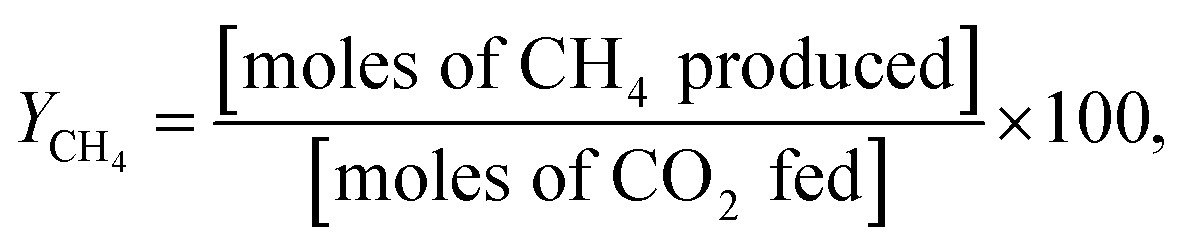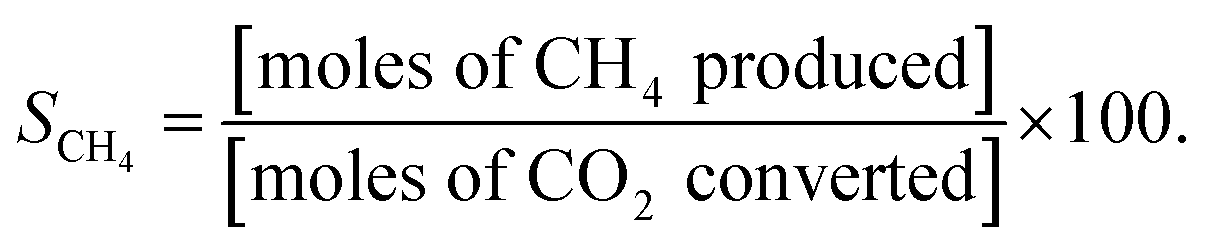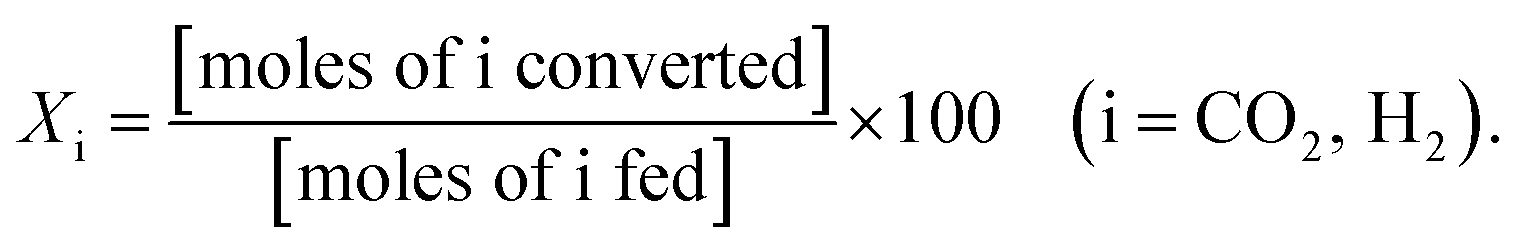DOI:
10.1039/C8RA06226A
(Paper)
RSC Adv., 2018,
8, 32095-32101
Collateral hydrogenation over proton-conducting Ni/BaZr0.85Y0.15O3−δ catalysts for promoting CO2 methanation†
Received
23rd July 2018
, Accepted 10th September 2018
First published on 17th September 2018
Abstract
Despite the importance of CO2 methanation for eco-friendly carbon-neutral fuel recycling, the current technologies, relying on catalytic hydrogenation over metal-based catalysts, face technological and economical limitations. Herein, we employ the steam hydrogenation capability of proton conductors to achieve collateral CO2 methanation over the Ni/BaZr0.85Y0.15O3−δ catalyst, which is shown to outperform its conventional Ni/Al2O3 counterpart in terms of CH4 yield (8% higher) and long-term stability (3% higher for 150 h) at 400 °C while exhibiting a CH4 selectivity above 98%. Moreover, infrared and X-ray photoelectron spectroscopy analyses reveal the appearance of distinct mobile proton-related OH bands during the methanation reaction.
Introduction
Global resource depletion and climate change are the most important ecological problems facing humanity at present, being both characterized by “carbon” as a common keyword. The combustion of fossil fuels has upset the natural balance maintained for many thousands of years, with the release of CO2 leading to climate change, e.g., global warming.1,2 Carbon neutrality focuses on the environmentally friendly recycling of excessive carbon emissions, providing a paradigm of solving the energy and environmental problems facing mankind.3,4 Among the technologies of carbon-neutral energy conversion, one of the most important ones is the methanation of CO2 with H2, or the Sabatier reaction (best performed at ∼400 °C), in which CO2 and H2 are exothermically converted to CH4 and H2O over a metal catalyst.| | |
CO2 (g) + 4H2 (g) → CH4 (g) + 2H2O (g), ΔG298 K = −113 kJ mol−1,
| (1) |
where G is the Gibbs free energy. Based on the results of studies conducted since the 1910s,5–17 the abovementioned methanation is most efficiently conducted at ∼400 °C using Ni metal-based catalysts (solely responsible for the dissociation of H2 gas) and Al2O3 supports.6–12,14,17 Recent studies have shown that the partial replacement of Ni with Ru can afford better-performing catalysts.5,8,9 However, Ni metal-based catalysts suffer from low selectivity and significant deactivation caused by the formation of nickel subcarbonyl species,5,9 with partial Ru alloying also being inappropriate for commercial applications due to the high cost of the latter metal.12 Compared to the extensively investigated metal catalysts, which play a key role in the dissociation of H2 gas, catalyst supports have been relatively underexplored due to the difficulty of utilizing catalytically active oxides at low oxygen partial pressures. Besides, the corresponding studies were merely focused on metal activity (improving reducibility6,10,14 and electron transfer by the redox couple17,18) and/or support effect (increasing metal dispersion,6,10,13 preventing metal coalescence,16,18 and improving reactant sorption7,12,17) enhancement, not being related to H2, which is the base material of the hydrogenation reaction. As a result, no new insights into and applications of the catalytic support contribution to CO2 methanation have been reported.
Herein, we propose a new multi-hydrogenation catalyst for CO2 methanation comprising Ni metal supported by proton-conductive 15 mol% Y-doped barium zirconate [BaZr0.85Y0.15O3−δ (BZY)], revealing that the above catalyst surpasses the conventionally used Ni/Al2O3 powder in terms of both CH4 yield (YCH4) and formation selectivity (SCH4, ∼100%). Using comprehensive surface analyses based on in situ Fourier transform infrared (FT-IR) and X-ray photoelectron spectroscopy (XPS) techniques, we clarify some aspects of mobile proton generation and its effect on CO2 methanation.
Experimental section
Synthesis and characterization of catalysts
Proton-conducting BZY powder (specific surface area = 12 m2 g−1) was synthesized by calcining a mixture of BaCO3 (Cerac), ZrO2 (Junsei), and Y2O3 (High Purity Chemicals) powders with the desired stoichiometric ratio at 1300 °C for 2 h, with detailed information provided in our previous paper.19,20 Spherical Al2O3 powder (Alfa Aesar, specific surface area = 28 m2 g−1) was chosen as an inert support for comparison. Ni(NO3)2·6H2O (Sigma-Aldrich) was used as a metal precursor. Ni-loaded catalysts (Ni/BZY and Ni/Al2O3) were prepared using a deposition–precipitation method followed by freeze-drying, and the obtained powders were calcined in an electric box furnace for 3 h at 600 and 400 °C, respectively. The crystal structures and morphologies of powders were examined by X-ray diffraction (XRD, D/Max-2500, Rigaku; Cu Kα) and transmission electron microscopy (TEM, Talos F200X, FEI; 200 kV) analyses. All diffraction patterns were obtained in the 2θ range of 20–80° with a scan step of 0.01° and a scan rate of 1° min−1. The specimens for TEM were prepared by dispersing the catalyst powder in ethanol and then drop-casting the solution onto a porous carbon film supported on a Cu grid. Before the installation of sample grid, it held on 10 minutes to evaporate liquid ethanol.
CO2 methanation tests of catalysts
All catalytic activity tests were performed using a gas chromatograph (7890B, Agilent) and an atmospheric-pressure fixed-bed tubular reactor. Typically, a 0.1 g powder sample was placed in the middle of the reactor held inside a tube furnace. The reactant gases were pre-mixed in the condition of H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2:Ar = 4:1:5, and the total feed flow rate was maintained at 100 mL min−1. Prior to being utilized in the methanation reaction, all catalysts were reduced by 10 mol% H2 gas balanced in Ar at 600 °C for 2 h, and their catalytic activities were evaluated in terms of YCH4, SCH4, XCO2, and XH2:
:CO2:Ar = 4:1:5, and the total feed flow rate was maintained at 100 mL min−1. Prior to being utilized in the methanation reaction, all catalysts were reduced by 10 mol% H2 gas balanced in Ar at 600 °C for 2 h, and their catalytic activities were evaluated in terms of YCH4, SCH4, XCO2, and XH2:| |
 | (2) |
| |
 | (3) |
| |
 | (4) |
Surface analyses of catalysts
In situ FT-IR spectra were recorded using a corresponding spectrometer (Nicolet 6700, Thermo Scientific) and a controlled atmospheric chamber (DiffusIR, PIKE Technologies) used to react the pre-mixed gases (H2:CO2:He = 4:1:5, total flow rate = 100 mL min−1). Additional technical details of the above FT-IR system can be found elsewhere.21 To stabilize the temperature of the reaction chamber up to 400 °C, we used He gas, which is an inert gas with the highest thermal conductivity. XPS (PHI 5000 VerasProbe, Ulvac-PHI) analyses were performed using monochromatic Al Kα (1486.6 eV) radiation, and spectral deconvolution was conducted through Gaussian–Lorentzian functions considering the chemistry of the powders.
Results and discussion
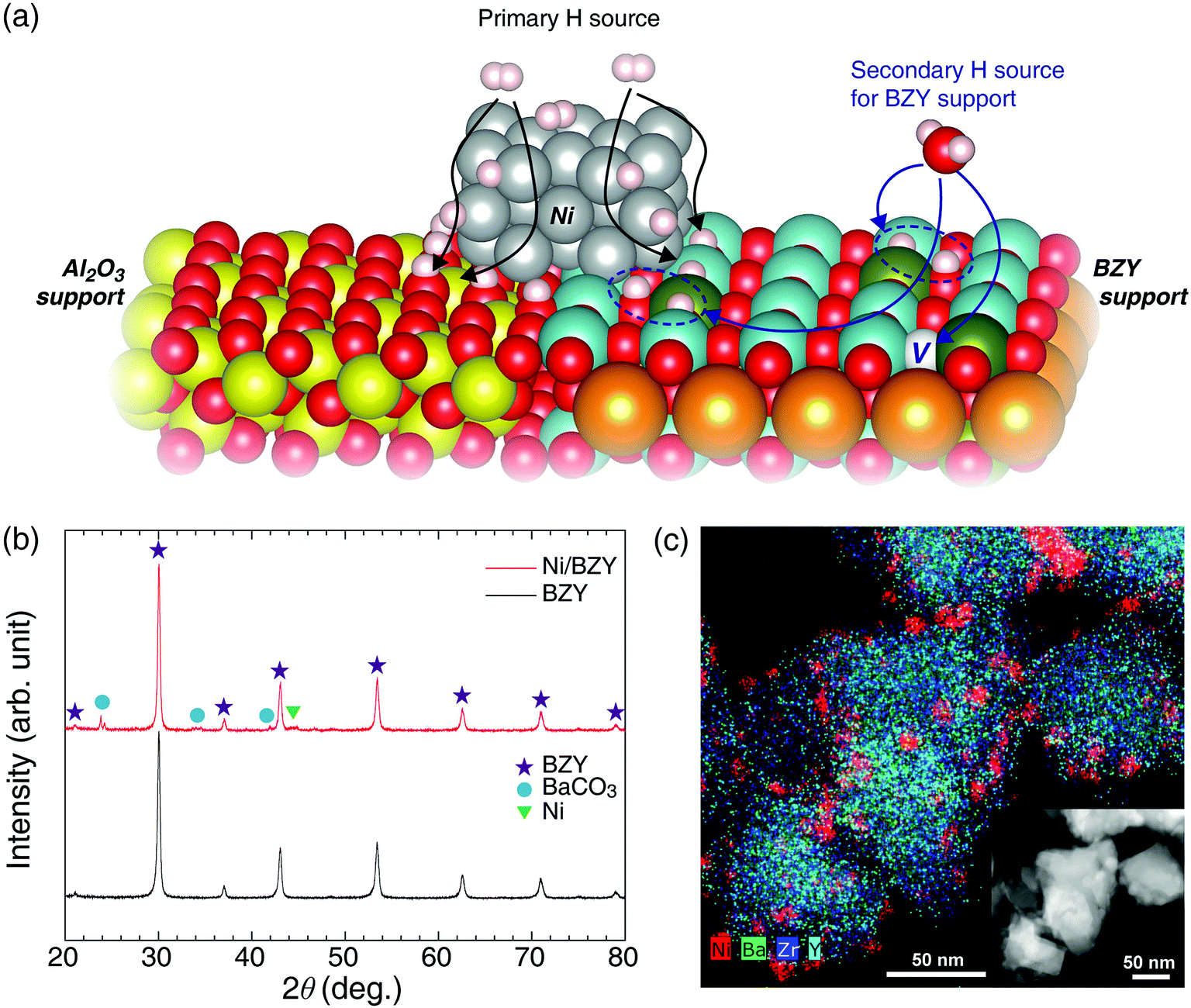
Proton-conductive ceramics are oxide-based inorganic materials capable of rapid proton transfer due to facilitating the dissociative adsorption of H2O via utilization of oxygen vacancies at 350–600 °C,22–24 with the best known examples including perovskites such as doped alkaline-earth cerates and zirconates. Based on the results of various studies conducted on electrolyte materials for low-temperature solid-oxide fuel cells,20,25 we concluded that the proton conduction process and the Sabatier reaction feature high similarity and compatibility. First, both of the above processes rely on a hydrogenation step for proton utilization. Second, in the temperature range of the Sabatier reaction, sufficient proton incorporation and conductivity are observed. Finally, both reactions take place in similar wet atmospheres. Considering that proton-conductive oxides adsorb H2O molecules to generate two mobile protons with hydroxyl group,22,23 we expected these functional supports to facilitate CO2 methanation by introducing a new hydrogenation reaction, as shown in Fig. 1(a). In previously reported studies on CO2 hydrogenation involving the Ni metal and Al2O3 support, the reaction mechanism was suggested with a formate (HCOO−) intermediate.12,21 The adsorbed CO on the catalyst is converted to CH4 due to hydrogen concentration and thermodynamic favorability at the transition state.21,26,27 In this step, sufficient hydrogenation process is very important to produce the target CH4 and to suppress the CO side reactions. Thus, in contrast to the inert Al2O3 support, BZY provides an additional hydrogenation pathway for CO2 methanation, along with the main one occurring on Ni metal, which results in the improvement of the overall reaction capacity.
 |
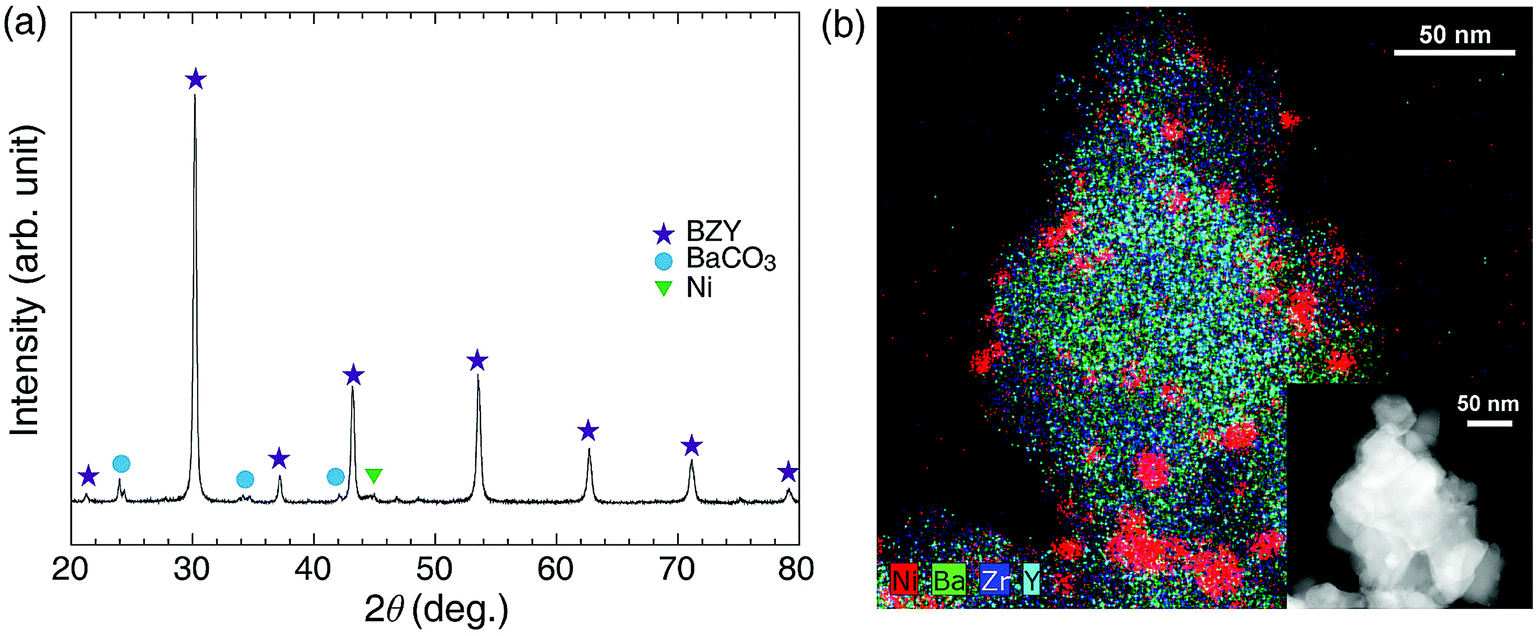
| | Fig. 1 (a) Schematic illustration of different hydrogenation processes occurring on Ni/BZY (right) and Ni/Al2O3 (left) surfaces. (b) XRD patterns of as-prepared BZY support and the Ni/BZY catalyst. (c) TEM and EDS mapping images of as-prepared Ni/BZY, with Ni, Ba, Zr, and Y elements indicated by red, green, blue, and cyan colors, respectively. | |
As shown in Fig. 1(b) and (c), the BZY support, one of the most reliable highly proton-conductive materials, was herein used to prepare a novel CO2 methanation catalyst. Fig. 1(b) presents the XRD patterns of the BZY support and 5 wt% Ni/BZY catalyst powders, revealing that the former pattern featured only a single perovskite (BaZrO3) phase and no secondary phases, whereas an additional Ni metal peak (at 44.5°) was observed for Ni/BZY, indicating that Ni was well compatible with the BZY support. Note that although BaCO3 peaks (at 23.9, 24.3, 34.6, and 42.0°) were detected in the XRD spectra of Ni/BZY prepared via urea hydrolysis, the effects of the above compounds on the overall CO2 methanation reaction were negligible (for both BZY and BaCO3/BZY, YCH4 = 0 at 400 °C; Fig. S1†). Fig. 1(c) shows the TEM and energy dispersive X-ray spectroscopy (EDS) images of as-prepared Ni/BZY powder, with the homogeneous distribution of BZY constituent elements indicating that no secondary phase was present in the as-prepared single-phase BZY, as confirmed by XRD results. Moreover, the BZY surface featured well distributed spherical Ni nanoparticles with an average diameter of ∼10.68 nm (standard deviation = 2.64 nm). ESI XRD and TEM data for comparison samples (Al2O3 support and Ni/Al2O3 powders) are provided in Fig. S2.†
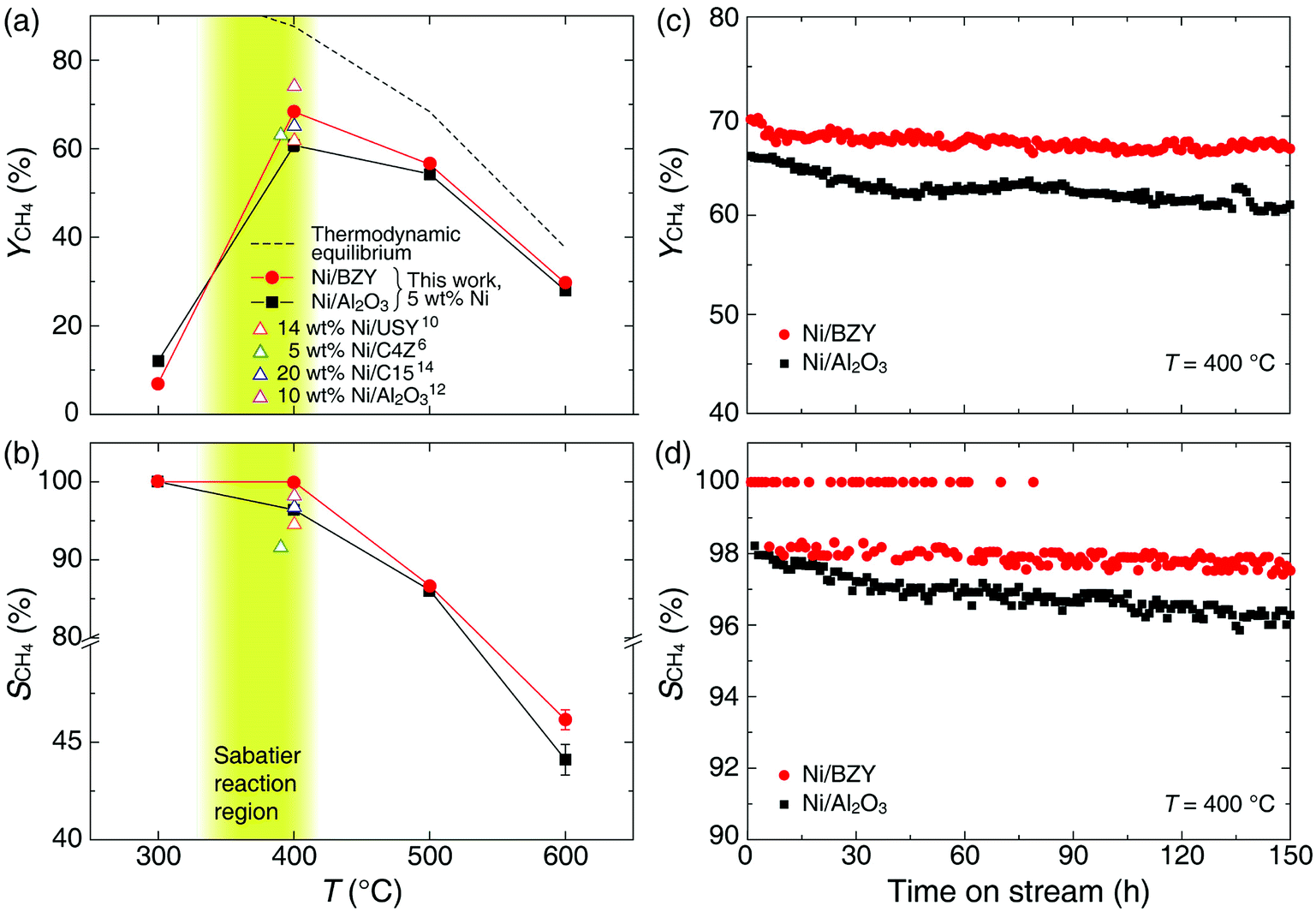
As is well known, low operating temperatures are desirable for achieving higher YCH4 and SCH4, since the Sabatier reaction is thermodynamic equilibrium-limited.15 Fig. 2(a) and (b) shows the temperature-dependent YCH4 and SCH4 values of Ni/BZY and Ni/Al2O3 obtained by gas chromatography analysis of reaction products formed in a fixed-bed tubular reactor, revealing that Ni/BZY achieved a maximum YCH4 = 68.33% at 400 °C, while the conventional Ni/Al2O3 featured a smaller value of YCH4 = 60.76%. Assuming that hydrogenation of Ni metal in the Ni/Al2O3 catalyst is the only hydrogen source, the increase in YCH4 at Ni/BZY is the results of new hydrogenation pathway by BZY support (about 12% of YCH4). Thus, the Ni/BZY system described herein achieved a substantially higher YCH4 than previously reported catalysts (only Ni metal and H2/CO2 ∼ 4 gas conditions are considered).6,10,12,14 The progress of CO2 methanation was gradually suppressed with increasing temperature due to the reverse water-gas shift (RWGS) reaction becoming thermodynamically dominant at T > 400 °C. In addition, Ni/BZY and Ni/Al2O3 exhibited different SCH4 behavior, i.e., the former system attained SCH4 = 100%, while the latter one achieved a relatively low SCH4 of 96.43% due to the production of CO gas by the RWGS reaction.15 ESI data of XCO2 and XH2 are also provided in Fig. S3.† The Ni/BZY catalyst showed higher performance than the Ni/Al2O3 catalyst: about 3.84% and 6.62% XCO2 and XH2, respectively. To investigate the long-term stability of both catalysts, we subjected them to 150 h CO2 methanation exposure at 400 °C, with the obtained results shown in Fig. 2(c) and (d). After 150 h, the YCH4 of Ni/BZY was reduced by ∼4% compared to the initial value, while that of Ni/Al2O3 was reduced by ∼7%. However, the corresponding long-term selectivity changes were more noticeable, i.e., for Ni/Al2O3, SCH4 gradually decreased to 96% of the original value after 150 h, whereas Ni/BZY maintained SCH4 ≈ 100% during the first 79 h, finally reaching SCH4 = 98% after 150 h.
 |
| | Fig. 2 Temperature-dependent (a) YCH4 and (b) SCH4 values of Ni/BZY and Ni/Al2O3. The dashed line represents the thermodynamic equilibrium performance of YCH4 under the chosen experimental conditions (H2/CO2 = 4.0). The yellow-green-colored area corresponds to the temperature range of the Sabatier reaction. USY, C4Z, and C15 denote ultra-stable Y zeolite, Ce0.8Zr0.2O2, and composite supports (55% γ-Al2O3 and 15% equivalent loading of ZrO2, TiO2, CeO2), respectively. Long-term stability test of Ni/BZY and Ni/Al2O3 performed at H2/CO2 = 4.0 and 400 °C: (c) YCH4 and (d) SCH4. | |
As shown in Fig. 3, XRD and TEM-EDS analyses of Ni/BZY catalysts subjected to 150 h exposure to CO2 methanation conditions demonstrated that no noticeable carbon deposition occurred, with performance degradation ascribed to the slight coarsening of Ni metal nanoparticles (∼1 nm diameter increase). ESI XRD and TEM data of a comparison sample (Ni/Al2O3 powders after 150 h CO2 methanation) are also provided in Fig. S4.† Combining the above results, we concluded that the CO2 methanation performance was significantly influenced by the proton conductivity of the support.
 |
| | Fig. 3 XRD pattern (a) and TEM-EDS images (b) of Ni/BZY subjected to 150 h CO2 methanation at 400 °C. The average particle diameter of Ni on the BZY support equaled ∼11.50 nm (standard deviation = 4.04 nm). The red, green, blue, and cyan colors correspond to the elements Ni, Ba, Zr, and Y, respectively. | |
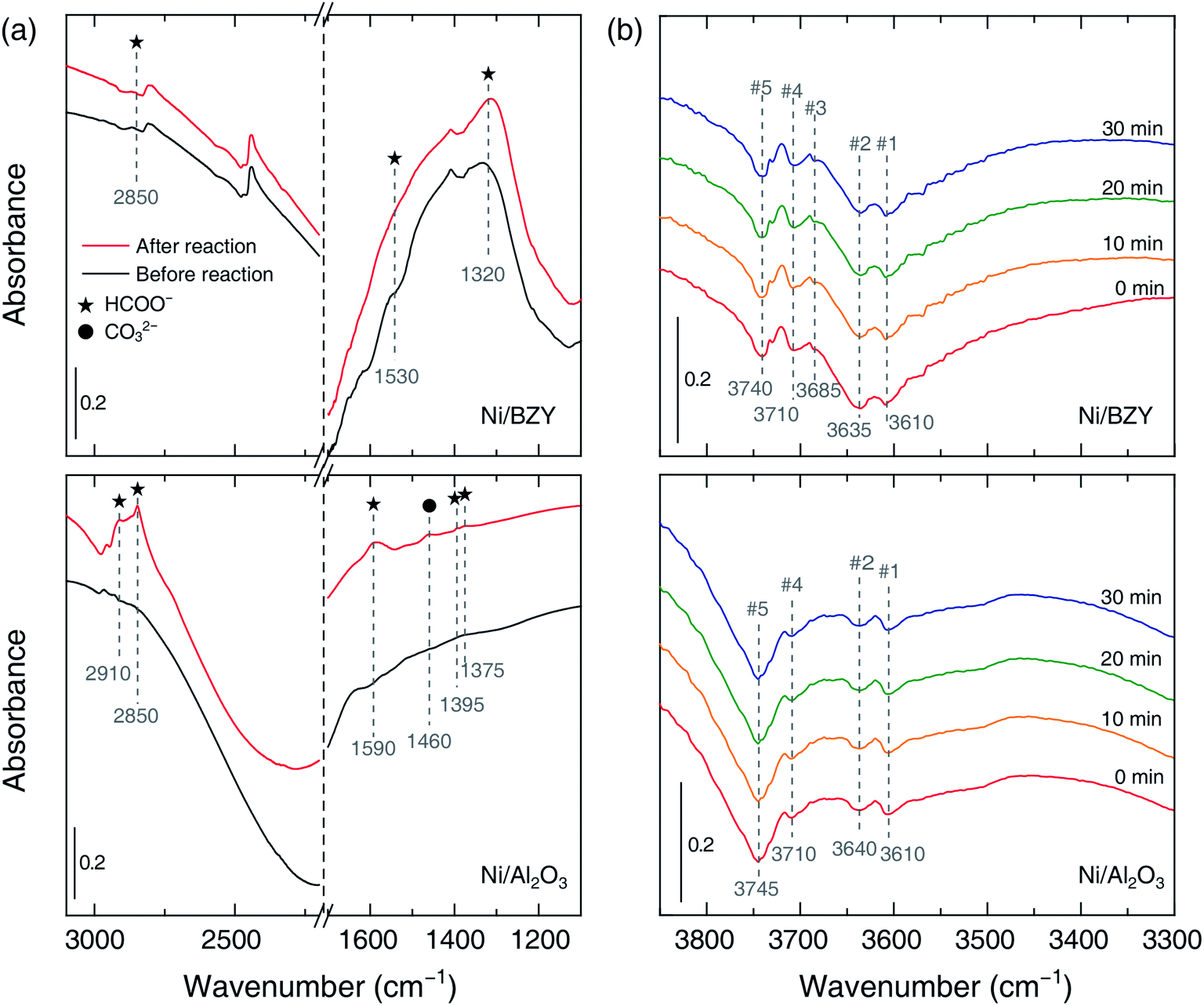
The role of the catalyst support was investigated by in situ FT-IR characterization of species adsorbed on the catalyst surface during methanation. Fig. 4(a) shows that the common IR bands at 2910–2850 and 1590–1320 cm−1 appearing in adsorbate spectra before and after the reaction can be ascribed to formate,21 which is suggested to be the main intermediate of CO2 methanation regardless of the support material. However, carbonate (CO32−, 1460 cm−1) was also observed on the surface of the Ni/Al2O3 catalyst after the reaction, suggesting that CO generation (resulting in lower SCH4) could be ascribed to a side reaction of the above species. Fig. 4(b) shows the time-dependent evolution of OH bands in the CO2 methanation mixture obtained using Ni/BZY and Ni/Al2O3 catalysts at 400 °C. Notably, four OH bands (#1, 2, 4, 5) at 3750–3600 cm−1 were observed for Ni/Al2O3, while a total of five OH bands (#1, 2, 3, 4, 5) were observed for Ni/BZY. As reported in literature,16,17 multiple OH bands are observed for Ni-loaded catalysts due to their use resulting in sufficient hydrogenation. Specifically, band #5 observed for all catalysts containing Ni metal was attributed to the hydroxyl moieties of the reaction sites generated at the Ni metal/support interface. In addition, it is well-known that low-wavenumber bands with low binding energies can be considered to have a multi-fold bond,28 i.e., a bond of a single OH species on the surface bound to a number of metal atoms. From the viewpoint of such reaction energy, band #5, exhibiting the largest wavenumber among the five OH bands, corresponded to species having the smallest number of surface bonds and thus most actively contributing to CO2 methanation. Therefore, the observation of the above band in the case of both Ni/BZY and Ni/Al2O3 was ascribed to the significant CO2 methanation progress achieved by these catalysts. However, Ni/BZY and Ni/Al2O3 could be discriminated by the presence/absence of the #3 band, which seemed to reflect the role of the proton-conductive support.
 |
| | Fig. 4 In situ FT-IR spectra of species adsorbed on Ni/BZY and Ni/Al2O3 during CO2 methanation at 400 °C. (a) Analysis of formate (star) and carbonate (filled circle) species before/after CO2 methanation. (b) Time-dependent analysis of OH groups contained in the flowing reactant gases at 400 °C. Red, yellow, green, and blue lines correspond to time steps of 0, 10, 20, and 30 min, respectively. | |
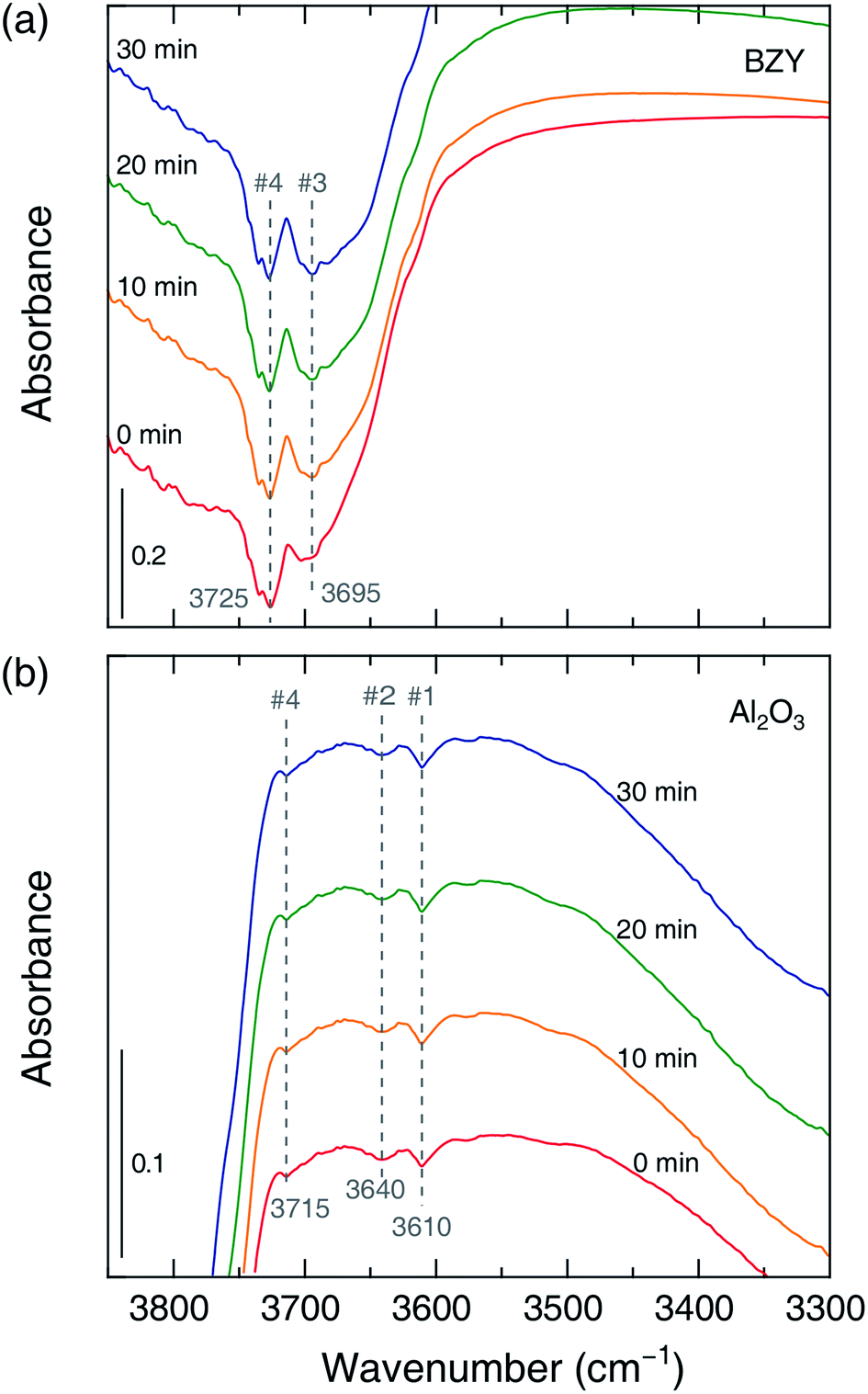
To confirm that hydrogenation process differences reflect the different roles of the proton-conductive support, we performed in situ FT-IR analysis during slow hydrogenation on the pure support. As shown in Fig. 4(b) and 5, the #3 band gained intensity with time in the case of the BZY support, in clear contrast to the results obtained for Al2O3, which allowed us to conclude that the above band corresponded to mobile protons produced due to the proton-conducting capability of BZY. Moreover, the additional OH production pathway was expected to facilitate CO2 methanation on Ni/BZY catalysts. Therefore, the difference of CO2 methanation performance between the two catalysts originated from the presence/absence of mobile protons, i.e., was related to the occurrence of additional hydrogenation processes on the proton-conducting support.
 |
| | Fig. 5 Time-dependent FT-IR spectra of OH groups recorded under the conditions of reactant gas flow at 400 °C [(a): BZY, (b): Al2O3]. Red, yellow, green, and blue lines correspond to times of 0, 10, 20, and 30 min, respectively. | |
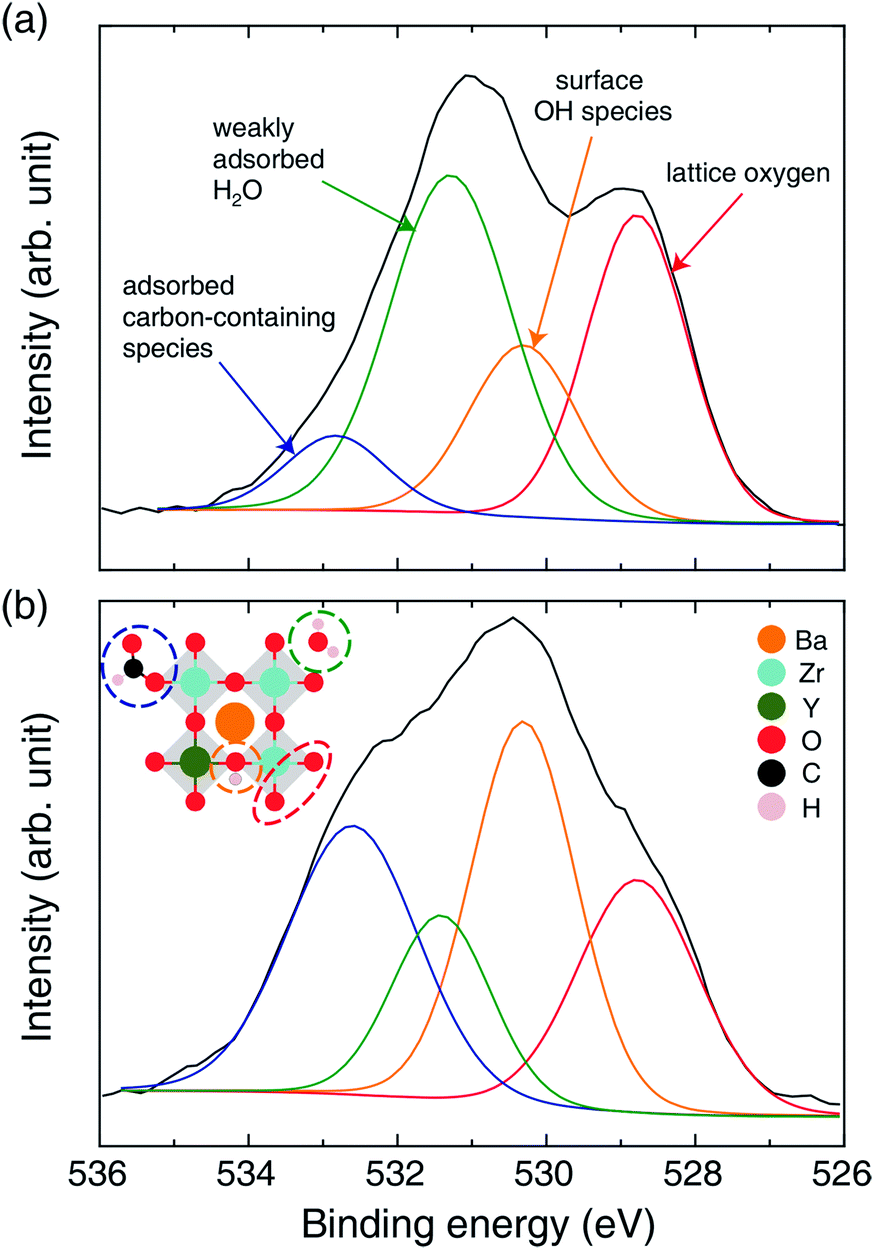
Tangible evidence of OH group formation from mobile protons on the BZY surface was provided by the O 1s XPS spectra of Ni/BZY (see Fig. 6). The peak centered at ∼528.5 eV was assigned to the lattice oxygen of BZY,29–31 whereas those at ∼531.5 and ∼533 eV were ascribed to weakly adsorbed H2O and adsorbed carbon-containing species such as BaCO3 and formate, respectively.32,33 Interestingly, the peak at ∼530.5 eV gained intensity with progressing CO2 methanation, thus probably corresponding to the surface hydroxyl groups associated with the mobile proton.33 In order to clarify the characteristics of OH groups in the XPS spectra of the proton-conductive support, Ni/Al2O3 was subjected to XPS analysis for comparison, with the results shown in Fig. S5.† In the above XPS spectra, the peak of Al–O bond lattice oxygen appeared at ∼531 eV, and ignorable OH peaks were observed, in agreement with the results of O 1s XPS analysis of conventional Al2O3 reported in a number of documents.34–36 Thus, the decreased amount of OH species observed for Ni/Al2O3 was in stark contrast to the large number of hydroxyl species observed on the surface of Ni/BZY under identical conditions. Thus, the proton-conductive BZY support clearly provided an alternative hydrogenation path, which improved the performance of Ni/BZY in the CO2 methanation reaction.
 |
| | Fig. 6 O 1s core level XPS spectra of Ni/BZY (a) before and (b) after methanation. Red, yellow, green, and blue lines correspond to lattice oxygen, surface OH species, weakly adsorbed H2O, and adsorbed carbon-containing species (contaminant, BaCO3 and formate), respectively. | |
Conclusions
In summary, we successfully demonstrated that the CO2 methanation performance of supported metal catalysts can be improved by collateral hydrogenation over a proton-conductive support. Since the proton-conductive BZY can incorporate H2O molecules and generate mobile protons in a wet atmosphere at 350–600 °C, the formation of mobile protons on this oxide support can provide an additional hydrogen source and influence the CO2 hydrogenation mechanism. Thus, Ni/BZY showed an 8% higher CH4 yield at 400 °C and a 3% higher long-term stability over 150 h than Ni/Al2O3. Interestingly, the distinct time-dependent FT-IR band of hydroxyl groups corresponding to mobile protons was only observed for BZY. In addition, according to XPS results, the amount of surface OH species increased during CO2 methanation, evidencing the generation of mobile protons in the proton-conductive support. Such understanding and exploitation of proton-conductive supports is expected to improve the performance of eco-friendly carbon-neutral CO2 methanation reactions and enhance their industrial applicability.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was conducted under the Global Frontier R&D Program on Center for Multiscale Energy Systems (Grant No. NRF-2015M3A6A7065442) the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT, Republic of Korea. This work also supported in part by the Institutional Research Program of the Korea Institute of Science and Technology (2E28040).
References
- G. A. Florides and P. Christodoulides, Environ. Int., 2009, 35, 390–401 CrossRef PubMed.
- D. Mattia, M. D. Jones, J. P. O'Byrne, O. G. Griffiths, R. E. Owen, E. Sackville, M. McManus and P. Plucinski, ChemSusChem, 2015, 8, 4064–4072 CrossRef PubMed.
- S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC.
- N. Yan and K. Philippot, Curr. Opin. Chem. Eng., 2018, 20, 86–92 CrossRef.
- D. J. Elliott and J. H. Lunsford, J. Catal., 1979, 57, 11–26 CrossRef.
- W. Cai, Q. Zhong and Y. Zhao, Catal. Commun., 2013, 39, 30–34 CrossRef.
- A. Borgschulte, N. Gallandat, B. Probst, R. Suter, E. Callini, D. Ferri, Y. Arroyo, R. Erni, H. Geerlings and A. Zuttel, Phys. Chem. Chem. Phys., 2013, 15, 9620–9625 RSC.
- S. Hwang, J. Lee, U. G. Hong, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, J. Ind. Eng. Chem., 2013, 19, 698–703 CrossRef.
- W. Zhen, B. Li, G. Lu and J. Ma, RSC Adv., 2014, 4, 16472–16479 RSC.
- I. Graça, L. V. González, M. C. Bacariza, A. Fernandes, C. Henriques, J. M. Lopes and M. F. Ribeiro, Appl. Catal., B, 2014, 147, 101–110 CrossRef.
- G. Garbarino, D. Bellotti, P. Riani, L. Magistri and G. Busca, Int. J. Hydrogen Energy, 2015, 40, 9171–9182 CrossRef.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178–184 CrossRef.
- H. H. Shin, L. Lu, Z. Yang, C. J. Kiely and S. McIntosh, ACS Catal., 2016, 6, 2811–2818 CrossRef.
- S. Abate, C. Mebrahtu, E. Giglio, F. Deorsola, S. Bensaid, S. Perathoner, R. Pirone and G. Centi, Ind. Eng. Chem. Res., 2016, 55, 4451–4460 CrossRef.
- X. Su, J. Xu, B. Liang, H. Duan, B. Hou and Y. Huang, J. Energy Chem., 2016, 25, 553–565 CrossRef.
- J. Xu, X. Su, H. Duan, B. Hou, Q. Lin, X. Liu, X. Pan, G. Pei, H. Geng, Y. Huang and T. Zhang, J. Catal., 2016, 333, 227–237 CrossRef.
- J. A. H. Dreyer, P. Li, L. Zhang, G. K. Beh, R. Zhang, P. H. L. Sit and W. Y. Teoh, Appl. Catal., B, 2017, 219, 715–726 CrossRef.
- Y. Zeng, H. Ma, H. Zhang, W. Ying and D. Fang, Fuel, 2014, 137, 155–163 CrossRef.
- S. M. Choi, J.-H. Lee, J. Hong, H. Kim, K. J. Yoon, B.-K. Kim and J.-H. Lee, Int. J. Hydrogen
Energy, 2014, 39, 7100–7108 CrossRef.
- S. M. Choi, J.-H. Lee, J. Hong, K. J. Yoon, J.-W. Son, B.-K. Kim, H.-W. Lee and J.-H. Lee, J. Power Sources, 2016, 332, 299–304 CrossRef.
- S. Choi, B.-I. Sang, J. Hong, K. J. Yoon, J.-W. Son, J.-H. Lee, B.-K. Kim and H. Kim, Sci. Rep., 2017, 7, 41207 CrossRef PubMed.
- Y. Yamazaki, P. Babilo and S. M. Haile, Chem. Mater., 2008, 20, 6352–6357 CrossRef.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC.
- J.-S. Kim and Y.-C. Kim, Solid State Ionics, 2017, 306, 137–141 CrossRef.
- K. Bae, D. Y. Jang, H. J. Choi, D. Kim, J. Hong, B.-K. Kim, J.-H. Lee, J.-W. Son and J. H. Shim, Nat. Commun., 2017, 8, 14553 CrossRef PubMed.
- D. E. Peebles, D. W. Goodman and J. M. White, J. Phys. Chem., 1983, 87, 4378–4387 CrossRef.
- A. Beuls, C. Swalus, M. Jacquemin, G. Heyen, A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 2–10 CrossRef.
- C. Binet, M. Daturi and J.-C. Lavalley, Catal. Today, 1999, 50, 207–225 CrossRef.
- W. Sun, M. Liu and W. Liu, Adv. Energy Mater., 2013, 3, 1041–1050 CrossRef.
- W. Sun, Z. Shi, M. Liu, L. Bi and W. Liu, Adv. Funct. Mater., 2014, 24, 5695–5702 CrossRef.
- C. Aruta, C. Han, S. Zhou, C. Cantoni, N. Yang, A. Tebano, T.-L. Lee, C. Schlueter and A. Bongiorno, J. Phys. Chem. C, 2016, 120, 8387–8391 CrossRef.
- D. Jampaiah, K. M. Tur, S. J. Ippolito, Y. M. Sabri, J. Tardio, S. K. Bhargava and B. M. Reddy, RSC Adv., 2013, 3, 12963–12974 RSC.
- P. Venkataswamy, D. Jampaiah, K. N. Rao and B. M. Reddy, Appl. Catal., A, 2014, 488, 1–10 CrossRef.
- B. V. Crist, Handbooks of Monochromatic XPS Spectra – 5 Volume Series, XPS International LLC, CA, 2005 Search PubMed.
- J. Peng, Q. Sun, Z. Zhai, J. Yuan, X. Huang, Z. Jin, K. Li, S. Wang, H. Wang and W. Ma, Nanotechnology, 2013, 24, 484010 CrossRef PubMed.
- A. Celebioglu, S. Vempati, C. Ozgit-Akgun, N. Biyikli and T. Uyar, RSC Adv., 2014, 4, 61698–61705 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06226a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ji-Won Son
a,
Ji-Won Son