DOI:
10.1039/C8RA08890J
(Paper)
RSC Adv., 2018,
8, 38919-38928
Vinylene and benzo[c][1,2,5]thiadiazole: effect of the π-spacer unit on the properties of bis(2-oxoindolin-3-ylidene)-benzodifuran-dione containing polymers for n-channel organic field-effect transistors†
Received
29th October 2018
, Accepted 5th November 2018
First published on 20th November 2018
Abstract
Two polymers based on (3E,7E)-3,7-bis(2-oxoindolin-3-ylidene)benzo[1,2-b:4,5-b′]difuran-2,6(3H,7H)-dione (BIBDF) coupled with (E)-2-(2-(thiophen-2-yl)vinyl)thiophene (TVT) or dithienylbenzothiadiazole (TBT), namely PBIBDF-TVT and PBIBDF-TBT were synthesized via the Stille coupling reaction. The effect of benzothiadiazole or vinylene-π spacer of the copolymers on optical properties, energy levels, electronic device performance and microstructure were studied. It was found that PBIBDF-TBT based OFET devices, annealed at 180 °C, showed better performance with the highest electron mobility of 2.9 × 10−2 cm2 V s−1 whereas PBIBDF-TVT polymer exhibited 5.0 × 10−4 cm2 V s−1. The two orders of magnitude higher electron mobility of PBIBDF-TBT over PBIBDT-TVT is a clear indicator of the better charge transport ability of this polymer semiconductor arising from its higher crystallinity and better donor–acceptor interaction.
Introduction
Organic field-effect transistors (OFETs) based on donor–acceptor incorporated π-conjugated polymeric semiconductors have attracted a great scientific interest in recent years due to their potential applications in low-cost, solution-processed and flexible organic electronics.1–4 OFET based on such flexible prototypes have been successfully demonstrated for a wide range of applications including logic circuits,5 chemical sensors,6 memory devices7 and light emitting transistors.8 In comparison with small molecules, polymers offer great advantages such as desirable solution processability, higher viscosity, better flexibility, superior elasticity, great mechanical strength and high thermal/electrochemical stabilities.9 Depending on charge transport properties, polymer semiconductors are generally classified as p-type (holes are the majority charge carriers), n-type (electrons are the majority charge carriers) and ambipolar (electron and holes are the majority charge carrier) materials. Most polymer semiconductors are p-type that have seen a significant increase in performances with very high hole mobility close to or greater than 10 cm2 V−1 s−1.10,11 In sharp contrast, n-type, electron transport polymer semiconductors have largely lagged behind their p-type counterparts. So far most efforts have been directed to the development of novel building blocks, particularly fused ring acceptor building blocks such as diketopyrrolopyrrole (DPP),12–15 naphthalene diimide (NDI)16–18 and perylene diimide (PDI)19,20 for high mobility polymers for OFETs. Among the most interesting electron-depleted cores used for n-channel polymer building blocks, (3E,7E)-3,7-bis(2-oxoindolin-3-ylidene)benzo[1,2-b:4,5-b′]difuran-2,6(3H,7H)-dione (BIBDF in Scheme 1) has demonstrated the greatest potential.3,21–24 BIBDF possesses a large fused aromatic ring with a symmetric and planar structure, which would enhance the π–π overlap and intermolecular interaction.25–27 Additionally, the electron withdrawing lactone groups in the central core combined with the lactam groups make the BIBDF unit strongly electron-deficient.26,27 On the other hand, a donor–acceptor (D–A) type conjugated polymer is one of the most successful and effective strategies to enhance the charge transporting capability of conjugated polymer. With this configuration, inter-/intra-molecular interactions of the polymer chains are easily tuned by varying the combination of different D and A units, and the strong inter chain D–A interaction will result in ordered chain packing and shorten the π–π stacking distance, facilitating the interchain charge transport.28–30 Following this approach, several BIBDF-based D–A conjugated polymers have been reported to demonstrate high charge-carrier mobilities with decent air stability. Recently, Zhang and co-worker have reported D–A polymer based on BIBDF as an acceptor and (E)-2-(2-(thiophen-2-yl)vinyl)thiophene (TVT) as a donor.30 The polymer PBIBDF-TVT showed close π–π distance and long-range ordered, lamellar crystalline structure leading to the high charge carrier mobility. This research also exhibited that the donor unit plays important roles in affecting the absorption bands, HOMO levels, lamellar packing and π–π stacking distances.
 |
| | Scheme 1 The synthetic route to BIBDF. | |
By taking inspiration from previous works,30 our investigation realizes the synthesis of a novel D–A conjugated polymer based on BIBDF as A unit employing dithienylbenzothiadiazole (TBT) as donor–acceptor–donor (D–A–D) unit along with their detailed characterizations. The dithienylbenzothiadiazole D–A–D is a weak electron-donating unit to reduce the energy levels which may enhance air stability of the material. In order to study the effect of stronger donor unit on the electron transport properties of polymer, we also synthesized the previously reported polymer PBIBDF-TVT for the comparison purpose. Benzothiadiazole-π spacer in TBT core is electron-deficient whereas vinylene π-linker in TVT core is electron donor. Such arrangement can lead to lower HOMO and LUMO energy levels of polymer PBIBDF-TBT compared to polymer PBIBDF-TVT. Vinyl linkage present in the backbone is electron rich and can be susceptible for easy oxidation so incorporating electron withdrawing block can stabilise the system and material can be more stable. HOMO/LUMO values of PBIBDF-TBT and PBIBDF-TVT were −5.62/−4.28 eV and −5.51/−4.16 eV, respectively. Both synthesized copolymers were successfully employed as active semiconductors in OFET devices and their electrical and morphological properties have been studied in detail. The PBIBDF-TBT polymer exhibits the highest electron mobility of 2.9 × 10−2 cm2 V s−1 whereas PBIBDF-TVT polymer exhibits 5.0 × 10−4 cm2 V s−1. Such higher mobility of PBIBDF-TBT is due to the strong donor–acceptor interaction. The comparative optoelectronic properties of both polymers and their OFET performance provide an important guideline for designing new polymer semiconductors.
Results and discussions
Synthesis and characterization
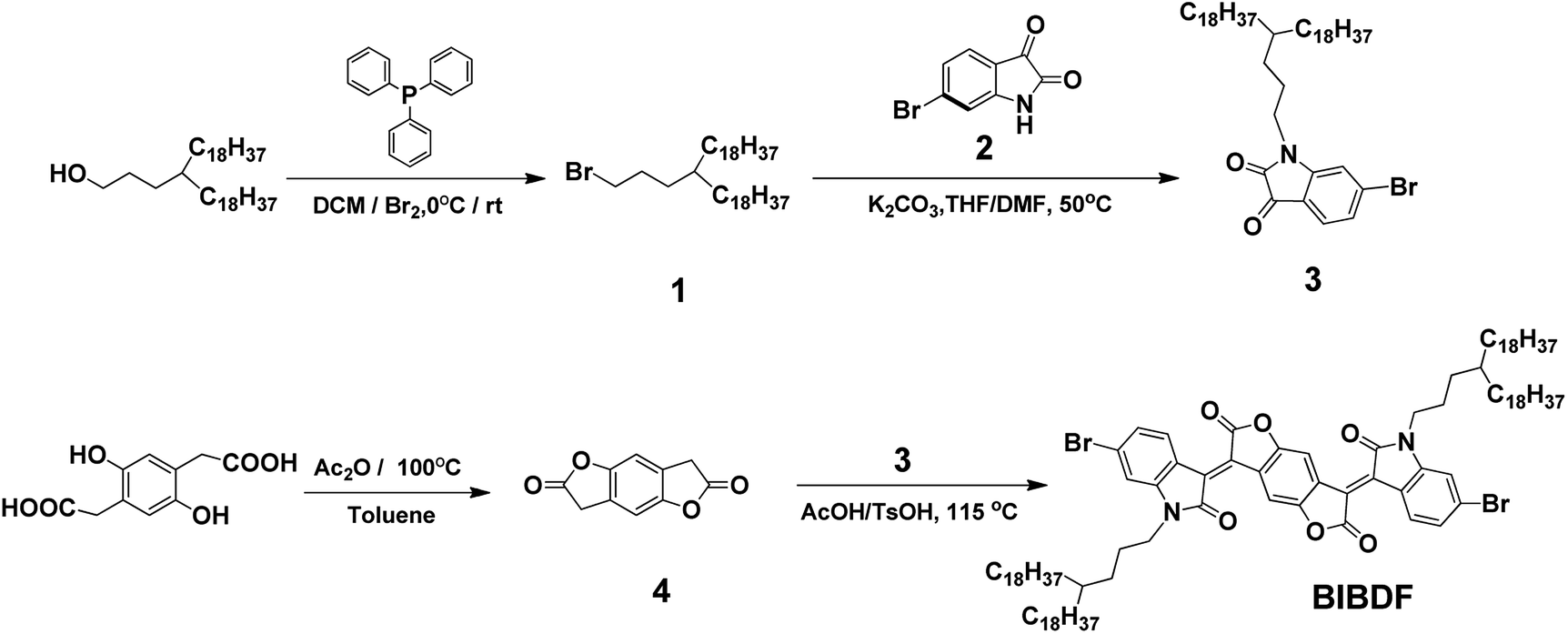
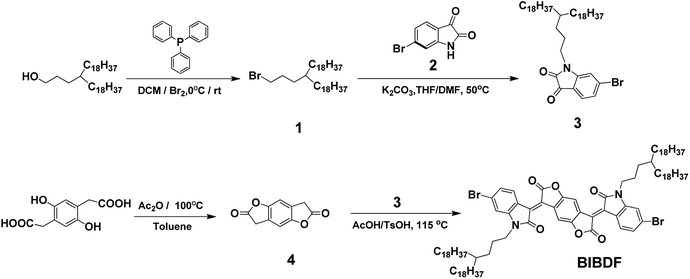
Scheme 1 shows the synthetic route to the electron-deficient fused aromatic ring (3E,7E)-3,7-bis(6-bromo-1-(4-octadecyldocosyl)-2-oxoindolin-3-ylidene)benzo[1,2-b:4,5-b′]difuran-2,6(3H,7H)-dione (BIBDF) as starting monomer. The synthesis started from the bromination of commercially available 3-octadecyl-henicosan-1-ol, giving compound (1) in a good yield. Compound 1 was then reacted with 6-bromo-1H-indole-2,3-dione (2) via N-alkylation to form its soluble derivative 6-bromo-1-(4-octadecyldocosyl)indoline-2,3-dione (3). The long branched alkyl chain 4-octadecyldocosyl was chosen to enhance the solubility of the final BIBDF polymers. The compound 3,7-dihydrobenzo[1,2-b:4,5-b′]difuran-2,6-dione (4) was synthesized from the dehydration reaction of 2,2′-(2,5-dihydroxy-1,4-phenylene)diacetic acid. Finally the target compound BIBDF was prepared from reacting compound 3 with compound 4 in acetic acid at about reflux temperature.
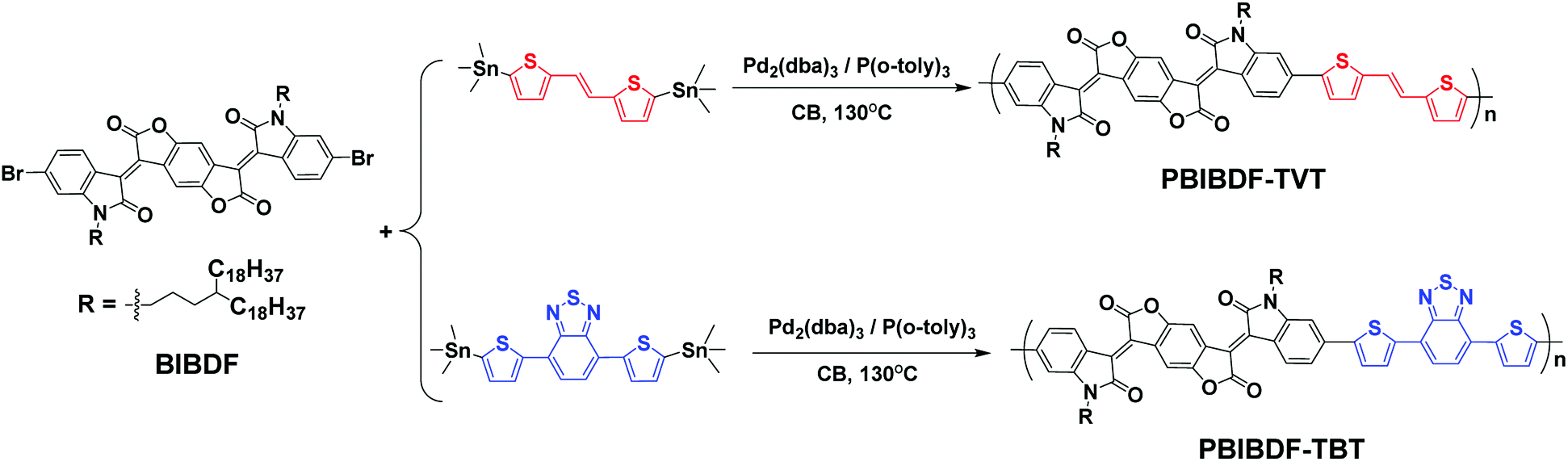
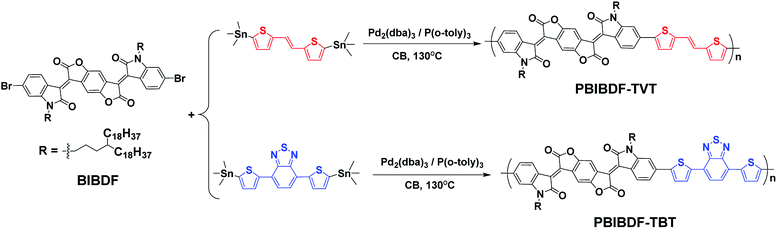
The synthetic routes of PBIBDF-TVT and PBIBDF-TBT are illustrated in Scheme 2. (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane (TVT), 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole (TBT) comonomers and BIBDF monomer were synthesized following the literature procedures.21,31,32 Two polymers were synthesized by Stille cross-coupling reaction using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 monomer ratio in the presence of tris(dibenzylidene-acetone)dipalladium (Pd2(dba)3) as a catalyst and tri(o-tolyl)phosphine (P(o-tol)3) as a ligand. The purification of the crude polymers was performed by Soxhlet extraction with methanol, hexane and acetone to remove the oligomers and other impurities present in the sample. At the last, chloroform was used to extract the pure polymer product from the crude sample. The finally extracted pure polymer solutions in chloroform were kept under rotavapor to remove solvent and the final pure polymer was finally precipitated using methanol as an anti-solvent. The purified polymers were investigated by X-ray photoelectron spectroscopy (XPS) (Fig. S11†) to confirm that polymers are metal-free. The precipitated polymer sample was then dried and used for further characterization and to study their comparative behaviour. All the two polymers were dark black solids that gradually dissolved in chloroform. The number (Mn) and the weight (Mw) average molecular weights of the polymers were determined by gel-permeation chromatography (GPC) with chloroform as an eluent at 30 °C. The Mn and Mw were 173.5 kDa and 870.2 kDa, respectively for PBIBDF-TVT, 360.6 kDa and 3064.9 kDa, respectively for PBIBDF-TBT.
:1 monomer ratio in the presence of tris(dibenzylidene-acetone)dipalladium (Pd2(dba)3) as a catalyst and tri(o-tolyl)phosphine (P(o-tol)3) as a ligand. The purification of the crude polymers was performed by Soxhlet extraction with methanol, hexane and acetone to remove the oligomers and other impurities present in the sample. At the last, chloroform was used to extract the pure polymer product from the crude sample. The finally extracted pure polymer solutions in chloroform were kept under rotavapor to remove solvent and the final pure polymer was finally precipitated using methanol as an anti-solvent. The purified polymers were investigated by X-ray photoelectron spectroscopy (XPS) (Fig. S11†) to confirm that polymers are metal-free. The precipitated polymer sample was then dried and used for further characterization and to study their comparative behaviour. All the two polymers were dark black solids that gradually dissolved in chloroform. The number (Mn) and the weight (Mw) average molecular weights of the polymers were determined by gel-permeation chromatography (GPC) with chloroform as an eluent at 30 °C. The Mn and Mw were 173.5 kDa and 870.2 kDa, respectively for PBIBDF-TVT, 360.6 kDa and 3064.9 kDa, respectively for PBIBDF-TBT.
 |
| | Scheme 2 The synthetic route to polymers PBIBDF-TVT and PBIBDF-TBT. | |
Thermal properties
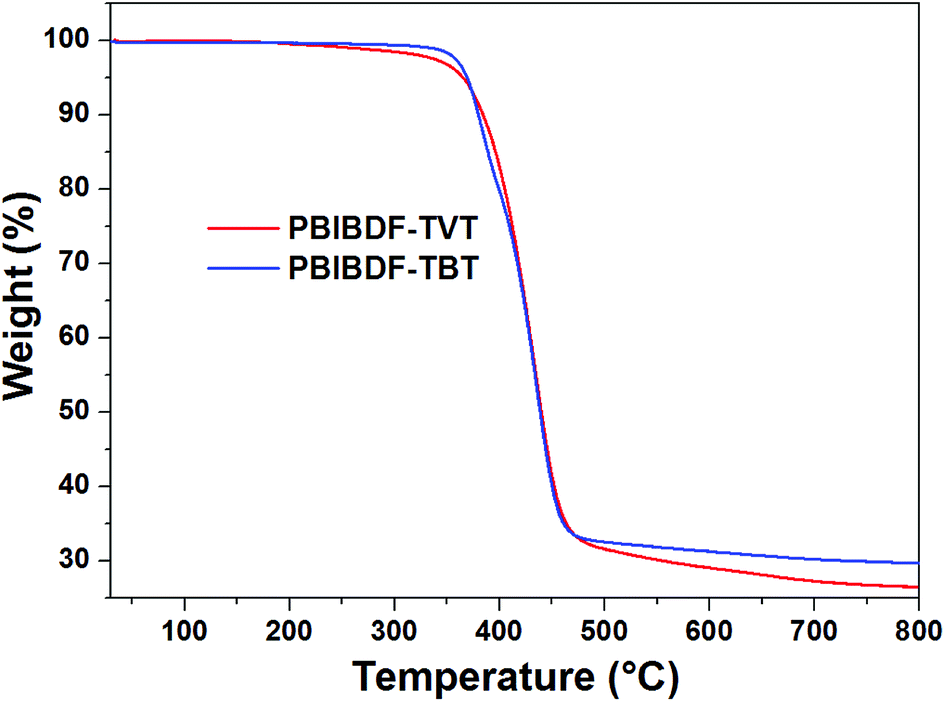
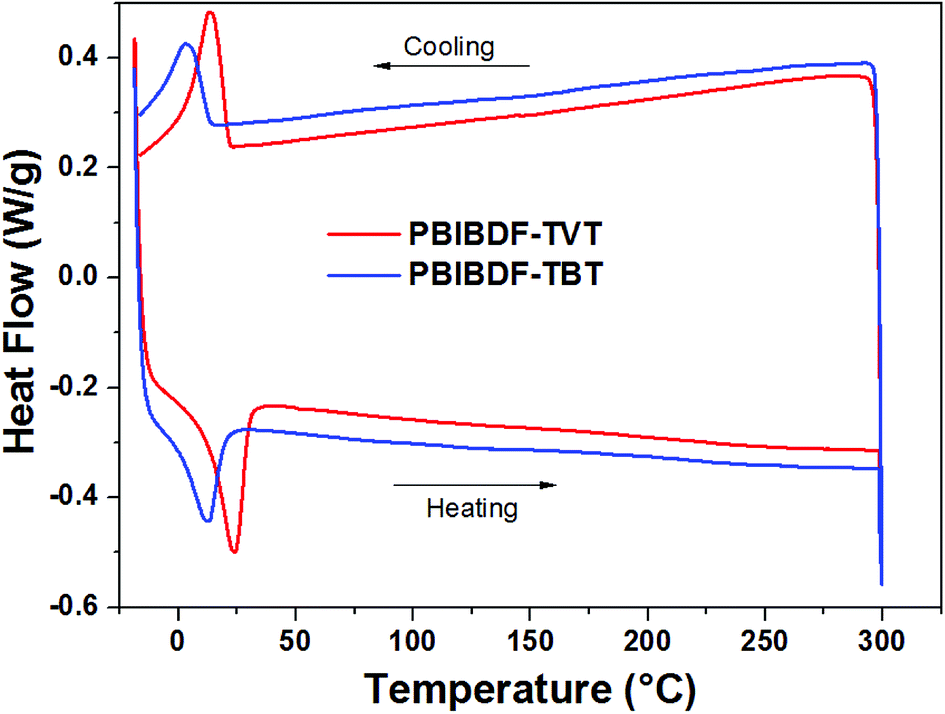
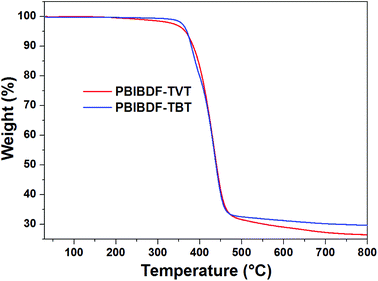
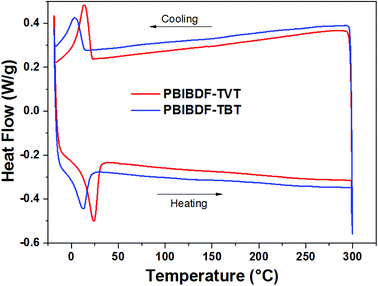
The thermal properties of PBIBDF-TVT and PBIBDF-TBT were investigated by thermogravimetric analysis (TGA) (Fig. 1) and differential scanning calorimetry (DSC) (Fig. 2) respectively. For the DSC measurement, polymer sample was employed for heating and cooling cycles using 10 °C per minute heating rate. As shown in Fig. 2, during DSC heating and cooling scans, PBIBDF-TVT and PBIBDF-TBT showed endothermic peaks at 12–24 °C and exothermic peaks at 2–13 °C. The similar transition temperatures of polymers based on BIBDF with similar alkyl chain was observed,3,23,30 which is possibly due to the phase transition temperature caused by the melting of the long alkyl chains. For the TGA measurement, both polymer samples were heated from room temperature to 800 °C using 10 °C per minute heating rate. Both polymers exhibited good thermal stabilities, with high decomposition temperature (at which a compound loses 5% of its weight): PBIBDF-TVT: 365 °C and PBIBDF-TBT: 370 °C. The thermally stable behaviour of polymers is beneficial for performing higher temperature annealing and thin film processing application in optoelectronic devices.
 |
| | Fig. 1 TGA thermograms of PBIBDF-TVT and PBIBDF-TBT under a nitrogen atmosphere at a heating rate of 10 °C min−1. | |
 |
| | Fig. 2 Differential Scanning Calorimetric (DSC) analysis of PBIBDF-TVT and PBIBDF-TBT with a scanning rate of 10 °C min−1. | |
Optical properties
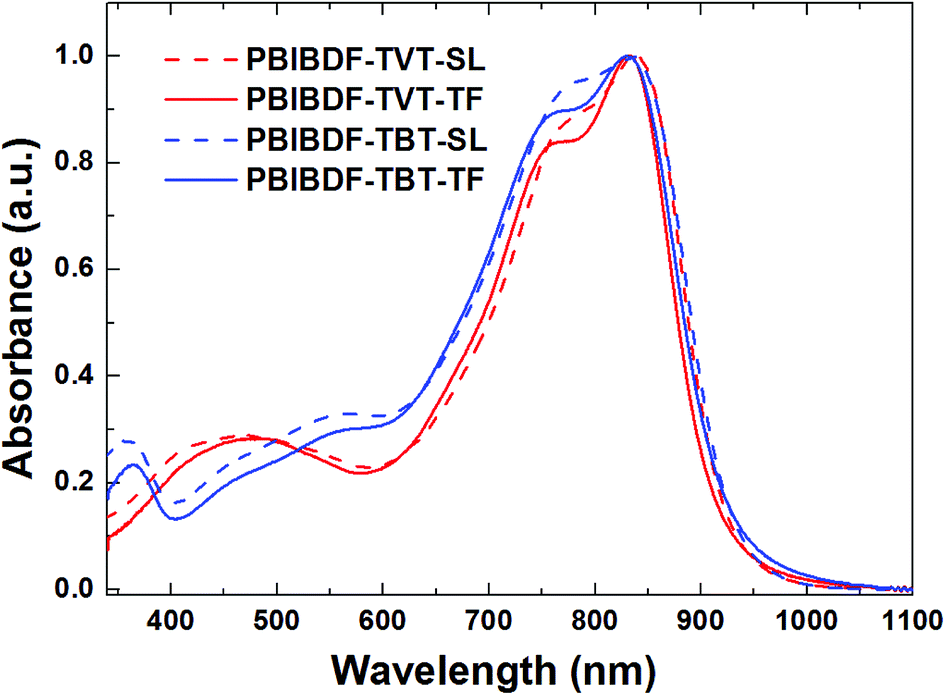
The optical properties of PBIBDF-TVT and PBIBDF-TBT were studied by ultraviolet-visible (UV-vis) absorption spectra in dilute chloroform solutions (around 10−4 M) and as thin films on glass substrate casted from chloroform (Fig. 3). In chloroform solution, both polymers showed broad and strong absorption at the range of 350 nm to 900 nm. The shorter absorption band at 350–600 nm corresponds to π–π* transitions, and longer wavelength band at 650–900 nm corresponds to intramolecular charge transfer (ICT). The longest wavelength absorption maxima (λmax) exhibited by PBIBDF-TVT and PBIBDF-TBT were similar at 837 nm. The absorption spectrum and absorption maximum of polymer PBIBDF-TVT in this study are similar to the same one reported by Zhang et al. group.30 In addition, the absorption spectrum of PBIBDF-TBT was quite broader than that of PBIBDF-TVT in the long wavelength band. Similar to some other polymers based on BIBDF,21 both PBIBDF-TVT and PBIBDF-TBT exhibited insignificant spectral shift upon going from the solution to the film, suggesting that polymers might form some pre-aggregates in solution due to their strong intermolecular interactions.33–35 Furthermore, the blue shift of absorption spectra and peaks of two polymers in the thin film state compared to those in the solution state may result from their unique H-aggregation-induced interchain packing in the solid state.25,36 The edges of the films absorption bands for PBIBDF-TVT and PBIBDF-TBT were at 920 and 925 nm, respectively, corresponding to optical bandgaps of 1.35 and 1.34 eV, respectively. The narrow bandgaps of polymers could be ascribed to the extension of π-conjugation as well as strong intramolecular and intermolecular interactions arising from the donor and acceptor moieties along the polymer backbone.26,30
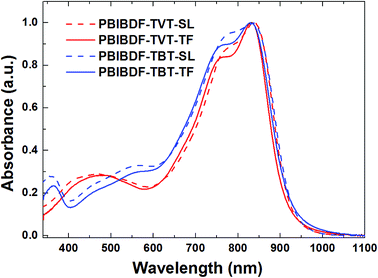 |
| | Fig. 3 UV-vis absorption spectra of PBIBDF-TVT and PBIBDF-TBT in chloroform solutions and as thin films. | |
Electron energy level determination
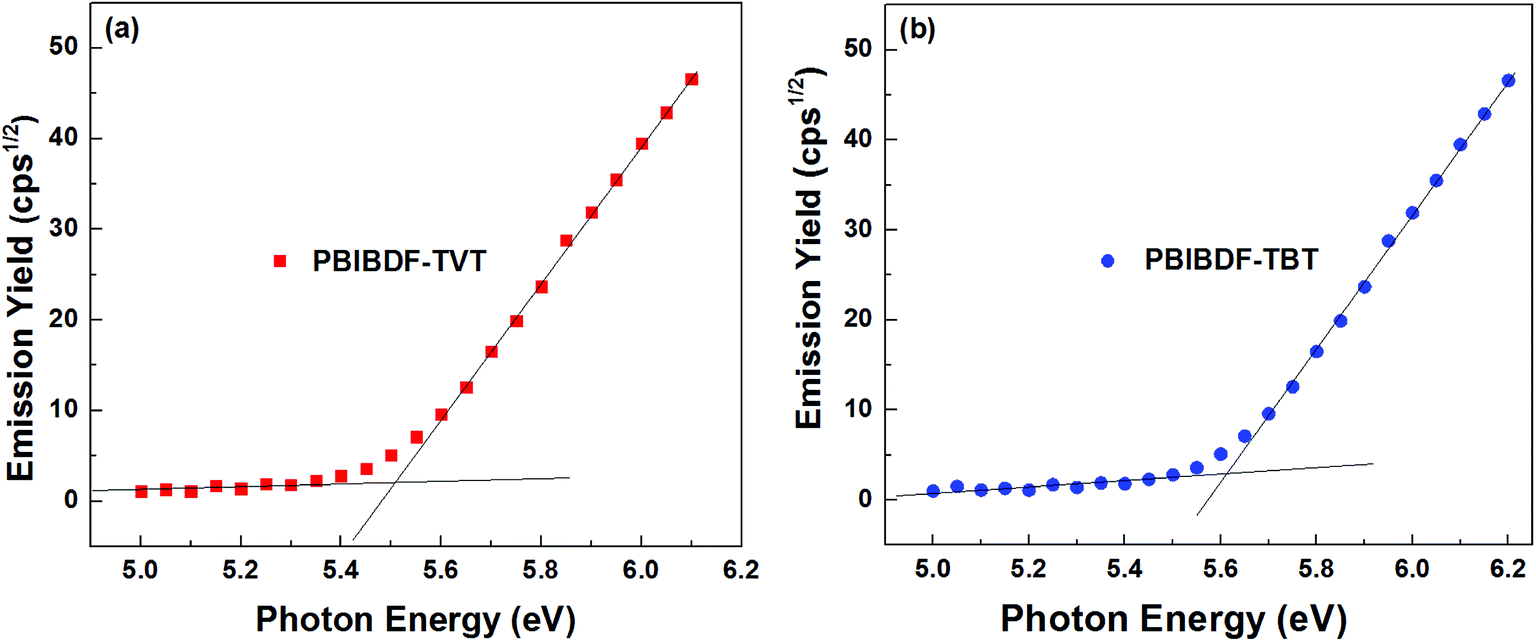
Photoelectron spectroscopy in the air (PESA) was used to determine HOMO energy levels of polymers. In our measurements, we spin-coated polymer thin films from chloroform solution on the glass substrates for PESA measurement (Fig. 4, data in Table 1). From the onset energy level using PESA data, we calculated HOMO values for PBIBDF-TVT and PBIBDF-TBT polymers and they were found to be −5.51 and −5.62 eV, respectively. It is clear from the observed energy levels that by introducing a stronger electron affinity spacer, the polymer PBIBDF-TBT possesses lower-lying HOMO levels. The LUMO energy levels of PBIBDF-TVT and PBIBDF-TBT, calculated from the optical band gap and HOMO value difference and they were determined to be −4.16 and −4.28 eV, respectively. The obtained LUMO values are appropriate and significantly lower for better air-stable electron transport.
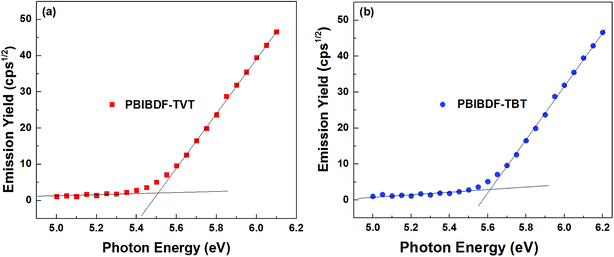 |
| | Fig. 4 Photoelectron spectroscopy in air (PESA) analysis of (a) PBIBDF-TVT and (b) PBIBDF-TBT in thin film. | |
Table 1 Optical and electrochemical data for the polymers PBIBDF-TVT and PBIBDF-TBT
| Polymer |
λmax (nm) |
Eoptg (eV) |
HOMOa (eV) |
LUMOb (eV) |
HOMOc (eV) |
LUMOc (eV) |
| Solution |
Film |
| The HOMO value determined by PESA. ELUMO = EPESAHOMO + Eoptg. DFT-estimated data. |
| PBIBDF-TVT |
837 |
834 |
1.35 |
−5.51 |
−4.16 |
−5.53 |
−3.85 |
| PBIBDF-TBT |
837 |
830 |
1.34 |
−5.62 |
−4.28 |
−5.67 |
−3.90 |
Furthermore, the cyclic voltammetry (CV) analysis with a glassy carbon working electrode, platinum-wire counter electrode and Ag/AgCl reference electrode using 0.1 M tetra(n-butyl)ammonium hexafluorophosphate (n-Bu4NPF6) in dichloromethane (DCM) solution as an electrolyte at a potential scan rate of 100 mV s−1 was also studied. Unfortunately, polymers are not completely soluble in DCM, then the mixture of chlorobenzene and DCM (7:3 v/v) was used, however polymers are aggregated in the solution (Fig. S12†), therefore good CV curves could not be obtained.
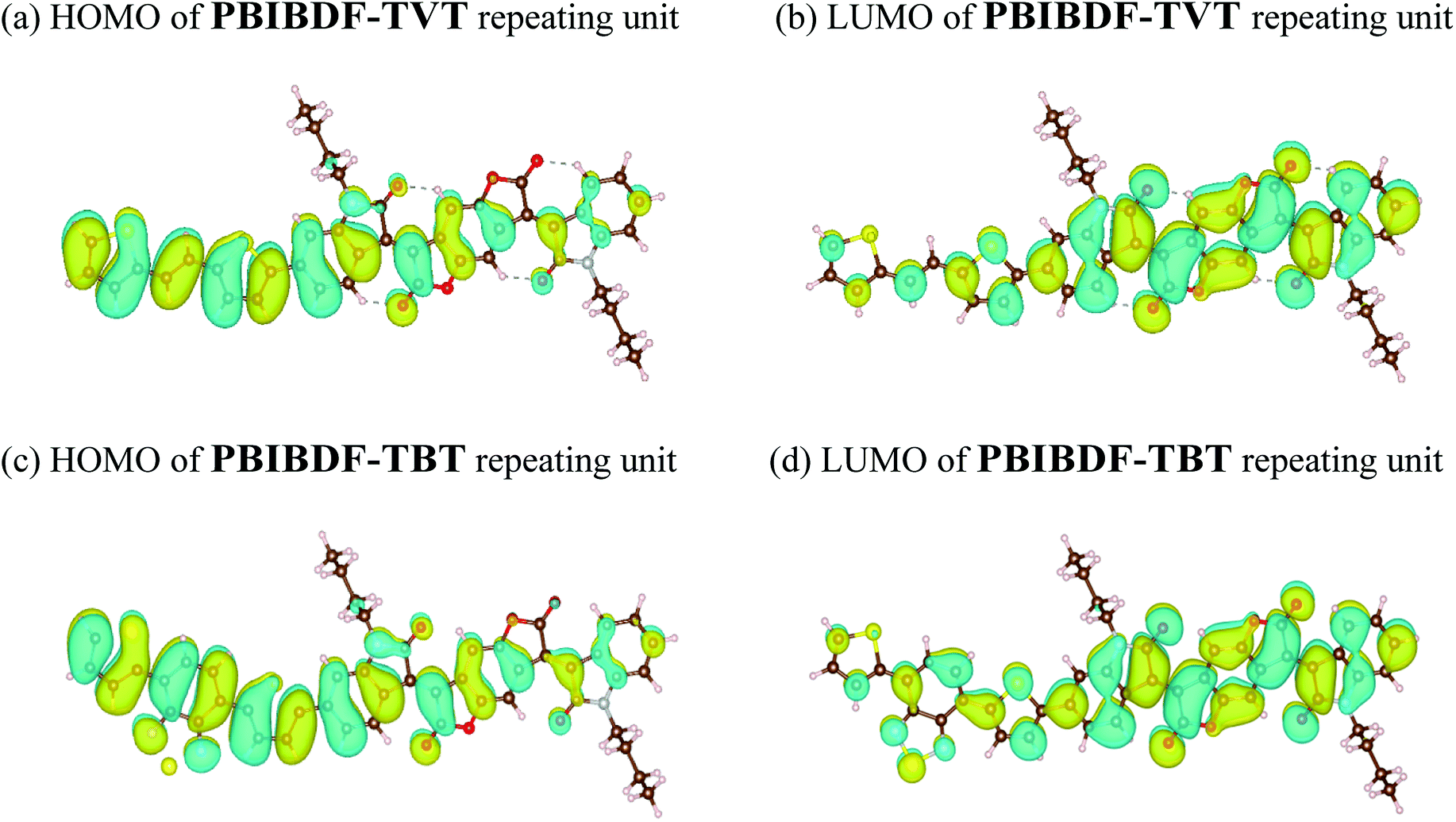
In addition, the structural and electric parameters of these two polymers were also investigated by density functional theory (DFT) calculations using the Gaussian 09 at the B3LYP/6-31g(d) level to study the electron density. As shown in Fig. 5, the HOMO electron densities of PBIBDF-TVT and PBIBDF-TBT are mainly located on the TVT and TBT moieties whereas their LUMO electron densities are mainly located on the BIBDF units, respectively. The calculated HOMO/LUMO energy levels of PBIBDF-TVT and PBIBDF-TBT are −5.53/−3.85 eV and −5.67/−3.90, respectively. The obtained theoretical HOMO and LUMO energy levels are in a good agreement with PESA measurements.
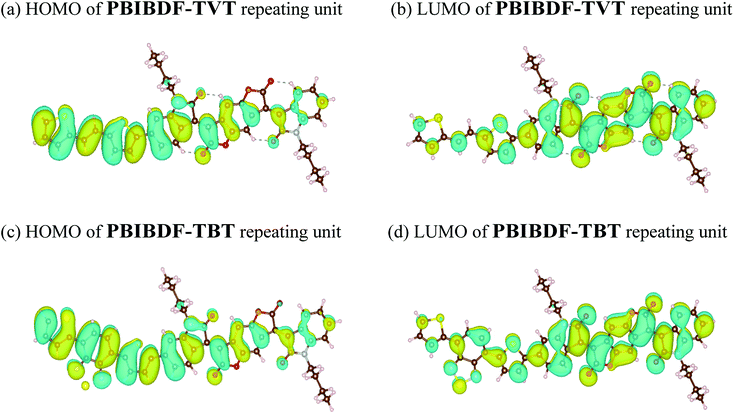 |
| | Fig. 5 The calculated molecular orbitals for PBIBDF-TVT and PBIBDF-TBT. | |
X-ray diffraction (XRD) analysis
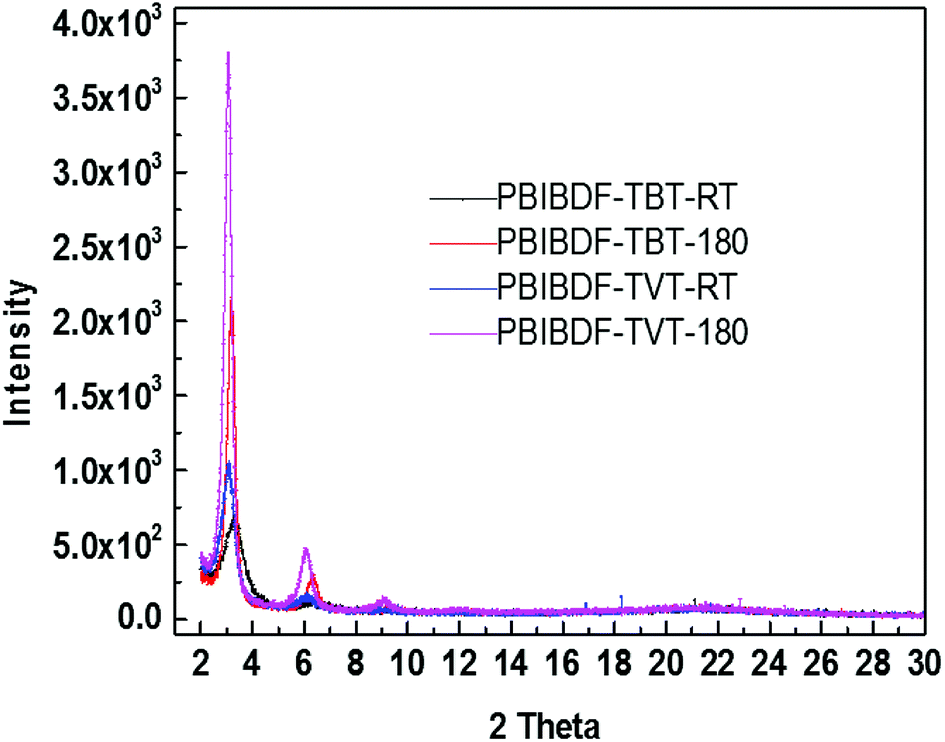
X-ray diffraction measurement was performed to examine the ordering and crystallinity of two polymers with the difference of π-space unit. The thin film XRD, for both polymers, was measured before- and after-annealing at 180 °C (this is the best optimized thin film annealing temperature condition which exhibits better charge transport-see next OFET device section) using exact device geometry (Si/SiO2 substrates) shown in Fig. 6. In case of PBIBDF-TVT, variation in the d-spacing and full width half maximum (FWHM) of annealed and non-annealed thin films were almost identical. We noticed the first diffraction peak for the non-annealed thin film at 2θ = 3.08°, corresponding to a d-spacing of 2.87 nm, with an FWHM of 0.58°. For the thin films annealed at 180 °C, the diffraction peak changed to 2θ = 3.18°, increasing the d-spacing slightly to 2.88 nm. However, in case of PBIBDF-TBT, the first peak was shifted from 3.29° to 3.18° upon annealing at 180 °C, leading to a d-spacing shift from 2.69 nm to 2.78 nm. Simultaneously, the FWHM was changed from 1.07 to 0.37 upon annealing. While comparing XRD, we clearly notice that an annealed thin film of PBIBDF-TBT exhibits higher crystallinity and lower d-spacing than that of the PBIBDF-TVT due to the presence of the strong electron withdrawing benzothiadiazole spacer moiety.
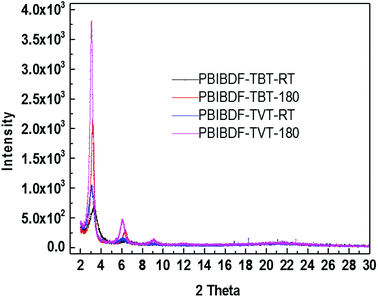 |
| | Fig. 6 X-ray diffraction (XRD) Patterns of Thin Film XRD at RT and after annealing at 180 °C for PBIBDF-TVT and PBIBBDF-TBT polymers. | |
OFET device characteristics and performance
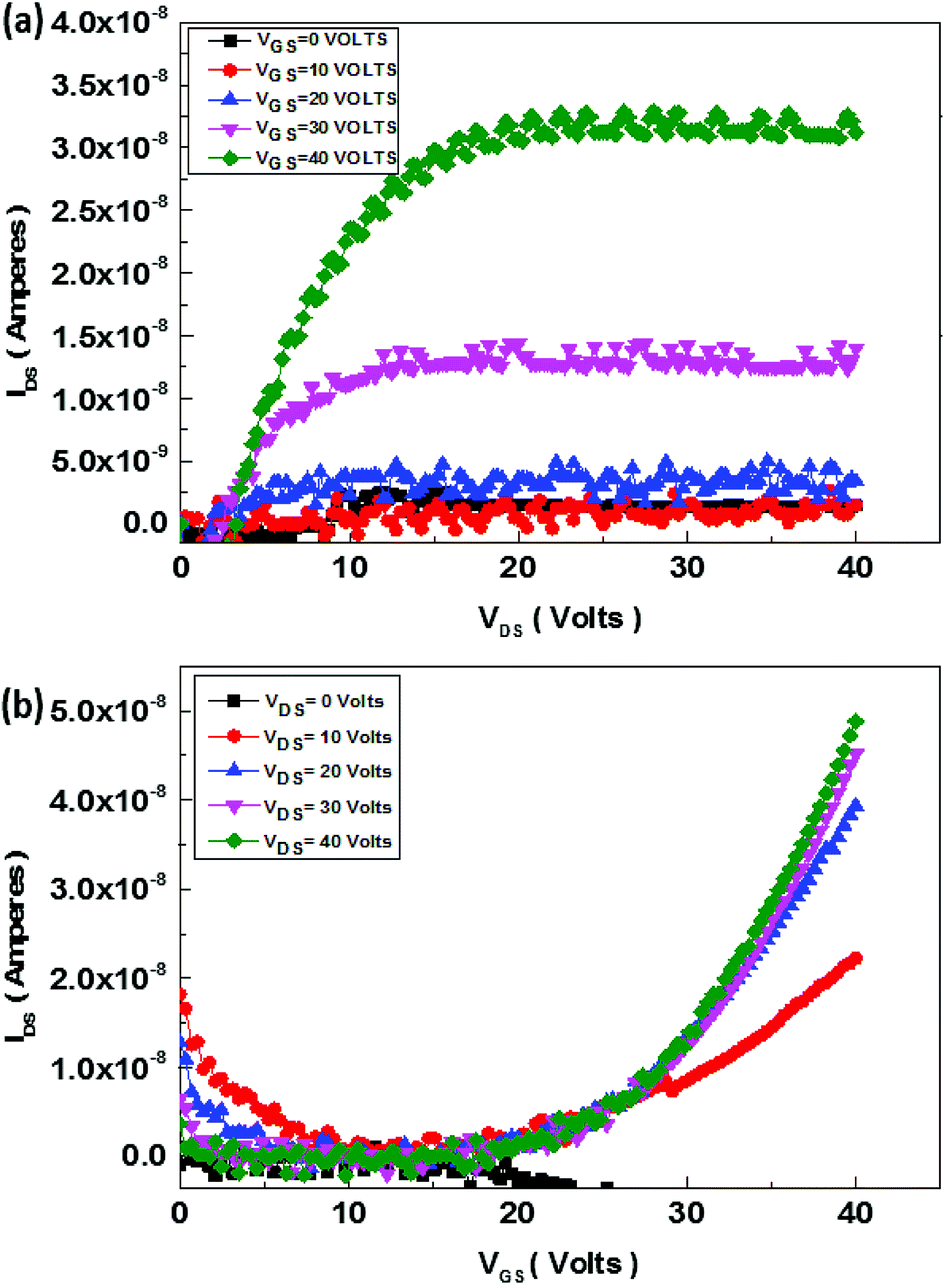
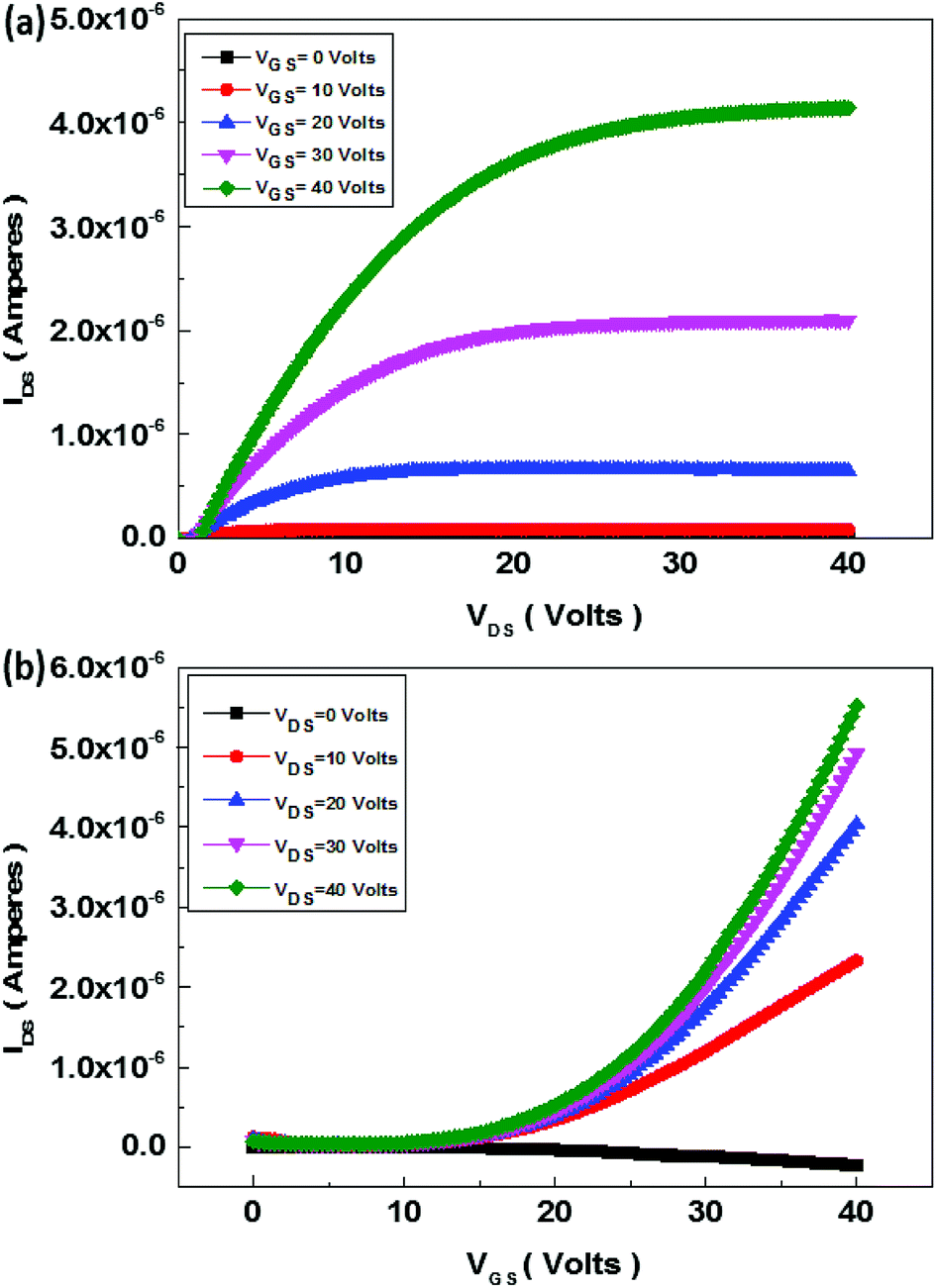
The schematics of OFET devices of PBIBDF-TVT and PBIBDF-TBT polymer semiconductors are shown in Fig. 7a and b, respectively. The output and transfer characteristics of PBIBDF-TVT and PBIBDF-TBT polymer semiconductors based OFETs were measured under an inert condition inside a glove box. The OFET devices of non-annealed thin films of both polymers did not exhibit any OFET characteristics. The devices with an annealed (at 180 °C) polymer thin film as an active layer, showed n-type behaviour. The output and transfer characteristics of PBIBDF-TVT and PBIBDF-TBT based OFETs are shown in Fig. 8 and 9, respectively. The electron mobilities, Ion/Ioff ratio and Vth for both polymers are summarised in Table 2. For PBIBDF-TVT based OFET, the electron mobility determined from the linear and saturation region, was found to be ∼4.2 × 10−4 cm2 V s−1 and ∼5.0 × 10−4 cm2 V s−1, respectively with an Ion/Ioff ratio of 102, and Vth of 16 volts. The electron mobility, determined from the transfer characteristics of PBIBDF-TBT polymer based OFETs, were 1.3 × 10−3 cm2 V s−1 (linear) and 2.9 × 10−2 cm2 V s−1 (saturation), respectively with an Ion/Ioff ratio of 102 and Vth of 12 volts. The two order of magnitude higher electron mobility of PBIBDF-TBT over PBIBDT-TVT in the saturation regime is a clear signature of better charge transport ability in this polymer. The high crystallinity of PBIBDF-TBT polymer thin films, annealed at 180 °C, can be attributed for high charge transport.
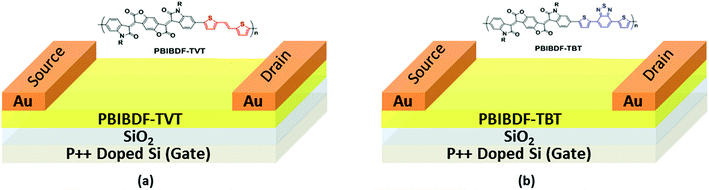 |
| | Fig. 7 Bottom-gate-top-contact OFET device structure using (a) PBIBDF-TVT and (b) PBIBDF-TBT based polymer semiconductors. | |
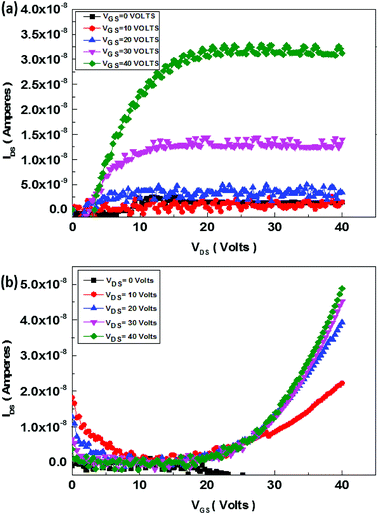 |
| | Fig. 8 Output (a) and transfer (b) characteristics of PBIBDF-TVT polymer based OFET devices after annealing at 180 °C. | |
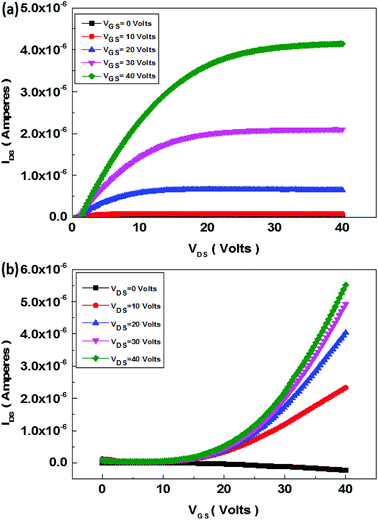 |
| | Fig. 9 Output (a) and transfer (b) characteristics of PBIBDF-TBT polymer based OFET devices after annealing at 180 °C. | |
Table 2 OFET device characteristics of PBIBDF-TVT and PBIBDF-TBT based polymers
| Polymers |
Anneal. temp. (°C) |
Vth (V) |
Ion/Ioff |
μe (Lin.) (cm2 V s−1) |
μe (Sat.) (cm2 V s−1) |
| PBIBDF-TVT |
180 |
16 |
102 |
4.2 × 10−4 |
5.0 × 10−4 |
| PBIBDF-TBT |
180 |
12 |
102 |
1.3 × 10−3 |
2.9 × 10−2 |
AFM thin film morphology
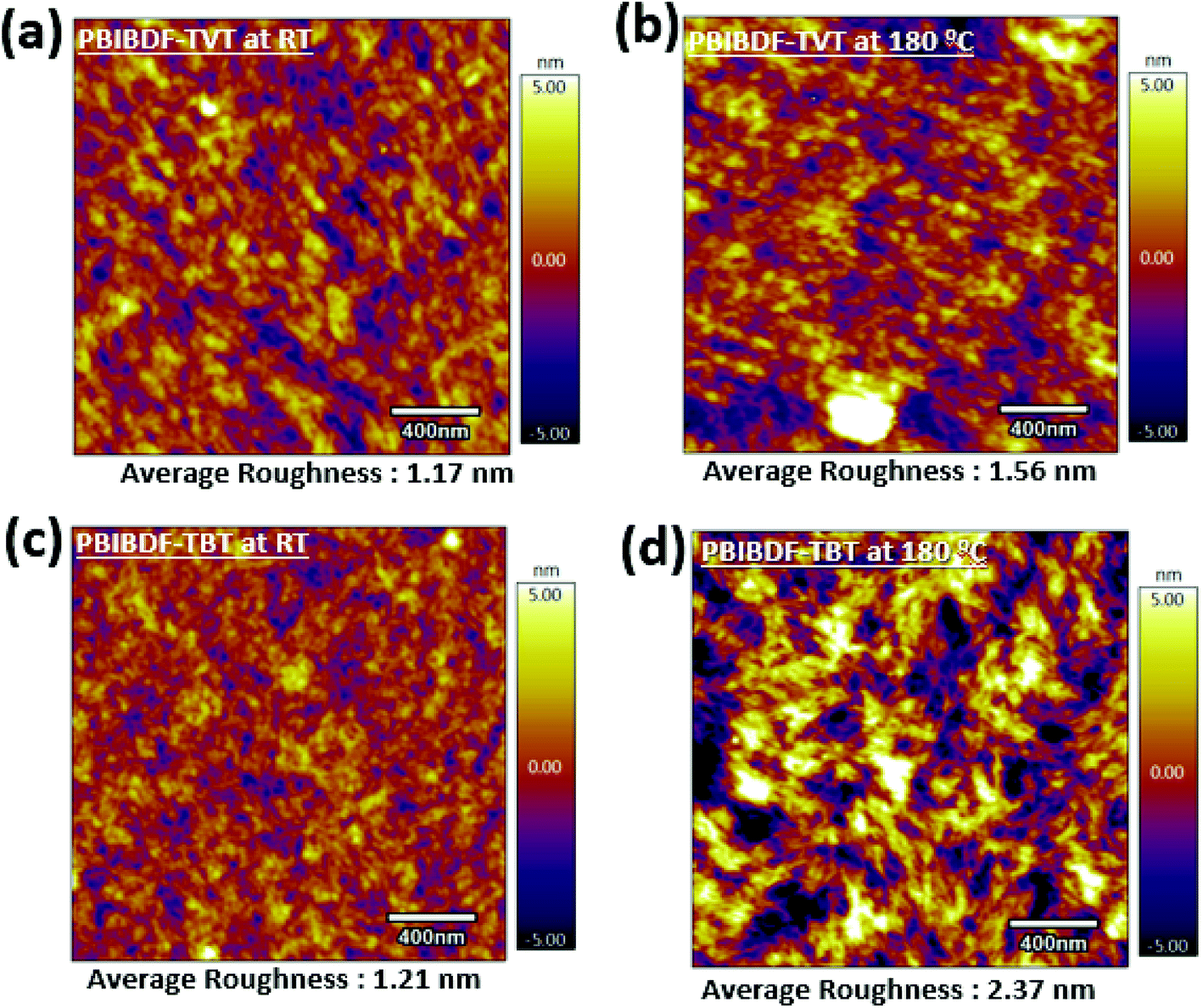
In order to further investigate the reasons for better charge transport in PBIBDF-TBT based OFETs as compared to the same of PBIBDF-TVT, we further studied the surface morphology and microstructures of the polymeric thin films by atomic force microscopy (AFM) of the thin films on the actual devices from which the characteristics were obtained. An AFM (area: 2 μm×2 μm) examination for both PIBDF-TVT and PIBDF-TBT thin films exhibits uniformly arranged grains, as shown in Fig. 10. It is clearly noticed that, upon annealing of both polymer thin films, the large domains with clear boundary areas emerge. It is also observed that the annealed thin films of both polymer exhibit large polycrystalline grains. This observation is consistent with previously reported PBIBDF-TVT based polymer thin film morphology in the literature.30 In annealed thin films, the extent of grain formation and their size is considerably higher in case of PIBDF-TBT than that of the PIBDF-TVT. This observation is further substantiated by estimating an average roughness of thin film surfaces. A non-annealed thin film of PBIBDF-TVT and PBIBDF-TBT polymer showed a surface roughness of 1.17 nm and 1.21 nm, respectively. Whereas, in case of 180 °C annealed polymer thin films, the surface roughness is enhanced from 1.56 nm (for PBIBDF-TVT) to 2.37 nm (for PBIBDF-TBT). The AFM image, Fig. 10d, shows evolution of higher interconnecting grains in annealed thin film of PBIBDF-TBT. Such microstructures in polymer thin films are known to facilitate charge transport in OFETs leading to high charge carrier mobility.37
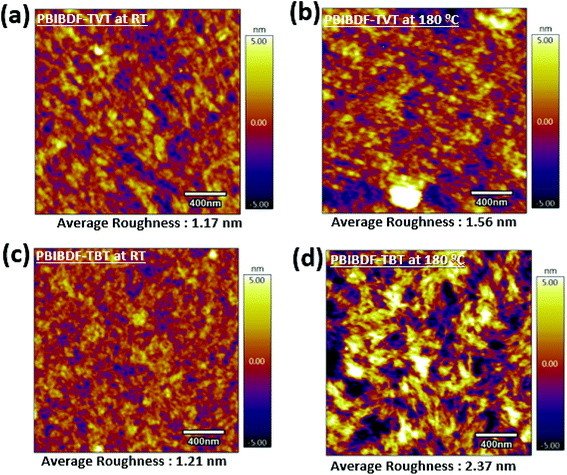 |
| | Fig. 10 AFM Images (2 μm by 2 μm) of (a) PBIBDF-TVT at room temperature and (b) at 180 °C, (c) PBIBDF-TBT at room temperature and (d) at 180 °C. | |
Conclusion
Two conjugated polymers PBIBDF-TVT and PBIBDF-TBT composed of BIBDF and (E)-2-(2-(thiophen-2-yl)vinyl)thiophene (TVT) or dithienylbenzothiadiazole (TBT) groups were prepared to investigate the influence of π-spacer on field-effect charge transport in OFETs, and microstructures in thin films. The HOMO energy levels, for PBIBDF-TVT and PBIBDF-TBT polymers, were found to be −5.51 and −5.62 eV, respectively. These results indicate that a stronger electron affinity spacer benzothiadiazole (compared to vinylene) lowers the HOMO energy level. Furthermore, the investigation of the two polymers for n-channel organic field-effect transistors was also performed. The PBIBDF-TBT polymer exhibits the highest electron mobility of 2.9 × 10−2 cm2 V s−1 whereas PBIBDF-TVT polymer exhibits electron mobility of 5.0 × 10−4 cm2 V s−1. The high crystallinity and interconnecting microstructures observed in a thin film of PBIBDF-TBT polymer, annealed at 180 °C, led to an improved electron mobility.
Experimental section
Materials and instruments
All starting materials were purchased commercially as analytical reagents and used directly without any further purification. 4-Octadecyl-henicosan-1-ol was purchased from Lymtech (Beijing) Technology Ltd, purity 95%. (3E,7E)-3,7-bis(2-oxoindolin-3-ylidene)benzo[1,2-b:4,5-b′]difuran-2,6(3H,7H)-dione (BIBDF) was prepared according to the literature. High resolution mass spectra were recorded on an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) equipped with an atmospheric pressure chemical ionisation (APCI) source, operating in the positive ion mode at a resolution of 120000 (at m/z 400). Gel Permeation Chromatography (GPC) against polystyrene standards was performed in chloroform at 30 °C and a flow rate of 1 mL min−1 on a Waters GPC assembly equipped with a Waters 1515 isocratic HPLC pump, Waters 2707 autosampler with a 100 mL injection loop, and a Waters 2487 dual wavelength absorbance detector analysed at 254 nm in series with a Waters 2414 refractive index detector at 30 °C. Three consecutive Waters Styragel columns (HR5, HR4, and HR1, all 7.8 × 300 mm, 5 μm particle size), preceded by a Waters Styragel guard column (WAT054405, 4.6 × 30 mm, 20 μm particle size) were used during analysis. A typical concentration of 1 mg polymer dissolved in 1 mL chloroform was used to run GPC samples. Thermal analysis was performed using a Pegasus Q500TGA thermogravimetric analyzer under a nitrogen atmosphere at a heating rate of 10 °C min−1. Differential scanning calorimeter (DSC) was conducted under nitrogen using a Chimaera instrument Q100 DSC. The sample was heated at 10 °C min−1 from −20 °C to 300 °C. Absorption spectra were recorded on Carry50 UV-vis Spectrophotometer. Photoelectron spectroscopies in air (PESA) measurements were conducted using on an AC-2 photoelectron spectrometer (Riken-Keiki Co.). The AFM images were taken on the sample on which the OFET devices where characterized. We used the Cypher AFM from (Oxford instrument, Asylum Research, Santa Barbara, CA). It was carried out by tapping mode in air using Etalon NT-MDT cantilever with force constant 3.5 N m−1.
Device fabrication
A heavily p-doped silicon wafer with a layer of ∼200 nm SiO2 on the surface was used as the substrate for bottom-gate, top-contact device geometry with channel length 80 μm and channel width 2 mm. The SiO2 functions as the gate insulator and the doped Si as the gate. The active polymer thin film was spin coated on top of the SiO2 surface using a PBIBDF-TVT or PBIBDF-TBT polymer solution in chloroform and dichlorobenzene (DCB) (6 mg mL−1) respectively at 1000 rpm for 60 seconds. The average thickness of films was around 50 nm. Annealing of polymer thin films was at different temperatures done for 20 min on a hot plate in a nitrogen atmosphere. Specifically, PBIBDF-TBT polymer thin films were annealed at 100, 180, 260 and 300 °C. PBIBDF-TVT polymer thin films were annealed at 62 and 180 °C. On top of PBIBDF-TVT or PBIBDF-TBT polymer thin film, an Au film of 100 nm was deposited as source and drain electrode by thermal evaporation. The Au electrodes were patterned via shadow masking method. The channel width (W) and length (L) of OFETs were 2 mm and 80 μm, respectively.
Synthesis
Synthesis of 19-(2-bromo-ethyl)-heptatriacontane (1). Triphenylphosphine (4.65 g, 17.70 mmol) was dissolved in DCM (100 mL) under argon for 15 minutes. Then the reaction mixture was cooled to 0 °C and followed by the addition of bromine (1.28 mL, 24.78 mmol). After 3-octadecyl-henicosan-1-ol (10.0 g, 17.70 mmol) in 100 mL of DCM was added dropwise via an additional funnel over 30 minutes, the reaction solution was stirred at room temperature for overnight under argon. The organic solvent was removed under the reduced pressure, and the residue was extracted with excess amount of hexane. The hexane was evaporated and the resulting crude yellow solid was purified by silica gel column eluting with hexane. The title compound was isolated as white-off solid (10 g, 90%). 1H NMR (600 MHz, CDCl3, ppm): δ 3.41 (t, J = 10.2 Hz, 2H), 1.85 (m, 2H), 1.57 (m, 1H), 1.28 (m, 68H), 0.90 (t, J = 9.6 Hz, 6H).
Synthesis of 6-bromo-1-(4-octadecyldocosyl)indoline-2,3-dione (3). 19-(2-Bromo-ethyl)-heptatriacontane (5.0 g, 7.96 mmol) was added under nitrogen atmosphere to a suspension of 6-bromo-1H-indole-2,3-dione (1.8 g, 7.96 mmol), potassium carbonate (2.2 g, 15.9 mmol) in a mixture of tetrahydrofuran (40 mL) and N,N-dimethylformamide (40 mL). The mixture was stirred at room temperature for 1 hour, and then stirred at 50 °C for overnight. After the reaction mixture was cooled to room temperature and extracted with DCM and water. The organic layer was washed with water, brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel using hexane/DCM (3:1, v/v) as eluent to give orange coloured oil (4.7 g, 75%). 1H NMR (600 MHz, CDCl3, ppm): δ 7.46 (d, J = 12 Hz, 1H), 7.28 (s, 1H), 7.06 (s, 1H), 3.67 (t, J = 11.4 Hz, 2H), 1.65 (m, 1H), 1.25 (m, 24H), 0.88 (m, J = 9.6 Hz, 6H).
Synthesis of 3,7-dihydrobenzo[1,2-b:4,5-b′]difuran-2,6-dione (4). In the mixture of 2,2′-(2,5-dihydroxy-1,4-phenylene)diacetic acid (2.0 g, 8.8 mmol) and anhydrous toluene (50 mL), acetic anhydride (20 mL) was added. The mixture was stirred at 100 °C for 5 h and then the solvent was removed under pressure. The residue was purified by silica gel chromatography with eluent (CHCl3), and recrystallized from toluene to give 3,7-dihydrobenzo[1,2-b:4,5-b′]difuran-2,6-dione as a white crystal (1.5 g, 90%). 1H NMR (600 MHz, CDCl3, ppm): δ 7.06 (s, 2H), 3.87 (s, 4H).
Synthesis of BIBDF. In the mixture of compound 3 (787 mg, 1.0 mmol) and acetic acid (20 mL), compound 4 (70 mg, 0.417 mmol) and p-toluenesulfonic acid monohydrate (TsOH, 23 mg, 0.12 mmol) was add under nitrogen atmosphere. The mixture was stirred at 115 °C under nitrogen for 17 h. The reaction mixture was then cooled to room temperature and filtered. The solid was washed with acetic acid and then methanol. The residue was purified by silica gel chromatography with eluent (hexane:DCM = 1:1 v/v) to give BIBDF as a black solid (430 mg, 60%). 1H NMR (600 MHz, CDCl3, ppm): δ 9.11 (s, 2H), 8.93 (d, J = 13.2 Hz, 2H), 7.21 (dd, J1 = 13.2 Hz, J2 = 2.4 Hz, 2H), 6.95 (s, 2H), 3.72 (t, J = 11.4 Hz, 4H), 1.67 (m, 4H), 1.22 (m, 24H), 0.86 (m, J = 9.6 Hz, 12H). 13C NMR (150 MHz, CDCl3, ppm): δ 167.13, 166.97, 151.85, 149.85, 135.88, 131.61, 128.98, 126.93, 126.64, 125.72, 119.83, 111.85, 111.06, 40.73, 37.07, 33.52, 31.94, 30.75, 30.12, 29.73, 29.68, 29.38, 26.69, 24.40, 22.71, 14.14. HRMS (EI, m/z) calcd: 1729.1512 (M+). Found: 1729.1589.
Synthesis of PIBDF-TVT. The following synthesis was based on a modified literature procedure.30 To a 20 mL dry Schlenk flask was added 7 (150 mg, 0.0868 mmol), (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethene (45.0 mg, 0.0868 mmol) and tri(o-tolyl)phosphine (4.3 mg, 16 mol%). After degassing and refilling argon for 3 times, a solution of tris(dibenzylideneacetone)-dipalladium (2.4 mg, 3 mol%) in dry chlorobenzene (10 mL) was added under an argon atmosphere. The flask was sealed and stirred for at 130 °C for 48 h. Four drops of bromobenzene was added and the reaction was kept at 130 °C for an additional 12 h. The reaction mixture was cooled to room temperature and added dropwise into methanol (100 mL) and the mixture was stirred for 1 h. Then the precipitated product was collected by filtration, and subjected to consecutive Soxhlet extractions with methanol, acetone, and hexane. The remaining solid was extracted with chloroform and precipitated with methanol, and filtered off to afford a black solid. Yield: 120 mg (80%).
Synthesis of PIBDF-TBT. The following synthesis was based on a modified literature procedure. To a 20 mL dry Schlenk flask was added 7 (150 mg, 0.0868 mmol), 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole (55 mg, 0.0868 mmol) and tri(o-tolyl)phosphine (4.3 mg, 16 mol%). After degassing and refilling argon for 3 times, a solution of tris(dibenzylideneacetone)-dipalladium (2.4 mg, 3 mol%) in dry chlorobenzene (10 mL) was added under an argon atmosphere. The flask was sealed and stirred for at 130 °C for 48 h. Four drops of bromobenzene was added and the reaction was kept at 130 °C for an additional 12 h. The reaction mixture was cooled to room temperature and added dropwise into methanol (100 mL) and the mixture was stirred for 1 h. Then the precipitated product was collected by filtration, and subjected to consecutive Soxhlet extractions with methanol, acetone, and hexane. The remaining solid was extracted with chloroform and precipitated with methanol, and filtered off to afford a black solid. Yield: 130 mg (80%).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There is no conflicts to declare.
Acknowledgements
T. T. Do is thankful to QUT for offering here QUTPRA scholarship to conduct her research work. B. B. Patil is thankful for Supervisor Bridging HDR Scholarship provided by QUT. We are thankful to CRC for Polymers, Central Analytical Research Facility (CARF), Institute of Future Environments, Queensland University of Technology (QUT) for providing equipment support. We are thankful to Advanced Nano Fabrication Facility (ANFF) located at University of Queensland (UQ) for providing support during Dicing of Substrates and during AFM Measurements. P. S. is thankful to QUT for the financial support from the Australian Research Council (ARC) for the Future Fellowship (FT130101337) and QUT core funding (QUT/ 322120-0301/07).
References
- O. Knopfmacher, M. L. Hammock, A. L. Appleton, G. Schwartz, J. Mei, T. Lei, J. Pei and Z. Bao, Nat. Commun., 2014, 5, 2954 CrossRef PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- Y. Q. Zheng, T. Lei, J. H. Dou, X. Xia, J. Y. Wang, C. J. Liu and J. Pei, Adv. Mater., 2016, 28, 7213–7219 CrossRef CAS PubMed.
- Z. Zhao, Z. Yin, H. Chen, L. Zheng, C. Zhu, L. Zhang, S. Tan, H. Wang, Y. Guo, Q. Tang and Y. Liu, Adv. Mater., 2017, 29, 1602410 CrossRef PubMed.
- M. Li, D. K. Mangalore, J. Zhao, J. H. Carpenter, H. Yan, H. Ade, H. Yan, K. Mullen, P. W. M. Blom, W. Pisula, D. M. de Leeuw and K. Asadi, Nat. Commun., 2018, 9, 451 CrossRef PubMed.
- B. Wang, T. P. Huynh, W. Wu, N. Hayek, T. T. Do, J. C. Cancilla, J. S. Torrecilla, M. M. Nahid, J. M. Colwell, O. M. Gazit, S. R. Puniredd, C. R. McNeill, P. Sonar and H. Haick, Adv. Mater., 2016, 28, 4012–4018 CrossRef CAS PubMed.
- W. Lv, H. Liu, W. Wang, E. Yang, H. Zhen and Q. Ling, RSC Adv., 2017, 7, 18384–18391 RSC.
- C. Soldano, R. D'Alpaos and G. Generali, ACS Photonics, 2017, 4, 800–805 CrossRef CAS.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- I. Kang, H. J. Yun, D. S. Chung, S. K. Kwon and Y. H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. Janssen and W. Li, Adv. Mater., 2016, 28, 943–950 CrossRef CAS PubMed.
- Y. Li, P. Sonar, L. Murphya and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 RSC.
- B. Sun, W. Hong, H. Aziz and Y. Li, Polym. Chem., 2015, 6, 938–945 RSC.
- J. Yang, H. Wang, J. Chen, J. Huang, Y. Jiang, J. Zhang, L. Shi, Y. Sun, Z. Wei, G. Yu, Y. Guo, S. Wang and Y. Liu, Adv. Mater., 2017, 29, 1606162 CrossRef PubMed.
- K. Guo, J. Bai, Y. Jiang, Z. Wang, Y. Sui, Y. Deng, Y. Han, H. Tian and Y. Geng, Adv. Funct. Mater., 2018, 28, 1801097 CrossRef.
- B. Kang, R. Kim, S. B. Lee, S. K. Kwon, Y. H. Kim and K. Cho, J. Am. Chem. Soc., 2016, 138, 3679–3686 CrossRef CAS PubMed.
- M. J. Sung, A. Luzio, W.-T. Park, R. Kim, E. Gann, F. Maddalena, G. Pace, Y. Xu, D. Natali, C. de Falco, L. Dang, C. R. McNeill, M. Caironi, Y.-Y. Noh and Y.-H. Kim, Adv. Funct. Mater., 2016, 26, 4984–4997 CrossRef CAS.
- Z. Chen, W. Zhang, J. Huang, D. Gao, C. Wei, Z. Lin, L. Wang and G. Yu, Macromolecules, 2017, 50, 6098–6107 CrossRef CAS.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS PubMed.
- W. Zhou, Y. Wen, L. Ma, Y. Liu and X. Zhan, Macromolecules, 2012, 45, 4115–4121 CrossRef CAS.
- J.-H. Dou, Y.-Q. Zheng, T. Lei, S.-D. Zhang, Z. Wang, W.-B. Zhang, J.-Y. Wang and J. Pei, Adv. Funct. Mater., 2014, 24, 6270–6278 CrossRef CAS.
- J. H. Dou, Y. Q. Zheng, Z. F. Yao, T. Lei, X. Shen, X. Y. Luo, Z. A. Yu, S. D. Zhang, G. Han, Z. Wang, Y. Yi, J. Y. Wang and J. Pei, Adv. Mater., 2015, 27, 8051–8055 CrossRef CAS PubMed.
- X. Zhou, N. Ai, Z.-H. Guo, F.-D. Zhuang, Y.-S. Jiang, J.-Y. Wang and J. Pei, Chem. Mater., 2015, 27, 1815–1820 CrossRef CAS.
- J. H. Dou, Y. Q. Zheng, Z. F. Yao, Z. A. Yu, T. Lei, X. Shen, X. Y. Luo, J. Sun, S. D. Zhang, Y. F. Ding, G. Han, Y. Yi, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 15947–15956 CrossRef CAS PubMed.
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Chem. Commun., 2015, 51, 13515–13518 RSC.
- Z. Yan, B. Sun and Y. Li, Chem. Commun., 2013, 49, 3790–3792 RSC.
- G. Zhang, P. Li, L. Tang, J. Ma, X. Wang, H. Lu, B. Kang, K. Cho and L. Qiu, Chem. Commun., 2014, 50, 3180–3183 RSC.
- X. Wang, H. H. Choi, G. Zhang, Y. Ding, H. Lu, K. Cho and L. Qiu, J. Mater. Chem. C, 2016, 4, 6391–6400 RSC.
- Y. He, C. Guo, B. Sun, J. Quinn and Y. Li, Polym. Chem., 2015, 6, 6689–6697 RSC.
- G. Zhang, J. Guo, M. Zhu, P. Li, H. Lu, K. Cho and L. Qiu, Polym. Chem., 2015, 6, 2531–2540 RSC.
- H. D. Pham, H. Hu, K. Feron, S. Manzhos, H. Wang, Y. M. Lam and P. Sonar, Sol. RRL, 2017, 1, 1700105 CrossRef.
- J. Eggert Carlé, J. Wenzel Andreasen, M. Jørgensen and F. Christian Krebs, Sol. Energy Mater. Sol. Cells, 2010, 94, 774–780 CrossRef.
- J. Kim and T. Swager, Nature, 2001, 411, 1030–1034 CrossRef CAS PubMed.
- N. Zhou, X. Guo, R. P. Ortiz, S. Li, S. Zhang, R. P. Chang, A. Facchetti and T. J. Marks, Adv. Mater., 2012, 24, 2242–2248 CrossRef CAS PubMed.
- J. Kim, A. R. Han, J. Hong, G. Kim, J. Lee, T. J. Shin, J. H. Oh and C. Yang, Chem. Mater., 2014, 26, 4933–4942 CrossRef CAS.
- J. Shin, H. A. Um, D. H. Lee, T. W. Lee, M. J. Cho and D. H. Choi, Polym. Chem., 2013, 4, 5688–5695 RSC.
- D. Gao, Z. Chen, J. Huang, W. Zhang, C. Wei, Z. Lin, D. Li and G. Yu, J. Mater. Chem. C, 2017, 5, 3568–3578 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-NMR and 13C-NMR spectra of all synthesized compounds, HRMS spectrum of BIBDF, GPC of polymers, UV-vis of thin films at annealing temperature of 180 °C, XRD of polymer powders and XPS spectra of polymers. See DOI: 10.1039/c8ra08890j |
| ‡ Both authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Samarendra P. Singhb,
Soniya D. Yambema,
Krishna Feroncd,
Kostya (Ken) Ostrikova,
John M. Bella and
Prashant Sonar
a,
Samarendra P. Singhb,
Soniya D. Yambema,
Krishna Feroncd,
Kostya (Ken) Ostrikova,
John M. Bella and
Prashant Sonar