
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ reactive coating of metallic and selenophilic Ag2Se on Se/C cathode materials for high performance Li–Se batteries†
Fugen Sun *,
Yahui Li‡
,
Zilong Wu‡,
Yu Liu,
Hao Tang,
Xiaomin Li,
Zhihao Yue and
Lang Zhou*
*,
Yahui Li‡
,
Zilong Wu‡,
Yu Liu,
Hao Tang,
Xiaomin Li,
Zhihao Yue and
Lang Zhou*
Institute of Photovoltaics, Nanchang University, 999 Qianhu Road, Nanchang 330031, China. E-mail: sunfugen@ncu.edu.cn; lzhou@ncu.edu.cn
First published on 21st September 2018
Abstract
A facile methodology to fabricate a metallic and selenophilic Ag2Se coating on a Se/nitrogen-doped mesoporous carbon composite, has been successfully developed based on the in situ redox reaction between Se and AgNO3 under ambient conditions. The in situ reactive growth of Ag2Se on Se ensures the complete encapsulation of Se by the Ag2Se coating, which endows the Ag2Se coating with the dual effects of physical entrapment and chemical binding to effectively confine polyselenide intermediates within the cathodes. With the further assistance of mesopore confinement of the nitrogen-doped carbons, the Ag2Se-coated Se/nitrogen-doped mesoporous carbon composites present much improved electrochemical performances with a high initial discharge capacity of 652 mA h g−1, a high coulombic efficiency of 95.4% and a high reversible capacity of 382 mA h g−1 after 100 cycles. These encouraging results suggest that the in situ reactive construction of metallic and chalcogenophilic coating layers on the chalcogen (e.g. S, Se and Te)-based electrode materials should be a promising and easy to scale-up method for practical applications of lithium batteries in light of the very simple in situ reaction processes involved.
1 Introduction
Rechargeable batteries with high energy density and long-lasting cycle performance are highly sought-after for electric vehicles and large-scale energy storage applications. Current Li-ion batteries using electrodes based on Li-ion insertion chemistry cannot satisfy this demand in terms of specific energy. Elemental chalcogens of Group 16 (VIA), such as O2, S and Se, are the most compelling cathode materials due to their wide availability and amazing energy density.1,2 However, Li–O2 batteries are far from commercialization because their cycling is seriously limited by electrolyte decomposition and large cell polarization. Li–S batteries are considered to be extremely promising due to their high specific capacity (1675 mA h g−1) and energy density (2600 W h g−1).3,4 Nevertheless, Li–S batteries suffer from low S utilization and fast capacity decay caused by the insulating nature of S and dissolution of polysulfide intermediates.5,6 As a solid congener of elemental S, Se rivals S in many aspects, including the much higher electronic conductivity (1 × 10−3 S m−1) than S (5 × 10−28 S m−1) and the comparable volumetric capacity (3254 mA h cm−3) with S.7,8 The high volumetric capacity of the Se cathode is particularly attractive for both EV and electronic device applications considering battery packs shall be installed in a limited space. These advantages of Se make it a new prospective cathode material for the high-energy rechargeable battery.9,10Analogous to Li–S batteries, Li–Se batteries also face the dissolution issue of high-order polyselenide intermediates, resulting in poor cycling performance and low coulombic efficiency.11,12 Motivated by the significant works on using carbon materials to encapsulate sulfur, improvements of the Li–Se batteries have been achieved by coupling the Se phases with various carbon materials, such as porous carbons,13 carbon nanotubes,14 and graphene15 (as listed in Table S2†). These carbon hosts not only act as electronic conduits to the Se encapsulated within, but also function as electrochemical reaction microchambers to physically confine the polyselenide intermediates, which results in enhanced utilization of Se.16,17 However, the weak interactions between non-polar carbon materials and polar polyselenides reduce the ability to bind and entrap these soluble polyselenides.18,19 As a consequence, the only use of non-polar carbon hosts can benefit the cell kinetics/reversibility and alleviate the capacity fading, but still fail to resolve the short cyclic problem of the Li–Se batteries.
From this aspect, using hybrid hosts of polar metal compounds with conductive carbons, has been proved to be a very promising approach for addressing the polyselenide dissolution issue in Li–Se batteries. Polar metal compounds including CoSe2,20 NbS2,21 Ni(OH)2 (ref. 22) and metal Co23 have been coupled with porous carbons for improving the performances of selenium cathodes. The relatively strong chemical binding between these polar compounds and polyselenides, combining with the physical adsorption of porous carbons, synergistically restrain the polyselenide shuttling. However, very few have concentrated on the rational integration of polar compounds with conductive carbons. Undoubtedly, there is still an urgent need to explore stronger encapsulating materials and to develop more facile synthetic procedures that yield optimized hybrid host structures for boosting the electrochemical performances of Se cathodes.
Herein, we demonstrate a simple methodology to fabricate an effective and multifunctional Ag2Se coating on the Se/nitrogen-doped mesoporous carbon particles, which is realized by an in situ redox reaction between Se and AgNO3 under ambient conditions. Unlike most of the insulated metal compounds used as polar hosts in the literature, the obtained orthorhombic Ag2Se coating possess bulk metallic conductivity as high as 2 × 103 S cm−1 at 298 K,24,25 which could interconnect with the conductive carbon frameworks for the fast electron transportation. Moreover, the in situ reactive growth of Ag2Se on Se ensures the complete encapsulation of Se by the Ag2Se coating with the dual effects of physical and chemical adsorption. The physical adsorption is created by the Ag2Se coating architectures, and chemical adsorption is affected by the selenophilic Ag–Se–Ag units, which lead to effective confinement of polyselenide intermediates within the cathodes. As a result, the Ag2Se coated Se/nitrogen-doped mesoporous carbon nanocomposites exhibit much improved cycling stability and rate performance. These encouraging results suggest that the in situ reactive coating of the metallic and selenophilic layers on the Se/C composite cathode materials would be a promising strategy to elevate the electrochemical performances of the advanced Li–Se batteries.
2 Experimental
2.1 Preparation of Se/nitrogen-doped mesoporous carbon nanocomposites
The nitrogen-doped mesoporous carbons (NMCs) were synthesized via a colloidal silica nanocasting method using phenol, melamine and formaldehyde as carbon precursors, as reported in our previous works.26,27 The Se/nitrogen-doped mesoporous carbon nanocomposites (Se/NMC) were prepared following a conventional melt-diffusion strategy. In a typical synthesis procedure, selenium and as-prepared NMCs were mixed homogeneously in a mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The mixture was degassed in a vessel and then sealed under vacuum. The melt infiltration was further carried out in the vacuum-sealed vessel at 260 °C for 10 h, and then Se/NMC was obtained.
:4. The mixture was degassed in a vessel and then sealed under vacuum. The melt infiltration was further carried out in the vacuum-sealed vessel at 260 °C for 10 h, and then Se/NMC was obtained.
2.2 Preparation of Ag2Se-coated Se/NMC nanocomposites
The Ag2Se coating was achieved by a facile in situ redox reaction between Se and AgNO3. Typically, the as-prepared Se/NMC was dispersed in an ethylene glycol (EG) solution. A controlled amount of AgNO3 in another EG solution was slowly added into the above EG solution under vigorous agitation, resulting in the redox reaction between Se and AgNO3. The reaction was allowed to proceed for 12 h under ambient conditions. The resulting Ag2Se-coated Se/NMC (Ag2Se@Se/NMC) composites were recovered by filtration, washed with water and dried.2.3 Material characterization
The morphologies of the samples were observed under SEM (JEOL 7100F) and TEM (JEOL 2100F). Nitrogen adsorption–desorption isotherms were measured at 77 K with a Quadrasorb SI analyzer. The Brunauer–Emmett–Teller method was utilized to calculate the specific surface area. The total pore volume was calculated using a single point at a relative pressure of 0.985. The pore size distributions were derived from the desorption branch by using the Barrett–Joyner–Halenda model. The XRD patterns were acquired on a Rigaku D/max 2550 diffractometer operating at 40 kV and 20 mA using Cu Kα radiation (λ = 1.5406 Å). Thermogravimetric analysis (TA Instrument Q600 Analyzer) of samples was carried out in a nitrogen flow. The samples were heated to 700 °C with a rate of 10 °C min−1. The surface chemistry of the samples was analyzed using an Axis Ultra DLD X-ray photoelectron spectroscopy. The X-ray source was operated at 15 kV and 10 mA. The working pressure was less than 2 × 10−8 torr (1 torr = 133.3 Pa). The N 1s, Se 3d and Ag 3d XPS spectras were measured at a step size of 0.1 eV. The binding energies were calibrated taking C 1s as a standard with a measured typical value of 284.6 eV. The N 1s, Se 3d and Ag 3d XPS signals were fitted with mixed Lorentzian–Gaussian curves, and a Shirley function was used to subtract the background using a XPS peak processing software. Powder electrical conductivity measurements (FZ-2010) were carried out at room temperature using the four-contact method. The samples were filled in a Teflon cylinder with inner diameter of 16 mm, and two stainless-steel plungers were used to deliver 4 MPa pressure through a hydraulic press device.2.4 Electrochemical tests
The Ag2Se@Se/NMC and Se/NMC samples were slurry-cast onto an aluminum current collector. Typically, 80 wt% Ag2Se@Se/NMC, 10 wt% carbon black (Super P conductive carbon black), and 10 wt% PVDF were mixed with N-methyl-2-pyrrolidone (NMP). The slurries were coated on aluminum current collectors and dried at 60 °C overnight. Electrochemical tests of these electrode materials were performed using coin cells with the selenium composite cathode and lithium metal as the counter electrode. The electrolyte was 1 M bis(trifluoromethane) sulfonamide lithium salt (LiTFSI) dissolved in a mixture of 1,3-dioxolane (DOL) and dimethoxymethane (DME) (1:1 by volume). The separator was a microporous membrane (Celgard 2400). The cell was assembled in an argon filled glovebox. The galvanostatic charge–discharge test and cyclic voltammetry measurements (CV) were conducted using an Arbin battery cycler (Arbin, BT2000, USA). All capacity values were calculated on the basis of selenium mass. Electrochemical impedance spectroscopy (EIS) was performed with an electrochemical working station PCI4/300 (Gamry Instrument, Warminster, PA, USA). The sinusoidal excitation voltage applied to the coin cells was 5 mV, with frequency range from 100 kHz to 0.01 Hz. All the electrochemical tests were performed at room temperature.
3 Results and discussion
The Ag2Se-coated Se/nitrogen-doped mesoporous carbon composites (Ag2Se@Se/NMC) were synthesized as shown schematically in Fig. 1. In brief, selenium was firstly infiltrated into the mesoporous carbon (NMCs) via the conventional melt-impregnation method. The obtained Se/NMC composite were then dispersed into ethylene glycol, followed by the addition of a controlled amount of AgNO3. Ethylene glycol reduce Ag+ cations to Ag atoms,28 which then are deduced to diffused quickly into the lattice of Se to form the Ag2Se coating on the surfaces of Se/NMC particles. The total preparation of Ag2Se@Se/NMC is an easy and feasible process without using sophisticated route, promising them real-world applications. In addition, it is well known that Ag2Se undergoes a reversible polymorphic-phase transformation between the low temperature orthorhombic and the high-temperature body centered cubic structure at about 130 °C; the high-temperature cubic Ag2Se phase is a superionic conductor that has been utilized as the solid electrolyte in photochargable secondary batteries; the low-temperature orthorhombic Ag2Se phase is a narrow bandgap semi-conductor with a high electronic conductivity of 2 × 103 S cm−1 (about threefold greater than graphite) that has been a promising candidate for thermoelectric applications.29,30 Thus, the in situ formed Ag2Se coating with a combination of high conductivity and selenophilic Ag–Se–Ag units which strong bind with Li2Sex species, could serve as a conductive blocker to ensure fast electronic/ionic transfer and prevent the polyselenides from diffusion into the electrolyte during discharge–charge cycling, as shown in Fig. 1b. | ||
| Fig. 1 Schematic illustration for the synthesis and cycling process of Se/NMC (a) and Ag2Se@Se/NMC (b). | ||
The Ag2Se coating on the Se/NMC composites could be observed from the SEM and TEM images, as shown in Fig. 2. The NMCs, which were obtained via a colloidal silica nanocasting process, has a well-developed spherical mesoporous structures (Fig. 2a) with a large specific surface area of 731 m2 g−1 and pore volume of 2.6 cm3 g−1 (more detailed characterizations on the porous structures and nitrogen-doped surface chemistry of the NMCs are shown in Fig. S1–S3†). After Se loading, the mesopores of NMCs were filled with Se, undoubtedly accompanying with a Se layer coating on the external surface of NMCs (Fig. 2b). The further reaction of AgNO3 with Se outside of NMCs results in a dense surface morphology which consists of interconnected Ag2Se nanoparticles (Fig. 2c). The scanning transmission electronic microscopy (STEM) elemental mapping images in Fig. S4† further indicate the position of Ag2Se coating on the surfaces of Se/NMC composites. Furthermore, the HRTEM image of the Ag2Se@Se/NMC composites in Fig. 2d shows the distinct lattice fringes with fringe spacings of 0.353 and 0.260 nm which are respectively corresponding to the (020) and (121) planes of orthorhombic Ag2Se phase, suggesting the interconnected orthorhombic Ag2Se nanocrystallites coating on the Se/NMC composites.
 | ||
| Fig. 2 SEM images of NMCs (a), Se/NMC (b) and Ag2Se@Se/NMC (c). HRTEM image of Ag2Se@Se/NMC (d). | ||
The crystal phases of the Ag2Se coating on the Se/NMC composites were further revealed by XRD analysis, as shown in Fig. 3a. After Ag2Se coating, the Ag2Se@Se/NMC composites contain typical P212121 orthorhombic Ag2Se diffraction peaks with strong peak intensity, further confirming the orthorhombic Ag2Se coating with high crystallinity. The in situ reactive crystallization of interconnected Ag2Se nanoparticles onto the Se ensures the complete enclosure of the Se by the Ag2Se coating. TGA analysis in Fig. 3b shows that the Se/NMC and Ag2Se@Se/NMC composites respectively exhibit the weight loss of approximately 73 and 67 wt% between 250 and 650 °C in a nitrogen flow, corresponding to the evaporation of Se in the composites during the heating process. Moreover, compared with the Se/NMC composites, the Se in the Ag2Se@Se/NMC composites evaporates at a significantly elevated temperature, indicating the strong encapsulation effect of selenophillic Ag2Se coating on the Se/NMC composites.
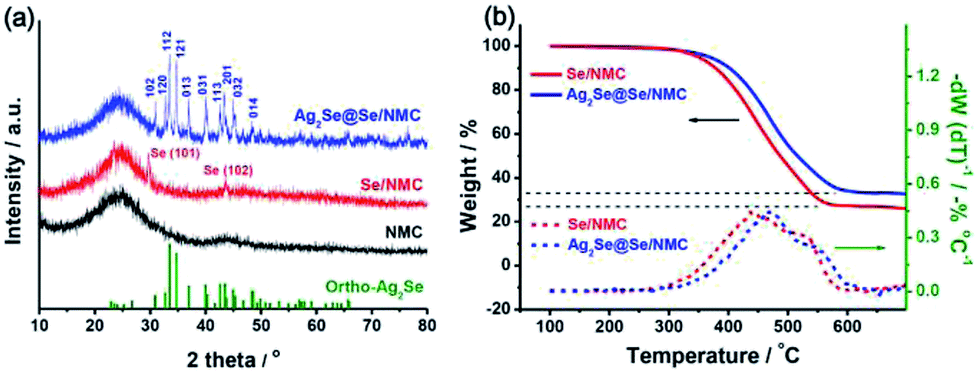 | ||
| Fig. 3 (a) X-ray diffraction patterns of NMCs, Se/NMC and Ag2Se@Se/NMC. (b) Thermogravimetric curves of Se/NMC and Ag2Se@Se/NMC. | ||
XPS survey scan of the Ag2Se@Se/NMC composites (Fig. 4a) reveals that the basic elemental composition consists of C, N, O, Se and Ag, without any other impurities. High-resolution Ag 3d XPS spectra of the Ag2Se@Se/NMC composites (Fig. 4b) displays two well-defined peaks at 373.60 and 367.55 eV which are corresponding to the Ag 3d3/2 and 3d1/2 of the Ag2Se coating, respectively. Due to the different chemical state of Se in the elemental Se and Ag2Se, the Se 3d XPS spectra of the Ag2Se@Se/NMC composites could be deconvoluted and curve-fitted into two doublet peaks with the binding energies of 55.88/55.03 and 54.28/53.43 eV, which are respectively attributed to the Se–Se bonds of the elemental Se and the Se–Ag bonds of the Ag2Se. Moreover, compared with the Se/NMC composites, the doublet Se 3d peaks of elemental Se in the Ag2Se@Se/NMC composites locate at the slightly reduced binding energy which shift toward that of selenides (Fig. 4c). This shift of binding energy indicates the change of electronic structures in elemental Se after Ag2Se coating, further suggesting the strong binding of the selenophillic Ag2Se with selenium. Furthermore, the digital image in Fig. S5† shows a significant decoloration of Li2Se6 solution after the adsorption of NMCs coated with Ag2Se, confirming the enhanced adsorption capability toward polyselenides after Ag2Se coating.
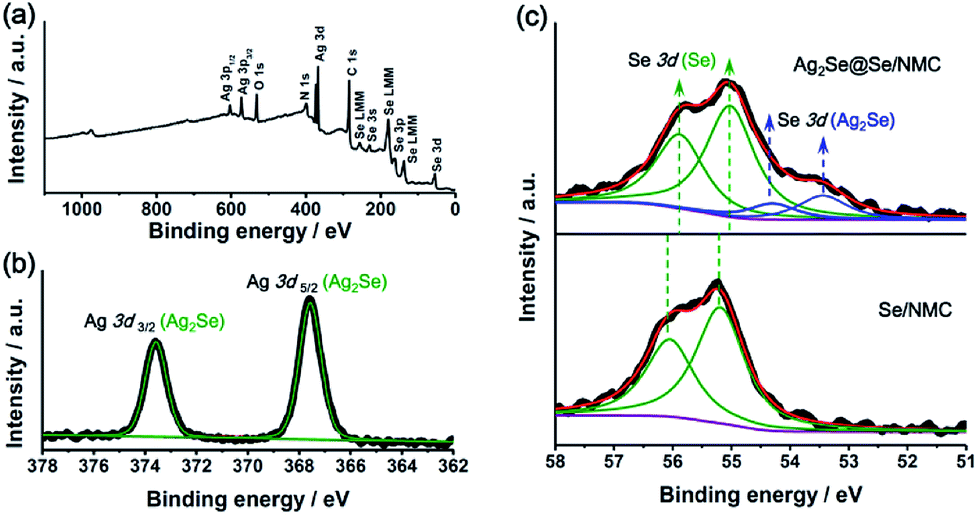 | ||
| Fig. 4 (a) Survey XPS spectra of Ag2Se@Se/NMC. (b) High-resolution Ag 3d XPS spectra of Ag2Se@Se/NMC. (c) High-resolution Se 3d XPS spectra of Se/NMC and Ag2Se@Se/NMC. | ||
The electrochemical performances of the Ag2Se@Se/NMC and Se/NMC composites were evaluated by cyclic voltammetry (CV), galvanostatic charge–discharge testing, and electrochemical impedance spectroscopy (EIS). As shown in the CV curves of the Ag2Se@Se/NMC and Se/NMC composites (Fig. 5a), two well-defined reduction peaks for both the composites exist distinctly, which are centered at 2.13 and 2.02 V corresponding to the two-step conversion of Se to soluble lithium polyselenides (Li2Sex, 4 ≤ x ≤ 8) and soluble lithium polyselenides to solid-state Li2Se2/Li2Se, respectively. Only one broad oxidation peak is observed, suggesting the transformation of Li2Se2/Li2Se to the polyselenides and then to Se with an overlapped delithiation process. Except for the intensity difference of these peaks, there are no other obvious differences observed. The higher peak intensity of the Ag2Se@Se/NMC composites than the Se/NMC composites indicates that the Se exhibit higher electrochemical activity after the metallic and selenophilic Ag2Se coating.
 | ||
| Fig. 5 Cyclic voltammograms (a) and the initial charge–discharge curve (b) of Se/NMC and Ag2Se@Se/NMC. | ||
The discharge and charge properties of the Ag2Se@Se/NMC and Se/NMC composites at a current density of 0.2C were further investigated, as shown in Fig. 5b. Two plateaus, corresponding to the formation of soluble polyselenides (Li2Sex, 4 ≤ x ≤ 8) at 2.1 V and solid-state Li2Se2 and Li2Se at 2.0 V, are observed for both the composites in the discharge process, which agree well with two apparent reduction peaks in the CV curves. The initial discharge and charge capacities of the Se/NMC composites are 620 and 691 mA h g−1, respectively, corresponding to an overcharge capacity of 71 mA h g−1 which is a typical feature of the polyselenide shuttling phenomenon.26 This implies that only confinement of Se in the nitrogen-doped mesoporous carbons cannot entirely prevent the shuttling phenomenon. Interestingly, after Ag2Se coating, the obtained Ag2Se@Se/NMC composites deliver a slightly higher discharge capacity of 652 mA h g−1 (approaching the theoretical capacity of 675 mA h g−1) than the Se/NMC composites, in accompany with a negligible overcharge capacity. Therefore, the multifunctional Ag2Se coating on the Se/NMC composites should provide physical confinement and chemical interaction, thus showing excellent efficiency for trapping the polyselenides.
The cycling performance of the Ag2Se@Se/NMC and Se/NMC composites at 0.2C were compared in Fig. 6. The Se/NMC sample delivers a considerable discharge capacity of 290 mA h g−1 after 100 cycles, which is comparable to the other carbon-selenium composites due to the mesopore confinement of nitrogen-doped carbons. On this basis, the Ag2Se coating further suppress the shuttle effect and facilitate the redox reaction kinetics, resulting in much improved cycling performances. As a result, The Ag2Se@Se/NMC sample exhibits better electrochemical performances with a higher reversible capacity of 382 mA h g−1 and a higher coulombic efficiency of 95.4% after 100 cycles than the Se/NMC counterpart (Fig. 6a). Moreover, the Ag2Se@Se/NMC sample presents better cycling responses to continuously varying current densities, as shown in Fig. 6b. At the maximum discharging rate of 5C (3.4 A g−1), the Ag2Se@Se/NMC delivers a reversible capacity of 294 mA h g−1, much higher than that of Se/NMC. After extended cycling for 400 cycles at 0.5 and 1C, the relatively high reversible capacities of 206 and 193 mA h g−1 have been retained for the Ag2Se@Se/NMC sample (Fig. 6c). The electrochemical performances of the Ag2Se@Se/NMC sample are among the best series of the carbon-selenium cathode materials (Table S2†). Such excellent kinetic behavior of the Ag2Se@Se/NMC composite is further supported by their much lower charge-transfer resistances and diffusion impedances than those of the Se/NMC composite from the EIS spectra (Fig. S6†) and powder electrical conductivity measurement results (Table S1†). There results further demonstrate the effective confinement of Li2Se and Li2Sex species via the combination of chemical and physical entrapment provided by the metallic and selenophilic Ag2Se coating nanostructure.
 | ||
| Fig. 6 (a) Cycling performance and coulombic efficiency of Ag2Se@Se/NMC and Se/NMC at 0.2C. (b) Rate performances of Ag2Se@Se/NMC and Se/NMC at different current densities. (c) Long-term cycling performance of Ag2Se@Se/NMC at 0.5 and 1C. | ||
4 Conclusion
In conclusion, a metallic and selenophilic Ag2Se protective coating was successfully constructed in situ on the Se/nitrogen-doped mesoporous carbon cathode materials via the direct redox reaction between Se and AgNO3. The total synthesis is an easy and feasible process without using sophisticated route, promising them real-world applications. Moreover, the obtained orthorhombic Ag2Se is a narrow bandgap semi-conductor with a high electronic conductivity, thus enabling fast electron transport through the coating. The combination of the physical Ag2Se coating structure and the intrinsic polyselenide binding by the selenophilic Ag–Se–Ag units lead to excellent polyselenide confinement. Resultantly, with the further assistance of nitrogen-doped mesoporous carbons, the Ag2Se-coated Se/nitrogen-doped mesoporous carbon composites deliver excellent reversibility, capacity stability, and rate capability at up to 5C. The present work paves new cost-effective avenues in the practical application of high-energy and long-life Li–Se batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (Grant No. 51502090), Natural Science Foundation of Jiangxi Province (Grant Nos. 20171BAB216007 and 20181BAB206006) and Innovation Fund Designated for Graduate Students of Nanchang University (Grant No. CX2017005).References
- H. Peng, J. Huang, X. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1700260 CrossRef.
- F. Sun, B. Zhang, H. Tang, Z. Yue, X. Li, C. Yin and L. Zhou, J. Mater. Chem. A, 2018, 6, 10104–10110 RSC.
- W. Tang, Z. Chen, B. Tian, H. Lee, X. Zhao, X. Fan, Y. Fan, K. Leng, C. Peng, M. Kim, M. Li, M. Lin, J. Su, J. Chen, H. Jeong, X. Yin, Q. Zhang, W. Zhou, K. Loh and G. Zheng, J. Am. Chem. Soc., 2017, 139, 10133–10141 CrossRef PubMed.
- F. Sun, H. Cheng, J. Chen, N. Zheng, Y. Li and J. Shi, ACS Nano, 2016, 10, 8289–8299 CrossRef PubMed.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef PubMed.
- F. Sun, J. Wang, H. Chen, W. Qiao, L. Ling and D. Long, Sci. Rep., 2013, 3, 2823 CrossRef PubMed.
- L. Zeng, W. Zeng, Y. Jiang, X. Wei, W. Li, C. Yang, Y. Zhu and Y. Yu, Adv. Energy Mater., 2015, 5, 1401377 CrossRef.
- C. Yang, S. Xin, Y. Yin, H. Ye, J. Zhang and Y. Guo, Angew. Chem., Int. Ed., 2013, 52, 8363–8368 CrossRef PubMed.
- K. Han, Z. Liu, J. Shen, Y. Lin, F. Dai and H. Ye, Adv. Funct. Mater., 2015, 25, 455–463 CrossRef.
- Y. J. Hong and Y. C. Kang, Carbon, 2017, 111, 198–206 CrossRef.
- Q. Cai, Y. Li, L. wang, Q. Li, J. Xu, B. Gao, X. Zhang, K. Huo and P. K. Chu, Nano Energy, 2017, 32, 1–9 CrossRef.
- C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. Wang, ACS Nano, 2013, 7, 8003–8010 CrossRef PubMed.
- Y. Qu, Z. Zhang, S. Jiang, X. Wang, Y. Lai, Y. Liu and J. Li, J. Mater. Chem. A, 2014, 2, 12255–12261 RSC.
- J. He, Y. Chen, W. Lv, K. Wen, P. Li, Z. Wang, W. Zhang, W. Qin and W. He, ACS Energy Lett., 2016, 1, 16–20 CrossRef.
- X. Peng, L. Wang, X. Zhang, B. Gao, J. Fu, S. Xiao, K. Huo and P. K. Chu, J. Power Sources, 2015, 288, 214–220 CrossRef.
- S. Xin, L. Yu, Y. You, H. Cong, Y. Yin, X. Du, Y. Guo, S. Yu, Y. Cui and J. B. Goodenough, Nano Lett., 2016, 16, 4560–4568 CrossRef PubMed.
- G. Xu, J. Liu, R. Amine, Z. Chen and K. Amine, ACS Energy Lett., 2017, 2, 605–614 CrossRef.
- T. Liu, C. Dai, M. Jia, D. Liu, S. Bao, J. Jiang, M. Xu and C. Li, ACS Appl. Mater. Interfaces, 2016, 8, 16063–16070 CrossRef PubMed.
- S. F. Zhang, W. P. Wang, S. Xin, H. Ye, Y. X. Yin and Y. G. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 8759–8765 CrossRef PubMed.
- J. Yang, H. Gao, D. Ma, J. Zou, Z. Lin, X. Kang and S. Chen, Electrochim. Acta, 2018, 264, 341–349 CrossRef.
- D. S. Choi, M. S. Yeom, Y.-T. Kim, H. Kim and Y. Jung, Inorg. Chem., 2018, 57, 2149–2156 CrossRef PubMed.
- R. Luo, Y. Lu, X. Hou, Q. Yu, T. Peng, H. Yan, X. Liu, J.-K. Kim and Y. Luo, J. Solid State Electrochem., 2017, 4, 1–8 Search PubMed.
- J. He, W. Lv, Y. Chen, J. Xiong, K. Wen, C. Xu, W. Zhang, Y. Li, W. Qin and W. He, J. Power Sources, 2017, 363, 103–109 CrossRef.
- B. Gates, Y. Wu, Y. Yin, P. Yang and Y. Xia, J. Am. Chem. Soc., 2001, 123, 11500–11501 CrossRef PubMed.
- C. Zeng, W. Zhang, S. Ding, Z. Yang, H. Zeng and Z. Li, CrystEngComm, 2013, 15, 5127–5133 RSC.
- F. Sun, J. Wang, H. Chen, W. Li, W. Qiao, D. Long and L. Ling, ACS Appl. Mater. Interfaces, 2013, 5, 5630–5638 CrossRef PubMed.
- F. Sun, J. Wang, D. Long, W. Qiao, L. Ling, C. Lv and R. Cai, J. Mater. Chem. A, 2013, 1, 13283–13289 RSC.
- U. Jeong and Y. Xia, Angew. Chem., Int. Ed., 2005, 44, 3099–3103 CrossRef PubMed.
- B. Gates, B. Mayers, Y. Wu, Y. Sun, B. Cattle, P. Yang and Y. Xia, Adv. Funct. Mater., 2010, 12, 679–686 CrossRef.
- Y. Gu, R. Cui, Z. Zhang, Z. Xie and D. Pang, J. Am. Chem. Soc., 2012, 134, 79–82 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06484a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |