
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally controlled biotransformation of glycyrrhizic acid via an asymmetric temperature-responsive polyurethane membrane†
Xiuhong Wu‡
 *ad,
Shaoyan Wanga,
Lina Zhanga,
Lidong Wubd and
Yi Chen‡cd
*ad,
Shaoyan Wanga,
Lina Zhanga,
Lidong Wubd and
Yi Chen‡cd
aSchool of Chemical and Engineering, University of Science and Technology Liaoning, Anshan, Liaoning 114051, PR China
bThe Key Laboratory of Control of Quality and Safety for Aquatic Products, Ministry of Agriculture, Chinese Academy of Fishery Sciences, Beijing, 100141, PR China
cThe Key Laboratory of Leather Chemistry and Engineering, Sichuan University, Ministry of Education, Chengdu 610065, PR China
dDepartment of Chemistry, Massachusetts of Institute of Technology, Cambridge, MA 02139, USA
First published on 10th October 2018
Abstract
Separating a target product from a relatively complex bioreaction system is often difficult. In this work, a “smart” bioreaction system was developed by using the special characteristic of temperature-responsive polyurethane (TRPU). By combining solvent evaporation with a wet phase inversion technique, an asymmetric membrane consisting of an integral and dense skin layer supported by a porous sublayer was prepared from a thermally responsive polyurethane that experiences a sudden free volume increase upon heating through a phase transition temperature of 56 °C. Subsequently, the asymmetric TRPU membrane served as the carrier of an immobilized enzyme, wherein β-glucuronidase was multipoint-conjugated by using biotin and streptavidin on the porous sublayer. Then, the material-asymmetric TRPU membrane served jointly as the antennae as well as the actuator, which reversibly responds to temperature to switch (on–off) the access of the reactant glycyrrhizic acid (GL). Under the optimal temperature (40 °C) and pH (7.0) conditions, the immobilized β-glucuronidase contributed to almost 33% yield of glycyrrhetinic acid 3-O-mono-β-D-glucuronide (GAMG) of the isolated counterpart for the same concentration of substrate (250 mg L−1) reaction for 24 h, while costing 1% of that of the isolated β-glucuronidase. Kinetic results showed that Vmax and Km values were 8.89 × 103 mg L−1 and 2.30 × 103 mg L−1 h−1, respectively. The specific functional polymer-immobilized β-glucuronidase design serves as a bioreactor of GL into GAMG, as well as a separator deliberately irritated and controlled by temperature. This “smart” support material presents a potential facilitator for the separation of complex biotransformation reactions.
1. Introduction
Synthetic polymers that exhibit a reversible response to temperature, pH, and light can be regarded as “smart” and hold great potential for applications in bioprocessing and pharmaceuticals.1–6 These polymers that undergo changes in size, conformation, or hydrophobicity in response to variations in temperature or pH are gaining increased attention. In particular, there is increasing interest in their potential application ranges, including drug delivery, bioprocessing, gas or liquid transport, and leather industry.7–11 Temperature-responsive polymers (TRPUs) prepared using various materials and procedures are generally characterized by a segmented structure with a partially miscible fixed phase and a reversible phase favorably imported with a typical switching temperature. When the environmental temperature is above the phase transition temperature, the free volume in the polymeric matrix dramatically expands.12,13 More advantageously, by manipulating the molecular structure or chemical composition, the transition of the reversible phase can also be interlinked to a temperature of interest for a particular application.From a TRPU, an asymmetric TRPU (ASTRPU) membrane can be prepared using a wet phase transfer approach, which consists of a porous sublayer enabling its contribution to various potential applications. In our previous studies, an asymmetric membrane was successfully synthesized and used in transport, medical, and environmental applications.14–16 In continuation, we intend to investigate its applications in the area of biotransformation by limiting the access of the substrate to the enzyme of interest, thereby controlling the reaction process.
Biotransformation is an effective means to obtain active components and the leading compounds of natural products in manufacturing biotherapeutic agents.17,18 Glycyrrhizic acid (GL) and glycyrrhetinic acid (GA) are important components extracted from the root of Glycyrrhiza glabra and have been widely used in traditional Chinese medicine. The biological actions of both compounds cover a wide spectrum, including anti-inflammatory, antiviral, antioxidative, and anticancer activities.19,20 When GL is converted to GA, the intermediate product glycyrrhetinic acid 3-O-mono-β-D-glucuronide (GAMG) is often produced. GAMG exhibits even higher antitumor and anti-inflammatory activities than GA. The most commonly used approach to acquire these effective components is extraction, despite its lower efficiency and higher cost. As an alternative method, chemical transformation or biotransformation has become increasingly attractive, particularly biotransformation as it is endowed with the benefit of higher selectivity and requires less use of organic solvents. Hence, it is regarded as a sort of “green chemistry” or “green technology”.21–24 Obviously, the enzymatic activity of either the whole cell or an isolated enzyme dominates the reaction rate, such as hydrolysis, esterification, oxidation, reduction, and asymmetric synthesis.25,26 As compared to the entire cell enzyme, the isolated enzymes contribute toward specific selectivity, although they may incur a higher cost. Therefore, the use of an immobilized enzyme is a considerably less expensive and convenient, as well as recyclable, means for the corresponding bioreaction. Among the general immobilized enzymes used, the catalytic-functions-(CFs)-immobilized enzyme is fairly promising, but few are obtained because of limitations of the carrier.27
In this work, a treated asymmetric polyurethane membrane served as a switch for biotransformation and a CFs-immobilized enzyme carrier because of its smart on–off characteristic. The multipoint immobilization using biotin and streptavidin referred to here has recently been regarded as the most rigid means of immobilization.28–31
2. Material and methods
2.1 Materials
A polycaprolactone diol with number-average molecular weight of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 4,4′-methylenebis(phenyl isocyanate) (MDI), 1,4-butanediol (BDO), and N,N′-dimethylformamide (DMF) were purchased from Sigma Chemical Co. β-Glucuronidase (EC.3.2.1.31, lyophilized powder from E. coli, 1000000–5000000 units per g), GL and GA (purity > 98%), and phosphate buffered saline (PBS) were purchased from Sigma Chemical Co. EZ-link Amine-PEG3-Biotin and EDC were purchased from Fisher, USA, and streptavidin was purchased from Invitrogen, USA. Hydrazide 4′-hydroxyazobenzene-2-carboxylic acid (HABA) was purchased from Pierce, USA.
000 g mol−1, 4,4′-methylenebis(phenyl isocyanate) (MDI), 1,4-butanediol (BDO), and N,N′-dimethylformamide (DMF) were purchased from Sigma Chemical Co. β-Glucuronidase (EC.3.2.1.31, lyophilized powder from E. coli, 1000000–5000000 units per g), GL and GA (purity > 98%), and phosphate buffered saline (PBS) were purchased from Sigma Chemical Co. EZ-link Amine-PEG3-Biotin and EDC were purchased from Fisher, USA, and streptavidin was purchased from Invitrogen, USA. Hydrazide 4′-hydroxyazobenzene-2-carboxylic acid (HABA) was purchased from Pierce, USA.
PBS: one tablet dissolved in 200 mL of deionized water yields 0.01 M phosphate buffer, 0.0027 M potassium chloride, and 0.137 M sodium chloride at a pH of 7.4 and temperature of 25 °C.
2.2 Synthesis of TRPU
A 250 mL round-bottom, three-necked separable flask equipped with a mechanical stirrer, nitrogen inlet thermometer, and condenser with a drying tube was used as the reactor. The TRPU resin was synthesized via solution polymerization in dimethylformamide (DMF) under an N2 atmosphere via two steps. During the first step, poly(epsilon-caprolactone) (PCL) 10000 and a two-fold molar ratio of MDI were charged into the reactor and stirred at 80 °C for 2 h to prepare an isocyanate-terminated prepolymer. The prepolymer was subsequently chain extended at 80 °C for another 2 h by successively adding a stoichiometric amount of MDI and chain extender, BDO. The final molar ratio of MDI to PCL10000 and BDO was maintained at 1:1 to yield a linear polymer. Occasionally, DMF was added into the reactor when the viscosity of the reaction mixture was too high. Finally, a viscous and transparent TRPU solution with 37.6 wt% solid content was obtained. The above synthesis procedure and molecular structure of TRPU are illustrated in Scheme 1.
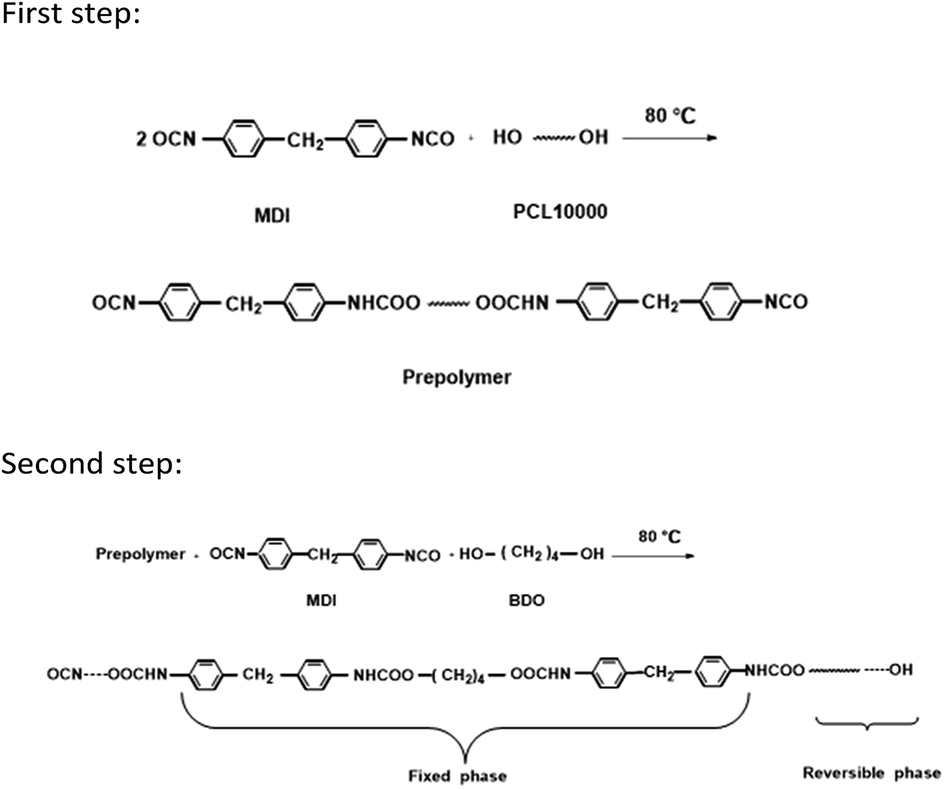 | ||
| Scheme 1 Synthesis procedure and molecular structure of TRPU. | ||
2.3 Synthesis of ASTRPU
ASTRPU was prepared as per our previous work.14 The reaction membrane was prepared using a wet phase inversion film formation process, i.e., casting the TRPU solution on a release paper and then heating the membrane for 15 min at 50 °C (near its crystal temperature). Subsequently, the membrane was immersed in water to saturate. Then, an asymmetric membrane was obtained by washing with water to exclude the solvent and trace metal ions. Finally, the membrane was dried at 40 °C under vacuum conditions for 10 h. The physical and chemical parameters of ASTRPU are listed in Table 1.2.4 Characteristic methods
The skin and sublayer porous structure of the synthesized ASTRPU were scanned using a scanning electron microscope (SEM; JSM-6100 scanning microscope, JEOL, Japan).Plasma (AutoGlow 100, Glow Research, USA) O2 treatment was applied to activate the porous sublayer surface of the ASTRPU membrane and was immediately available to conjugate with EDC and biotin hydrazide. Using X-ray photoelectron spectroscopy (XPS; PHI Versaprobe II, Japan), the elemental composition changes in the ASTRPU membrane before and after the plasma treatment were explored. Peak analysis was performed using CasaXPS version 2.1.9 software, in which the Shirley background was subtracted from all the spectra prior to fitting.
2.5 Analytical methods
To determine the transformational temperature of the synthesized ASTRPU, a differential scanning calorimeter (DSC; Discovery, USA) was used. The temperature range was measured from −80 °C to 270 °C at an increasing temperature rate of 10 °C min−1. Triplicate scanning was performed; finally, an average temperature was obtained.To measure the surface area of the ASTRPU membrane, the Brunauer–Emmett–Teller (BET; ASAP2020, USA) method was applied. The sample was pretreated by drying for 12 h. Then, the sample was measured and analyzed using the instrument software.
To test the molecular weight, the gel permeation chromatography (GFC) method was used.
To determine the effect of biotinylating on the structure of β-glucuronidase, circular dichroism (CD; Jasco J-1500, Japan) was used. The spectra were measured using a 0.1 cm path length cell and recorded at 25 °C in the range of 250–195 nm at a scanning speed of 50 nm min−1, step resolution of 0.1 nm, bandwidth of 1 nm, response of 4 s, and sensitivity of 200 mdeg.
To measure the bioreaction of GL, the product GA and the intermediate product GAMG were analyzed using a high-pressure liquid chromatography system (HPLC; Waters, 2487 series, MA, USA). The column was Extend C18 (150 × 4.6 mm; I.D.: 5 μm) with a flow rate of 1.00 mL min−1, detection wavelength of 254 nm, and at ambient temperature. The solvent system consisted of methanol and 3.6% acetic acid at a ratio of 85:15 (v/v).
To verify the degree of biotinylating, an ultraviolet visible spectrometer (UV-vis; Thermo 2010, USA) was used with HABA assay.
All the experiments were performed at the Massachusetts Institute of Technology, USA.
3. Results and discussion
3.1 Characteristics of ASTRPU membranes
DCS analysis (Fig. 1) reveal that the prepared ASTRPU undergoes a sharp phase transition at 56 °C. The BET specific area of the membrane was 4.70 m2 g−1. Since the porous sublayer would make direct contact with the reactant GL and β-glucuronidase, the morphology of the sublayer surface was evident in Fig. 2a. Contrary to the skin layer surface (Fig. 2c), the entire sublayer appeared to be replete with interconnected perforations. The SEM image involving the morphologies of the cross-section are shown in Fig. 2b. Fig. 2 shows that the abundant porous structure of the sublayer was available for the following enzymatic conjugation.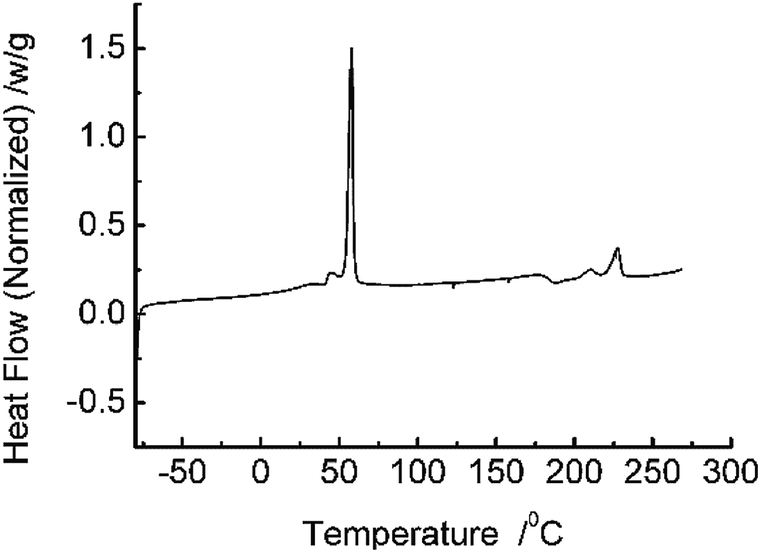 | ||
| Fig. 1 DSC curve of synthesized TRPU. | ||
 | ||
| Fig. 2 SEM images of the morphologies of (a) porous sublayer surface; (b) cross-section; (c) skin layer surface of ASTRPU. | ||
3.2 Switching or “on–off” characteristics of ASTRPU
The microstructure of the multiple finger-like pores in the porous sublayer indicated a larger area for β-glucuronidase loading. The transportability of TRPU was determined via the concentration change in GL initiated by the temperature change. By repeatedly switching the release assembly between two shaking water baths at different temperatures, the dynamic transport behaviors of GL through the ASTRPU membrane over four cycles of temperature oscillations between 25 °C and 45 °C were investigated, and the results are shown in Fig. 3.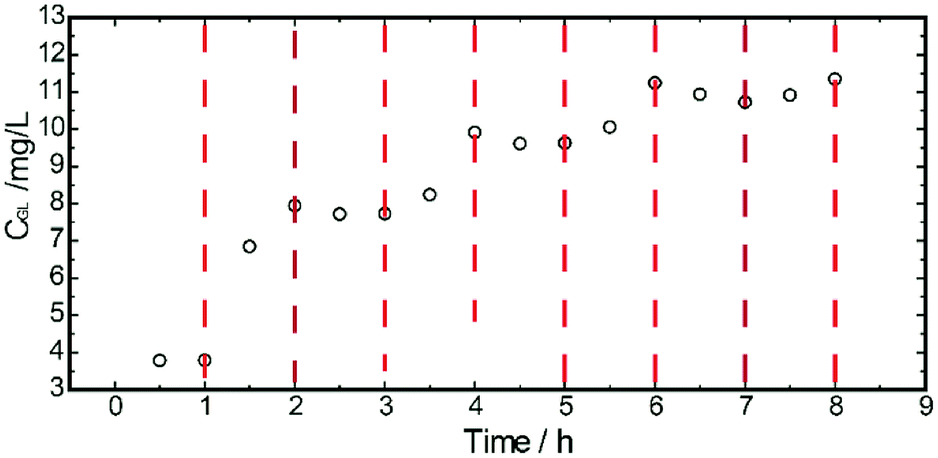 | ||
| Fig. 3 Accumulative release profile of the ASTRPU membrane. | ||
As shown, the transport of GL through the ASTRPU membrane abruptly accelerated upon heating to 45 °C and then leveled off once the temperature reached 25 °C. Over four cycles of temperature oscillations, the on–off switching transport of GL was maintained, indicating good reproducibility and stability as the response to temperature.
3.3 Plasma treatment of ASTRPU membranes
To facilitate the conjugation of β-glucuronidase onto the membrane surface, the membrane surface was first treated by O2 plasma (AutoGlow 100, Glow Research, USA) to alter the hydrophobic surface into a hydrophilic one. The compositional change in the porous sublayer was illustrated using XPS (PHI 5000 VersaProbe II, Japan) data obtained from the MIT Center for Materials Science and Engineering. Peak analysis was performed using the CasaXPS version 2.1.9 software with the Shirley background subtracted from all the spectra prior to fitting. The HR envelopes were analyzed and peak-fitted after the subtraction of the Shirley background using the Gaussian–Lorentzian peak shapes obtained from the CasaXPS software package. The chemical compositions of the porous surface of ASTRPU before and after plasma O2 treatment are listed in Table 2. It is obvious that the most significant change is the O content, while the N content seldom changes. As shown in Fig. 4, after treatment by O2 plasma, the asymmetrical membrane became activated with the additional introduction of –C–O and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (binding energy, BE: 286.5 eV and 289.2 eV, respectively). The O 1s lines also show an increase in –C–O (BE: 533.2 eV).32
O groups (binding energy, BE: 286.5 eV and 289.2 eV, respectively). The O 1s lines also show an increase in –C–O (BE: 533.2 eV).32
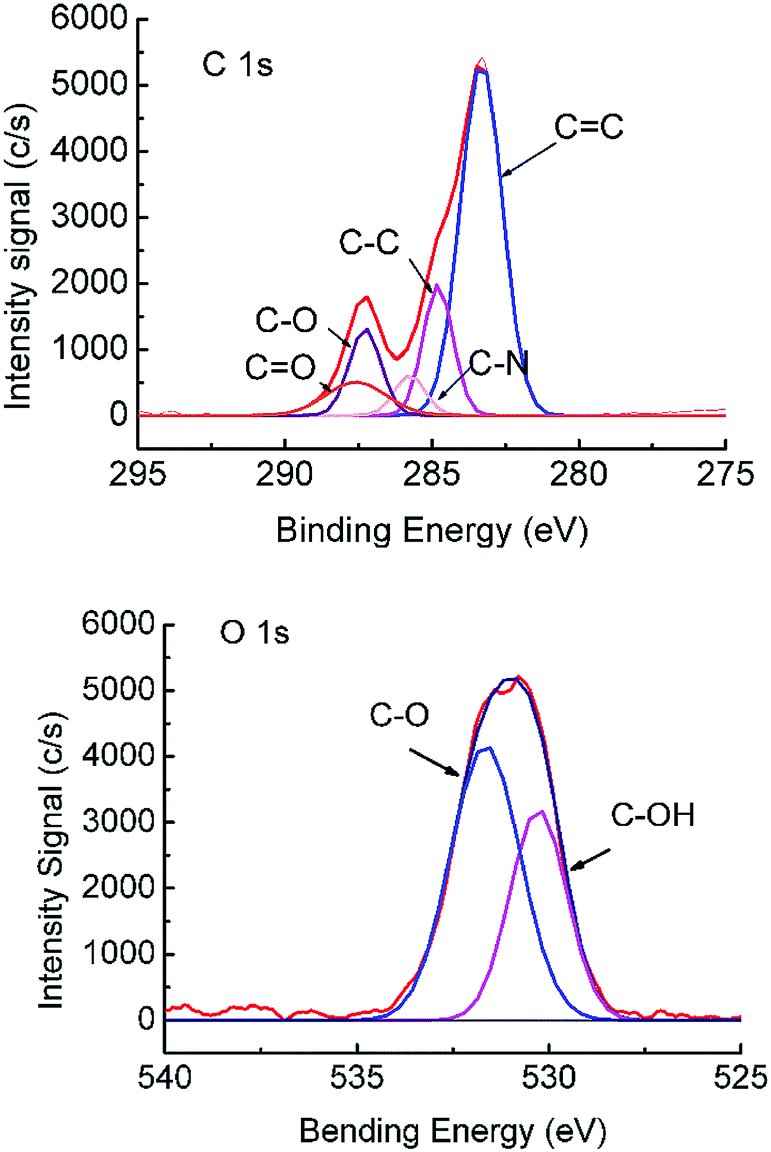 | ||
| Fig. 4 XPS spectra of the C 1s and O 1s peaks of the O2-plasma-treated asymmetric TRPU. Treatment time: 1 min. | ||
3.4 Biotinylated ASTRPU and β-glucuronidase
Immediately, the ASTRPU membrane and the target β-glucuronidase were both biotinylated using EZ-link Amine-PEG3-Biotin and EDC following the Fisher protocol. The biotinylate β-glucuronidase was quantified using a Pierce Biotin Quantitation Kit supplied by Thermo Scientific. The kit contained the reagent HABA, which enables a rapid estimation of the mole-to-mole ratio of biotin to protein. Because biotin is a relatively small molecule, it can be conjugated to many proteins without altering the biological activity of the protein. A protein can be conjugated with several biotin molecules, each of which can bind one molecule of avidin, thereby considerably increasing the sensitivity of many assays. The bond formation between biotin and avidin is rapid; once formed, it remains unaffected by the most extremes pH values, organic solvents, and other denaturing agents. With regard to quantitative analysis, a solution containing biotinylated protein was added to a mixture of HABA and avidin. Because of the higher affinity toward avidin, biotin displaces the HABA and the absorbance at 500 nm proportionately decreases. Using this method, an unknown amount of biotin present in a solution can be quantified in a single cuvette by measuring the absorbance of the HABA–avidin solution before and after the addition of the biotin-containing sample. The change in absorbance is related to the amount of biotin in the sample via the extinction coefficient of the HABA–avidin complex.3.5 Conjugate β-glucuronidase onto the ASTRPU membrane
A schematic of the immobilization process is shown in Fig. 5. The quantitative number of both biotinylate and isolated β-glucuronidase conjugated to the porous sublayer membrane was determined using the HABA assay: 0.31 mg cm−2 and 1.50 mmol mL−1, respectively. To the best of our knowledge, such an approach of immobilized β-glucuronidase has not yet been reported. The means of immobilizing β-glucuronidase presented here was that of multipoint crosslinking (one streptavidin molecule offers four binding sites for trapping) and it behaves rather rigidly than that in a traditional binding approach.28 To determine the effect of biotin on the structure of β-glucuronidase, a circular dichroism (CD; Jasco J-1500, Japan) was used. Solutions of biotinylate and original β-glucuronidase were prepared at a concentration of 0.50 and 0.49 mg mL−1 in PBS, respectively. The structural changes in the biotinylated β-glucuronidase was established by comparing the CD spectra. Typically, three scans were accumulated and subsequently averaged.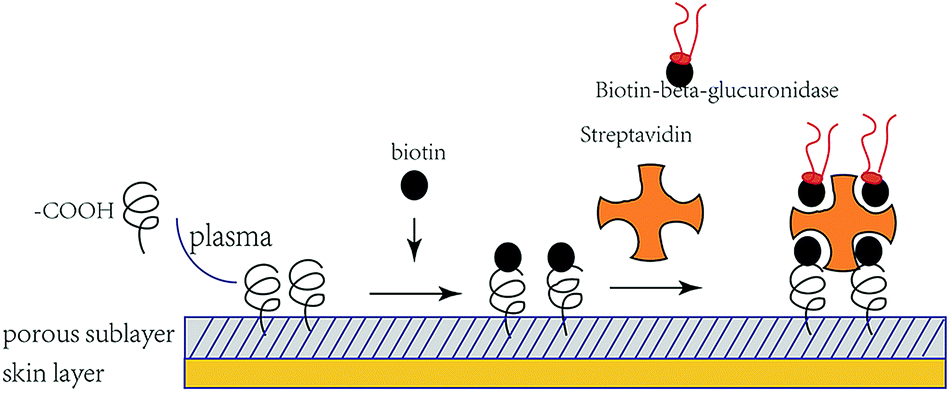 | ||
| Fig. 5 Schematic of ASTRPU–β-glucuronidase binding. | ||
3.6 Comparison of the before and after structures of biotinylated β-glucuronidase
CD was used to monitor the β-glucuronidase structure's alternation, as shown in Fig. 6. Thus far, we know that the far-UV spectra for isolated β-glucuronidase and biotinylated β-glucuronidase exhibited similar spectra with strong negative ellipticity near 220 nm, and most of the spectra of the biotinylated β-glucuronidase overlapped with the spectra of the original type, indicating that the secondary structure of the biotinylated β-glucuronidase did not change during the course of biotinylating.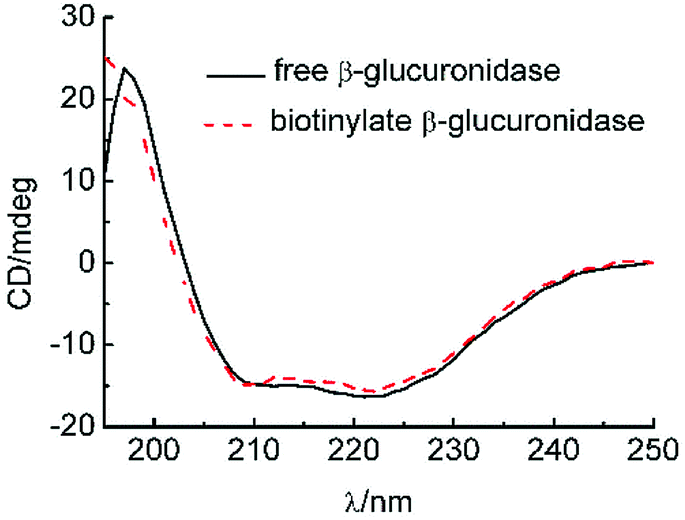 | ||
| Fig. 6 CD graphs for isolated and biotinylated β-glucuronidase. | ||
3.7 Reaction of GL into GAMG on biotinylated β-glucuronidase ASTRPU membranes
With regard to isolated β-glucuronidase, under suitable reaction conditions, namely, a pH of 7.0 (PBS modulated) and a temperature of 35 °C, the hydrolysis of GL catalyzed by β-glucuronidase served as the probe model reaction to investigate the integral design of the temperature-responsive ASTRPU–β-glucuronidase bioreaction assembly. The reaction occurred in a circular membrane surface (I.D.: 0.68 cm; area: 1.45 cm2). One microliter of 100 mg mL−1 GL solution was added into a 1.5 mL bottle, and the porous, sublayer ASTRPU membrane served as the cover with binding β-glucuronidase apposite to the GL solution (i.e., on the outside of the cover) and the edges of the membrane were thoroughly sealed to prevent leakage. This assembly was then hung in an inverted manner in a 50 mL conical tube containing 1.2 mL of PBS (pH 7.0) in advance. Thereafter, the 50 mL conical tube was incubated in a shaker at a certain temperature for 24 h. The products of the hydrolysis reaction in the PBS were analyzed using HPLC. The HPLC results showed that a different amount of GAMG was produced as a function of temperature, wherein the irradiated membrane released different concentrations of GL to access the β-glucuronidase conjugated in the porous sublayer. Fig. 7 shows the detailed results of such a tendency. The maximum GAMG product concentration was obtained at a temperature of 50 °C. At a higher temperature, (e.g., 55 °C), less GAMG was produced, which might have been caused by a decrease in or loss of enzymatic activity as compared to that at 50 °C. Within the model reaction, the ASTRPU membrane played the role of a switch for GL approaching the β-glucuronidase irradiated by the temperature alteration. To investigate the kinetic behavior of the system, the reaction assembly was incubated in a shaker at 40 °C for 24 h. The Michaela constant, Km, and the maximum reaction rate, Vmax, were measured using the Lineweaver–Burk plotting method as 8.89 × 103 mg L−1 and 2.3 × 103 mg L−1 h−1, respectively. For isolated β-glucuronidase, the Km and Vmax values were 131.07 mg L−1 and 1.64 × 102 mg L−1 h−1, respectively. Km ≫ C[GL] indicated that both the isolated and immobilized enzymatic reactions were at a first-order magnitude. Although the Km value for the immobilized version was greater than that for the isolated version (representing less affinity ability toward the substrate), the Vmax value was nearly 10 times that of the isolated version, indicating that more product was produced more rapidly. Under the optimal temperature (40 °C) and pH (7.0), the immobilized ASTRPU–β-glucuronidase contributed to almost 33% of the GAMG of the isolated counterpart for the same concentration of substrate (i.e., 250 mg L−1) for a reaction time of 24 h. However, the β-glucuronidase used was just 1% that of the isolate. The lost yield could form a considerable compromise given the lower consumption of β-glucuronidase.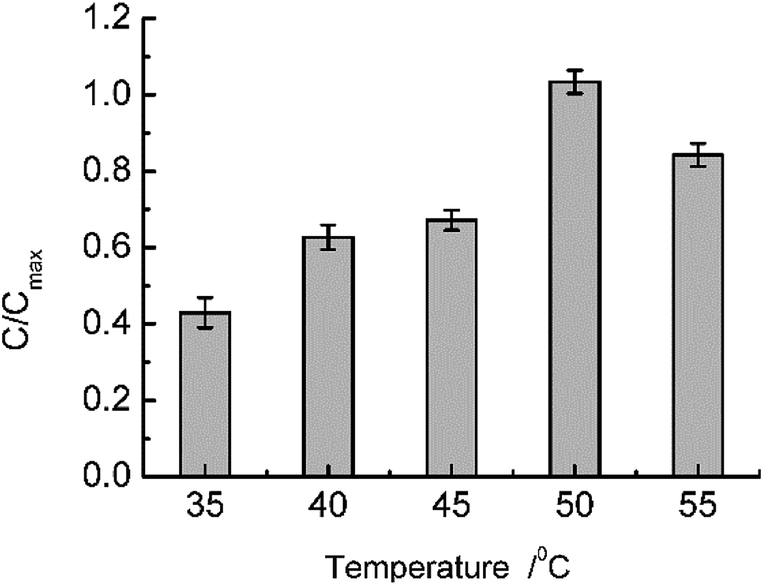 | ||
| Fig. 7 Temperature-dependent GL biotransformation on an immobilized ASTRPU membrane. | ||
4. Conclusions
In summary, from the perspective of controlling the bioconversion process, or alternatively, to obtain intermediate products of interest (e.g., GAMG) in this work, the characteristic of TRPU was used. A catalytic bioreaction assembly coupled with the smart “on–off” switch was designed. The β-glucuronidase multipoint was rigidly immobilized onto ASTRPU via biotin and streptavidin. The ASTRPU–β-glucuronidase reactor succeeded in controlling the access of the reactant GA toward β-glucuronidase by deliberately alternating the temperature, thereby controlling the bioconversion.The developed reactor exhibited the potential advantage of obtaining important intermediate products or some of the steps during the reaction. Meanwhile, the reactant, product, and catalytic enzyme used were independent of each other, benefiting from the separation. Moreover, in addition to the immobilized enzyme that could be repeatedly used in most of the reaction system, the reactant could be repeatedly used in this system, which seldom occurs in most cases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Professor Alexander M. Klibanov for help applying the experimental condition for all the experiments at the Massachusetts Institute of Technology. They gratefully acknowledge the National Nature Science Foundation of China [Grant Number 21576127] for support. They also thank Professor Chun Li for applying the extract GAMG to the chemical standard and Sappi North America for applying for the release paper.References
- A. Carolina de las Heras, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- Y. L. Tai, T. Chen and G. Lubineau, ACS Appl. Mater. Interfaces, 2017, 9, 32184–32191 CrossRef CAS PubMed.
- T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci., 2002, 99(26), 16593–16597 CrossRef PubMed.
- Y. L. Tai, T. K. Bera, Z. G. Yang and G. Lubineau, Nanoscale, 2017, 9, 7888–7894 RSC.
- N. Nath and A. Chikou, Adv. Mater., 2003, 14(17), 1243–1248 CrossRef.
- A. Kikuchi and T. Okano, Prog. Polym. Sci., 2002, 27, 1165–1193 CrossRef CAS.
- L. J. Zhou, D. Liang, X. L. He, J. H. Li, H. Tan, J. S. Li, Q. Fu and Q. Gu, Biomaterials, 2012, 33, 2734–2745 CrossRef CAS PubMed.
- Y. L. Tai and G. Lubineau, Sci. Rep., 2016, 6, 19632–19639 CrossRef CAS PubMed.
- H. Zhou, H. H. Shi, H. J. Fan, J. Zhou and J. X. Yuan, Macromol. Res., 2009, 17(7), 528–532 CrossRef CAS.
- Y. L. Tai and G. Lubineau, Small, 2017, 3, 1603486–1603493 CrossRef PubMed.
- H. Zhou, J. Zhou, H. J. Fan, Y. Chen, F. F. Yang, J. X. Yuan and R. W. Liu, Desalination, 2009, 249, 843–849 CrossRef CAS.
- H. H. Shi, Y. Chen, H. J. Fan, J. Xiang and B. Shi, J. Appl. Polym. Sci., 2010, 117, 1820–1827 CAS.
- Y. Chen, Y. Liu, H. J. Fan, H. Li, B. Shi, H. Zhou and B. Y. Peng, J. Membr. Sci., 2007, 287, 192–197 CrossRef CAS.
- H. Xu, J. M. Chang, Y. Chen, H. J. Fan and B. Shi, J. Mater. Sci., 2013, 48, 6625–6639 CrossRef CAS.
- J. M. Chang, X. Y. Guan, Y. Chen and H. J. Fan, Polym. Chem., 2016, 7, 3398–3405 RSC.
- J. M. Chang, Y. Chen, S. Y. Zhao, X. Y. Guan and H. J. Fan, Polym. Chem., 2015, 6, 8150–8160 RSC.
- R. G. Mehta, G. Murrillo, R. Naithani and X. J. Peng, Pharm. Res., 2010, 27, 950–961 CrossRef CAS PubMed.
- J. C. Kehr, D. G. Picchi and E. Dittmann, Beilstein J. Org. Chem., 2011, 7, 1622–1635 CrossRef CAS PubMed.
- A. Mahmud, J. Mohammad and P. P. Bibhu, Ann. Microbiol., 2014, 64, 683–688 CrossRef.
- A. Morana, A. D. Lazzaro, I. D. Lernia, C. Ponzone and M. D. Rosa, Biotechnol. Lett., 2002, 24, 1907–1911 CrossRef CAS.
- W. J. Tang, Y. A. Yang, H. Xu, J. B. Shi and X. H. Liu, Sci. Rep., 2014, 4, 4706–4711 Search PubMed.
- S. J. Feng, C. Li, X. L. Xu and X. Y. Wang, J. Mol. Catal. B: Enzym., 2006, 43, 63–67 CrossRef CAS.
- H. A. Amin, H. A. El-Menoufy, A. A. El-Mehalawy and E. E. Mostafa, Malays. J. Microbiol., 2010, 6(2), 209–216 Search PubMed.
- I. Kaleem, H. Shen, B. Lv, B. Wei, A. Rasool and C. Li, Chem. Eng. Sci., 2014, 106, 136–143 CrossRef CAS.
- S. Wenda, S. Illner, A. Mell and U. Kragal, Green Chem., 2011, 13, 3007–3047 RSC.
- J. M. Woodley, Trends Biotechnol., 2008, 26(6), 321–327 CrossRef CAS PubMed.
- L. Q. Cao, Curr. Opin. Chem. Biol., 2005, 9, 217–226 CrossRef CAS PubMed.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- A. S. Hoffman, Clin. Chem., 2000, 46(9), 1478–1486 CAS.
- J. M. Hannink, J. L. Jeroen, M. Cornelissen, J. A. Farrera, P. Foubert, F. C. De Schryver, A. J. Nico, M. Sommerdijk, J. Roeland and M. Nolte, Angew. Chem., Int. Ed., 2001, 40(24), 4732–4734 CrossRef CAS PubMed.
- J. Hyun, Y. J. Zhu, A. Liebmann-Vinson, T. P. Beebe and A. Chilkoti Jr, Langmuir, 2001, 17, 6358–6367 CrossRef CAS.
- X. Fu, M. J. Jenkins, G. M. Sun, I. Bertoti and H. S. Dong, Surf. Coat. Technol., 2012, 206, 4799–4807 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC assay of biotransformation results. See DOI: 10.1039/c8ra06202a |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |