless, the lattice parameters (Table 1), calculated from the Le Bail refinement of the XRD spectrums (Fig. 5 and 6), remained constant among MOF nanosheets, nanocubes and the International Centre for Diffraction Data (ICDD) database.28 Therefore, the staring coordination complexes of different copper(II)-ligands determine the PO tendency toward crystallization of MOF in the solvothermal method without affecting unit cell structure.
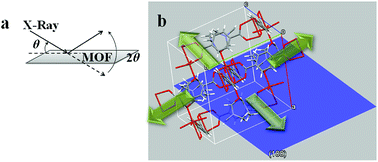 |
| | Fig. 4 (a) Illustration of out-of-plane XRD diffraction on MOF nanosheet; (b) hypothetical view of a MOF crystal unit with (100) plan (blue). | |
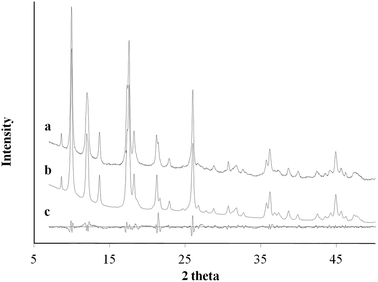 |
| | Fig. 5 XRD pattern profile of MOF nanocubes synthesized via Cu2+–Ac− coordination complex (a); the calculated Le Bail refinement fitting (b); and the difference plot (c). | |
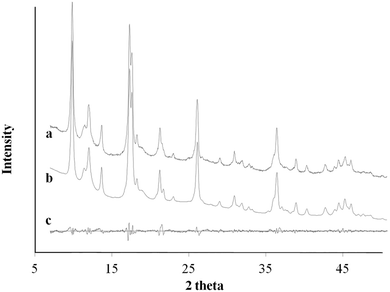 |
| | Fig. 6 XRD pattern profile of MOF nanosheets synthesized via Cu2+–Cl− coordination complex (a); the calculated Le Bail refinement fitting (b); and the difference plot (c). | |
Table 1 Le Bail refinement and lattice parameters of MOF nanocubes and nanosheets
| Le Bail refinement |
MOF nanocubes |
MOF nanosheets |
ICDD database |
| ‘Rwp’ is R-value in weighted pattern; ‘Rp’ is R-value in pattern; background subtracted and peak only contributions. |
| R-value (Rwp)a |
4.25 |
2.95 |
N/A |
| R-value (Rp)a |
2.78 |
2.10 |
N/A |
| Specimen displacement correction (mm) |
−0.3858 (16) |
−0.1848 (17) |
N/A |
| Cell volume (Å3) |
1211.11 (18) |
1216.1 (2) |
1204.275 (1) |
| a (Å) |
11.3862 (9) |
11.447 (3) |
11.4143 (3) |
| b (Å) |
14.4948 (12) |
14.482 (5) |
14.2687 (4) |
| c (Å) |
7.7670 (6) |
7.757 (2) |
7.7800 (2) |
| β (°) |
109.126 (6) |
108.92 (2) |
108.119 (1) |
Porosity and functionalities
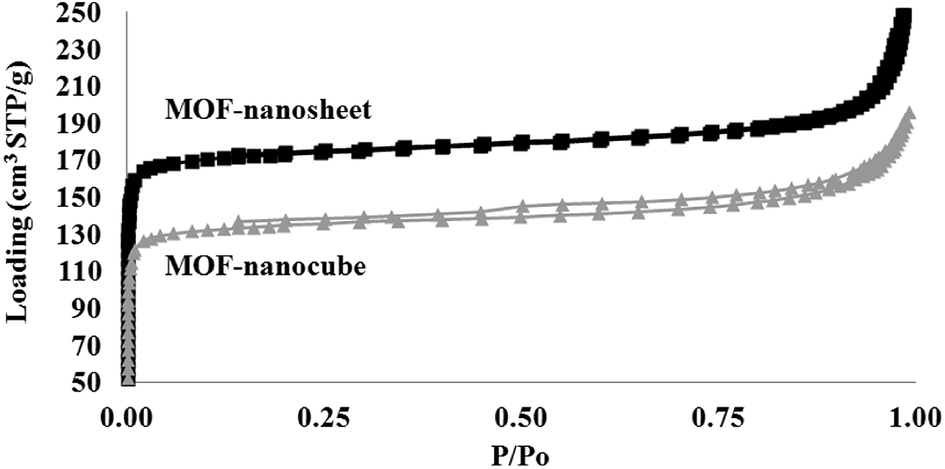
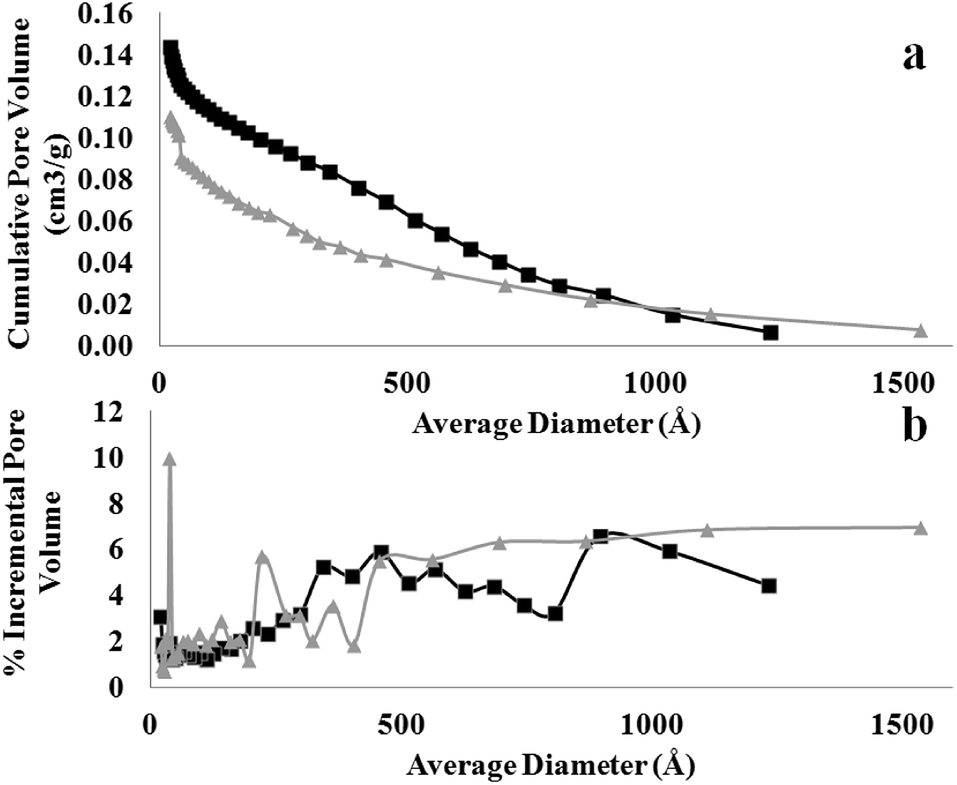
The differences in crystal packing and MOF morphology also determine the porosity and functionality of the as-obtained MOF nanostructures. Fig. 7 shows the N2 isotherm, from which the Brunauer–Emmett–Teller (BET) surface area and total pore volume (TOPV). The sharp rise in the isotherms at low values of P/Po is indicative of a large population of micropores (<20 Å). A comparison of the micropore volume to the total pore volume evaluated from the loading at P/Po of ∼1.0 reveals that ∼63% of the pore volume, of both samples, comes from the micropores (Fig. 8). This is also consistent with the high BET surface area values evaluated for these materials, i.e., 708 and 544 m2 g−1 for the MOF nanosheets and nanocubes, respectively. The cupric-TPA nanocubes have a similar BET surface area compared to a previous report,28 but the cupric-TPA nanosheet showed a 30.2% increase in BET surface area. The improvement of the surface area in MOF nanosheet could be attributed to the involvement of mesoporous and macroporous cavities (Fig. 2b), especially inside the agglomerated nanosheets structures. The cumulative and percent incremental pore volume plots are provided in Fig. 8a and b, respectively. The data suggests that a broad pore size distribution exists in the mesoporous and macroporous regions. The MOF nanocubes also have a small population of pores centered at ∼40 Å and above 500 Å, whereas MOF nanosheets have an extensive amount of mesoporous and macroporous cavities above 400 Å.
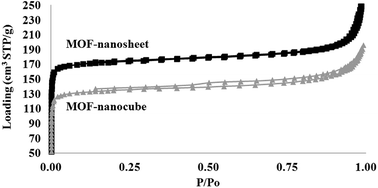 |
| | Fig. 7 Comparison of N2 adsorption–desorption isotherms measured at 77 K as a function of relative pressure between MOF nanosheet (top) and MOF nanocubes (bottom). | |
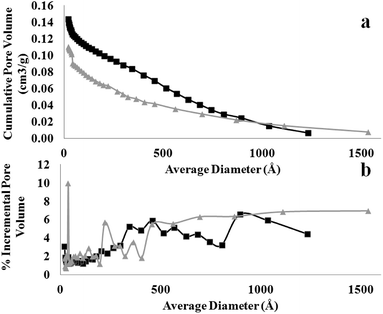 |
| | Fig. 8 Comparison of (a) cumulative and (b) incremental pore size distribution from desorption measured at 77 K between MOF nanosheet (square) and nanocube (triangle). | |
Conclusions
A facile and template-free solvothermal method has been developed for mesoporous and macroporous MOF nanosheets. It was found that starting coordination complexes of different copper(II)-ligand mediated the controlled growth and morphology of MOF crystals. By controlling the size and shape of the MOF crystals, it shows the possibility to adjust and tailor the structure and performances of the assemblies. This work provides a bottom-up approach to synthesize MOF films and nanosheets in a simple and scalable way, which may have potential applications for energy and biomedical applications.
Conflicts of interest
The authors declare no conflicting interests.
Acknowledgements
This work is supported by USDA-NIFA Specialty Crop Research Initiative Award No. 2016-51181-25403 and USDA-ARS Specific Corporative Agreement No. S51600000035794. We acknowledge the support of Dr John Zielinski at Intertek Chemicals & Pharmaceuticals service to conduct the work of N2 isotherms, BET and TOPV measurements. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA; USDA is an equal opportunity provider and employer.
References
- H. C. Streit, M. Adlung, O. Shekhah, X. Stammer, H. K. Arslan, O. Zybaylo, T. Ladnorg, H. Gliemann, M. Franzreb, C. Woell and C. Wickleder, ChemPhysChem, 2012, 13, 2699–2702 CrossRef PubMed.
- A. Ahmed, M. Forster, R. Clowes, P. Myers and H. Zhang, Chem. Commun., 2014, 50, 14314–14316 RSC.
- A. Ahmed, N. Hodgson, M. Barrow, R. Clowes, C. M. Robertson, A. Steiner, P. McKeown, D. Bradshaw, P. Myers and H. Zhang, J. Mater. Chem. A, 2014, 2, 9085–9090 RSC.
- R. Adams, C. Carson, J. Ward, R. Tannenbaum and W. Koros, Microporous Mesoporous Mater., 2010, 131, 13–20 CrossRef.
- J. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabres I Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef PubMed.
- Y. Zhang, X. Bo, C. Luhana, H. Wang, M. Li and L. Guo, Chem. Commun., 2013, 49, 6885–6887 RSC.
- X. Wang, Q. Wang, Q. Wang, F. Gao, F. Gao, Y. Yang and H. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 11573–11580 CrossRef PubMed.
- M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056–6058 RSC.
- L. Li, S. Xiang, S. Cao, J. Zhang, G. Ouyang, L. Chen and C. Su, Nat. Commun., 2013, 4, 1774 CrossRef PubMed.
- M. A. Green, Nat. Mater., 2010, 9, 539–540 CrossRef PubMed.
- L. Peng, J. Zhang, Z. Xue, B. Han, X. Sang, C. Liu and G. Yang, Nat. Commun., 2014, 5, 4465 CrossRef PubMed.
- Y. Liu, S. Liu and Z. Yue, RSC Adv., 2015, 5, 31742–31745 RSC.
- A. Kondo, C. C. Tiew, F. Moriguchi and K. Maeda, Dalton Trans., 2013, 42, 15267–15270 RSC.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- K. A. Cychosz, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2009, 131, 14538–14543 CrossRef PubMed.
- Y. Wu, F. Li, W. Zhu, J. Cui, C. Tao, C. Lin, P. M. Hannam and G. Li, Angew. Chem., Int. Ed., 2011, 50, 12518–12522 CrossRef PubMed.
- R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565–571 CrossRef PubMed.
- G. Xu, T. Yamada, K. Otsubo, S. Sakaida and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 16524–16527 CrossRef PubMed.
- S. Hermes, M. K. Schroter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef PubMed.
- E. Biemmi, C. Scherb and T. Bein, J. Am. Chem. Soc., 2007, 129, 8054–8055 CrossRef PubMed.
- C. Munuera, O. Shekhah, H. Wang, C. Woell and C. Ocal, Phys. Chem. Chem. Phys., 2008, 10, 7257–7261 RSC.
- D. Bradshaw, S. El-Hankari and L. Lupica-Spagnolo, Chem. Soc. Rev., 2014, 43, 5431–5443 RSC.
- L. Sun, J. Li, J. Park and H. Zhou, J. Am. Chem. Soc., 2012, 134, 126–129 CrossRef PubMed.
- J. Liu, B. Lukose, O. Shekhah, H. K. Arslan, P. Weidler, H. Gliemann, S. Braese, S. Grosjean, A. Godt, X. Feng, K. Muellen, I.-B. Magdau, T. Heine and C. Woell, Sci. Rep., 2012, 2, 921 CrossRef PubMed.
- Y. Hu, X. Dong, J. Nan, W. Jin, X. Ren, N. Xu and Y. M. Lee, Chem. Commun., 2011, 47, 737–739 RSC.
- P. Li, Y. Maeda and Q. Xu, Chem. Commun., 2011, 47, 8436–8438 RSC.
- C. G. Carson, K. Hardcastle, J. Schwartz, X. Liu, C. Hoffmann, R. A. Gerhardt and R. Tannenbaum, Eur. J. Inorg. Chem., 2009, 2338–2343 CrossRef.
- C. G. Carson, K. Hardcastle, J. Schwartz, X. Liu, C. Hoffmann, R. A. Gerhardt and R. Tannenbaum, Eur. J. Inorg. Chem., 2009, 2338–2343 CrossRef.
- Z. Zhuang, Q. Peng, B. Zhang and Y. Li, J. Am. Chem. Soc., 2008, 130, 10482–10483 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yaguang Luo*b,
Kelsey Kanyuckc,
Natalie Saenzb,
Kevin Reeda,
Peter Zavalijc,
Joseph Moweryb and
Gary Bauchanb
*a,
Yaguang Luo*b,
Kelsey Kanyuckc,
Natalie Saenzb,
Kevin Reeda,
Peter Zavalijc,
Joseph Moweryb and
Gary Bauchanb