
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmmonia decomposition over Ni catalysts supported on perovskite-type oxides for the on-site generation of hydrogen†
Kaname Okura,
Kazunari Miyazaki,
Hiroki Muroyama *,
Toshiaki Matsui and
Koichi Eguchi*
*,
Toshiaki Matsui and
Koichi Eguchi*
Department of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: eguchi@scl.kyoto-u.ac.jp; muroyama.hiroki.5c@kyoto-u.ac.jp
First published on 17th September 2018
Abstract
Ammonia decomposition has attracted increasing attention as a promising process for the on-site generation of hydrogen. In this study, Ni catalysts supported on perovskite-type oxides (ABO3) were prepared and the activity for ammonia decomposition was examined. The Ni/ANbO3 (A = Na and K) and Ni/AEMnO3 (AE = Ca, Sr, and Ba) catalysts were less effective for this reaction. Meanwhile, the Ni/REAlO3 (RE = La, Sm, and Gd) catalysts exhibited relatively high activity. For Ni/AETiO3 and Ni/AEZrO3, the performance strongly depended on the A-site element of the perovskite-type oxides, and the Sr and Ba elements were more effective than the Ca one in the respective series. The catalytic activity for Ni/AEZrO3 was higher than Ni/AETiO3 in the case of the same alkaline earth element, and Ni/BaZrO3 was the most active among the samples investigated in this work. For these series, the order in the performance corresponded well with that in the basic property. The nitrogen desorption profiles revealed that the evolution of nitrogen atoms, which is one of the kinetically slow steps, effectively proceeded for Ni/SrZrO3 and Ni/BaZrO3 compared with the conventional Ni catalysts. This promotion effect would be ascribed to the strong basic properties of the SrZrO3 and BaZrO3 supports, resulting in the high activity of Ni/SrZrO3 and Ni/BaZrO3 for ammonia decomposition.
Introduction
Nowadays, the establishment of fuel cell systems is strongly required because of concerns about serious environmental issues and the depletion of fossil fuels. Fuel cells are power generation devices operating without harmful emissions, and their energy conversion efficiency is higher than that of conventional internal-combustion engines. Hydrogen is utilized as a primary fuel source for fuel cells. However, the direct storage and transportation of hydrogen are major obstacles for the commercialization of fuel cells due to its low volumetric density (81.8 g m−3) and boiling point (−252.8 °C). Thus, the feasibility of the compounds incorporating hydrogen as a hydrogen storage material has been discussed, and is called as a hydrogen carrier. These carriers can be transported to the consumption area, and reformed or decomposed to generate hydrogen on the spot.1,2Among the candidates for the hydrogen carriers, ammonia has been a subject of attention owing to several interesting features. Ammonia has significantly high gravimetric hydrogen density (17.6 wt%) and energy density (3000 W h kg−1) as compared with other hydrogen carriers. In addition, the liquefaction of ammonia proceeds under mild conditions (−33.4 °C at atmospheric pressure or 8.46 atm at 20 °C). Moreover, the hydrogen generation via ammonia decomposition does not emit any CO and CO2. The produced gas from this reaction is preferable as a fuel for fuel cells operative at low temperatures such as polymer electrolyte fuel cells (PEFCs) since the Pt electrode is readily poisoned by CO. Furthermore, the handling and the infrastructure for ammonia are well established because it is one of the most fundamental industrial chemicals.3–7
The ammonia decomposition reaction endothermically proceeds as described in eqn (1). According to thermodynamic calculation, the equilibrium ammonia conversion achieves above 99% at 400 °C under a pressure of 1 atm.7
| NH3 → 1/2N2 + 3/2H2 ΔH° = +46 kJ mol−1 | (1) |
The catalytic ammonia decomposition involves the adsorption of ammonia, the stepwise hydrogen dissociation of adsorbed ammonia, and the desorption of nitrogen and hydrogen. The density functional theory (DFT) calculations and kinetic analysis supposed that the first and second dehydrogenation of ammonia and/or the combinative desorption of nitrogen atoms were the kinetically slow steps.8–13
The previous studies have demonstrated that various metals (Ru,14–28 Ir,15 Ni,14,15,28–39 Rh,14 Pt,14 Pd,14 Co,40–42 and Fe14,43), alloys (Mo–Fe,44 Ni–Fe,45 and Ni–Pt46), nitrides (MoNx,47–50 FeMoNx,49 NiMoNx,48,49,51 and CoMoNx49,50,52,53), and carbides (WC54 and VCx55) served as the catalysts for ammonia decomposition. Among them, the Ru metal is widely known as the most active catalyst. Until now, much effort has been devoted for the development of the highly active Ru catalysts.16–28 However, the utilization of precious metal catalyst is unfavorable due to its limited availability. Thus, in recent years, the Ni metal has attracted much interest as the alternative catalysts owing to its low cost and the highest activity for ammonia decomposition among non-noble metals.4–7
Various materials have been investigated as supports of Ni catalyst for ammonia decomposition. The supports with high surface area such as mesoporous silica,30 Ce0.8Zr0.2O2,38 and multi-wall carbon nanotube (MWCNT)39 were preferable for the Ni-based catalytic system. In our study, the rare-earth oxides were effective as the support materials although their surface areas were significantly small (<10 m2 g−1).36 This was because hydrogen, which is well known as the inhibitive species for ammonia decomposition, readily desorbed from the Ni metal supported on these oxide materials. Moreover, the promotion effect of the additive species has been also studied for Ni catalysts. The modification by Sr and Ba species or rare-earth oxides improved the performance of supported Ni catalysts.32–35,37 Based on these results, the presence of basic components is expected to promote ammonia decomposition over Ni catalysts.
Thus, in this work, we focused on the perovskite-type oxides including alkaline, alkaline earth, and rare-earth elements, and employed them as the support material to develop highly active Ni catalysts. The perovskite-type oxide is generally expressed as a chemical formula of ABO3, in which two types of sites, A site and B site, are occupied by cations. These materials are known to exhibit interesting properties as the support of catalysts for various chemical reactions. Wang et al. reported that the BaZrO3-supported Ru catalyst showed the outstanding performance for ammonia synthesis.56 Urasaki et al. showed that the Ni catalysts supported on the perovskite-type oxides had the superior long-term stability as compared with the conventional catalysts in the steam reforming of hydrocarbons.57,58 In this study, the effective species in the perovskite-type oxides for ammonia decomposition were discussed by systematically changing the A-site and B-site elements.
Experimental
Sample preparation
The perovskite-type oxides were fabricated by the solid state reaction method or the citric acid complex method. ANbO3 (A = Na and K) and AETiO3 (AE = Ca, Sr, and Ba) were prepared by the former method, while REAlO3 (RE = La, Sm, and Gd), AEMnO3, and AEZrO3 by the latter one. In the solid state reaction method, the following carbonates or metal oxides were used as the starting materials; Na2CO3, K2CO3, CaCO3, SrCO3, BaCO3, Nb2O5 (Wako Pure Chemical Industries, Ltd.), and TiO2 (Sigma-Aldrich, Co.). These compounds were stoichiometrically mixed and ground in ethanol overnight. After the evaporation of ethanol, the obtained powder was calcined at 1100 °C for 5 h in air. In the case of the citric acid complex method, the corresponding nitrate salts were employed as the starting compounds; Ca(NO3)2·6H2O, Sr(NO3)2, Ba(NO3)2, La(NO3)3·6H2O, Sm(NO3)3·6H2O, Gd(NO3)3·6H2O, Al(NO3)3·9H2O, Mn(NO3)2·6H2O, and ZrO(NO3)2·2H2O (Wako Pure Chemical Industries, Ltd.). The metal nitrates with stoichiometric ratios were dissolved in distilled water and the solution was stirred at ca. 60 °C for 1 h. Then, citric acid (Wako Pure Chemical Industries, Ltd.) was added to this solution at a 1.5 molar times of total metal cations. Subsequently, the pH in the solution was adjusted to 8.0 by the 28% NH3 aqueous solution (Wako Pure Chemical Industries, Ltd.). After the evaporation of water at 90 °C, the precursor was heated at 350 °C for 3 h. The resulting powder was calcined at 1100 °C for 5 h in air.The supported Ni catalysts were prepared by the impregnation method. As the support material, Nb2O5, TiO2, Al2O3 (AKP G-015, Sumitomo Chemical Co., Ltd.), MnO2 (Wako Pure Chemical Industries, Ltd.), ZrO2 (Sigma-Aldrich, Co.), and the prepared perovskite-type oxides were used. Ni(NO3)2·6H2O (Wako Pure Chemical Industries, Ltd.) was employed as a nickel source. The nickel nitrate was dissolved in the distilled water and mixed with the support oxide. After the nickel nitrate solution was evaporated on a steam bath at 80 °C, the obtained precursor was calcined at 600 °C for 5 h in air. The Ni metal loading was fixed at 40 wt%.
Characterization
X-ray diffraction (XRD) measurement was carried out for the as-calcined and reduced catalysts on X-ray diffractometer (Rigaku, Ultima IV) at a setting of 40 kV and 40 mA with a scanning rate of 2° min−1. The BET surface area of the support materials was examined by N2 physisorption at −196 °C (BEL Japan, Bellsorp-miniII). Before the measurement, the samples were treated at 300 °C for 30 min under vacuum. The Ni surface area was estimated by the CO pulse measurement (BEL Japan, BELCAT-B) based on the assumption that CO molecules adsorbed on Ni surface atoms in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Prior to the CO adsorption, the catalysts were reduced at 600 °C for 2 h. To clarify the basic property of the Ni catalysts, the CO2 temperature-programmed desorption (CO2-TPD) measurement was performed (BEL Japan, BELCAT-B). The catalyst was pretreated in pure O2 at 800 °C for 1 h, and subsequently in pure H2 at the same temperature for 2 h. After the exposure to CO2 at 50 °C for 1 h, the samples were heated in the He flow at a heating rate of 5 °C min−1 (flow rate: 30 ml min−1). The desorbed CO2 was confirmed by a thermal conductive detector (TCD). To discuss the desorption process of nitrogen in ammonia decomposition, the ammonia temperature-programmed surface reaction (NH3-TPSR) measurement was carried out (BEL Japan, BELCAT-A). The pretreatment condition was the same as that in the CO2-TPD measurement except for the reactant adsorption step. Ammonia was supplied at 50 °C for 1 h, followed by the gas replacement by Ar for 5 h. The desorption behavior of N2 in a heating process at a rate of 5 °C min−1 in Ar (flow rate: 30 ml min−1) was monitored by an on-line mass spectrometer (Pfeiffer Vacuum, OmniStar GSD320).
:1. Prior to the CO adsorption, the catalysts were reduced at 600 °C for 2 h. To clarify the basic property of the Ni catalysts, the CO2 temperature-programmed desorption (CO2-TPD) measurement was performed (BEL Japan, BELCAT-B). The catalyst was pretreated in pure O2 at 800 °C for 1 h, and subsequently in pure H2 at the same temperature for 2 h. After the exposure to CO2 at 50 °C for 1 h, the samples were heated in the He flow at a heating rate of 5 °C min−1 (flow rate: 30 ml min−1). The desorbed CO2 was confirmed by a thermal conductive detector (TCD). To discuss the desorption process of nitrogen in ammonia decomposition, the ammonia temperature-programmed surface reaction (NH3-TPSR) measurement was carried out (BEL Japan, BELCAT-A). The pretreatment condition was the same as that in the CO2-TPD measurement except for the reactant adsorption step. Ammonia was supplied at 50 °C for 1 h, followed by the gas replacement by Ar for 5 h. The desorption behavior of N2 in a heating process at a rate of 5 °C min−1 in Ar (flow rate: 30 ml min−1) was monitored by an on-line mass spectrometer (Pfeiffer Vacuum, OmniStar GSD320).
The Ni particle size distribution and the morphology were examined by a transmission electron microscope (TEM, JEM-2100F, JEOL) equipped with an energy dispersive X-ray spectrometer (EDS, JED-2300T, JEOL). Prior to the analysis, the Ni catalysts were reduced at 600 °C for 2 h in 50% H2/Ar. The average Ni particle size of each sample was estimated from 150–180 particles.
Catalytic activity
The catalytic ammonia decomposition reaction was performed in a fixed-bed flow reactor. The catalyst (10–18 mesh, 300 mg) was placed in the center of quartz tube, and reduced at 600 °C for 2 h in 50% H2/Ar (flow rate: 80 ml min−1). After the cooling down to 350 °C in Ar, pure NH3 was supplied at the weight/flow (W/F) ratio of 0.60 g s cm−3 under an atmospheric pressure. The catalytic activity was evaluated in a heating process. The stability test was carried out for 35 h at 550 °C and W/F = 0.18 g s cm−3. The flow rate of outlet gas was measured by a soap-film flow meter (HORIBA STEC) after removing unconverted ammonia by diluted sulfuric acid. The conversion of ammonia was calculated by the following equation with an assumption that the ratio of produced nitrogen to hydrogen was 1:3.| NH3 conversion/% = (Fout/2Fin) × 100 | (2) |
Results and discussion
Perovskite-type oxide support
The crystal structure of the Ni catalysts supported on the perovskite-type oxide was examined by XRD analysis. As the representative samples in each system, the XRD patterns of the as-calcined and reduced Ni/NaNbO3, Ni/LaAlO3, Ni/CaMnO3, Ni/SrTiO3, and Ni/BaZrO3 catalysts are shown in Fig. 1(a) and (b), respectively. For the as-calcined Ni catalysts, the characteristic peaks assigned to the phases of the corresponding support oxide and nickel oxide were confirmed. After the reduction treatment at 600 °C, nickel oxide was changed to metallic nickel. As for the perovskite-type oxides, NaNbO3, LaAlO3, SrTiO3, and BaZrO3 were stable under the reducing atmosphere, while CaMnO3 decomposed to various compounds. The similar tendency was observed in the XRD patterns of the other Ni/ANbO3, Ni/REAlO3, Ni/AEMnO3, Ni/AETiO3, and Ni/AEZrO3 catalysts (see Fig. S1 and S2 in ESI†). This result suggests that the AEMnO3 oxides were inappropriate as the support material for ammonia decomposition.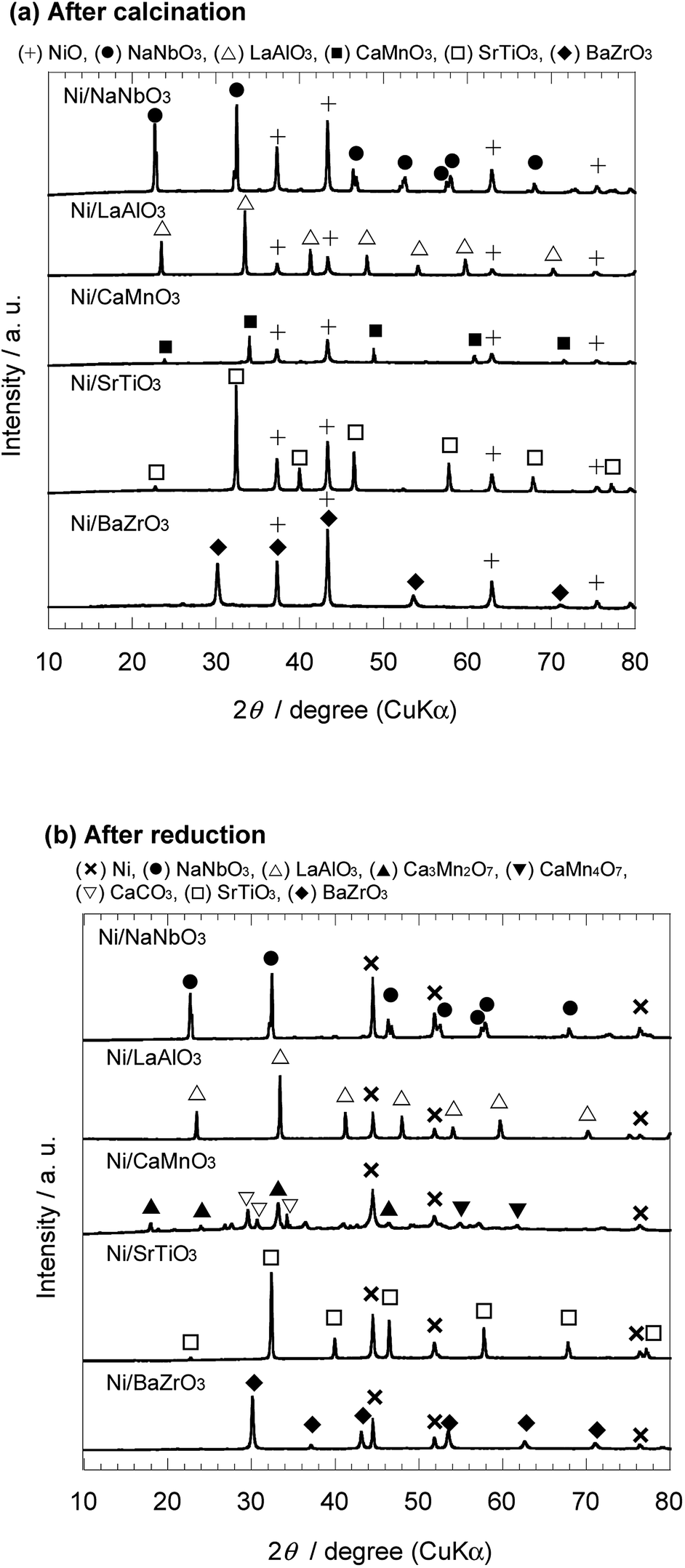 | ||
| Fig. 1 XRD patterns of 40 wt% Ni/NaNbO3, Ni/LaAlO3, Ni/CaMnO3, Ni/SrTiO3, and Ni/BaZrO3 (a) after calcination at 600 °C and (b) after subsequent reduction at 600 °C. | ||
The physical properties of the Ni catalysts were studied. Table 1 summarizes the BET surface area of each support oxide determined by N2 adsorption/desorption measurement. The specific surface area of all the perovskite-type oxides was considerably small (<10 m2 g−1) probably because the sintering of support material remarkably proceeded during the calcination at 1100 °C in the preparation step. The support with small surface area should lead to the low dispersion of Ni particles. Actually, in most of the prepared Ni catalysts, the Ni surface area estimated by the CO pulse chemisorption measurement was significantly low (see Table 1). This suggests that the Ni particle size for these samples was highly large. Besides, the inhibition of CO adsorption on Ni metal by the strong metal-support interaction (SMSI) effect would be partly responsible for this result.59 For example, it has been reported that BaTiO3 and BaZrO3 were the SMSI oxides.56,60
| Catalyst | Support surface areaa (m2 gsample−1) | Ni surface areab (m2 gsample−1) |
|---|---|---|
| a Determined by N2 physisorption measurement.b Determined by CO pulse measurement after reduction at 600 °C. | ||
| Ni/Nb2O5 | 7.4 | Trace |
| Ni/NaNbO3 | 4.1 | Trace |
| Ni/KNbO3 | 8.4 | Trace |
| Ni/Al2O3 | 130 | 4.9 |
| Ni/LaAlO3 | 9.9 | 0.83 |
| Ni/SmAlO3 | 8.1 | 1.4 |
| Ni/GdAlO3 | 4.6 | 1.3 |
| Ni/MnO2 | 2.4 | Trace |
| Ni/CaMnO3 | 6.6 | Trace |
| Ni/SrMnO3 | 7.9 | Trace |
| Ni/BaMnO3 | 7.2 | Trace |
| Ni/TiO2 | 12 | Trace |
| Ni/CaTiO3 | 6.5 | Trace |
| Ni/SrTiO3 | 5.5 | Trace |
| Ni/BaTiO3 | 4.9 | Trace |
| Ni/ZrO2 | 15 | Trace |
| Ni/CaZrO3 | 6.0 | Trace |
| Ni/SrZrO3 | 6.1 | Trace |
| Ni/BaZrO3 | 7.4 | Trace |
Catalytic activity
Fig. 2 depicts the ammonia conversion at 550 °C for the Ni catalysts prepared in this study. In the Ni/niobate series, the performances of Ni/Nb2O5, Ni/NaNbO3, and Ni/KNbO3 were almost the same. Among the samples investigated, the conversions for these catalysts were considerably low. The Ni/REAlO3 catalysts exhibited relatively high activity, and were more active than the Ni/Al2O3 catalyst. As reported in the previous literatures,32–36 the presence of rare-earth elements led to the enhancement of performance for ammonia decomposition. The activity of Ni/REAlO3 was scarcely affected depending on the rare-earth element. The conversions of Ni/AEMnO3 were higher than those of Ni/ANbO3, while lower than those of Ni/REAlO3. As mentioned in Fig. 1, the AEMnO3 oxides decomposed to various species under the reducing atmosphere. A collapse of the support structure would give rise to the sintering of the loaded metal. Additionally, a part of the Ni/AETiO3 and Ni/AEZrO3, their performances strongly depended on the A-site element of the perovskite-type oxides. In series, the Sr and Ba elements were more effective than the Ca one. Hereafter, the support effect of AETiO3 and AEZrO3 was discussed in detail.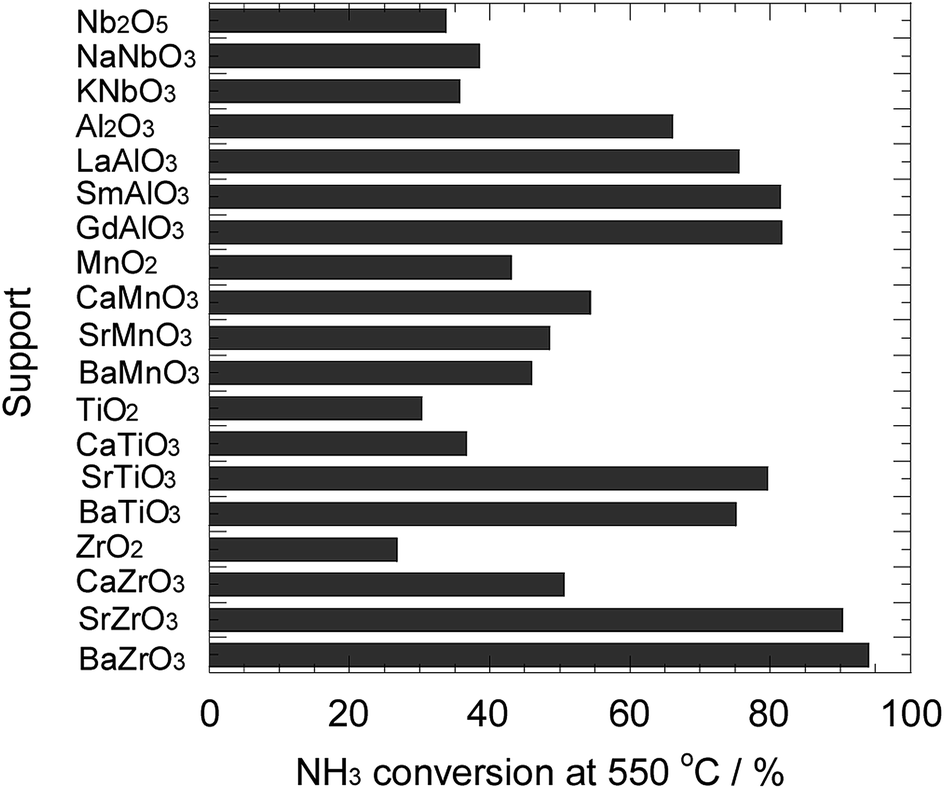 | ||
| Fig. 2 Ammonia conversion for ammonia decomposition at 550 °C for the Ni catalysts supported on various oxides. Reaction condition; NH3: 100 vol%, W/F = 0.60 g s cm−3. | ||
Ni/titanate and Ni/zirconate
As mentioned above, the support effect of AETiO3 and AEZrO3 depended on the alkaline earth element of the perovskite-type oxides. The temperature dependencies of ammonia conversion for the Ni/titanate and Ni/zirconate series are indicated in Fig. 3 and 4, respectively. The decomposition reaction started from ca. 350 °C and the ammonia conversion increased with a rise in reaction temperature. In the Ni/titanate series, Ni/SrTiO3 and Ni/BaTiO3 were more active than Ni/TiO2 and Ni/CaTiO3, and the reaction almost completed at ca. 600 °C over the former catalysts. On the other hand, in the case of the Ni/zirconate catalysts, the order of the performance in this temperature range was as follows; Ni/BaZrO3 > Ni/SrZrO3 > Ni/CaZrO3 > Ni/ZrO2. The behavior of activity against the difference in the alkaline earth element was similar in both series; the Sr and Ba species were more preferable as the constituent elements of support material. Besides, the Ni/AEZrO3 catalysts exhibited higher ammonia conversion than the Ni/AETiO3 ones for the same AE element, and the Ni/BaZrO3 catalyst was the most active in this work. As for Ni/BaZrO3, the catalytic stability was studied at 550 °C and W/F = 0.18 g s cm−3. As shown in Fig. 5, the ammonia conversion at this reaction condition was ca. 62% and remained unchanged over 30 h. This result clarified that the Ni/BaZrO3 catalyst was highly stable.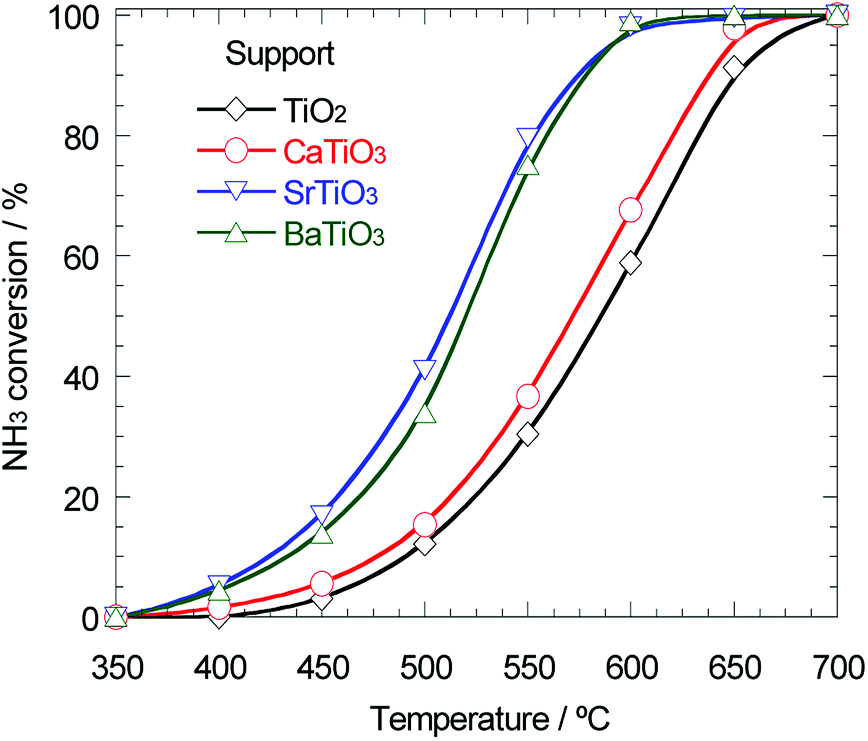 | ||
| Fig. 3 Ammonia conversion for ammonia decomposition against reaction temperature over 40 wt% Ni/TiO2, Ni/CaTiO3, Ni/SrTiO3, and Ni/BaTiO3. Reaction condition; NH3: 100 vol%, W/F = 0.60 g s cm−3. | ||
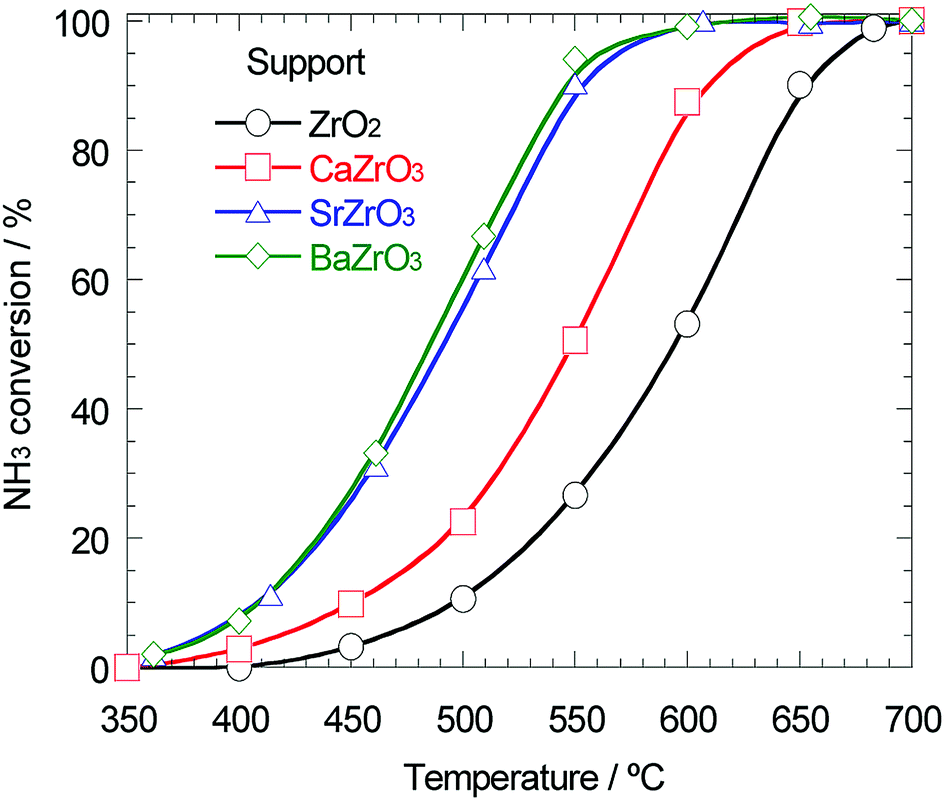 | ||
| Fig. 4 Ammonia conversion for ammonia decomposition against reaction temperature over 40 wt% Ni/ZrO2, Ni/CaZrO3, Ni/SrZrO3, and Ni/BaZrO3. Reaction condition; NH3: 100 vol%, W/F = 0.60 g s cm−3. | ||
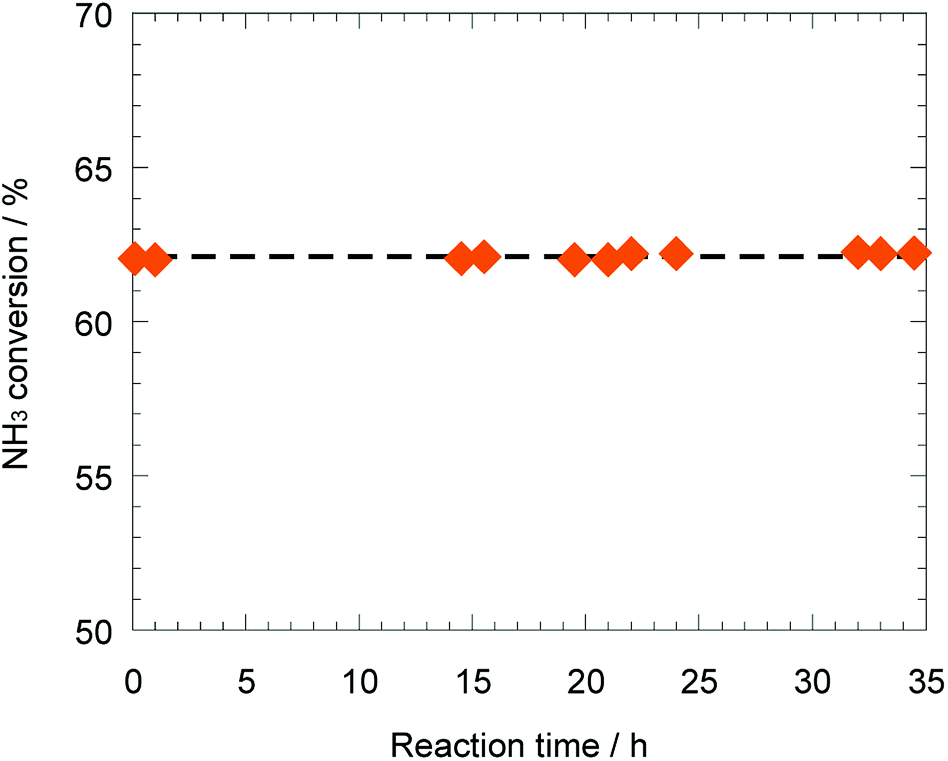 | ||
| Fig. 5 Stability test over 40 wt% Ni/BaZrO3 at 550 °C. Reaction condition; NH3: 100 vol%, W/F = 0.18 g s cm−3. | ||
The microstructure and the average Ni particle size of the Ni/AEZrO3 series were examined by TEM observation to evaluate the influence of alkaline earth element on the morphology of catalysts. Fig. 6 illustrates the TEM images and the Ni particle size distributions for Ni/ZrO2, Ni/CaZrO3, Ni/SrZrO3, and Ni/BaZrO3 after the reduction at 600 °C. For comparison, the result of Ni/Al2O3 is also depicted. It is expected that the Ni dispersion for Ni/Al2O3 is relatively high judging from the large Ni surface area as listed in Table 1. In all the images, some Ni particles are indicated by white arrows based on the result of EDS analysis. The enormous particles (>ca. 1000 nm) observed in the Ni/zirconate series were assigned to the support oxides. The average Ni particle size was 37.4 nm for Ni/ZrO2. The Ni/CaZrO3 and Ni/SrZrO3 catalysts had larger Ni particles as compared with Ni/ZrO2, and the average Ni size was 48.4 nm and 57.6 nm, respectively. In the case of Ni/BaZrO3, the ratio of Ni particles above 100 nm was high, and the average Ni particle size was 99.2 nm. In comparison with Ni/Al2O3 (average Ni size; 23.7 nm), the Ni particles for the Ni/AEZrO3 catalysts were significantly larger and the particle size distribution range was wider regardless of alkaline earth elements in the perovskite-type oxides. These results implied that the AEZrO3 supports were ineffective for the dispersion of Ni particles, leading to the smaller Ni surface area. These dispersion states should be ascribed to the small surface area of the zirconate supports. Considering that Ni/SrZrO3 and Ni/BaZrO3 were highly active, these supports would remarkably enhance the turnover frequency (TOF). Although the Ni surface and/or Ni/support material interface serve as the reaction sites in ammonia decomposition, in this catalyst system the differences in the activity could not be explained in terms of the size and surface area of Ni particle. Thus, the chemical properties of support surface and the electronic state of nickel species could be expected to affect the catalytic activity.
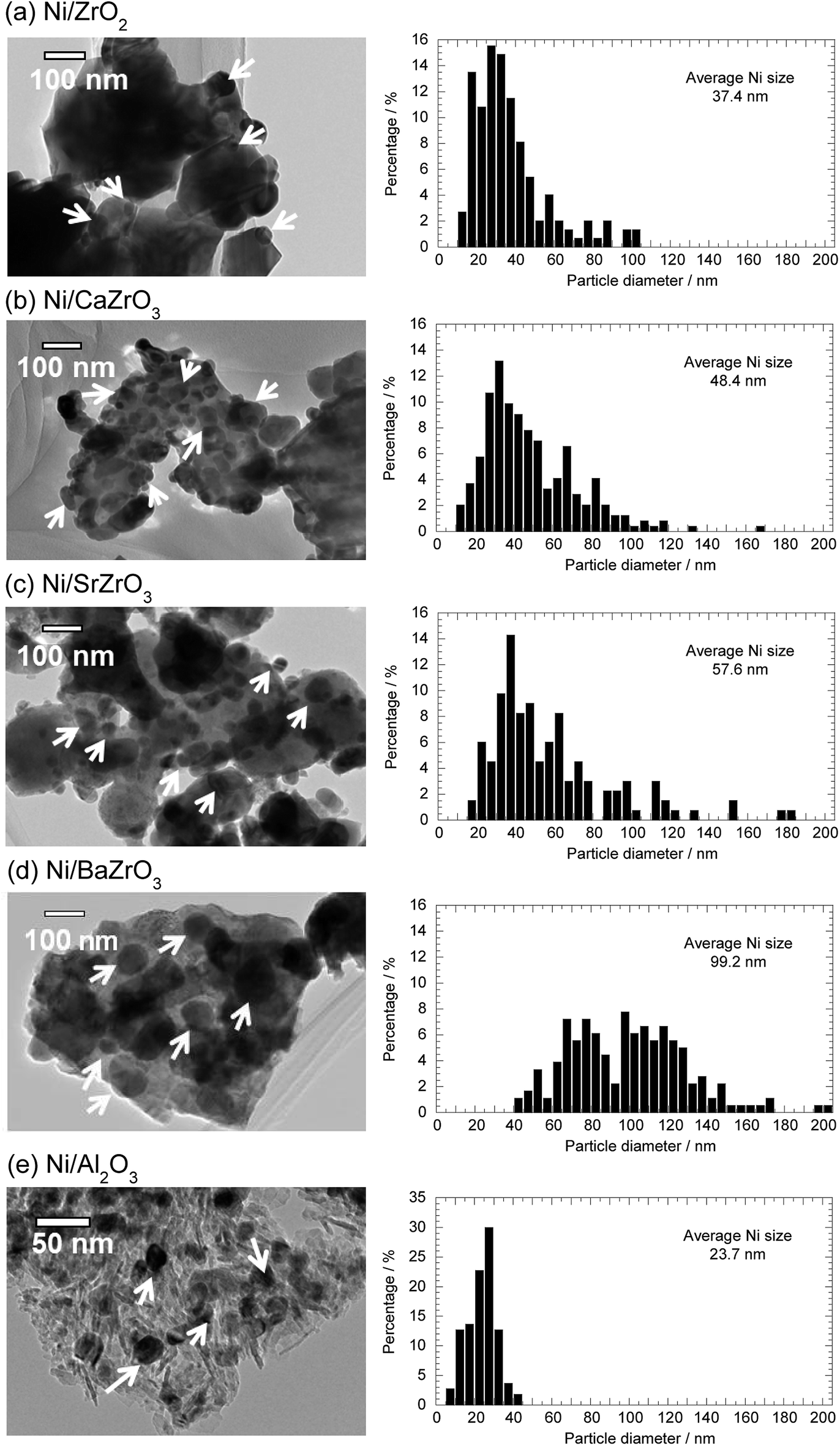 | ||
| Fig. 6 TEM images and Ni particle size distribution histograms of 40 wt% (a) Ni/ZrO2, (b) Ni/CaZrO3, (c) Ni/SrZrO3, (d) Ni/BaZrO3, and (e) Ni/Al2O3. | ||
Next, the basic property of the Ni/AETiO3 and Ni/AEZrO3 catalysts was evaluated by the CO2-TPD measurement since it is known that the catalysts with the strong basic sites promote ammonia decomposition.19 Fig. 7 describes the CO2 desorption profiles for the samples reduced at 800 °C. In the Ni/AETiO3 series, the CO2 desorption peak initiated from ca. 50 °C and the peak shape was similar to each other. On the other hand, the evolution of CO2 was detected at relatively high temperatures for Ni/AEZrO3 as compared with Ni/AETiO3 at the same A-site element. This means that the basic property of Ni/AEZrO3 was stronger than that of Ni/AETiO3. In particular, Ni/SrZrO3 and Ni/BaZrO3 should possess highly strong basic sites judging from the desorption above 700 °C. The CO2 desorption amount per the sample weight in the range of 50–800 °C is presented in Fig. 8. From the desorption amount for Ni/AETiO3 and Ni/AEZrO3 in the case of the same A-site element, the amount of basic sites was larger for the latter series. The electronegativity of Zr smaller than that of Ti might be one of the reasons for this result. Moreover, when the B-site element was fixed, the CO2 desorption amount was larger for the Sr and Ba species than the Ca one in both series. This result indicated that the Sr or Ba components were more preferable for an increase in the basic sites. The catalytic test for Ni/AETiO3 and Ni/AEZrO3 (see Fig. 2–4) revealed that the latter sample was more active for the same AE element, and that the Sr and Ba species were more effective than the Ca species in the respective series. This trend corresponded to that of basic property in Ni/AETiO3 and Ni/AEZrO3. Therefore, the strong basic property of the perovskite-type oxide would be responsible for the high catalytic activity for ammonia decomposition.
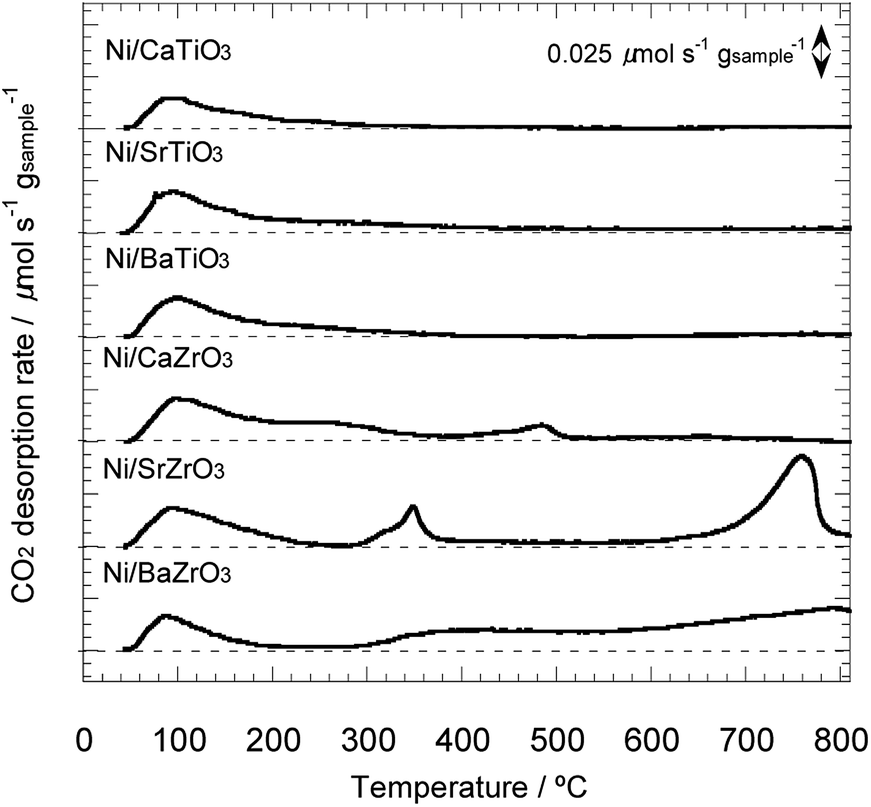 | ||
| Fig. 7 CO2–TPD profiles for 40 wt% Ni/CaTiO3, Ni/SrTiO3, Ni/BaTiO3, Ni/CaZrO3, Ni/SrZrO3, and Ni/BaZrO3 in He under a heating rate of 5 °C min−1. | ||
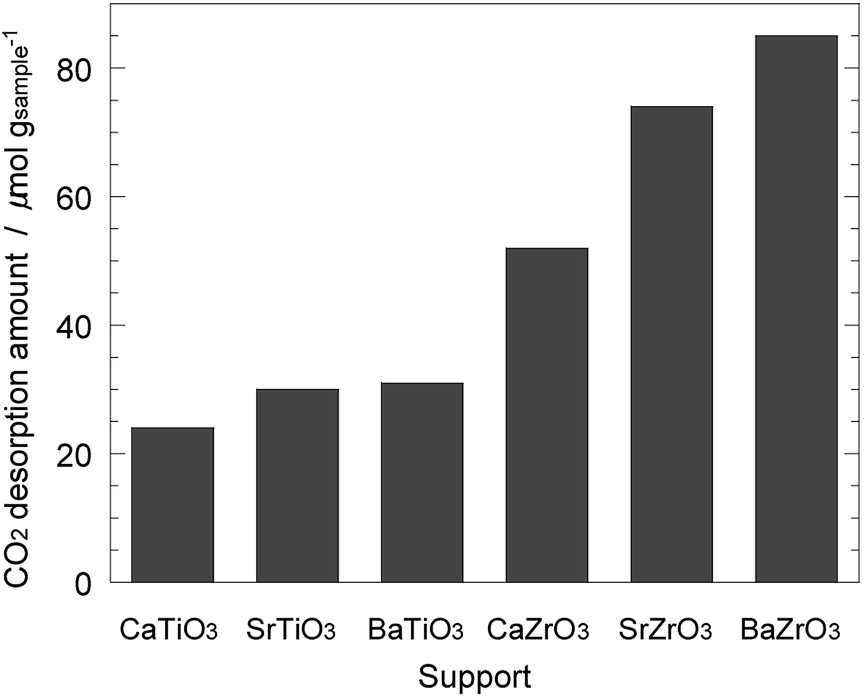 | ||
| Fig. 8 CO2 desorption amount in the range of 50–800 °C obtained by CO2–TPD measurement in Fig. 7 for 40 wt% Ni/CaTiO3, Ni/SrTiO3, Ni/BaTiO3, Ni/CaZrO3, Ni/SrZrO3, and Ni/BaZrO3. | ||
In previous literatures, the electron donation from the additive or support to the active metal promoted the nitrogen desorption step, which is kinetically slow.19,37,61 Thus, the NH3–TPSR measurement was carried out to discuss the nitrogen desorption behavior for the Ni/AEZrO3 series. The NH3 adsorption was conducted at 50 °C. Fig. 9 shows the nitrogen (m/z = 28) desorption behavior for the Ni/ZrO2 and Ni/AEZrO3 catalysts. In the case of Ni/ZrO2, which was low active for ammonia decomposition, the nitrogen desorption was confirmed above ca. 500 °C. For Ni/CaZrO3, the desorption initiated at ca. 180 °C and continuously proceeded even above 600 °C. Considering that the peak area at ca. 550 °C was large, the ratio of nitrogen atoms strongly-interacting with Ni metal would be relatively high. These desorption temperature range were higher than those for the conventional Ni catalysts supported on alumina or rare-earth oxide,35–37 suggesting that Ni/ZrO2 and Ni/CaZrO3 were ineffective for the nitrogen evolution. On the other hand, in the profiles of Ni/SrZrO3 and Ni/BaZrO3, the nitrogen desorption started at ca. 120–130 °C, and terminated at relatively low temperatures as compared with Ni/ZrO2 and Ni/CaZrO3. These onset desorption temperatures were significantly low as compared with those of various Ni catalysts.35–37 This result means that the SrZrO3 and BaZrO3 supports were effective for the nitrogen desorption, leading to the higher performance of Ni/SrZrO3 and Ni/BaZrO3. Considering that the electron donation from the support to active metal could promote the nitrogen desorption step,61 this support effect would be mainly ascribed to the strong basic property of SrZrO3 and BaZrO3.
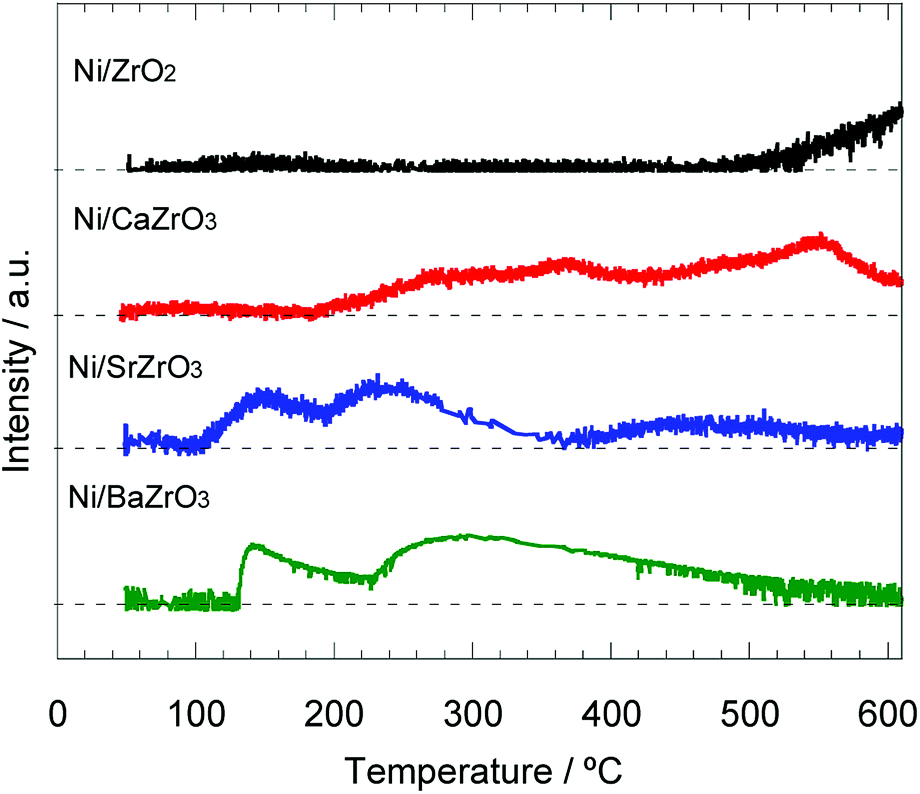 | ||
| Fig. 9 N2 desorption profiles for 40 wt% Ni/ZrO2, Ni/CaZrO3, Ni/SrZrO3, and Ni/BaZrO3 obtained by NH3–TPSR measurement in Ar flow under a heating rate of 5 °C min−1. | ||
Conclusions
The ammonia decomposition was investigated over the Ni catalysts supported on the perovskite-type oxides. The Ni/ANbO3 and Ni/AEMnO3 catalysts were less effective for this reaction, while the Ni/REAlO3 ones exhibited relatively high activity. For Ni/AETiO3 and Ni/AEZrO3, the performance strongly depended on the alkaline earth metal in the perovskite-type oxides, and the Sr and Ba species were superior to the Ca species in the respective series. The activity was higher for the Ni/AEZrO3 catalyst than the Ni/AETiO3 one in the case of the same AE element, and the Ni/BaZrO3 catalyst was the most active among the samples investigated. In the Ni/AETiO3 and Ni/AEZrO3 series, the trend in the catalytic activity corresponded with that in the basic property. The NH3-TPSR measurement for the Ni/AEZrO3 catalysts revealed that the nitrogen desorption effectively proceeded over Ni/SrZrO3 and Ni/BaZrO3. This promotion effect would be ascribed to the strong basic property of SrZrO3 and BaZrO3 supports, and lead to the high activity of Ni/SrZrO3 and Ni/BaZrO3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Council for Science, Technology and Innovation (CSTI), Cross-ministerial Strategic Innovation Promotion Program (SIP), “energy carrier” (Funding agency: JST).Notes and references
- A. J. Appleby, Int. J. Hydrogen Energy, 1994, 19, 175–180 CrossRef.
- D. K. Ross, Vacuum, 2006, 80, 1084–1089 CrossRef.
- T. V. Choudhary and D. W. Goodman, Catal. Today, 2002, 77, 65–78 CrossRef.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Energy Environ. Sci., 2012, 5, 6278–6289 RSC.
- E. García-Bordejé, S. Armenise and L. Roldán, Catal. Rev., 2014, 56, 220–237 CrossRef.
- T. E. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef.
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, Appl. Catal., A, 2004, 277, 1–9 CrossRef.
- S. Stolbov and T. S. Rahman, J. Chem. Phys., 2005, 123, 204716 CrossRef PubMed.
- X. Duan, G. Qian, C. Fan, Y. Zhu, X. Zhou, D. Chen and W. Yuan, Surf. Sci., 2012, 606, 549–553 CrossRef.
- X. Duan, J. Ji, G. Qian, C. Fan, Y. Zhu, X. Zhou, D. Chen and W. Yuan, J. Mol. Catal. A: Chem., 2012, 357, 81–86 CrossRef.
- D. A. Hansgen, D. G. Vlachos and J. G. Chen, Nat. Chem., 2010, 2, 484–489 CrossRef PubMed.
- Z. Ulissi, V. Prasad and D. G. Vlachos, J. Catal., 2011, 281, 339–344 CrossRef.
- A. Takahashi and T. Fujitani, J. Chem. Eng. Jpn., 2016, 49, 22–28 CrossRef.
- S. F. Yin, Q. H. Zhang, B. Q. Xu, W. X. Zhu, C. F. Ng and C. T. Au, J. Catal., 2004, 224, 384–396 CrossRef.
- T. V. Choudhary, C. Svadinaragana and D. W. Goodman, Catal. Lett., 2001, 72, 197–201 CrossRef.
- W. Raróg, Z. Kowalczyk, J. Sentek, D. Składanpwski, D. Szmigiel and J. Zielinski, Appl. Catal., A, 2001, 208, 213–216 CrossRef.
- D. Szmigiel, W. Raróg-Pilecka, E. Miśkiewicz, Z. Kaszkur and Z. Kowalczyk, Appl. Catal., A, 2004, 264, 59–63 CrossRef.
- S. F. Yin, B. Q. Xu, W. X. Zhu, C. F. Ng, X. P. Zhou and C. T. Au, Catal. Today, 2004, 93–95, 27–38 CrossRef.
- S. F. Yin, B. Q. Xu, S. J. Wang and C. T. Au, Appl. Catal., A, 2006, 301, 202–210 CrossRef.
- L. Li, Z. H. Zhu, Z. F. Yan, G. Q. Lu and L. Rintoul, Appl. Catal., A, 2007, 320, 166–172 CrossRef.
- K. Nagaoka, K. Honda, M. Ibuki, K. Sato and Y. Takita, Chem. Lett., 2010, 39, 918–919 CrossRef.
- A. Klerke, S. K. Klitgaard and R. Fehrmann, Catal. Lett., 2009, 130, 541–546 CrossRef.
- W. Raróg-Pilecka, D. Szmigiel, Z. Kowalczyk, S. Jodzis and J. Zielinski, J. Catal., 2003, 218, 465–469 CrossRef.
- S. J. Wang, S. F. Yin, L. Li, B. Q. Xu, C. F. Ng and C. T. Au, Appl. Catal., B, 2004, 52, 287–299 CrossRef.
- K. Nagaoka, T. Eboshi, N. Abe, S. Miyahara, K. Honda and K. Sato, Int. J. Hydrogen Energy, 2014, 39, 20731–20735 CrossRef.
- A. M. Karim, V. Prasad, G. Mpourmpakis, W. W. Lonergan, A. I. Frenkel, J. G. Chen and D. G. Vlachos, J. Am. Chem. Soc., 2009, 131, 12230–12239 CrossRef PubMed.
- F. R. Garciá-Garciá, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Top. Catal., 2009, 52, 758–764 CrossRef.
- X. K. Li, W. J. Ji, J. Zhao, S. J. Wang and C. T. Au, J. Catal., 2005, 236, 181–189 CrossRef.
- C. Plana, S. Armenise, A. Monzón and E. G. Bordejé, J. Catal., 2010, 275, 228–235 CrossRef.
- H. Liu, H. Wang, J. Shen, Y. Sun and Z. Liu, Appl. Catal., A, 2008, 337, 138–147 CrossRef.
- H. Muroyama, C. Saburi, T. Matsui and K. Eguchi, Appl. Catal., A, 2012, 443–444, 119–124 CrossRef.
- A. M. Karim, V. Prasad, G. Mpourmpakis, W. W. Lonergan, A. I. Frenkel, J. G. Chen and D. G. Vlachos, J. Am. Chem. Soc., 2009, 131, 12230–12239 CrossRef PubMed.
- J. Zhang, H. Xu, X. Jin, Q. Ge and W. Li, Appl. Catal., A, 2005, 290, 87–96 CrossRef.
- W. Zheng, J. Zhang, Q. Ge, H. Xu and W. Li, Appl. Catal., B, 2008, 80, 98–105 CrossRef.
- K. Okura, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, Appl. Catal., A, 2015, 505, 77–85 CrossRef.
- K. Okura, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, ChemCatChem, 2016, 8, 2988–2995 CrossRef.
- K. Okura, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, RSC Adv., 2016, 6, 85142–85148 RSC.
- Q. F. Deng, H. Zhang, X. X. Hou, T. Z. Ren and Z. Y. Yuan, Int. J. Hydrogen Energy, 2012, 37, 15901–15907 CrossRef.
- H. Zhang, Y. A. Alhamed, Y. Kojima, A. A. Al-Zahrani, H. Miyaoka and L. A. Petrov, Int. J. Hydrogen Energy, 2014, 39, 277–287 CrossRef.
- Ł. Czekajło and Z. Lendzion-Bielun, Chem. Eng. J., 2016, 289, 254–260 CrossRef.
- S. Podila, H. Dris, S. F. Zaman, Y. A. Alhamed, A. A. AlZahrani, M. A. Daous and L. A. Petrov, J. Mol. Catal. A: Chem., 2016, 414, 130–139 CrossRef.
- S. Podila, Y. A. Alhamed, A. A. AlZahrani, M. A. Daous and L. A. Petrov, Int. J. Hydrogen Energy, 2015, 40, 15411–15422 CrossRef.
- R. Pelka, I. Moszyńska and W. Arabczyk, Catal. Lett., 2009, 128, 72–76 CrossRef.
- B. Lorenzut, T. Montini, M. Bevilacqua and P. Fornasiero, Appl. Catal., B, 2012, 125, 409–417 CrossRef.
- S. B. Simonsen, D. Chakraborty, I. Chorkendorff and S. Dahl, Appl. Catal., A, 2012, 447–448, 22–31 CrossRef.
- A. S. Chellappa, C. M. Fischer and W. J. Thomson, Appl. Catal., A, 2002, 227, 231–240 CrossRef.
- W. Zheng, T. P. Cotter, P. Kaghazchi, T. Jacob, B. Frank, K. Schlichte, W. Zhang, D. S. Su, F. Schüth and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 3458–3464 CrossRef PubMed.
- C. Liang, W. Li, Z. Wei, Q. Xin and C. Li, Ind. Eng. Chem. Res., 2000, 39, 3694–3697 CrossRef.
- A. Srifa, K. Okura, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, Catal. Sci. Technol., 2016, 6, 7495–7504 RSC.
- S. Podila, S. F. Zaman, H. Driss, Y. A. Alhamed, A. A. Al-Zahrani and L. A. Petrov, Catal. Sci. Technol., 2016, 6, 1496–1506 RSC.
- D. V. Leybo, A. N. Baiguzhina, D. S. Muratov, D. I. Arkhipov, E. A. Kolesnikov, V. V. Levina, N. I. Kosova and D. V. Kuznetsov, Int. J. Hydrogen Energy, 2016, 41, 3854–3860 CrossRef.
- Z. Zhao, H. Zou and W. Lin, J. Rare Earths, 2013, 31, 247–250 CrossRef.
- A. Srifa, K. Okura, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, Appl. Catal., B, 2017, 218, 1–8 CrossRef.
- S. S. Pansare, W. Torres and J. G. Goodwin Jr, Catal. Commun., 2007, 8, 649–654 CrossRef.
- J. G. Choi, J. Catal., 1999, 182, 104–116 CrossRef.
- Z. Wang, B. Liu and J. Lin, Appl. Catal., A, 2013, 458, 130–136 CrossRef.
- K. Urasaki, Y. Sekine, S. Kawabe, E. Kikuchi and M. Matsukata, Appl. Catal., A, 2005, 286, 23–29 CrossRef.
- K. Urasaki, K. Tokunaga, Y. Sekine, S. Kawabe, M. Matsukata and E. Kikuchi, Catal. Commun., 2008, 9, 600–604 CrossRef.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef.
- Z. Wang, J. Lin, R. Wang and K. Wei, Catal. Commun., 2013, 32, 11–14 CrossRef.
- F. Hayashi, Y. Toda, Y. Kanie, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2013, 4, 3124–3130 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06100a |
| This journal is © The Royal Society of Chemistry 2018 |