
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBufalin suppresses colorectal cancer cell growth through promoting autophagy in vivo and in vitro
Zhe
Pan
a,
Yunpeng
Xie†
 a,
Jie
Bai
b,
Qiuyue
Lin
a,
Xiaonan
Cui
*a and
Ningning
Zhang
*a
a,
Jie
Bai
b,
Qiuyue
Lin
a,
Xiaonan
Cui
*a and
Ningning
Zhang
*a
aThe First Affiliated Hospital, Dalian Medical University, 116021, Dalian, Liaoning, China. E-mail: zhangningning@dmu.edu.cn; Xiaonan_cui@163.com; Fax: +8641183635963; Fax: +8641186110312; Tel: +8618098873517 Tel: +8618604266169
bSchool of Public Health, Dalian Medical University, 116044, Dalian, Liaoning, China
First published on 19th November 2018
Abstract
Specific groups in Asia, including the Chinese, are more susceptible to colorectal cancer (CRC). The best strategy for anticancer drug action is to induce cancer cell apoptosis and autophagy. Bufalin is a potent inducer of apoptosis in some human cancer cell lines, but bufalin has barely been evaluated in colorectal cancer cells as a potent autophagy inducing agent. The aim of this study was to investigate the roles and interactions of bufalin in autophagy and the effects of the drug on human colorectal cancer. We applied bufalin and autophagy inhibitors (CQ and 3-MA) in LoVo cells to investigate their potential anticancer bioactivity under certain concentrations of bufalin to monitor autophagy and cell proliferation in vivo and in vitro. Bufalin induced autophagy of LoVo and inhibited proliferation of LoVo cells. Bufalin inhibited the expression of autophagy-related (ATG) proteins and tumor growth in vivo. Our studies identified that bufalin could potentially be a small molecule inhibitor for cancer therapy.
1. Introduction
Colorectal cancer (CRC) is a worldwide public health challenge with the fourth-highest cancer death rate, with an estimated 694![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in 2012. According to various reports, certain ethnic groups in Asia, including the Chinese, are more susceptible to CRC.1 The best strategy for anti-tumor drug action is to induce apoptosis and autophagy in tumor cells; therefore, induction of autophagy may be a promising strategy to increase tumor cell death.2
000 deaths in 2012. According to various reports, certain ethnic groups in Asia, including the Chinese, are more susceptible to CRC.1 The best strategy for anti-tumor drug action is to induce apoptosis and autophagy in tumor cells; therefore, induction of autophagy may be a promising strategy to increase tumor cell death.2
“Huachansu” in saline is an injectable drug, which has been officially approved for the cancer treatment program in China.2 Bufalin is the main immune response component of digoxin and is one of the components of “Huachansu”.3 The results showed that in patients with liver cancer and pancreatic cancer, the dose of Huachansu was eight times that of the standard dose in China (20 mL per m per day or 20–25 mL per adult patient per day, containing bufalin at 14.3 ± 0.03 ng mL−1).4 This dose is much smaller than the maximum dose recommended by the American Medical Board (<100 μg),5 suggesting that bufalin may display an effective anti-cancer biological activity at a certain concentration. It has been confirmed that bufalin has a significant inhibitory effect on the proliferation of transplanted liver cancer in nude mice.6 In addition, results from in vitro studies have shown that bufalin plays a powerful role as an apoptosis-inducing agent in various human tumor cell lines, including those of lung cancer,7 hepatocellular carcinoma,8 colorectal cancer,9 leukemia,10 prostate cancer11 and gastric cancer.12 It has been demonstrated that bufalin inhibits tumor cell proliferation through cell cycle arrest12 and apoptosis, but its exact mechanism of action remains unknown. It has been shown that bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation.13 However, there have been no detailed studies on the effects of autophagy on human colon cancer in vivo, which is an important way to identify the effect of bufalin on colon cancer.
Autophagy is a physiological process that allows the degradation of cytoplasmic contents, including unfolded proteins and membrane organelles under certain stress conditions. This is a temporary survival mechanism. Certain cellular stresses, such as oxidative stress, nutrient starvation, misfolded protein accumulation, and radiation, induce autophagy. Once initiated, the process delivers cytosolic material to a lysosome through a double-membrane organelle called autophagosome, which contains a portion of the cytoplasm and intracellular organelles.14 However, recent studies have shown that autophagy is also a cell death mechanism and the response from various cancer cells to different anticancer therapies.15–17 Autophagy is a highly regulated process involving the activity of autophagy-associated proteins (ATGs). They can be divided into 4 functional groups. ATG proteins that regulate autophagy include ATG1 or ULK1, ATG13, and ATG17. The vesicle-nucleated ATG proteins include ATG6 or BECLN 1 and ATG9. Other ATG proteins, such as ATG5, ATG12 and LC3, are involved in the formation of autophagosomes. Finally, ATG proteins are responsible for autophagy-related proteins, including ATG2, ATG9, and ATG18.18–20 3-MA, LY294002 and wortmannin can inhibit the PI3K activity,21 which is one of the major upstream regulators of autophagy. Chloroquine (CQ) can inhibit autophagosome fusion with lysosomes and autophagosome degradation.22
On this basis, LoVo cells and nude mice were used as experimental models to verify the hypothesis that bufalin could reduce proliferation by promoting autophagy. The results suggest that bufalin has potential application prospects in the treatment of colorectal cancer.
2. Materials and methods
2.1 Materials and cell culture
Bufalin was obtained from Sigma (St. Louis, MO) and dissolved in DMSO. LoVo cells were obtained from the American Type Culture Collection. LoVo cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (HyClone, Logan, UT, USA). Cells were supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT, USA) and maintained in a humidified atmosphere with 5% CO2 at 37 °C.2.2 Cell proliferation assay
Cell proliferation was measured by a Cell Counting Kit-8 assay. The cells were suspended in 10% heat-inactivated bovine serum, plated in 96-well plates, incubated for 24 hours, and divided into treatment groups of different concentrations. Then, the plates were incubated for 1, 2, 3 and 4 days. After incubation, 10 μL of CCK-8 solution was added to each well and incubated at 37 °C for additional 2 hours. Optical density (OD) value of absorbance at 450 nm was measured with a Thermo Scientific Fluoroskan Ascent FL. The results were plotted as the means ± SD of three independent experiments with three determinations per sample for each experiment.2.3 Western blotting
Cells were washed in PBS before incubation with lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM Na3VO4, 0.2 mM phenylmethylsulfonyl fluoride, 0.5% Nonidet P-40) on ice for 10 min. The cell lysates were centrifuged at 9000 g for 15 min and then, the supernatant was collected. The protein concentration was determined with the Coomassie Protein Assay reagent using bovine serum albumin (BSA) as a standard. An equal amount of protein extract (30 μg) was separated by 12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose filter (NC) membrane. The membrane was blocked in 5% skim milk for 2 hours at room temperature in Tris-buffered saline containing 0.1% Tween 20 (TBST) and probed with primary antibodies overnight at 4 °C. The membrane was washed three times with TBST. Then, the antibody was incubated with horseradish peroxidase-binding antibody for 1 hour at room temperature. After washing four times with TBST, the membrane was visualized using ECL from BIO RAD Laboratories. The Western blots shown represent at least three independent experiments. Density analysis was performed using Sigon Image 4.03 to quantitatively determine the density of each band of the target protein. Mean ± SD of each group of proteins and intensities were determined by GAPDH/β-actin densitometry in these three experiments.2.4 Xenograft tumor models of human CRC
Animal experiments were conducted with the approval of the Animal Research Committee of Dalian Medical University. Male nude mice were taken from Dalian Medical University. These animals (4–6 weeks old, 18–25 grams) were placed in a specific pathogen free (SPF) environment and provided with sterile water and food. Mice were tested after 1 week of adaptation. LoVo cells (5 × 106) suspended in 200 μL of PBS were subcutaneously injected to the right side of the mice. Mice were randomized into three groups with a tumor volume of 100 m3: (a) 0 mg per kg per day, (b) 0.5 mg per kg per day, and (c) 1.0 mg per kg per day. Two different concentrations of bufalin were injected twice a week for 3 weeks. Systemic weight and tumor volume were measured every 2 days after tumor inoculation. Tumor volume was calculated according to the following formula: tumor volume (MM3) = 1/2 × A (tumor length) × B2 (tumor width). At the end of the experiment, all animals were sacrificed, tumor tissue was aseptically removed, weight was recorded and further studies were performed.2.5 Immunohistochemistry
Serial sections of 6 μm were prepared from paraffin-embedded tissues. The sections were fixed at 60 °C for 3 h, demethylated, and reconstituted in alcohol. The antigen was removed by microwave defrosting in citrate buffer for 25 min and washed with phosphate buffered saline (PBS) after cooling for 1 hour. Slides were incubated in 3% H2O2 for 20 min to block endogenous peroxidase activity. After washing in PBS, blockade was blocked with goat serum at 37 °C for 15 min, and non-specific binding of the conjugated secondary antibody was eliminated after incubation with LC3 antibody (1:100) overnight at 4 °C. After washing with PBS, the sections were incubated with biotinylated secondary antibody for 40 min at 37 °C. Next, the sections were washed with PBS, and then incubated with streptavidin-horseradish peroxidase for 40 min at 37 °C. Positive reactions were visualized with a diaminobenzidine (DAB)-peroxidase substrate and counterstaining with haematoxylin for 30 s. Photomicrographs were taken using an Olympus TH4-200 microscope (Tokyo, Japan).
2.6 H&E staining
Serial sections of 6 μm were prepared from paraffin-embedded tissues. The sections were fixed at 60 °C for 3 h, demethylated, and reconstituted in alcohol. Then they were soaked in filtered Harris hematoxylin for 2 min and rinsed with water twice for 1 min each. The slide was immersed in 0.3% ammonium hydroxide and rinsed with water twice for 1 min each. Then, the slide was soaked in Eosin-Y and washed with water 3 times for 1 min each. The slide was dehydrated for 70% 1 min, 80% ethanol for 1 min, 95% ethanol for 1 min, 100% ethanol twice for 1 min, and xylene twice for 1 min each. Then, Permount was added and the slips were covered.2.7 Statistical analyses
Data were expressed as the mean ± SD for three independent experiments with GraphPad Prism software. The Student's t-test was used to make a statistical comparison between groups. One-way analysis of variance (ANOVA) was employed to determine the statistical significance between different groups. *P < 0.05 was considered statistically significant (Fig. 1).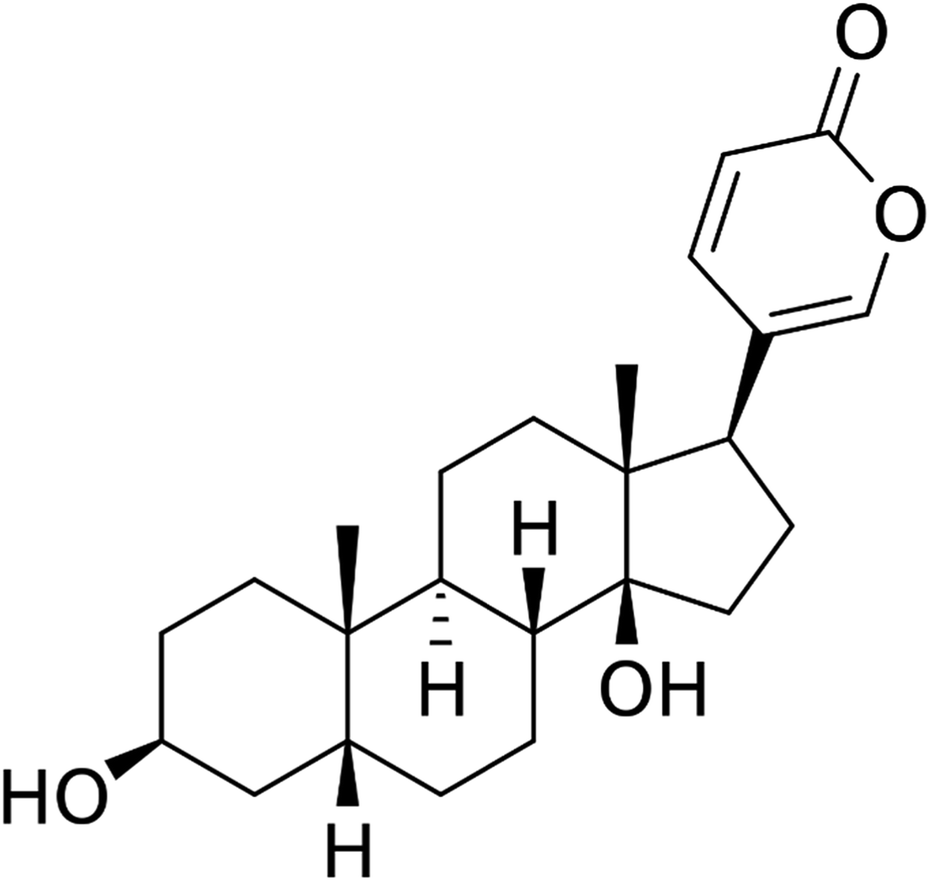 | ||
| Fig. 1 Structure of bufalin. | ||
3. Results
3.1 Bufalin promoted the expression of autophagy-related proteins (ATGs) in LoVo cells
Previous experiments have determined the concentration of bufalin in treated tumor cells.23 To determine whether bufalin induced autophagy-related protein (ATG) expression, LoVo cells were treated without or with bufalin (10 nM) for 0 h, 12 h and 24 h, and the expression of ATGs was detected by Western Blot. The results showed that bufalin induced ATGs expression in a time-dependent manner in LoVo cells (Fig. 2A). We also studied the dose-dependent response of bufalin (24 h) at 0 nM, 5 nM and 10 nM. The result showed that the protein expression of ATGs increased in a dose-dependent manner (Fig. 2B).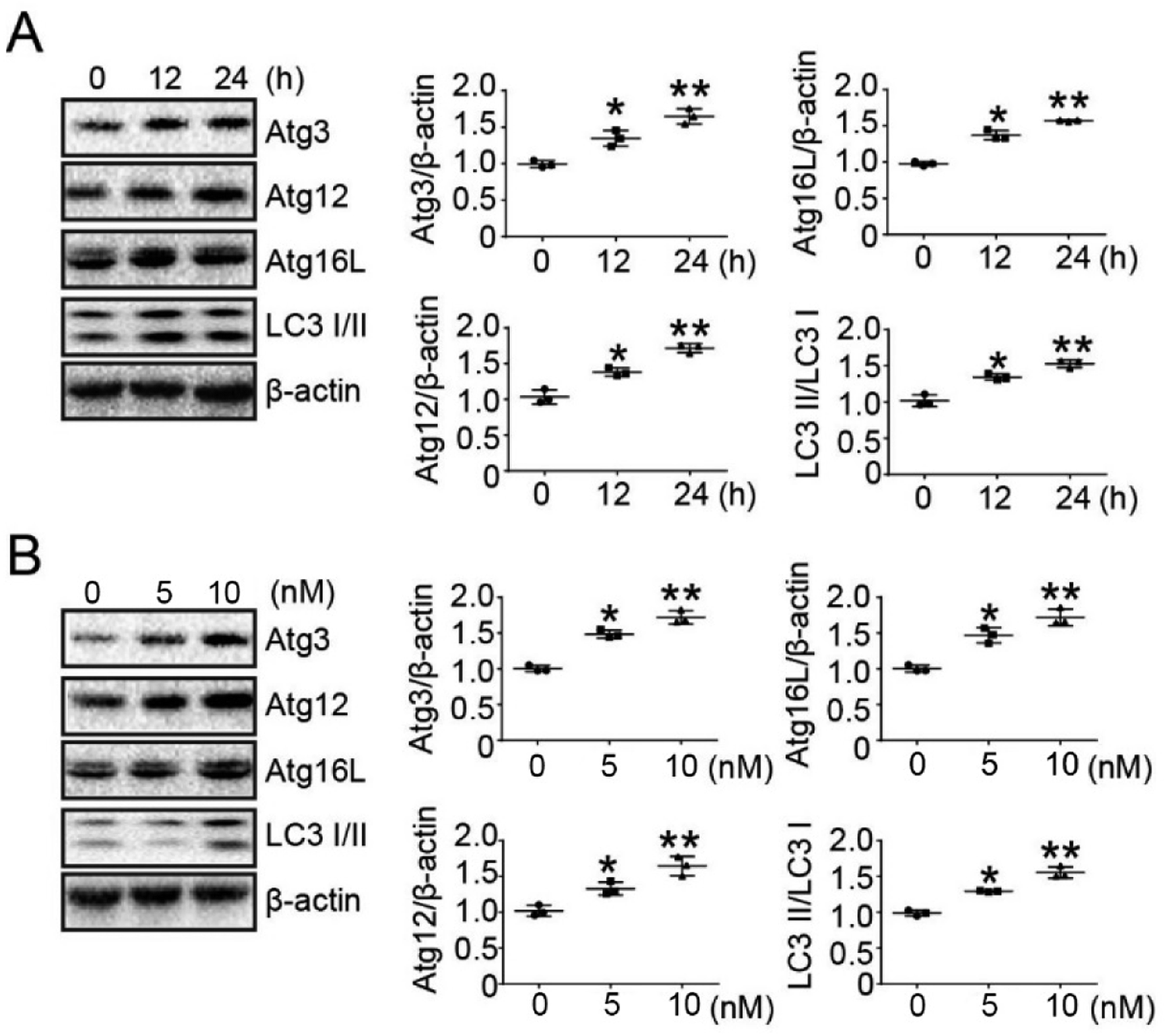 | ||
| Fig. 2 Bufalin promoted the expression of autophagy-related proteins (ATGs) in LoVo cells. (A) Western blot analysis of Atg3, Atg12, Atg16L and LC3 I/II expression in LoVo cells treated with bufalin at 10 nM for 0 h, 12 h and 24 h. The amount of β-actin served as the loading control. (B) Western blot analysis of Atg3, Atg12, Atg16L and LC3 I/II expression in LoVo cells treated with bufalin for 24 h at 0 nM, 5 nM and 10 nM. The amount of β-actin served as the loading control. | ||
3.2 Autophagy inhibitor (CQ and 3-MA) abated the ability of proliferation and expression of ATGs induced by bufalin in LoVo cells
To confirm that bufalin regulation of LoVo proliferation occurs through autophagy, we used Western Blot and Cell Counting Kit-8 (CCK-8) to detect the proliferating ability of LoVo. As shown in Fig. 3A and C, the expressions of Atg3, Atg5, Beclin 1 and Atg12 decreased compared with those observed in control (*P < 0.05), and reversed by treatment with 3-MA (5 mM) or CQ (25 μM) (#P < 0.05). As shown in Fig. 3B and D, the bufalin-treated groups exhibited drastically decreased cell proliferation compared with control (*P < 0.05), and was reversed by treatment with 3-MA or CQ (#P < 0.05). | ||
| Fig. 3 Autophagy inhibitor (CQ and 3-MA) abated the ability of proliferation and the expression of ATGs induced by bufalin in LoVo cells. (A) The expression of Atg3, Atg5, Beclin 1 and Atg12 was detected by Western blot, being treated with bufalin and 3-MA. (B) The ability of LoVo cell proliferation was detected by CCK-8 being treated with bufalin and 3-MA. (C) The expression of Atg3, Atg5, Beclin 1 and Atg12 was detected by Western blot being treated with bufalin and CQ. (D) The ability of LoVo cell proliferation was detected by CCK-8. (*P < 0.05, compared with control), (#P < 0.05, compared with bufalin group). | ||
3.3 Inhibition of tumor growth by bufalin in mice
The effect of bufalin on tumor growth was further examined in nude mice with human colorectal tumor xenografts. LoVo cells were injected subcutaneously into the armpit of nude mice. When the tumors were ∼100 mm3 in size, the animals were treated with different concentration of bufalin. The volume of each tumor was measured by sliding calipers at 2 day intervals. As shown in Fig. 4A, the periodic measurement of the tumor xenograft volume indicated that the tumor volume in nude mice decreased significantly in the high-concentration bufalin group (1.0 mg per kg per body) compared with that observed in the vehicle group (P < 0.01) after 7 days of the experiment. The tumor volume also decreased significantly in the low-dose bufalin group (0.5 mg per kg per body) compared with the vehicle group (P < 0.01) after 11 days of the experiment. After three weeks, we executed the mice humanely and found that the weight of the tumor in the bufalin-treated (0.5, 1.0 mg kg−1 day) group significantly decreased compared with that of the group treated with 0 mg kg−1 day (Fig. 4B and C).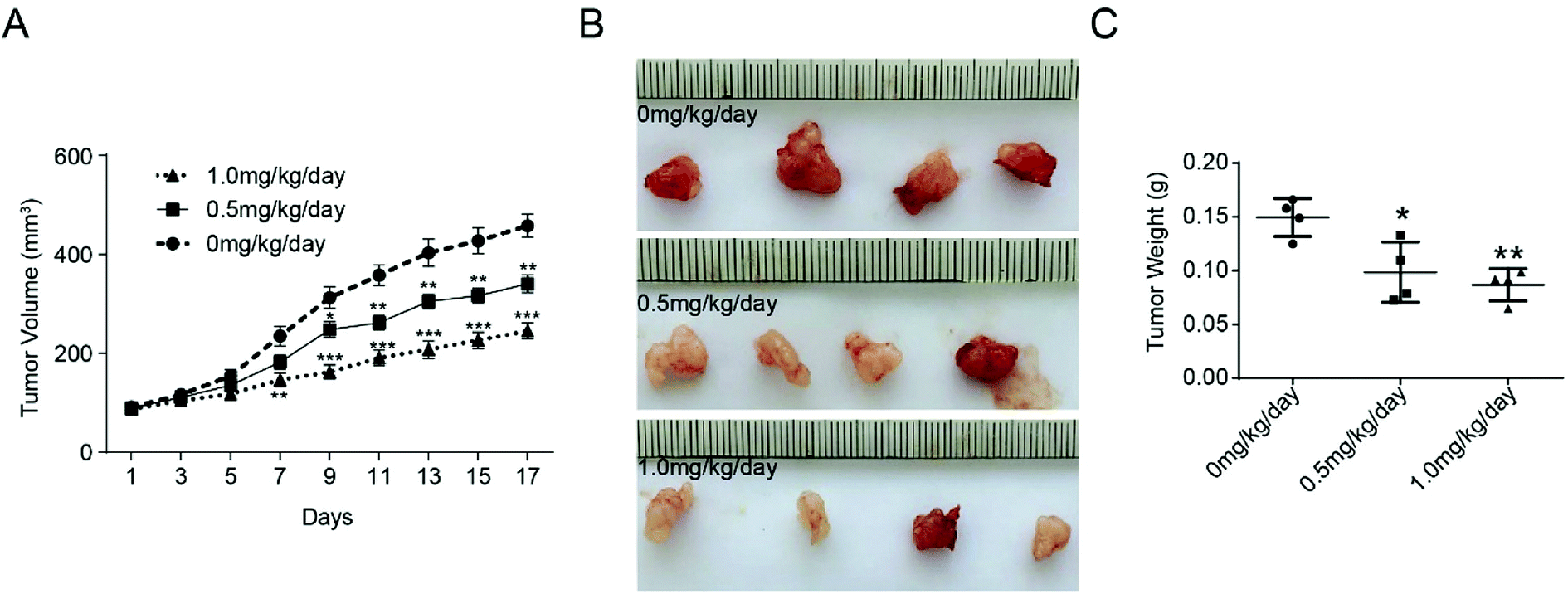 | ||
| Fig. 4 Inhibition of tumor growth by bufalin in mice. (A) The tumor volume of each group of nude mice was measured and calculated as V = (width2 × length)/2, **P < 0.01, ***P < 0.001. (B) The morphology of the tumor xenografts in each nude mouse after 17 days of treatment. (C) The weight of tumor grafts 17 days after the intratumoral injection of 0 mg per kg per day, 0.5 mg per kg per day and 1.0 mg per kg per day, *P < 0.05, **P < 0.01. | ||
3.4 Immunohistochemistry analysis of LC3 expression
We examined tumor xenograft samples from each treatment group for H&E and expression of LC3 using IHC analysis to further determine the mechanisms involved in bufalin-mediated induction of autophagy of LoVo cells in vivo. As shown in Fig. 5A, in the 0 mg per kg per day group, the surface of the cancer cells was irregular, the nucleus was round, the chromatin levels increased, the free ribosome levels were increasing and abundant mitochondria and endoplasmic reticulum were found in the cytoplasm. However, cells in 0.5 mg per kg per day and 1.0 mg per kg per day treatment groups showed cytoplasmic condensation, chromatin condensation in the inner nuclear membrane in clumps or crescent, and cytoplasm foaming. As shown in Fig. 5B and Table 1, the relative expression of LC3 in the tumors of nude mice in the high bufalin dose group increased significantly compared with that in the vehicle group.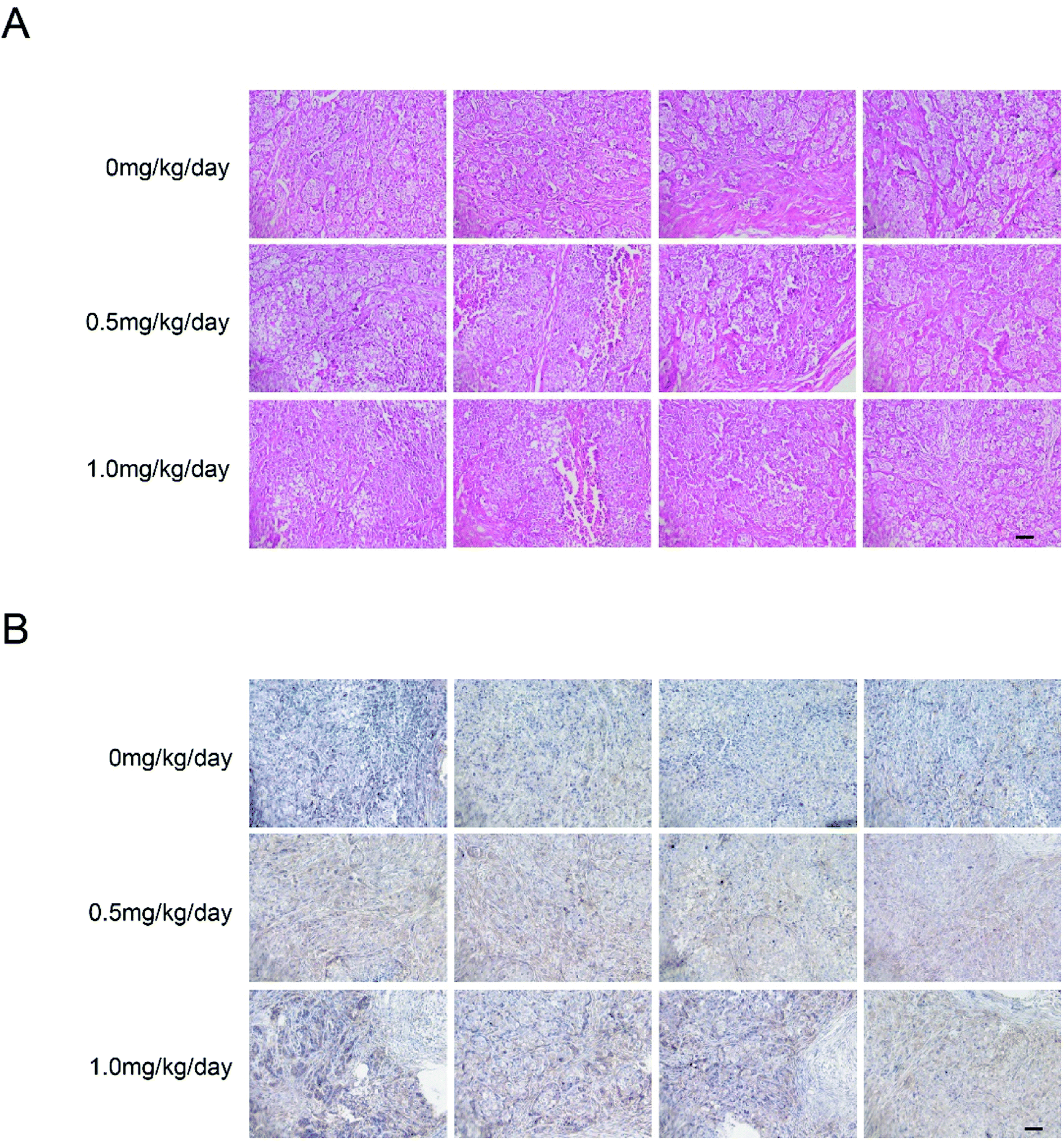 | ||
| Fig. 5 Immunohistochemical detection of LC3 protein in tumor xenograft samples. H&E staining (A) and immunohistochemistry (B) were performed after the intratumoral injection of 0 mg per kg per day, 0.5 mg per kg per day and 1.0 mg per kg per day of bufalin for 17 days. Immunodetectable LC3 protein is indicated by brown staining; nuclei are counterstained blue. Original magnification, ×200. | ||
| Vehicle group | Bufalin (0.5 mg per kg per day) group | Bufalin (1.0 mg per kg per day) group | |
|---|---|---|---|
| a **P < 0.01, ***P < 0.001. | |||
| H-score | 0.22 ± 0.03 | 0.43 ± 0.04** | 0.61 ± 0.06*** |
3.5 Effects of bufalin on protein expression of ATGs in xenograft model
We used a LoVo cells xenograft model to further study the antitumor effect of bufalin on the protein expression of autophagy in vivo. The relative protein expression levels of Atg3, Atg5, Beclin 1, Atg7, Atg12, Atg 16 L and LC3 I/II in tumor tissues were determined by Western Blot. These results showed that bufalin treatment significantly upregulated the protein expression of Atg3, Atg5, Beclin 1, Atg7, Atg12, Atg 16L and LC3 I/II in a dose-dependent manner compared with the vehicle group (P < 0.05 to 0.01, Fig. 6). Thus, bufalin might exert an anti-tumor effect via the autophagy pathway.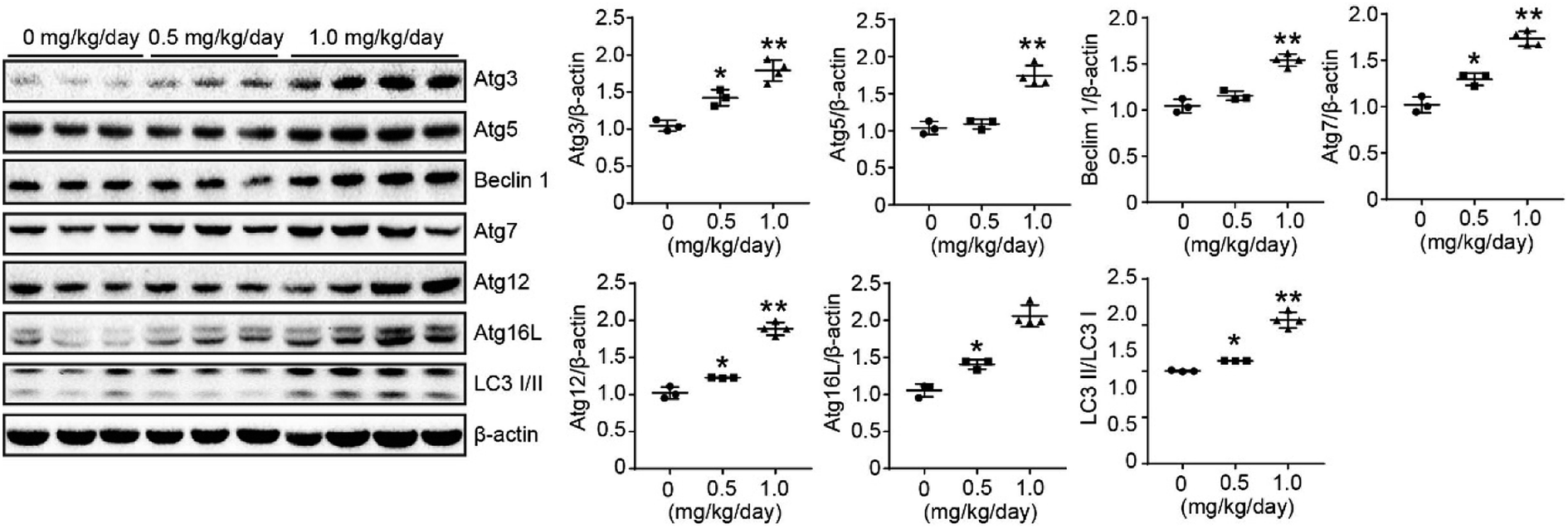 | ||
| Fig. 6 Effects of bufalin on protein expression of ATGs in xenograft model. The proteins were extracted from tumor xenografts. The Atg3, Atg5, Beclin 1, Atg7, Atg12, Atg16L, and LC3 I/II expression levels were measured by Western blot analysis. *P < 0.05, **P < 0.01. | ||
4. Discussion
Over the past decade, a combination of multiple anticancer drugs has significantly improved survival rates in patients with CRC in Phases III and IV.24 However, 30% of CRC patients experience disease recurrence in stage III, even though radical resection of the primary tumor has been performed.25 Therefore, a new strategy is urgently needed to minimize residual tumors.Bufalin has been reported to play a key role in cancer cell death and differentiation in ovarian and prostate cancers and is less toxic to normal cells at low doses.26 Bufalin can also induce ROS production in lung cancer cells.27 However, the effect of bufalin on autophagy in colorectal cancer in the null mice model has not been thoroughly investigated.
In this study, the dose-dependent and time-dependent expression of autophagy-related proteins was monitored by Western blotting when cells were treated with bufalin. CCK-8 and Western blot analysis showed that CQ and 3-MA could reverse the proliferation and autophagy-related protein expression of LoVo cells induced by bufalin. In addition, we demonstrated that bufalin can inhibit tumor growth by promoting autophagy in vivo.
Autophagy is involved in promoting or inhibiting cancer cell death, depending on the cellular background and the intensity and duration of stress stimuli. However, the molecular mechanism of the dual role of autophagy remains unclear. Typically, autophagy promotes a portion of the cytoplasm and organelles to enter autophagic vesicles as part of a stress response. Moreover, under continuous autophagy stimulation, excessive autophagy depletion of organelles and key proteins eventually leads to caspase-independent cell death. In this regard, autophagy may be used as a therapeutic target only if it is highly activated in cancer cells. Our data demonstrated that induction of autophagy by bufalin causes cell death in colon cancer cells.
In previous studies in this laboratory, we have reported that bufalin promotes apoptosis in colon cancer cells. Many apoptosis-inducing stimuli often induce autophagy, such as ceramide treatment in breast cancer and colon cancer, and both apoptosis and autophagy are simultaneously upregulated.28 In the clinical trial of arsenic trioxide treatment of T lymphocytes, it was also found that both apoptosis and autophagy were activated simultaneously,29 and the antibacterial drug chloroiodoquinoline induced autophagic death and apoptosis in leukemia and myeloma cells by disrupting the mTOR signaling pathway.30 Targeting knockout of the autophagy-associated protein ATG7 or autophagy inhibitor 3-methyladenine inhibits caspase activation and reduces apoptosis.31 In many cases, the potential for autophagy-induced cell death is apoptotic inhibition, but it plays a key role in the defect of apoptotic function. Cell autophagy was up-regulated in mouse embryonic fibroblasts with apoptosis-deficient Bax/Bak−/− treated with etoposide, toxic carrot lactone, etc., and specific inhibitors inhibited cell survival after autophagy.32 In these cases, autophagy and apoptosis together trigger cell death through synergistic, complementary, or alternative mechanisms.
In the present study, we found that bufalin as an effective agent for inducing autophagy in human colon cancer cells and nude mice will pave the way for further development of the clinical treatment of colorectal cancer.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by the National Natural Science Foundation of China (81800142, 81600197); the funds from Basic Scientific Research Projects of Colleges and Universities in Liaoning Province (LQ2017021).References
- S. C. Ng and S. H. Wong, Colorectal cancer screening in Asia, Br. Med. Bull., 2013, 105, 29–42 CrossRef CAS.
- Q. Huang, Y. S. Ou, Y. Tao, H. Yin and P. H. Tu, Apoptosis and autophagy induced by pyropheophorbide-α methyl ester-mediated photodynamic therapy in human osteosarcoma MG-63 cells, Apoptosis, 2016, 21, 749–760 CrossRef CAS PubMed.
- Z. Meng, P. Yang, Y. Shen, W. Bei, Y. Zhang, Y. Ge, R. A. Newman, L. Cohen, L. Liu, B. Thornton, D. Z. Chang, Z. Liao and R. Kurzrock, Pilot study of huachansu in patients with hepatocellular carcinoma, nonsmall-cell lung cancer, or pancreatic cancer, Cancer, 2009, 115, 5309–5318 CrossRef PubMed.
- N. Takai, N. Kira, T. Ishii, T. Yoshida, M. Nishida, Y. Nishida, K. Nasu and H. Narahara, Bufalin, a traditional oriental medicine, induces apoptosis in human cancer cells, Asian Pac. J. Cancer Prev., 2012, 13, 399–402 CrossRef.
- S. Marchetti and J. H. Schellens, The impact of FDA and EMEA guidelines on drug development in relation to Phase 0 trials, Br. J. Cancer, 2007, 97, 577–581 CrossRef CAS.
- K. Q. Han, G. Huang, W. Gu, Y. H. Su, X. Q. Huang and C. Q. Ling, Anti-tumor activities and apoptosis-regulated mechanisms of bufalin on the orthotopic transplantation tumor model of human hepatocellular carcinoma in nude mice, World J. Gastroenterol., 2007, 13, 3374–3379 CrossRef CAS PubMed.
- X. H. Kang, Z. Y. Xu, Y. B. Gong, L. F. Wang, Z. Q. Wang, L. Xu, F. Cao and M. J. Liao, Bufalin Reverses HGF-Induced Resistance to EGFR-TKIs in EGFR Mutant Lung Cancer Cells via Blockage of Met/PI3k/Akt Pathway and Induction of Apoptosis, J. Evidence-Based Complementary Altern. Med., 2013, 2013, 243859 Search PubMed.
- F. Hu, J. Han, B. Zhai, X. Ming, L. Zhuang, Y. Liu, S. Pan and T. Liu, Blocking autophagy enhances the apoptosis effect of bufalin on human hepatocellular carcinoma cells through endoplasmic reticulum stress and JNK activation, Apoptosis, 2014, 19, 210–223 CrossRef CAS PubMed.
- N. Zhang, Y. Xie, Y. Tai, Y. Gao, W. Guo, W. Yu, J. Li, X. Feng, J. Hao, Y. Gao, X. Zhao, Y. Liao, W. Jiang, G. Liu, W. Deng and X. Cui, Bufalin Inhibits hTERT Expression and Colorectal Cancer Cell Growth by Targeting CPSF4, Cell. Physiol. Biochem., 2016, 40, 1559–1569 CrossRef CAS.
- M. Watabe, K. Ito, Y. Masuda, S. Nakajo and K. Nakaya, Activation of AP-1 is required for bufalin-induced apoptosis in human leukemia U937 cells, Oncogene, 1998, 16, 779–787 CrossRef CAS PubMed.
- J. Y. Yeh, W. J. Huang, S. F. Kan and P. S. Wang, Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells, Prostate, 2003, 54, 112–124 CrossRef CAS.
- D. Li, X. Qu, K. Hou, Y. Zhang, Q. Dong, Y. Teng, J. Zhang and Y. Liu, PI3K/Akt is involved in bufalin-induced apoptosis in gastric cancer cells, Anticancer Drugs, 2009, 20, 59–64 CrossRef CAS PubMed.
- C. M. Xie, W. Chan, S. Yu, J. Zhao and C. Cheng, Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation, Free Radical Biol. Med., 2011, 51, 1365–1375 CrossRef CAS.
- C. He and D. J. Klionsky, Regulation mechanisms and signaling pathways of autophagy, Annu. Rev. Genet., 2009, 43, 67–93 CrossRef CAS PubMed.
- Y. L. Liu, P. M. Yang, C. T. Shun, M. S. Wu, J. R. Weng and C. C. Chen, Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma, Autophagy, 2010, 6, 1057–1065 CrossRef CAS.
- H. W. Chiu, S. Y. Ho, H. R. Guo and Y. J. Wang, Combination treatment with arsenic trioxide and irradiation enhances autophagic effects in U118-MG cells through increased mitotic arrest and regulation of PI3K/Akt and ERK1/2 signaling pathways, Autophagy, 2009, 5, 472–483 CrossRef CAS.
- D. K. Kim, J. S. Yang, K. Maiti, J. I. Hwang, K. Kim, D. Seen, Y. Ahn, C. Lee, B. C. Kang, H. B. Kwon, J. Cheon and J. Y. Seong, A gonadotropin-releasing hormone-II antagonist induces autophagy of prostate cancer cells, Cancer Res., 2009, 69, 923–931 CrossRef CAS PubMed.
- T. Shintani and D. J. Klionsky, Autophagy in health and disease: a double-edged sword, Science, 2004, 306, 990–995 CrossRef CAS PubMed.
- B. Levine and D. J. Klionsky, Development by self-digestion: molecular mechanisms and biological functions of autophagy, Dev. Cell, 2004, 6, 463–477 CrossRef CAS PubMed.
- B. Levine and J. Yuan, Autophagy in cell death: an innocent convict?, J. Clin. Invest., 2005, 115, 2679–2688 CrossRef CAS.
- D. Liu, Y. Yang, Q. Liu and J. Wang, Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells, Med. Oncol., 2011, 28, 105–111 CrossRef CAS PubMed.
- C. Bellodi, M. R. Lidonnici, A. Hamilton, G. V. Helgason, A. R. Soliera, M. Ronchetti, S. Galavotti, K. W. Young, T. Selmi, R. Yacobi, R. A. Van Etten, N. Donato, A. Hunter, D. Dinsdale, E. Tirro, P. Vigneri, P. Nicotera, M. J. Dyer, T. Holyoake, P. Salomoni and B. Calabretta, Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells, J. Clin. Invest., 2009, 119, 1109–1123 CrossRef CAS.
- N. Zhang, Y. Xie, Y. Tai, Y. Gao, W. Guo, W. Yu, J. Li, X. Feng, J. J. Hao, Y. Gao, X. R. Zhao, Y. N. Liao, W. Jiang, G. Liu, W. G. Deng and X. N. Cui, Bufalin inhibits htert expression and colorectal cancer cell growth by targeting cpsf4, Cell. Physiol. Biochem., 2016, 40, 1559–1569 CrossRef CAS PubMed.
- J. J. Marin, D. M. F. Sanchez, B. Castano, L. Bujanda, M. R. Romero, O. Martinez-Augustin, R. D. Moral-Avila and O. Briz, Chemoprevention, chemotherapy, and chemoresistance in colorectal cancer, Drug Metab. Revi., 2012, 44, 148–172 CrossRef CAS PubMed.
- T. Andre, C. Boni, M. Navarro, J. Tabernero, T. Hickish, C. Topham, A. Bonetti, P. Clingan, J. Bridgewater, F. Rivera and A. de Gramont, Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial, J. Clin. Oncol., 2009, 27, 3109–3116 CrossRef CAS PubMed.
- N. Takai, T. Ueda, M. Nishida, K. Nasu and H. Narahara, Bufalin induces growth inhibition, cell cycle arrest and apoptosis in human endometrial and ovarian cancer cells, Int. J. Mol. Med., 2008, 21, 637–643 CAS.
- L. Sun, T. Chen, X. Wang, Y. Chen and X. Wei, Bufalin Induces Reactive Oxygen Species Dependent Bax Translocation and Apoptosis in ASTC-a-1 Cells, J. Evidence-Based Complementary Altern. Med., 2011, 2011, 249090 Search PubMed.
- S. Pattingre, C. Bauvy, S. Carpentier, T. Levade, B. Levine and P. Codogno, Role of jnk1-dependent bcl-2 phosphorylation in ceramide-induced macroautophagy, Autophagy, 2009, 284, 2719–2728 CAS.
- W. Qian, J. Liu, J. Jin, W. Ni and W. Xu, Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of beclin-1, Leuk. Res., 2007, 31, 329 CrossRef CAS.
- B. Cao, J. Li, X. Zhou, J. Juan, K. Han, Z. Zhang, Y. Kong, J. Wang and X. Mao, Clioquinol induces pro-death autophagy in leukemia and myeloma cells by disrupting the motor signaling pathway, Sci. Rep., 2014, 4, 5749 CrossRef CAS PubMed.
- L. Yu, A. Alva, H. Su, P. Dutt, E. Freundt, S. Welsh, E. H. Baehrecke and M. J. Lenardo, Regulation of an ATG7 beclin 1 program of autophagic cell death by caspase-8, Science, 2004, 304, 1500–1502 CrossRef CAS PubMed.
- S. Shimizu, T. Kanaseki, N. Mizushima, T. Mizuta, S. Arakawa-Kobayashi, C. B. Thompson and Y. Tsujimoto, Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes, Nat. Cell Biol., 2004, 6, 1221–1228 CrossRef CAS PubMed.
Footnote |
| † Zhe Pan and Yunpeng Xie contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |