
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cation dynamics by 1H and 13C MAS NMR in hybrid organic–inorganic (CH3CH2NH3)2CuCl4
Ae Ran Lim *ab and
Yong Lak Jooc
*ab and
Yong Lak Jooc
aAnalytical Laboratory of Advanced Ferroelectric Crystals, Jeonju University, Jeonju 55069, South Korea. E-mail: aeranlim@hanmail.net; arlim@jj.ac.kr; Tel: +82-63-220-2514
bDepartment of Science Education, Jeonju University, Jeonju 55069, South Korea
cSchool of Chemical and Biomolecular Engineering, Cornell University, Ithaca, New York 14853, USA
First published on 4th October 2018
Abstract
To understand the dynamics of the cation in layered perovskite-type (CH3CH2NH3)2CuCl4, the temperature-dependent chemical shifts and spin–lattice relaxation times T1ρ in the rotating frame have been measured using 1H magic angle spinning nuclear magnetic resonance (MAS NMR) and 13C cross-polarization (CP)/MAS NMR techniques. Each proton and carbon in the (CH3CH2NH3)+ cation is distinguished in MAS NMR spectra. The Bloembergen–Purcell–Pound (BPP) curves for 1H T1ρ in CH3CH2 and NH3, and for the 13C T1ρ in CH3 and CH2 are revealed to have minima at low temperatures. This implies that the curves represent the CH3 and NH3+ rotational motions. The amplitude of the cationic motion is enhanced at the C-end, that is, the N-end of the organic cation is fixed to the inorganic layer through N–H⋯Cl hydrogen bonds, and T1ρ becomes short with larger-amplitude molecular motions.
I. Introduction
Metal–organic hybrids, which consist of organic and inorganic components, have recently attracted much attention because these materials have many possibilities for the tailoring of their functionalities and physical properties including optical, electrical and magnetic properties by adjusting the organic and/or metal building blocks. Hybrid metal–organic compounds based on the perovskite structures are of increasing interest due to their potential use for solar cells.1,2 However, toxicity and chemical instability issues of halide perovskites still remain as the main drawbacks for use in solar cells. The crystalline structure of compounds of the type (CnH2n+1NH3)2MCl4, where n = 1, 2, 3 … and M represents divalent metals (M = Cu, Cd, …), may be described as a sequence of alternating organic–inorganic layers.3–6 Many compounds in this family have been extensively investigated and have demonstrated successive phase transitions. This family of materials crystallizes in the layered perovskite structure, which consists of infinite, staggered layers of corner-sharing MCl6 octahedra interleaved by alkylammonium cations.7 Because of the layered character of their structure, these crystals become appropriate substances for investigations of two-dimensional electronic systems. The cavities between the octahedra are occupied by the ammonium heads of the organic cations, which, importantly, form strong N–H⋯Cl hydrogen bonds to any of the eight chloride ions.8Ethylammonium copper chloride (CH3CH2NH3)2CuCl4 is a layered perovskite-type compound that undergoes a complicated sequence of phase transitions. Differential scanning calorimetry (DSC) data indicates several phase transitions, at 236 K (=TC4), 330 K (=TC3), 357 K (=TC2), and 371 K (=TC1), as temperature increases.9–14 The peaks at 236 K, 330 K, and 371 K are very weak and can perhaps correspond to second-order transformations.13 The phase transitions in this crystal are mostly connected with changes in the arrangement of the alkylammonium chains. Fig. 1 shows the room-temperature orthorhombic crystal structure of (CH3CH2NH3)2CuCl4.8,15 The hybrids have the orthorhombic crystal structure with the space group Pbca, and the lattice constants are a = 7.47 Å, b = 7.35 Å, and c = 21.18 Å at room temperature.16 The CuCl6 octahedra are strongly distorted with elongated Cu–Cl bonds orthogonal to each other on adjacent octahedra. The CuCl6 sheets are sandwiched between two layers of alkylammonium. The structure of the organic component consists of a double layer of alkylammonium ions with their charged ends, the nitrogen atoms, oriented to the nearest CuCl6 plane.4 The complete structure is constituted by corner-sharing CuCl6 octahedra, forming the inorganic layers, and bilayers of organic cations attached to the octahedra by their NH3 heads.17,18
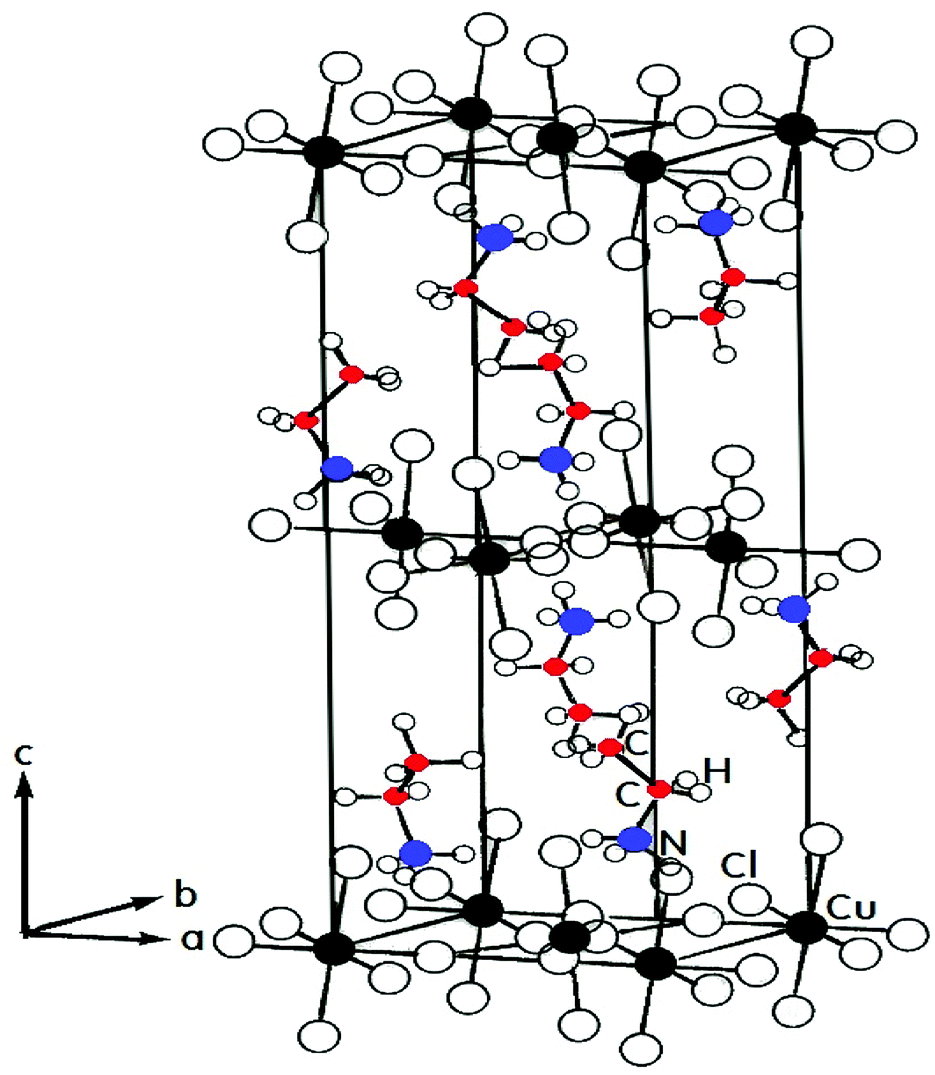 | ||
| Fig. 1 Orthorhombic structure of a (CH3CH2NH3)2CuCl4 crystal at room temperature. | ||
The structural geometry and molecular motions of the organic molecules within the layered hybrid structure is important for determining the influence of temperature on the evolution of the structural phase transitions in the perovskite structure. Physical properties in particular depend on the characteristics of metallic anion and the organic cation.
In the present study, the crystal structure and thermal stability for (CH3CH2NH3)2CuCl4 was observed by means of conventional X-ray, thermogravimetric analysis (TGA), and optical polarizing microscopy. In order to clarify the structural geometry and dynamics of the cation in the organic–inorganic (CH3CH2NH3)2CuCl4, we investigated the chemical shifts and the spin–lattice relaxation time T1ρ in the rotating frame using 1H magic angle spinning nuclear magnetic resonance (MAS NMR) and 13C cross-polarization (CP)/MAS NMR. The CH3CH2 and NH4 groups of the CH3CH2NH3 cation are distinguishable in 1H MAS NMR spectra, and the CH3 and CH2 groups are distinguished by 13C CP/MAS NMR spectra. We investigated the 1H and 13C dynamics in the (CH3CH2NH3)+ cation near the phase-transition temperatures.
II. Experimental method
Crystals of (CH3CH2NH3)2CuCl4 were obtained by slow evaporation at 25 °C from an aqueous solution of C2H5NH2·HCl and CuCl2·2H2O in the stoichiometric 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 proportion. The obtained crystals were yellow square plates, typically 5 mm × 5 mm in area and 0.5 mm in thickness.
:1 proportion. The obtained crystals were yellow square plates, typically 5 mm × 5 mm in area and 0.5 mm in thickness.
The structure of the (CH3CH2NH3)2CuCl4 crystals was determined at room temperature with an X-ray diffraction system (PANalytical, X'pert pro MPD) with a Cu-Kα (λ = 1.5418) radiation source. Measurements were taken in a θ–2θ geometry from 10° to 60° at 45 kV and with a tube power of 40 mA. And, the TGA curve at a heating rate of 10 °C min−1 was measured under N2 atmosphere, and the mass of the powdered sample used in the TGA experiment was 11.41 mg.
The chemical shifts and the T1ρ values for (CH3CH2NH3)2CuCl4 were obtained by 1H MAS NMR and 13C CP/MAS NMR at Larmor frequencies of ω0/2π = 400.13 and 100.61 MHz, respectively, using Bruker 400 MHz NMR spectrometers at the Korea Basic Science Institute, Western Seoul Center. Crystalline powdered samples were placed within a 4 mm CP/MAS probe, and the MAS rate for 1H and 13C measurements, to minimize spinning sideband overlap, was set to 10 kHz. The 1H T1ρ values were determined using a π/2−t sequence by varying the duration of spin-locking pulses. 13C T1ρ values were measured by varying the duration of the spin-locking pulse applied after the CP preparation period. The width of the π/2 pulse used for measuring T1ρ for 1H and 13C was 3.7 μs, with the spin-locking field at 67.56 kHz. The chemical shifts and T1ρ were measured over a temperature range of 180–430 K.
III. Experimental results
The measured structure at room temperature exhibited orthorhombic symmetry with cell parameters of a = 7.480 Å, b = 7.375 Å, c = 21.254 Å for (CH3CH2NH3)2CuCl4 crystal. This result is consistent with the results reported by Steadman and Willett.16The TGA curve of (CH3CH2NH3)2CuCl4 is shown in Fig. 2 for measuring thermal stability. The first occurrence of mass loss begins at approximately 430 K (Td), which is the onset of partial thermal decomposition. The second weight loss of 25.1% near 530 K is due to the removal of the CH3CH2NH3Cl from the compound, leaving intermediate CH3CH2NH3CuCl3 that belongs to another known class of compounds ABX3. Near 560 K, CuCl2 remains as the residue and when it reaches 580 K, the total weight loss becomes 65.55%. The color of the crystal is dark yellow at room temperature although it has slightly inhomogeneous hue due to surface roughness. As the temperature increases, the color of the crystal varies from dark yellow (300 K, 350 K), brown (400 K), to dark brown (450 K, 500 K), and then they start melting at 530 K as shown in the inset in Fig. 2. The TGA and optical polarizing microscopy results show that the crystal above 430 K allows CH3 to partially escape by the breaking the weak C–N bond.
 | ||
| Fig. 2 Thermogravimetric analysis of (CH3CH2NH3)2CuCl4 (inset: color changes of a (CH3CH2NH3)2CuCl4 crystal according to the temperature): (a) 300 K, (b) 350 K, (c) 400 K, (d) 450 K, (e) 500 K, and (f) 530 K. | ||
The 1H NMR spectra at a frequency of 400.13 MHz were obtained by MAS NMR. The 1H spectrum recorded at room temperature is shown in the inset in Fig. 3; the spectrum shows two peaks at chemical shifts of δ = 0.23 and 12.12 ppm, which are assigned to the protons of the CH3CH2 and NH3 groups, respectively. The spinning sidebands for CH3CH2 are marked with asterisks and those for NH3 are marked with open circles. However, the different 1H signals from CH3 and CH2 cannot be resolved, and therefore the combined CH3CH2 peak is very broad and has a larger intensity due to the overlap of the CH3 and CH2 peaks. The peak with the lower chemical shift is attributed to the protons in CH3CH2, and that of the higher chemical shift is attributed to the protons in NH3. The 1H chemical shifts for the alkyl and ammonium groups slowly and monotonously vary with temperature, indicating that the surrounding environments of the protons in the alkyl and ammonium groups change continuously, as shown in Fig. 3; here, the chemical shifts for protons in CH3CH2 and NH3 near TC1, TC2, and TC3 are nearly constant with temperature, whereas those for protons in CH3CH2 and NH3 below TC4 change more abruptly.
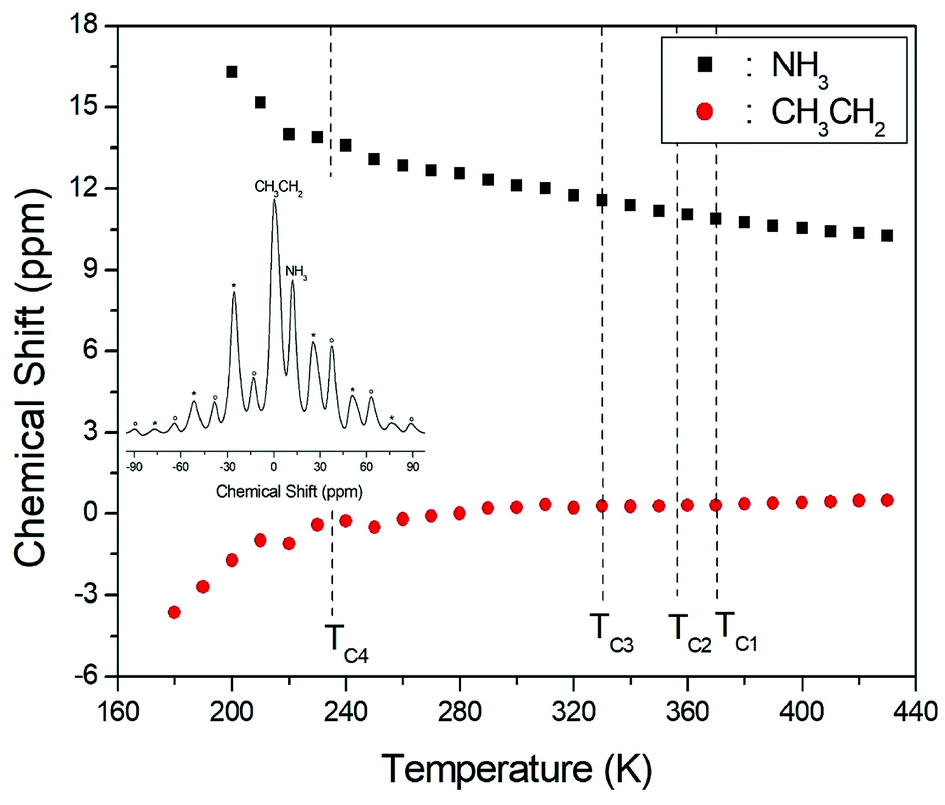 | ||
| Fig. 3 Chemical shifts for 1H MAS NMR of (CH3CH2NH3)2CuCl4 as a function of temperature (inset: 1H MAS NMR spectrum of (CH3CH2NH3)2CuCl4 at 300 K with spinning sidebands indicated by asterisks and open circles). | ||
The T1ρ values for the CH3CH2 and NH3 protons in (CH3CH2NH3)2CuCl4 were obtained as a function of temperature. The magnetization traces of both the alkyl and ammonium protons may be described by a single exponential function19–21
| S(t)/S0 = exp(−t/T1ρ), | (1) |
| T1ρ−1 = (N/20)(γHγCħ/rH–C3)2{4τC/(1 + ω12τC2) + τC/[1 + (ωH − ωC)2τC2] + 3τC/[1 + (ωC2τC2)] + 6τC/[1 + (ωH + ωC)2τC2] + 6τC/[1 + ωH2τC2]}. | (2) |
 | ||
| Fig. 4 1H spin–lattice relaxation times T1ρ in the rotating frame for the CH3CH2 and NH3 groups of (CH3CH2NH3)2CuCl4 as a function of inverse temperature. | ||
Here, γH and γC are the gyromagnetic ratios for the 1H and 13C nuclei, respectively; N is the number of directly bound protons; rH–C is the H–C internuclear distance; ħ is the reduced Planck constant; ωH and ωC are the Larmor frequencies of 1H and 13C, respectively; and ω1 is the frequency of the spin-locking field. We analyzed our data assuming that T1ρ would show a minimum when ω1τC = 1, and that the BPP relation between T1ρ and the characteristic frequency ω1 could be applied. We sensitively controlled the minima in the T1ρ temperature variations and the slopes around the minima. From these results, the value of (γHγCħ/rH–C3)2 for the proton in eqn (2) was obtained. We then calculated the temperature dependences of the τC values for protons by using the obtained values of (γHγCħ/rH–C3)2. The temperature dependence of τC follows a simple Arrhenius equation:
|
τC = τ0exp(−Ea/RT),
| (3) |
τC vs. 1000/T curve shown in Fig. 5; we obtained Ea = 12.19 ± 1.30 kJ mol−1 and Ea = 8.33 ± 0.50 kJ mol−1 for CH3CH2 and NH3, respectively. The rotational motion for alkyl groups is activated, whereas the rotational motion for ammonium groups at the end of the organic cation is less strongly activated.
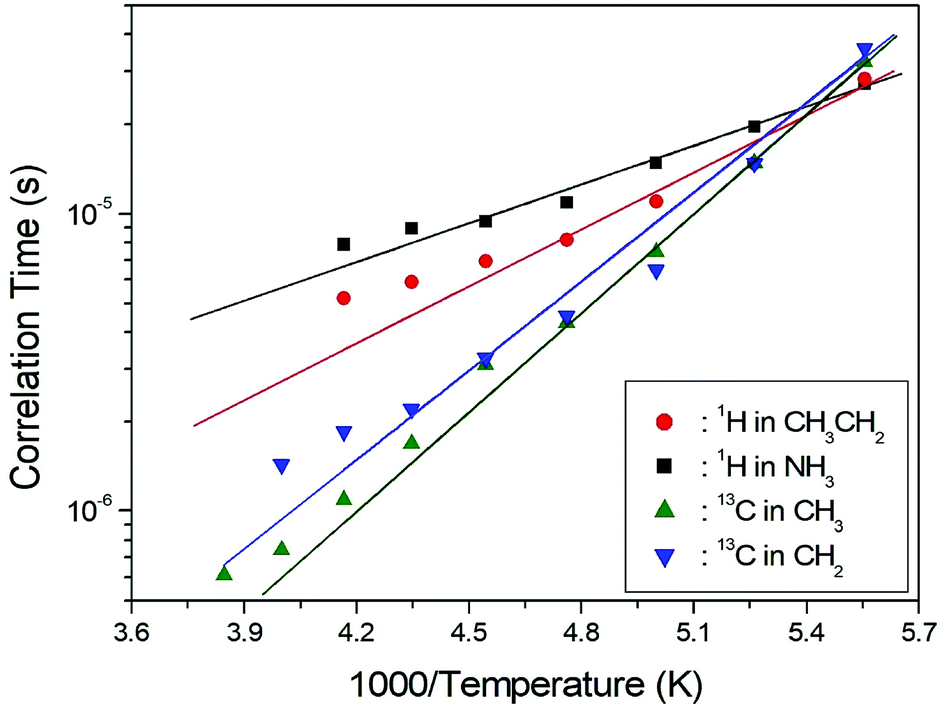 | ||
| Fig. 5 Arrhenius plots of the natural logarithm of the correlation time for each 1H and 13C of (CH3CH2NH3)2CuCl4 as a function of inverse temperature. | ||
The structural analysis of the carbons in (CH3CH2NH3)2CuCl4 was performed by 13C CP/MAS NMR, and the corresponding spectrum is shown in Fig. 6, as a function of temperature; the 13C CP/MAS NMR spectrum at room temperature shows two signals at chemical shifts of δ = 50.77 ppm and δ = 113.50 ppm with respect to tetramethysilane (TMS), which can be assigned to CH3 and CH2, respectively. The 13C chemical shift of CH2 abruptly shifts with temperature, whereas that of CH3 changes only much less with temperature. The full width at half maximum (FWHM) linewidths for the 13C of CH3 and CH2 in Fig. 7 showed a monotonic decrease with increasing temperature, with no particular anomalies attributable to the phase transitions. The linewidth of the 13C signal assigned to CH3 is broad compared to that of CH2, and the linewidth narrows significantly with increasing temperature. This narrowing of the 13C linewidths is attributed to internal motions that the line widths follow the same temperature dependence as some internal motions, hence the motions are responsible for the line widths.
 | ||
| Fig. 6 13C CP/MAS NMR spectra of (CH3CH2NH3)2CuCl4 measured at different temperatures. | ||
 | ||
| Fig. 7 Temperature dependences of line widths of 13C NMR spectra of CH3 and CH2 in (CH3CH2NH3)2CuCl4. | ||
To obtain the 13C T1ρ values, the nuclear magnetization was also measured at several temperatures as a function of delay time. The signal intensity of the nuclear magnetization recovery curves for 13C is described by a single exponential function as in eqn (1) at all temperatures. The 13C T1ρ values for CH3 and CH2 in (CH3CH2NH3)2CuCl4 are plotted as a function of inverse temperature in Fig. 8. The temperature dependences of the 13C MAS NMR T1ρ values seem to be similar. The T1ρ values for CH3 and CH2 both increase with temperature in the same manner; whereas, the 13C T1ρ values near the phase-transition temperatures are approximately continuous. The T1ρ values for CH3 and CH2 at room temperature are 33.85 ms and 109.40 ms, respectively. The amplitude of the cationic motion is enhanced at its CH3 end, and the central CH2 moiety is fixed to the NH3 group in the organic cation. The T1ρ curve below TC4 can be reproduced by BPP theory. The BPP curves for CH3 and CH2, showing minima at low temperatures, is almost the same as those of the CH3CH2 and NH3 shifts of the 1H MAS NMR measurements. Ea for the rotational motion of CH3 and CH2 can be obtained from the logτC vs. 1000/T curve shown in Fig. 5; we obtained Ea = 21.35 ± 0.45 kJ mol−1 for CH3 and Ea = 19.72 ± 1.76 kJ mol−1 for CH2, respectively, which, considering their error ranges, are the same values.
 | ||
| Fig. 8 13C spin–lattice relaxation times T1ρ in the rotating frame for CH3 and CH2 in (CH3CH2NH3)2CuCl4 as a function of inverse temperature. | ||
IV. Conclusion
We discuss the molecular motions for cation of Cu-based hybrid materials, where we replace Pb with nontoxic Cu metal for lead-free perovskite solar cells, and investigate their potential toward solar cell applications based on ionic dynamics of the cation in hybrid organic–inorganic (CH3CH2NH3)2CuCl4 by NMR studies. The cation dynamics and interionic interactions through hydrogen bonds are expected to be closely related with the physical properties due to the potential applications. The cation dynamics in a layered perovskite-type (CH3CH2NH3)2CuCl4 were investigated as a function of temperature by 1H MAS NMR and 13C CP/MAS NMR experiments. The CH3CH2 and NH4 units in the CH3CH2NH3 cation were distinguished by the 1H MAS NMR spectra, and the CH3 and CH2 units in the CH3CH2NH3 cation were also clearly distinguished in the 13C CP/MAS NMR spectra. To obtain detailed information about the cation dynamics of this crystal, the spin–lattice relaxation time T1ρ in the rotating frame for both 1H and 13C were measured, revealing that these atoms undergo rotational motions at low temperatures. The BPP curves for the 1H T1ρ in CH3CH2 and NH3, and for the 13C T1ρ in CH3 and CH2, were shown to have a minimum at low temperatures; the T1ρ of 1H and 13C showed a minimum and is governed by the tumbling motion of the CH3CH2 and NH3 groups, indicating that the 1H and 13C atoms in the CH3CH2NH3+ groups exhibit high mobility at low temperatures. The molecular motions for 1H and 13C in the CH3CH2NH3+ cation were very free at low temperatures. T1ρ provides insight into the changes in the cation reorientation rates at low temperature.The 13C T1ρ values in CH3 increased with temperature, a trend that has been observed in alkyl chains attached to the (CH3CH2NH3) cation due to its greater mobility toward its free end. The CH3CH2NH3 cationic motion is enhanced at the opposing end of the cation to the NH4+ group probably because this group is bound to the inorganic layer through the N–H⋯Cl hydrogen bonds. The 13C T1ρ is usually dominated by the fluctuation of the anisotropic chemical shift, and it becomes shorter with larger-amplitude molecular motions. This implies that the amplitude of the cationic motion is enhanced at the C-end, that is, the N-end of the organic cation is fixed at the inorganic layer through N–H⋯Cl hydrogen bonds. The cationic motion, being associated with the fluctuation of the molecular axis, is expected to be gradually excited with increasing temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science, and Technology (2018R1D1A1B07041593 and 2016R1A6A1A03012069).References
- A. M. Elseman, A. E. Shalan, S. Sajid, M. M. Rashad, A. M. Hassan and M. Li, ACS Appl. Mater. Interfaces, 2018, 10, 11699 CrossRef CAS PubMed.
- J. A. Aramburu, P. Garcia-Fernandez, N. R. Mathiesen, J. M. Garcia-Lastra and M. Moreno, J. Phys. Chem. C, 2018, 122, 5071 CrossRef CAS.
- A. Caretta, M. C. Donker, A. O. Polyakov, T. T. M. Palstra and P. H. M. van Loosdrecht, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 20405 CrossRef.
- E. Suprayoga, A. A. Nugroho, A. O. Polyakov, T. T. M. Palstra and I. Watanabe, J. Phys.: Conf. Ser., 2014, 551, 12054 CrossRef.
- J. Ding, H. Li, L. Wen, X. Kang, H. Li and J. Zhang, J. Magn. Magn. Mater., 2013, 346, 91 CrossRef CAS.
- R. Lefi, F. B. Naser and H. Guermazi, J. Alloys Compd., 2017, 696, 1244 CrossRef CAS.
- K. Ohwada, K. Ishii, T. Inami, Y. Murakami, T. Shobu, H. Ohsumi, N. Ikeda and Y. Ohishi, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 14123 CrossRef.
- B. Kundys, A. Lappas, M. Viret, V. Kapustianyk, V. Rudyk, S. Semak, Ch. Simon and I. Bakaimi, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 224434 CrossRef.
- V. Kapustianyk, V. Rudyk and M. Partyka, Phys. Status Solidi B, 2007, 244, 2151 CrossRef CAS.
- V. B. Kapustianik, V. V. Bazhan and Yu. M. Korchak, Phys. Status Solidi B, 2002, 234, 674 CrossRef CAS.
- W. Kleemann, F. J. Schafer, E. Karajamaki, R. Laiho and T. Levola, Physica B, 1983, 119, 269 CrossRef CAS.
- V. Kapustianik, Yu. Korchak, I. Polovinko, R. Tchukvinskyi, Z. Czapla and S. Dacko, Phys. Status Solidi B, 1998, 207, 95 CrossRef CAS.
- C. B. Mohamed, K. Karoui, F. Jomni, K. Guidara and A. B. Rhaiem, J. Mol. Struct., 2015, 1082, 38 CrossRef CAS.
- I. R. Jahn, K. Knorr and J. Ihringer, J. Phys.: Condens. Matter, 1989, 1, 6005 CrossRef CAS.
- P. Zolfaghari, G. A. de Wijs and R. A. de Groot, J. Phys.: Condens. Matter, 2013, 25, 295502 CrossRef CAS PubMed.
- J. P. Steadman and R. D. Willett, Inorg. Chim. Acta, 1970, 4, 367 CrossRef CAS.
- A. O. Polyakov, A. H. Arkenbout, J. Baas, G. R. Blake, A. Meetsma, A. Caretta, P. H. M. van Loosdrecht and T. T. M. Palstra, Chem. Mater., 2012, 24, 133 CrossRef CAS.
- A. Caretta, R. Miranti, R. W. A. Havenith, E. Rampi, M. C. Donker, G. R. Blake, M. Montagnese, A. O. Polyakov, R. Broer, T. T. M. Palstra and P. H. M. van Loosdrecht, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 24301 CrossRef.
- J. L. Koenig, Spectroscopy of Polymers, Elsevier, New York, 1999 Search PubMed.
- V. J. Mcbrierty and K. J. Packer, Nuclear Magnetic Resonance in Solid Polymers, Cambridge University Press, Cambridge, 1993 Search PubMed.
- A. R. Lim, RSC Adv., 2018, 8, 18656 RSC.
- A. R. Lim, RSC Adv., 2017, 7, 55276 RSC.
| This journal is © The Royal Society of Chemistry 2018 |