DOI:
10.1039/C8RA06548A
(Paper)
RSC Adv., 2018,
8, 35395-35402
Synthesis, antitumor activity and molecular mechanism of doxorubicin conjugated trimethyl-chitosan polymeric micelle loading Beclin1 siRNA for drug-resisted bladder cancer therapy
Received
3rd August 2018
, Accepted 28th August 2018
First published on 16th October 2018
Abstract
Herein, we describe a convenient approach for the preparation of a polymeric micelle using doxorubicin (DOX) conjugated trimethyl-chitosan (TMC) with Beclin-1 siRNA (Si-Beclin1/DOX-TMC). This micelle displayed a potent capacity for autophagy inhibition and reversed drug-resistance to DOX in BIU-87/ADR cell lines. The Si-Beclin1/DOX-TMC micelle was highly cytotoxic to both drug-sensitive BIU-87 and drug-resistant BIU-87/ADR cells. Its capacity to reverse drug-resistance was dependent upon upregulation of autophagy levels in BIU-87/ADR cells. DOX was conjugated to TMC via a pH-sensitive Schiff base, which responded to the acidic lysosome microenvironment and resulted in the cytoplasmic release of DOX. The structure of DOX conjugation to the TMC polymeric micelle was characterized by NMR, GPC, TEM and DLS. DOX release profiles in different pH environment were determined by HPLC. Cellular uptake, changes to nuclei morphology and formation of autophagosomes were observed using a fluorescence microscope. Finally, in vivo antitumor activity of systemic Si-Beclin1/DOX-TMC micelle administration was evaluated in BIU-87/ADR xenograft models and Si-Beclin1/DOX-TMC micelles showed significantly suppressed tumor growth.
Introduction
Bladder cancer is one of the ten most common neoplasms worldwide, with an annual incidence of over 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases and an estimated 17000 deaths each year in the United States.1 In China, the estimated incidence and mortality due to bladder cancer are mostly due to common tumor of the urinary system.2 Urothelial carcinoma (also called transitional cell carcinoma) is the predominant histopathologic subtype of bladder cancers.3–5 Bladder urothelial carcinoma is usually sensitive to combined chemotherapy, e.g. methotrexate, vinblastine, doxorubicin (DOX) and cisplatin (MVAC), or gemcitabine plus cisplatin (GC), which are standard therapies for advanced bladder cancer.6 However, over half of bladder cancer patients are likely to have a recurrence within five year of cystoscope cutting and chemotherapy. The main cause for the bladder cancer recurrence after chemotherapy is attributed to the presence of drug-resistant urothelial carcinoma cells, however, the detailed mechanisms involved in such drug-resistance and cancer recurrence are still unclear.7–9
000 new cases and an estimated 17000 deaths each year in the United States.1 In China, the estimated incidence and mortality due to bladder cancer are mostly due to common tumor of the urinary system.2 Urothelial carcinoma (also called transitional cell carcinoma) is the predominant histopathologic subtype of bladder cancers.3–5 Bladder urothelial carcinoma is usually sensitive to combined chemotherapy, e.g. methotrexate, vinblastine, doxorubicin (DOX) and cisplatin (MVAC), or gemcitabine plus cisplatin (GC), which are standard therapies for advanced bladder cancer.6 However, over half of bladder cancer patients are likely to have a recurrence within five year of cystoscope cutting and chemotherapy. The main cause for the bladder cancer recurrence after chemotherapy is attributed to the presence of drug-resistant urothelial carcinoma cells, however, the detailed mechanisms involved in such drug-resistance and cancer recurrence are still unclear.7–9
Autophagy is an evolutionarily conserved biological process whereby intracellular organelles and compositions are encapsulated and degraded by autophagosomes and autolysosomes, respectively.10–12 Autophagy can be triggered by various extracellular signals and stresses including starvation, hypoxia, chemotherapeutic drugs, among others. Numerous studies have shown that autophagy acts as a double-edged sword that varies in different stages of tumor progression.13,14 In the carcinogenesis stage, autophagy has been shown to clean cells with harmful mutations or damaged organelles. In contrast, cancer cells within established tumors resist radiation and chemotherapy using autophagy. Regulation of autophagy-related genes by RNA interference has been reported to synergistically reverse multidrug resistance of cancer cells. Recently, Ojha et al. reported that drug-resistant bladder cancer cells have higher basal autophagic flux levels than that of sensitive ones.15–17 Chemotherapeutic treatment leads to increased levels of autophagy whereas the addition of autophagy inhibitors synergistically enhances cytotoxicity with such drugs on drug-resistant bladder cancer cells.18–22
Chitosan is a carbohydrate polymer comprised of N-glucosamine or N-acetylglucosamine. There are many reports on the applications of chitosan as drug or gene carrier because of its biocompatibility, low toxicity and versatility for chemical modifications.23 However, chitosan must be maintained in an aqueous solution of neutral or alkaline media, which remarkably limits its biomedical applications. Numerous studies have reported the preparation and biomedical properties of modified chitosan such as graft-copolymerization, alkylation or multiple alkylation, carboxymethylation, michael addition and direct conjugation to chemotherapeutics as well as peptides or proteins.24–34 Chitosan derivatives, N-quaternization modified chitosan, i.e. N,N,N-trimethyl chitosan (TMC) achieves considerable attention as a drug and gene carrier because of its physiochemical stability, biocompatibility, synthetic feasibility and controllable cationic charge as well as its water-solubility via regulation of its hydrophilic and hydrophobic groups.35–38
Herein, we synthesized covalently conjugated DOX to TMC scaffold with siRNA Beclin-1 to form a polymeric micelle. We aimed to further find an efficient method for the establishment of a carbohydrate polymer-based drug/siRNA codelivery nanocarrier with both chemotherapeutic and autophagy inhibitory functions. Our co-delivery micelle was demonstrated to be effective against drug-resistance of bladder cancer cells in vitro and in vivo by regulating the balance between autophagy and apoptosis.
Results and discussion
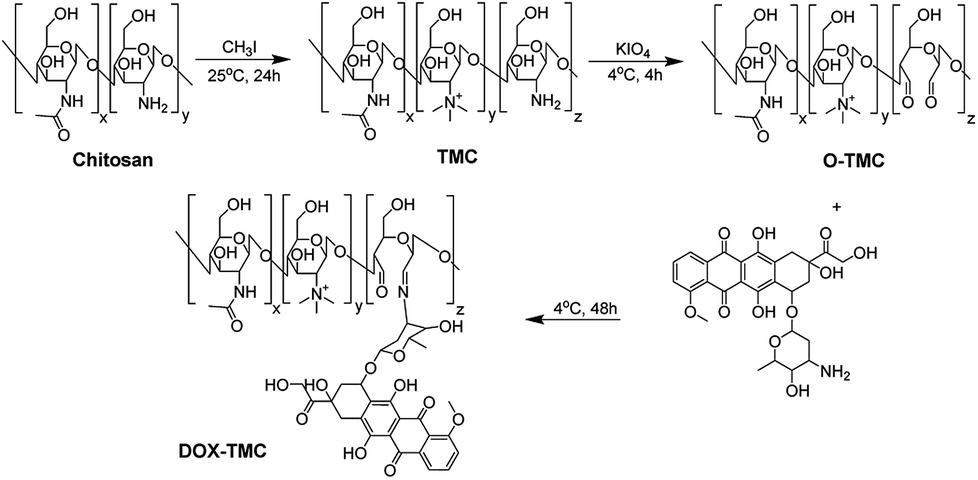
Synthesis of doxorubicin conjugated trimethyl chitosan (DOX-TMC)
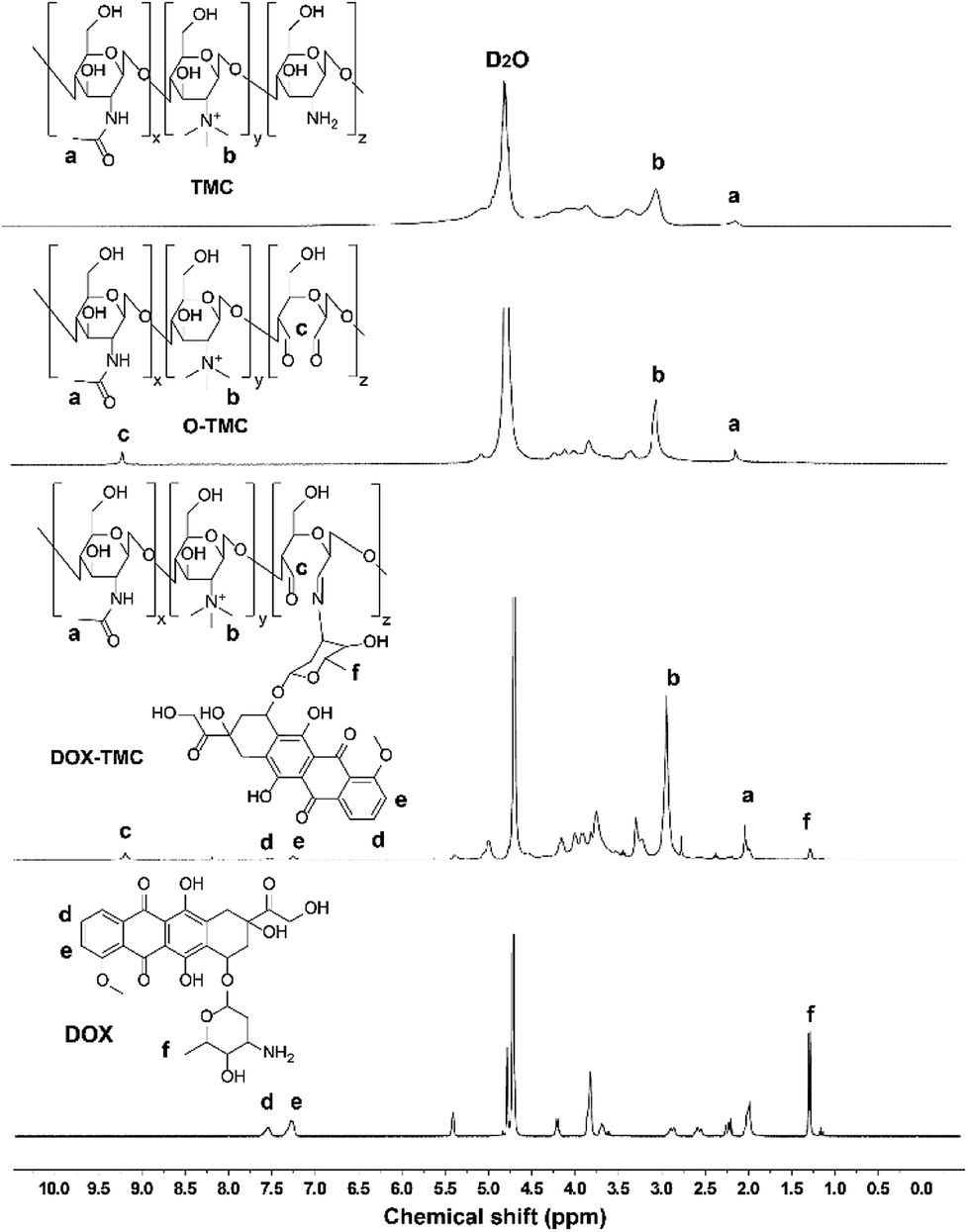
The synthetic process of DOX-TMC is shown in Fig. 1, and the molecular weight distributions of TMC and DOX-TMC were determined by GPC (gel permeation chromatography). As shown in Table 1, Mn, Mw and polydispersity index (PDI = Mw/Mn) of DOX-TMC were 15 kDa, 22 kDa and 1.47, respectively. These results positively correlated to the molecular weight distribution of corresponding TMC or O-TMC. First, certain amine groups of the aminoglucose fragments in chitosan were quaternized by CH3I to form TMC. Then, aminoglucose fragments were oxidized to form dialdehyde by KIO4. DOX-TMC were synthesized by forming the Schiff's base between the amino groups of DOX and the aldehyde group of oxidized TMC. The chemical structure of DOX-TMC was confirmed by 1H NMR (Fig. 2). As reported in previous studies, a single peak at 2.1 ppm was attributed to the hydrogen of acetyl group within TMC; signals at about 2.9 ppm were attributed to the methyl hydrogen of the trimethylamine group within TMC. Additionally, the peaks at about 7.3 and 7.5 ppm were attributed to the aromatic hydrogens of the anthracene group within DOX. The broad single peak around 9.1 ppm was attributed to the aldehyde hydrogen of O-TMC and DOX-TMC. The multiple peaks between 2.8 and 4.0 were attributed to the oligosaccharide on TMC and DOX, respectively.
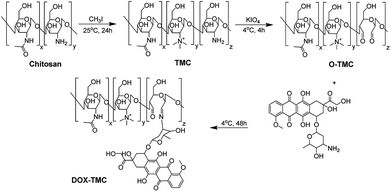 |
| | Fig. 1 Synthesis route of DOX-TMC. | |
Table 1 Molecular weights of various TMC derivatives
| |
Mn (kDa) |
Mw (kDa) |
PDI (Mw/Mn) |
| TMC |
29 |
51 |
1.75 |
| O-TMC |
11 |
17 |
1.54 |
| DOX-TMC |
15 |
22 |
1.47 |
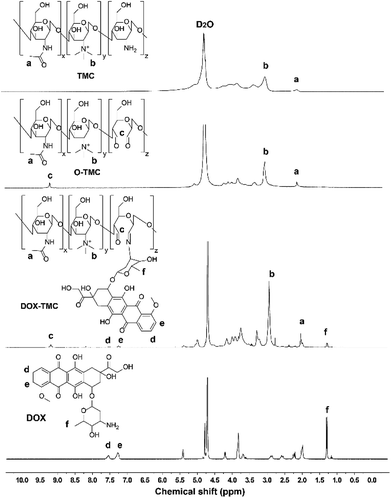 |
| | Fig. 2 1H NMR spectra of TMC, O-TMC, DOX and DOX-TMC. | |
Preparation and characterization of siRNA Beclin-1-loaded DOX-TMC polymeric micelle
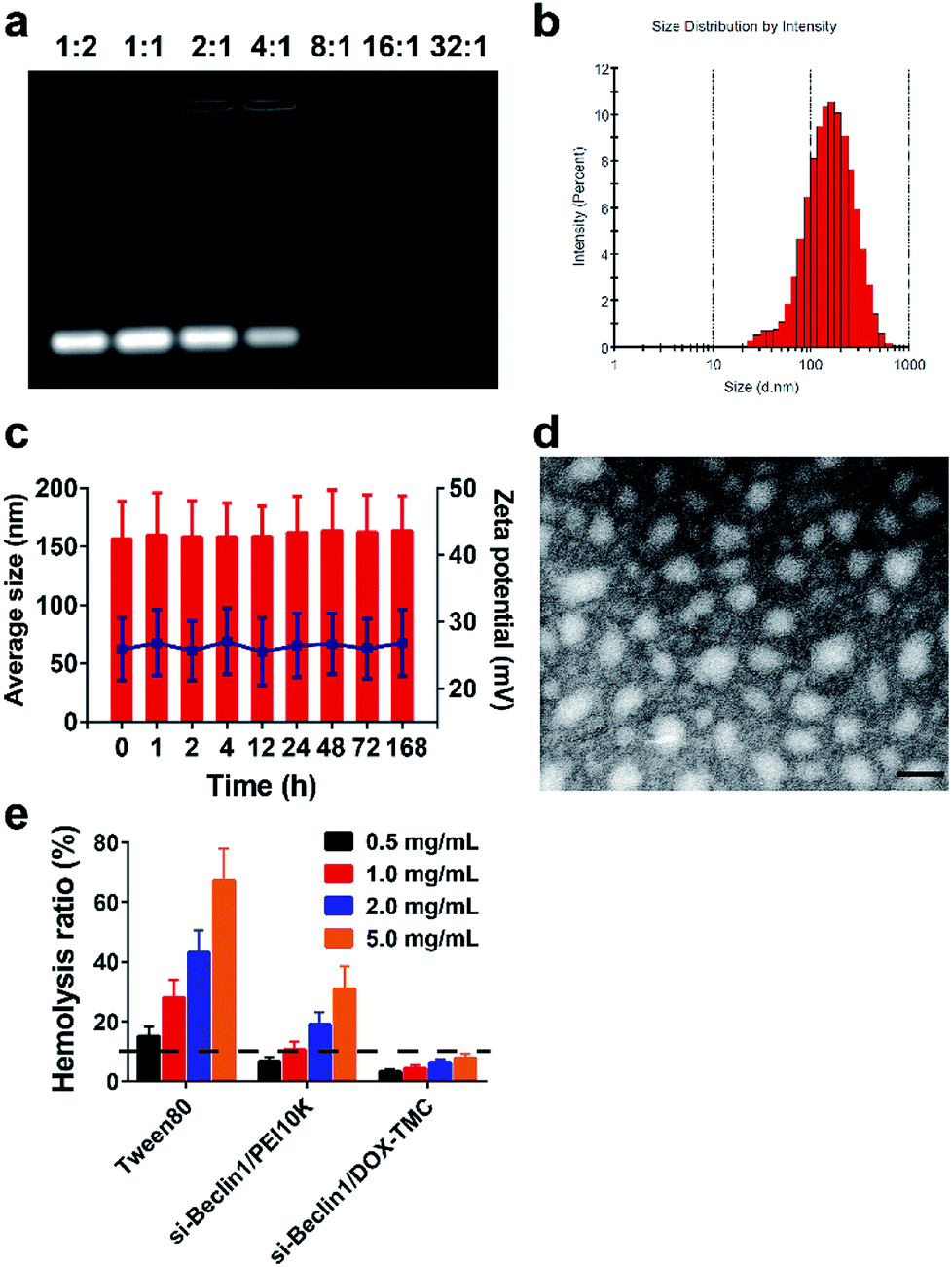
SiRNA Beclin-1-loaded DOX-TMC polymeric micelles were formed by electrostatic interactions between the positively charged TMC backbone and negatively charged siRNA. Fluctuations in particle size as well as the zeta potential of the micelles were plotted against the weight ratios of DOX-TMC/siRNA in Fig. 3. SiRNA-loaded DOX-TMC polymeric micelle particle size increased with the ascending N/P ratio from 0.5 to 16 in the range of 110 to 450 nm. At an N/P ratio of 8.0, the micelles had the lowest mean particle size at 112.3 nm. When the N/P ratio increased from 0.5 to 16.0, the zeta potential increased correspondingly, however, the zeta potential did not rise considerably when the N/P ratio was above 4.0.
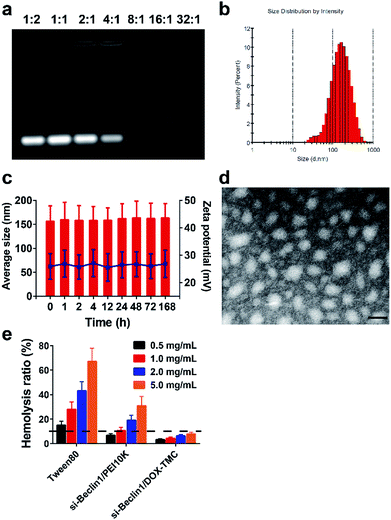 |
| | Fig. 3 Characterization of siRNA loaded DOX-TMC micelles. (a) Agarose gel electrophoresis of siRNA loaded by DOX-TMC; (b) the size distribution of siRNA loaded DOX-TMC micelles; (c) stability of siRNA loaded DOX-TMC micelles in 10% serum; (d) transmission electron microscopy images of siRNA loaded DOX-TMC micelles (scale bar indicates 100 nM); (e) hemolysis test results different concentrations of siRNA loaded DOX-TMC micelles after incubation with red blood cells suspension for 3 h. | |
Average particle size, size distribution and zeta potential of siRNA-loaded DOX-TMC polymeric micelles were determined by DLS (Dynamic Lighting Scattering) method using a Malvern ZS90 Zetasizer (Malvern Instruments Ltd., Malvern, UK) at room temperature. As shown in Fig. 3, mean micelle size was 152.3 ± 30.4 nm, with a PDI value of 0.237 ± 0.19 (Fig. 3b), and a mean zeta potential of about 28.6 ± 5.0 mV. The siRNA-loaded DOX-TMC polymeric micelle particle size was around 150 nm with a relatively narrow distribution, which provided the potential for accumulation at tumor sites via the EPR effect. Furthermore, the size distribution and zeta potentials of siRNA loaded DOX-TMC micelles displayed little change with incubation of 10% serum, suggesting the serum stability of DOX-TMC micelles (Fig. 3c). The morphology of siRNA-loaded DOX-TMC micelles were visualized with TEM. These micelles displayed a generally homogenous and nearly spherical morphology (Fig. 3d). The hemolysis tests also suggested the good biocompatibility of DOX-TMC micelles. There were less than 5% hemolysis caused by DOX-TMC micelles even at a concentration of 5.0 mg mL−1, which were far less than that of Tween80 or PEI10K (Fig. 3e).
In vitro evaluation of siRNA-loaded DOX-TMC polymeric micelles
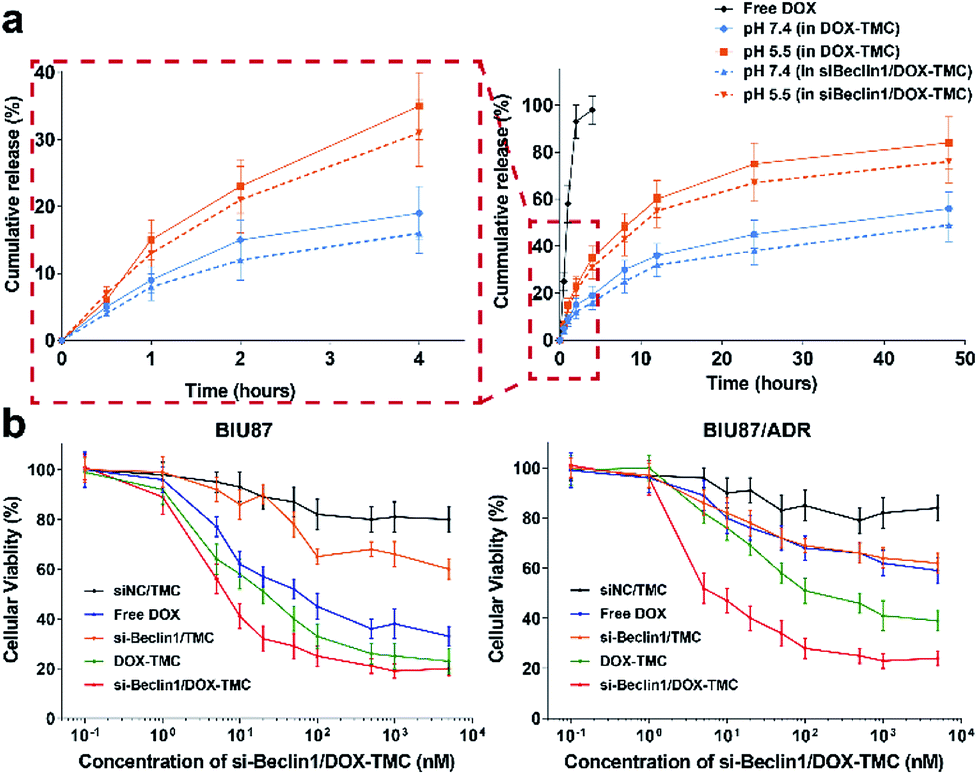
The release profiles of DOX from micelles behaved in a pH-sensitive manner. Over 85% of free DOX molecules were released within 2 hours and almost all were released within 4 hours. In contrast, only about 40% of total DOX as part of DOX-TMC micelles were released at pH 7.4 and around 70% at pH 5.5 (Fig. 4a), regardless of whether siRNA Beclin-1 was loaded into the micelles. These results suggest that siRNA-loaded DOX-TMC micelles could release DOX faster in an acidic microenvironment. In cytotoxicity assays, treating drug sensitive BIU-87 cells and resistant BIU87/ADR cells with blank TMC only caused marginal cytotoxicity (Fig. 4b), while treating these cells with siRNA Beclin-1 complex and TMC only slightly inhibited proliferation. Moreover, both free DOX and DOX-TMC displayed distinct cytotoxicity in BIU-87 cells but had nominal inhibitory effect on proliferation in BIU-87/ADR cells. Compared to free DOX, DOX-TMC caused higher cytotoxicity in BIU-87 cells, probably because the conjugation ability of TMC promoted cellular uptake. Finally, siRNA Beclin-1-loaded DOX-TMC micelles were observed to significantly inhibit cell proliferation in both BIU-87 and BIU-87/ADR cells.
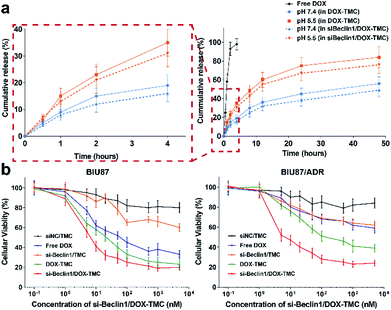 |
| | Fig. 4 (a) The accumulative release profiles of doxorubicin from DOX-TMC and si-Beclin1/DOX-TMC in different pH environment; (b) cell viability curves of sensitive BIU87 cells and resistant BIU87/ADR cells incubated with siNC/TMC, si-Beclin1/TMC, free DOX, DOX-TMC and si-Beclin1/DOX-TMC, respectively. | |
Intracellular distribution, autophagy and apoptosis induced by siRNA-loaded DOX-TMC polymeric micelle
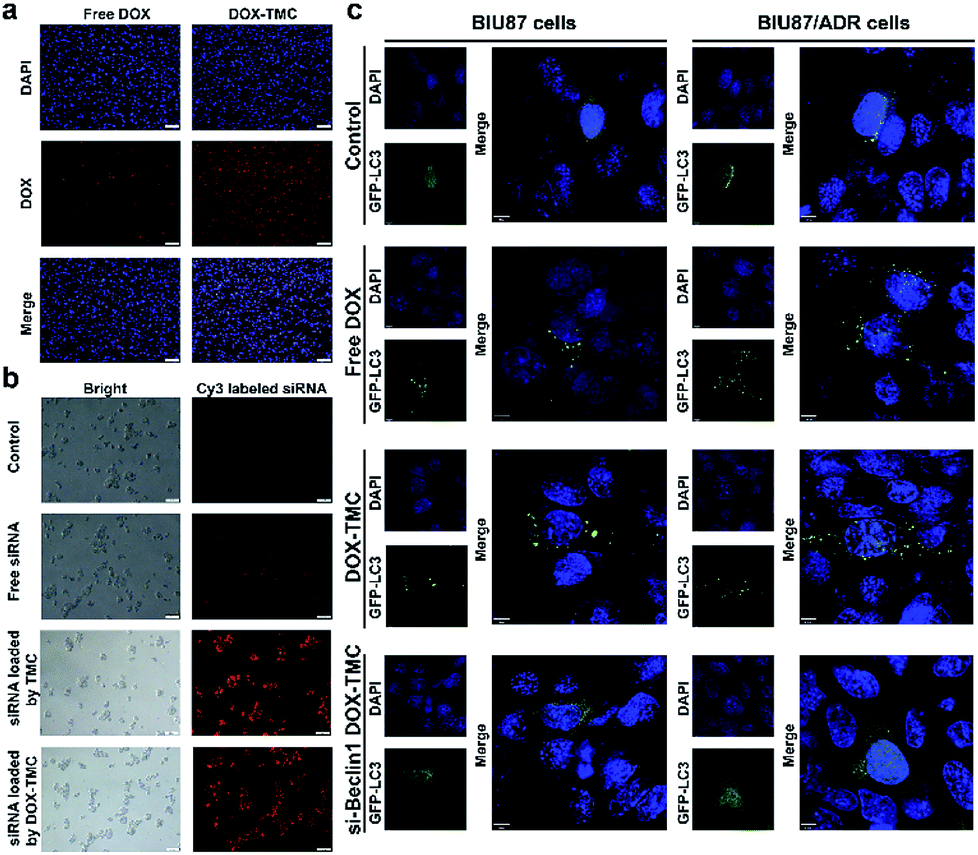
The cellular uptake and intracellular distribution of free DOX, DOX-TMC and FTIC-siRNA-loaded DOX-TMC micelles in BIU-87/ADR cells were detected with a scanning confocal microscope. Both DOX and FITC-siRNA in siRNA-loaded DOX-TMC micelles were mostly taken up by free DOX or FITC-siRNA, respectively (Fig. 5a and b). Quantitative analysis indicated that the fluorescence intensity of FITC-siRNA, when delivered in DOX-TMC micelles, was 7.6-fold higher than when delivered as free siRNA. Fluorescence intensity of DOX when delivered in siRNA-loaded DOX-TMC micelles was 3.9-fold higher than when delivered as free DOX. Intracellular distribution analysis of FITC-siRNA and DOX in different groups demonstrated that siRNA was mainly localized to the cytoplasm and DOX was generally distributed in both the cytoplasm and nuclei.
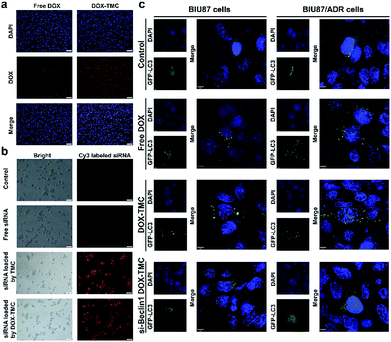 |
| | Fig. 5 (a) The cellular uptake of doxorubicin of free DOX or DOX-TMC by incubated with BIU87/ADR cells for 24 h (scale bar indicates 200 μM); (b) cellular uptake of Cy3-labeled siRNA of free siRNA, complexed by TMC or complexed by DOX-TMC after incubation with BIU87/ADR cells for 24 h (scale bar indicates 20 μM); (c) observation of GFP-LC3 puncta stimulated by free DOX, DOX-TMC and si-Beclin1/DOX-TMC in BIU87 or BIU87/ADR cells (scale bar indicates 6 μM). | |
Basal autophagy levels of BIU-87 and BIU-87/ADR cells were detected after transfection with the GFP-CL3 plasmid. As shown in Fig. 5c, the average puncta in BIU-87/ADR cells were remarkably higher than that of BIU-87 cells. With free DOX treatment, the average autophagy levels in both BIU-87 and BIU-87/ADR cells were significantly boosted. As expected, the GFP-LC3 puncta induced by DOX treatment in BIU-87/ADR cells were significantly more than that of BIU-87 cells.
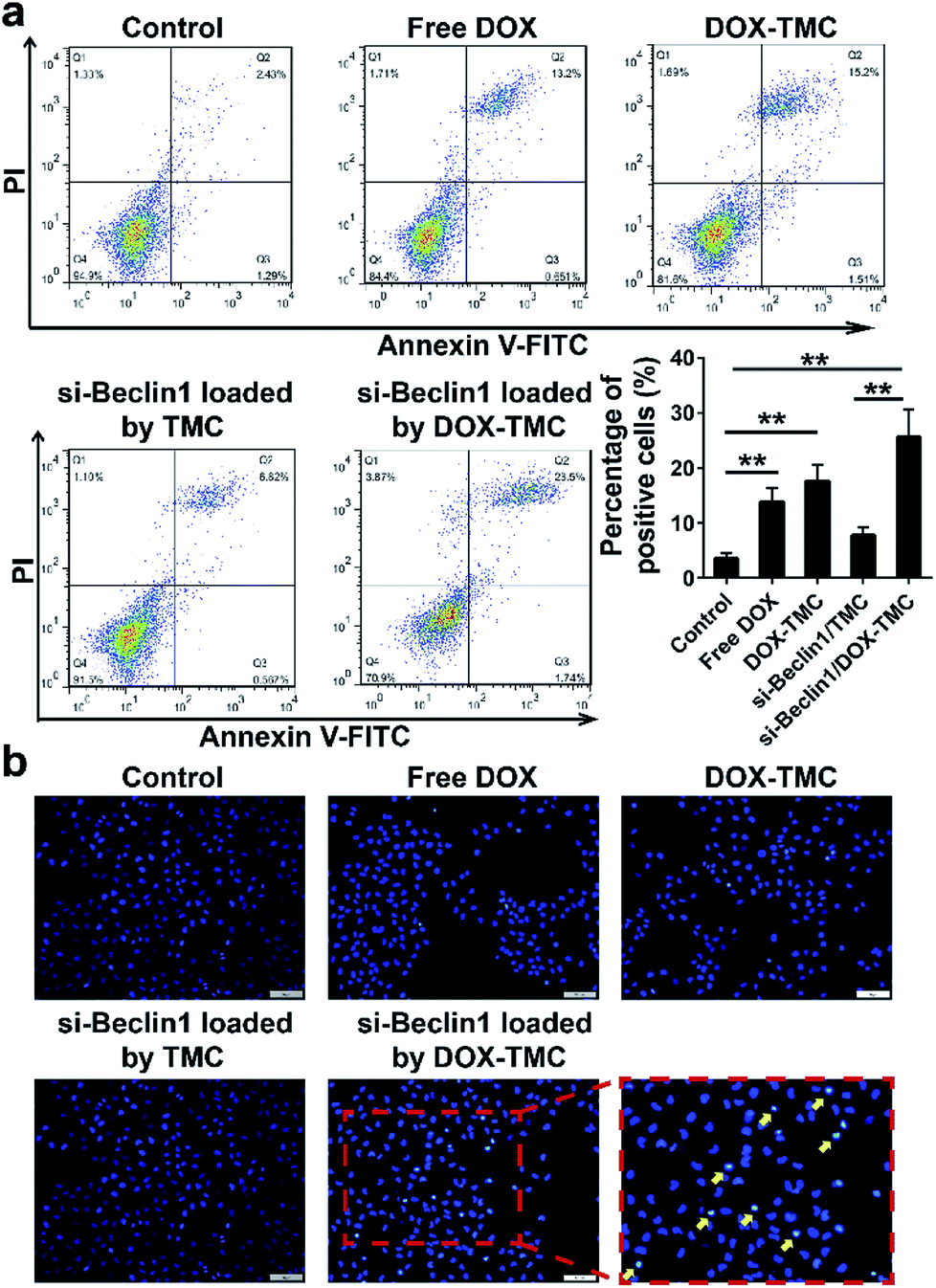
In order to suppress the drug resistance related autophagy in BIU-87/ADR cells, siRNA Beclin-1 was delivered using TMC or DOX-TMC as non-viral carriers. As shown in Fig. 5c, both basal autophagy and DOX-induced autophagy were remarkably declined. For further study of the relationship between autophagy and apoptosis in drug-resistant bladder cancer cells, apoptosis by BIU-87/ADR cells induced by free DOX, siRNA Beclin-1, DOX-TMC and siRNA Beclin-1-loaded DOX-TMC were analysed by flow cytometry with Annexin V/PI dual staining and nuclei morphological changes with Hoechst-33258 staining (Fig. 6). We observed that si-Beclin1/TMC, free DOX and DOX-TMC only induced marginal to moderate apoptosis in BIU-87/ADR cells, respectively. The siRNA Beclin-1-loaded DOX-TMC notably boosted apoptosis in drug-resistant BIU-87/ADR cells. These results suggested that the addition of siRNA Beclin-1 may overcome the autophagy-related resistance of BIU-87/ADR cells and synergistically enhance the cytotoxicity of DOX. Beclin-1 is one of the most important subunits of the PI3K complex III (PI3KC3), which plays a vital role in the initiation of the autophagosome. Deficiency in Beclin-1 directly inhibits the formation of autophagosome, which reverses the autophagy-dependent drug resistance of BIU-87/ADR cells.
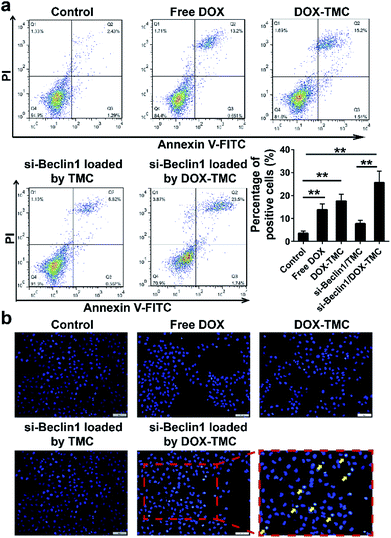 |
| | Fig. 6 The apoptosis assays of BIU87/ADR cells treated by free DOX, DOX-TMC, si-Beclin1/TMC and si-Beclin1/DOX-TMC, respectively; (a) flow cytometry assay stained by Annexin V/PI dual-staining; (b) the nuclei morphological changes stained by Hochest33258 (scale bar indicates 50 μM; ** p < 0.01). | |
siRNA Beclin-1 synergistically suppresses proliferation and reverses drug-resistance in BIU-87/ADR cells by inhibiting autophagy and inducing mitochondrial apoptosis
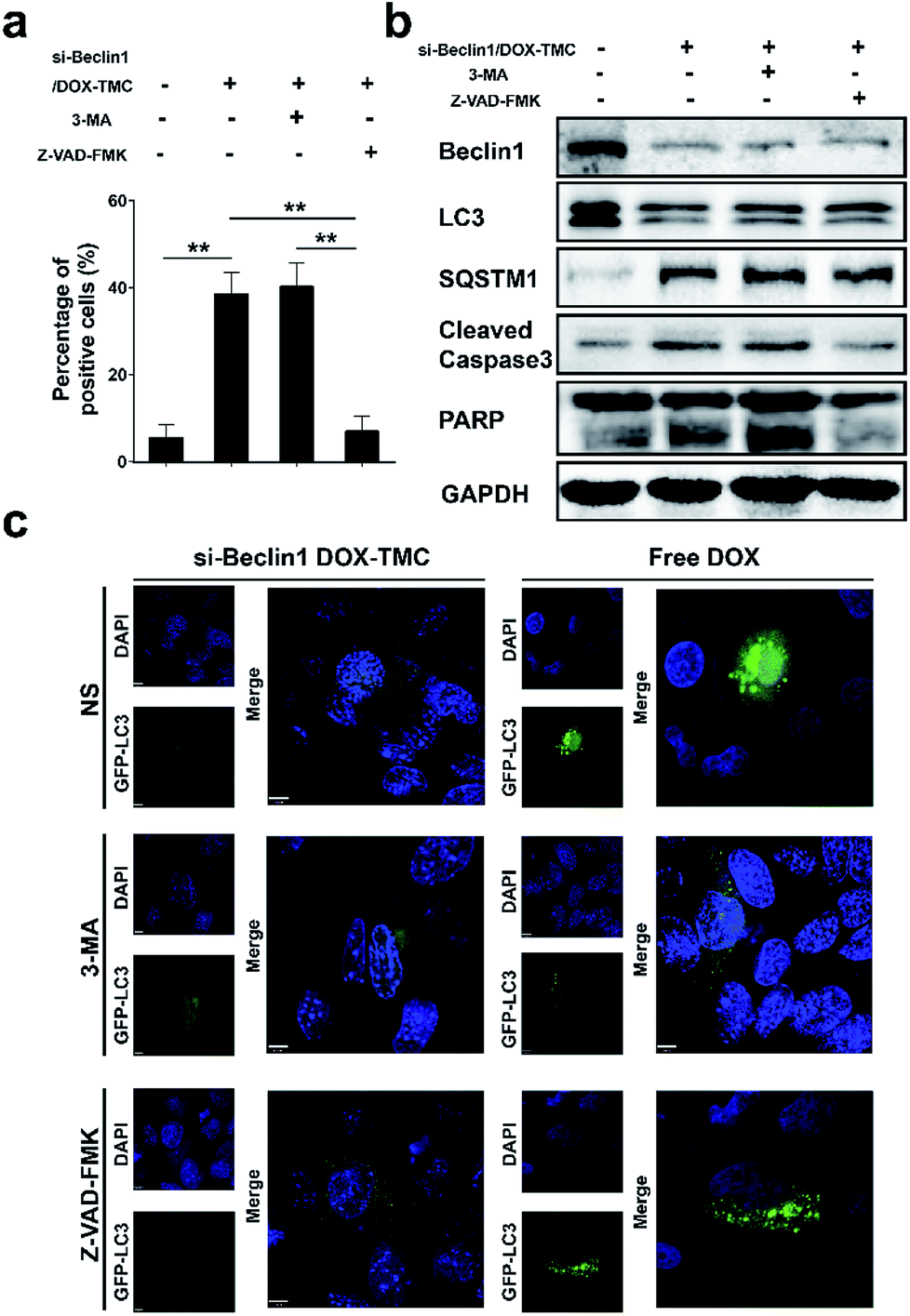
To identify the mechanism by which the autophagy and apoptosis and their interactions via siBeclin-1/DOX-TMC are responsible for its anti-cancer effects, we treated BIU-87/ADR cell lines with siBeclin-1/DOX-TMC alone or siBeclin1/DOX-TMC together with the autophagy inhibitor 3-MA or apoptosis inhibitor Z-VAD-FMK, and then assessed the cell proliferation ratio by MTT assays and apoptosis level with Annexin V-FITC/PI staining followed by flow cytometry. The apoptotic BIU-87/ADR cells induced by siBeclin-1/DOX-TMC could be reversed by 3-MA or Z-VAD-FMK (Fig. 7a and b), respectively. Similarly, western blotting showed that both 3-MA and Z-VAD-FMK suppressed the cleavage of caspase-9 and PARP induced by siBeclin1/DOX-TMC, but the siBeclin-1/DOX-TMC promoted SQSTM1 degradation was only reversed by 3-MA rather than Z-VAD-FMK (Fig. 7b). These results suggested that siBeclin-1/DOX-TMC induced BIU-87/ADR cell apoptosis depended on inhibition of protective autophagy.
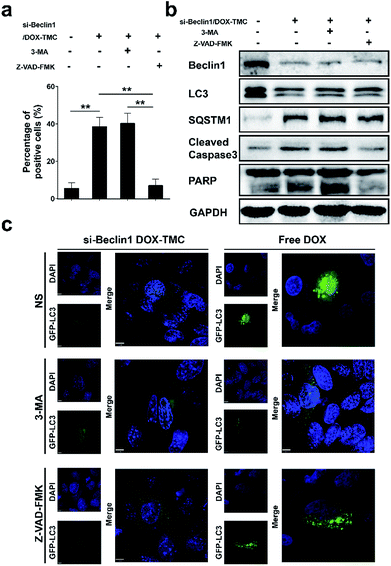 |
| | Fig. 7 (a) The changes on total apoptosis levels following si-Beclin1/DOX-TMC combined with autophagy inhibitor 3-MA or apoptosis inhibitor Z-VAD-FMK; (b) the changes on autophagy or apoptosis related proteins following si-Beclin1/DOX-TMC combined with 3-MA or Z-VAD-FMK; (c) the changes on autophagy levels followed by si-Beclin1/DOX-TMC or free DOX combined with 3-MA or Z-VAD-FMK (scale bar indicates 6 μM; ** p < 0.01). | |
siBeclin-1/DOX-TMC polymeric micelle suppresses drug-resistant bladder cancer cells in xenograft models
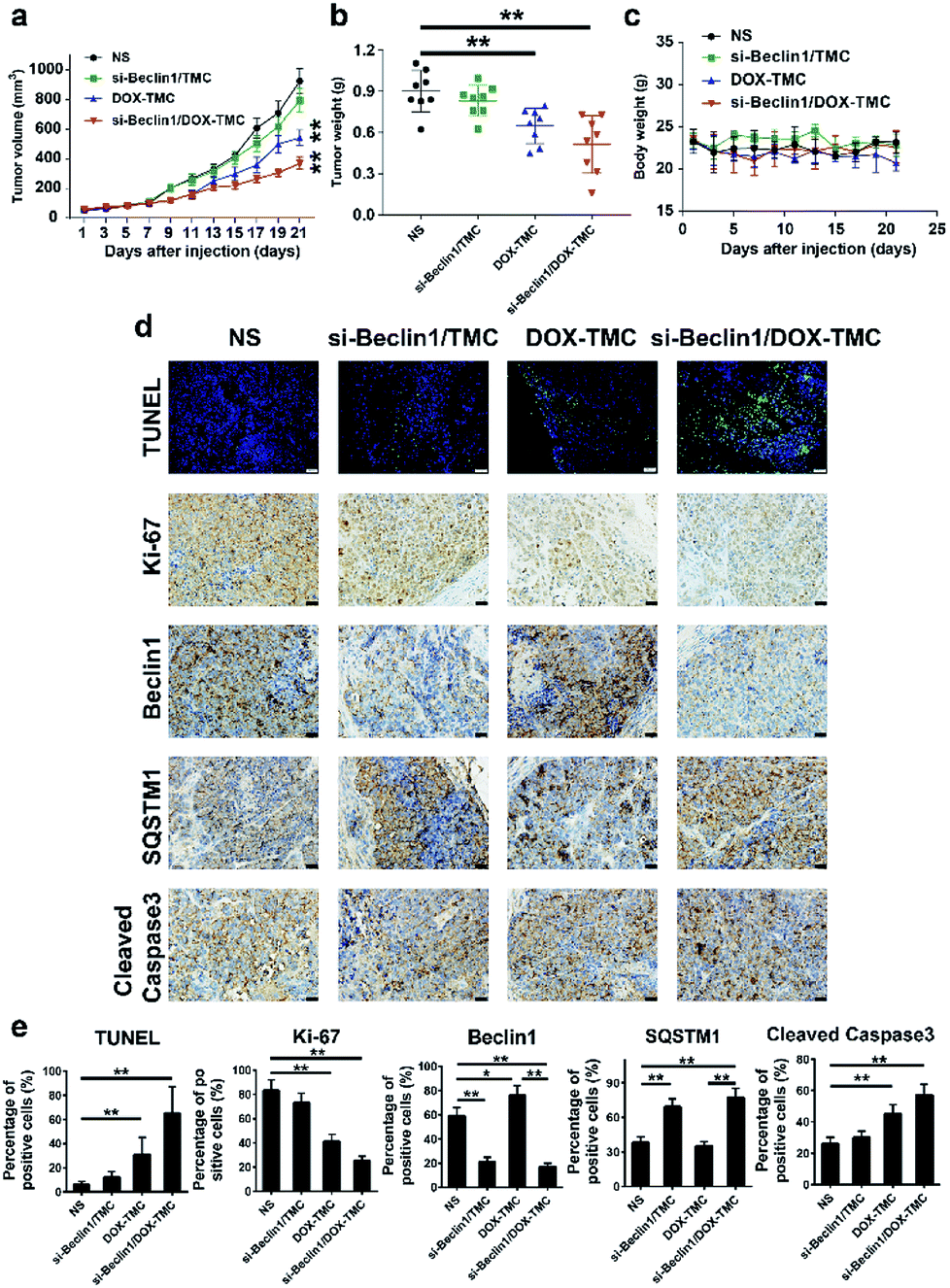
In vivo anti-tumor effects of siBeclin-1/DOX-TMC micelles and its potential molecular mechanisms were validated in a BIU-87/ADR bladder cancer xenograft model. The siBeclin-1/DOX-TMC micelles displayed a higher tumor inhibition capacity than free DOX, DOX-TMC and siBeclin-1 lipoplex (Fig. 8a). The greatest tumor proliferation ratio (T/C) of the siBeclin-1/DOX-TMC micelles was 36.7%. The results based on tumor volume changes were also confirmed by evaluation of excised tumor weights (Fig. 8b). The IHC (immunohistochemistry) analysis demonstrated that both siBeclin-1/DOX-TMC micelles and siBeclin-1 lipoplex knocked down the expression of Beclin-1 and up-regulated SQSTM1 expression, but the apoptosis marker cleaved caspase-3 was only activated in siBeclin-1/DOX-TMC micelles group (Fig. 8c–e). Treatment of tumor-bearing mice with free DOX or DOX-TMC only showed marginal therapeutic effect, partially because of the protective autophagy induced by DOX in BIU-87/ADR cells. We also assessed the changes of apoptosis markers TUNEL and proliferation marker Ki-67 in all groups (Fig. 8e), they were consistent with measurements of tumor volume and weight. These results indicate that DOX induced protective autophagy either as free drug or conjugated form, which could be reversed by co-delivery with siBeclin-1. In brief, siBeclin-1 suppressed protective autophagy in BIU-87/ADR cells and reversed their drug resistance. These in vitro and in vivo results may better describe the mechanism by which the combination of siBeclin-1 and DOX-TMC achieves the synergistic anti-tumor activity.
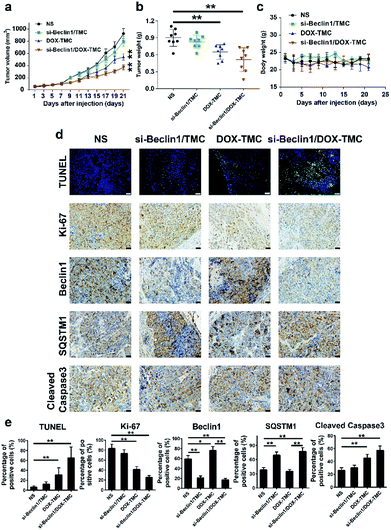 |
| | Fig. 8 Antitumor effects of si-Beclin1/DOX-TMC in vivo, compared to effects observed with NS, si-Beclin1/TMC and DOX-TMC. (a) curves of tumor volumes in each group; (b) statistics of tumor weight in each group; (c) curves of body weight changes; (d) immunohistological staining against TUNEL (scale bar indicates 50 μM), Ki67, Beclin1, SQSTM1 and cleaved caspase3 (scale bar indicates 20 μM); (e) quantification of each marker in panel C (*p < 0.05 to NS group, **p < 0.01 to NS group, respectively). | |
Experimental
Materials and regents
Chitosan (with the acetylation degree of 10%) was purchased from Sigma-Aldrich Chemical Co. (Burlington, MA, USA). Doxorubicin hydrochloride was purchased from Energy Chem. Co. (Shanghai, China). The other chemicals obtained were of analytical grade and directly used. BIU87 and BIU87/ADR cells were obtained from National Infrastructure of Cell Line Resource (Wuhan, China) and maintained in Dulbecco's Modified Eagle's Medium containing 10% fetal bovine serum and 1% penicillin–streptomycin (Invitrogen, Waltham, USA) in 5% CO2 at 37 °C.
Synthesis and characterization of DOX-TMC
The experimental procedures to synthesize O-methylation free TMC is depicted in Fig. 1 and is according to the method reported by Muzzarelli, Verheul and our previous studies, with some modifications.35,39,40 In brief, chitosan was suspended in the mixture of formic acid and 37% formaldehyde; after refluxing for 12 hours the solvent was removed by vacuum distillation. The residue was neutralized by 1 mol L−1 NaOH solution, then filtered and washed by deionized water. The iodomethane and NMP (N-methylpiperidine) were added and the reaction mixture was kept at room temperature for 24 hours. The reaction mixture was poured into ethanol/diethyl ether (v/v = 50:50), and the precipitated TMC was collected by centrifugation and washed twice by diethyl ether. The TMC was oxidized according to the procedure reported by Vold et al. and Bao et al. with few modifications.41,42 The potassium periodate was added into acetate buffer solution of TMC under nitrogen atmosphere at 4 °C. The reaction was performed for 4 hours at 4 °C and quenched by 10% ethylene glycol. The oxidative TMC (O-TMC) was purified by dialysis (MWCO 3500) and then lyophilization. DOX-TMC was prepared by coupling the aldehyde groups of O-TMC with the primary amine groups of doxorubicin. The TMC dissolved in deionized water was added into aqueous solution of doxorubicin, the reaction mixture was kept at 4 °C for 48 h in the dark. The crude product was purified by dialysis to remove unreacted doxorubicin and then lyophilized to obtain the final product DOX-TMC.
The chemical structures of TMC, O-TMC and DOX-TMC were determined by 1H NMR at 400 MHz (AvanceIII, Bruker, Germany) by using D2O as solvent. The relative molecular weight and molecular weight distributions of TMC, O-TMC and DOX-TMC were determined by GPC method (gel permeation chromatography, USA) by a Waters 510 instrument and Waters 2410 refractive index detector.
Preparation and characterization of siRNA/DOX-TMC complexes
The siRNA/DOX-TMC complexes were prepared in various w/w (weight/weight) ratios of DOX-TMC to siRNA. In brief, both siRNA and DOX-TMC were dissolved in HEPES buffer (20 mM, pH 7.4), the DOX-TMC stock solutions were added to the siRNA solution with expected proportions, the siRNA/DOX-TMC complexes were prepared by vortex-mixing and self-assembly. The hydrodynamic diameter and zeta potential of siRNA/DOX-TMC micelles were measured using dynamic light scattering (DLS) (Zetasizer Nano ZS90, Malvern, UK) at room temperature while the morphology of siRNA/DOX-TMC micelles was further analysed by TEM (Hitachi, JEM-2000, Japan). The RNA binding capacity of DOX-TMC was analysed by gel electrophoresis, as described in our previous reports.43 The haemolysis ability of DOX-TMC was evaluated by the method described in previously published procedure.18 The DTX release profiles from siRNA/DOX-TMC micelles were investigated using dialysis method as described before.35
In vitro cellular toxicity analysis
The cell viability assay was performed by a MTT assay; BIU87 cells and BIU87/ADR cells were plated in 1 × 104 cells per well into 96-well plates and incubated overnight at 37 °C. Then, the cells were treated with free drugs or micelles at various concentrations for 24 h. The MTT assay was measured at 570 nm by a Thermo Fisher Multiskan MK3 microplate reader (Thermo Fisher Scientific, MA, USA).
In vitro intracellular uptake and distribution
To observe the intracellular uptake and distribution of micelles, BIU87/ADR cells were plated onto a glass Petri dish in 1 × 104 cells per cm2 overnight. Subsequently, cells were incubated with culture medium containing free DOX, DOX-TMC, free cy3-siRNA, cy3-siRNA/TMC and cy3-siRNA/DOX-TMC (DOX 10 ng mL−1, cy3-siRNA 100 nM). After incubating for another 2 hours, cells were rinsed with PBS, and then fixed with 4% paraformaldehyde. The nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) and observed by fluorescence microscopy (Olympus FV-1000, Olympus, Japan).
Apoptosis and autophagy assays
The cell apoptosis-inducing effects of micelles in BIU87/ADR cells were determined using flow cytometry. BIU87/ADR cells were seeded in six-well plates under 5% CO2 at 37 °C, and then co-cultured with free DOX, DOX-TMC, siBeclin-1/TMC and siBeclin-1/DOX-TMC, respectively (10 nM DOX and 100 nM siBeclin-1) at desired pH value for 24 h. Cells were stained with Annexin V-APC and propidium iodide (PI), then analyzed by flow cytometry. Untreated cells were used as negative controls. For autophagy assay, BIU87 and BIU87/ADR cells were transfected with GFP-LC3 plasmid, then added with free DTX, free DOX, DOX-TMC and siBeclin-1/DOX-TMC, respectively for 48 h. The formation of autophagosome puncta were observed by confocal laser scanning microscopy (CLSM) (Leica TCS SP2, Lecia microsystems, German). Untreated groups were used as negative controls.
Western blot analysis
BIU87/ADR cells were plated into six-well plates and incubated overnight, then treated with siBeclin1/DOX-TMC with or without 3-MA, Z-VAD-FMK. The cells were harvested and re-suspended on ice for 10 min with RIPA lysis buffer, followed by centrifugation for 5 min. The following steps for western blot analysis is described in our previous reports.35
Xenograft models and immunohistochemistry analysis
The in vivo antitumor activity of siBeclin1/DOX-TMC were performed according to the Guidelines for the Care and Use of Laboratory Animals (National Standard of the P. R. China, no. GB/T 35982-2018), and study procedures were approved by the Committee on Ethics of Animal Experimentation of West China Hospital of Sichuan University. The BIU87/ADR tumor-bearing mice were obtained by subcutaneous injection of BIU87/ADR cells (6 × 105 cells per mice) into the right forelimb region of male BALB/C nude mice. When the tumors grew to about 100 mm3 in volume, the mice were randomly divided into four groups with eight mice in each group as follows: NS, free DOX, DOX-TMC and siBeclin1/DOX-TMC. The mice were intravenously injected at the equivalent dose of 5 mg kg−1 DTX and 5 mg kg−1 siBeclin1. The tumor volume and body weight of mice were monitored every two days for 21 days. After the mice were euthanized, a 4 μm section was cut from tumor tissues. All sections were fixed in 4% paraformaldehyde overnight and subjected to paraffin embedding. Immunostaining for Ki-67, Beclin1, SQSTM1 and cleaved caspase3 were performed according to standard protocols. The primary antibody was an anti-mouse monoclonal antibody against Ki-67, Beclin1, SQSTM1 and cleaved caspase3, respectively. The sections were incubated with the primary antibody at RT for 2–4 hours and then washed with the secondary antibody for 30 min, the reaction was finally visualized using a VECTASTAIN ABC Kit and detected with diaminobenzidine (DAB) as the chromogen. The immunofluorescent staining of TUNEL were performed according to our previously reported method.35
Conclusions
In the current study, we report a convenient approach to prepare a polymeric micelle based on doxorubicin conjugated trimethyl-chitosan with Beclin-1 siRNA. The prepared Si-Beclin1/DOX-TMC micelle silences the Beclin-1 protein expression to inhibit protective autophagy of BIU-87/ADR cells, while simultaneously inducing apoptosis. Our study presents a novel strategy to overcome chemotherapy resistance by nanomedicines, in which regulating the autophagy and apoptosis are integrated by rational design and preparation of multi-functional biocompatible carriers of siRNA and drug co-delivery. These results indicate that co-delivery of Beclin-1 siRNA and DOX by a biocompatible carbohydrate based nano-carrier can effectively suppress drug-resistant bladder cancer in preclinical studies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (81672552 and 81603065), the Science & Technology Department of Sichuan Province (2017JY0226) and the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZY2016104).
Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, CA, 2018, 68, 7–30 Search PubMed
 .
. - W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, A. Jemal, X. Q. Yu and J. He, CA, 2016, 66, 115–132 Search PubMed .
- Y. Du, L. Wang, W. Wang, T. Guo, M. Zhang, P. Zhang, Y. Zhang, K. Wu, A. Li, X. Wang, J. He and J. Fan, J. Biomed. Nanotechnol., 2018, 14, 161–167 CrossRef CAS PubMed .
- S. Antoni, J. Ferlay, I. Soerjomataram, A. Znaor, A. Jemal and F. Bray, Eur. Urol., 2017, 71, 96–108 CrossRef PubMed .
- S. Chavan, F. Bray, J. Lortet-Tieulent, M. Goodman and A. Jemal, Eur. Urol., 2014, 66, 59–73 CrossRef PubMed .
- S. K. Sweeney, Y. Luo, M. A. O'Donnell and J. G. Assouline, J. Biomed. Nanotechnol., 2017, 13, 232–242 CrossRef CAS PubMed .
- M. Katoh and H. Nakagama, Med. Res. Rev., 2014, 34, 280–300 CrossRef CAS PubMed .
- M. A. Tabrizi, P. G. Baraldi, S. Baraldi, S. Gessi, S. Merighi and P. A. Borea, Med. Res. Rev., 2017, 37, 936–983 CrossRef PubMed .
- J. W. Tessmann, J. Buss, K. R. Begnini, L. M. Berneira, F. R. Paula, C. M. P. de Pereira, T. Collares and F. K. Seixas, Biomed. Pharmacother., 2017, 94, 37–46 CrossRef CAS PubMed .
- Z. P. Pan, Y. J. Chen, J. Y. Liu, Q. L. Jiang, S. Y. Yang, L. Guo and G. He, Eur. J. Med. Chem., 2018, 144, 517–528 CrossRef CAS PubMed .
- N. Zhang, Y. Huang, F. B. Wu, Y. B. Zhao, X. Li, P. F. Shen, L. Yang, Y. Luo, L. Yang and G. He, Mol. Pharmaceutics, 2016, 13, 2466–2483 CrossRef CAS PubMed .
- B. W. Ke, M. Tian, J. J. Li, B. Liu and G. He, Med. Res. Rev., 2016, 36, 983–1035 CrossRef PubMed .
- K. A. Bauckman and I. U. Mysorekar, Autophagy, 2016, 12, 850–863 CrossRef CAS PubMed .
- Y. G. Liang, J. L. Zhu, H. S. Huang, D. M. Xiang, Y. Li, D. Y. Zhang, J. X. Li, Y. L. Wang, H. L. Jin, G. S. Jiang, Z. Y. Liu and C. S. Huang, Autophagy, 2016, 12, 1229–1239 CrossRef CAS PubMed .
- M. Ishaq, R. Ojha, K. Sharma, G. Sharma, S. K. Singh and S. Majumdar, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2560–2573 CrossRef CAS PubMed .
- R. Ojha, V. Jha and S. K. Singh, Bba-Mol Cell Res, 2016, 1863, 347–359 CrossRef CAS PubMed .
- R. Ojha, S. K. Singh and S. Bhattacharyya, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 2484–2497 CrossRef CAS PubMed .
- L. L. Chen, M. Qian, L. W. Zhang, J. Xia, Y. M. Bao, J. Y. Wang, L. Y. Guo and Y. C. Li, RSC Adv., 2018, 8, 17710–17722 RSC .
- Y. G. Chen, X. X. Li, H. Xiao, J. P. Xiao, B. Li, X. Y. Chen, Y. Wang, D. Cheng and X. T. Shuai, RSC Adv., 2018, 8, 2082–2091 RSC .
- N. Xu, X. B. Huang, G. F. Yin, M. J. Bu, X. M. Pu, X. C. Chen, X. M. Liao and Z. B. Huang, RSC Adv., 2018, 8, 15604–15612 RSC .
- J. L. Zhang, Y. Luo, X. F. Zhao, X. W. Li, K. X. Li, D. W. Chen, M. X. Qiao, H. Y. Hu and X. L. Zhao, RSC Adv., 2016, 6, 113173–113184 RSC .
- M. Zhang, X. Zhao, Z. Fang, Y. Niu, J. Lou, Y. Wu, S. Zou, S. Xia, M. Sun and F. Du, RSC Adv., 2017, 7, 3369–3375 RSC .
- M. Amidi, E. Mastrobattista, W. Jiskoot and W. E. Hennink, Adv. Drug Delivery Rev., 2010, 62, 59–82 CrossRef CAS PubMed .
- Y. Lu, X. Y. Li, X. D. Zhou, Q. Wang, X. W. Shi, Y. M. Du, H. B. Deng and L. B. Jiang, RSC Adv., 2014, 4, 33355–33361 RSC .
- F. Y. Ding, H. B. Deng, Y. M. Du, X. W. Shi and Q. Wang, Nanoscale, 2014, 6, 9477–9493 RSC .
- X. Q. Zhang, H. Zhang, L. Q. Yin, R. Hu, T. Qiu, Y. H. Yin, X. Xiong, H. Zheng and Q. Wang, J. Biomed. Nanotechnol., 2016, 12, 1688–1698 CrossRef CAS PubMed .
- S. J. Xin, X. Y. Li, Q. Wang, R. Huang, X. L. Xu, Z. J. Lei and H. B. Deng, J. Biomed. Nanotechnol., 2014, 10, 803–810 CrossRef CAS PubMed .
- R. Hu, H. Zheng, J. Cao, Z. Davoudi and Q. Wang, J. Biomed. Nanotechnol., 2017, 13, 1097–1105 CrossRef CAS .
- R. Hu, H. Zheng, J. Cao, Z. Davoudi and Q. Wang, J. Biomed. Nanotechnol., 2017, 13, 1058–1068 CrossRef CAS .
- W. Li, X. Y. Li, Q. Wang, Y. J. Pan, T. Wang, H. G. Wang, R. Song and H. B. Deng, Carbohydr. Polym., 2014, 99, 218–225 CrossRef CAS PubMed .
- Y. Yang, S. P. Wang, Y. T. Wang, X. H. Wang, Q. Wang and M. W. Chen, Biotechnol. Adv., 2014, 32, 1301–1316 CrossRef CAS PubMed .
- Y. W. Cai, C. F. Wu, Z. Y. Liu, L. J. Zhang, L. H. Chen, J. Q. Wang, X. K. Wang, S. T. Yang and S. Wang, Environ. Sci.: Nano, 2017, 4, 1876–1886 RSC .
- D. Q. Chen, C. Wang, F. Jiang, Z. Liu, C. Y. Shu and L. J. Wan, J. Mater. Chem. B, 2014, 2, 4726–4732 RSC .
- Y. X. Wang, X. Niu, X. L. Guo, H. Yu, Z. H. Liu, Z. Q. Zhang and S. Yuan, Glycobiology, 2018, 28, 318–332 CrossRef PubMed .
- B. Li, Q. Ma, G. He, X. R. Song, F. B. Wu, Y. Zheng, S. Zeng, C. Liu and W. Ren, Colloid Polym. Sci., 2013, 291, 1319–1327 CrossRef CAS .
- Y. Zheng, X. R. Song, G. He, Z. Cai, Y. Zhou, B. Yu, J. P. Xu, Y. Q. Wei and S. X. Hou, J. Drug Targeting, 2011, 19, 647–656 CrossRef CAS PubMed .
- B. Xu, Q. S. Jin, J. Zeng, T. Yu, Y. Chen, S. Z. Li, D. Q. Gong, L. L. He, X. Y. Tan, L. Yang, G. He, J. H. Wu and X. R. Song, ACS Appl. Mater. Interfaces, 2016, 8, 25753–25769 CrossRef CAS PubMed .
- Y. B. Zhao, D. Y. Lin, F. B. Wu, L. Guo, G. He, L. Ouyang, X. R. Song, W. Huang and X. Li, Int. J. Mol. Sci., 2014, 15, 17565–17576 CrossRef PubMed .
- R. A. A. Muzzarelli and F. Tanfani, Carbohydr. Polym., 1985, 5, 297–307 CrossRef CAS .
- R. J. Verheul, M. Amidi, S. van der Wal, E. van Riet, W. Jiskoot and W. E. Hennink, Biomaterials, 2008, 29, 3642–3649 CrossRef CAS PubMed .
- I. M. Vold and B. E. Christensen, Carbohydr. Res., 2005, 340, 679–684 CrossRef CAS PubMed .
- X. Bao, W. Wang, C. Wang, Y. Wang, J. Zhou, Y. Ding, X. Wang and Y. Jin, Biomaterials, 2014, 35, 8450–8466 CrossRef CAS PubMed .
- X. Wang, F. Wu, G. Li, N. Zhang, X. Song, Y. Zheng, C. Gong, B. Han and G. He, Acta Biomater., 2018, 74, 414–429 CrossRef CAS PubMed .
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac
*ac