
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of C, S, Se and P donor ligands in copper(I) mediated C–N and C–Si bond formation reactions†
Katam Srinivas and
Ganesan Prabusankar *
*
Department of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, Telangana, India-502 285. E-mail: prabu@iith.ac.in; Fax: +91 40 2301 6032; Tel: +91 40 2301 6089
First published on 18th September 2018
Abstract
The first comparative study of C, S, Se and P donor ligands-supported copper(I) complexes for C–N and C–Si bond formation reactions are described. The syntheses and characterization of eight mononuclear copper(I) chalcogenone complexes, two polynuclear copper(I) chalcogenone complexes and one tetranuclear copper(I) phosphine complex are reported. All these new complexes were characterized by CHN analysis, FT-IR, UV-vis, multinuclear NMR and single crystal X-ray diffraction techniques. The single crystal X-ray structures of these complexes depict the existence of a wide range of coordination environments for the copper(I) center. This is the first comparative study of metal–phosphine, metal–NHC and metal–imidazolin-2-chalcogenones in C–N and C–Si bond formation reactions. Among all the catalysts, mononuclear copper(I) thione, mononuclear copper(I) N-heterocyclic carbene and tetranuclear copper(I) phosphine are exceedingly active towards the synthesis of 1,2,3-triazoles as well as for the cross-dehydrogenative coupling of alkynes with silanes. The cross-dehydrogenative coupling of terminal alkynes with silanes represents the first report of a catalytic process mediated by metal–imidazolin-2-chalcogenones.
Introduction
The chemistry of “soft” Lewis donors such as imidazolin-2-chalcogenones (NHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E, E = S and Se)-supported metal complexes have received much attention in catalysis during the past two decades due to their tuneable σ-donor and π-accepting properties.1–3 In fact, the electron cloud at the metal centre is responsible for the activities associated with the metal complexes. Thus, the metal will be more electrophilic when attached to more π-acceptor ligands (PPh3), while, it becomes relatively less electrophilic when attached to weak π-acceptors such as NHC and NHCE (Chart 1).4 Notably, the stronger σ-donor abilities over the π-accepting nature of NHCE compared to both phosphine and NHC is due to the existence of a larger contribution (66%) of the zwitterionic form (NHC+–E−).5
E, E = S and Se)-supported metal complexes have received much attention in catalysis during the past two decades due to their tuneable σ-donor and π-accepting properties.1–3 In fact, the electron cloud at the metal centre is responsible for the activities associated with the metal complexes. Thus, the metal will be more electrophilic when attached to more π-acceptor ligands (PPh3), while, it becomes relatively less electrophilic when attached to weak π-acceptors such as NHC and NHCE (Chart 1).4 Notably, the stronger σ-donor abilities over the π-accepting nature of NHCE compared to both phosphine and NHC is due to the existence of a larger contribution (66%) of the zwitterionic form (NHC+–E−).5
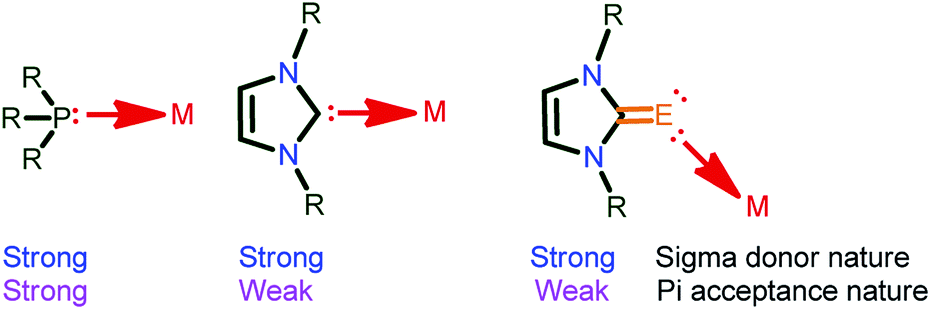 | ||
| Chart 1 σ-donor and π-accepting nature of PPh3, NHC and NHCE ligands.2 | ||
Although, the coordination chemistry of transition metals with NHCE is well known,6 the catalytic applications of these complexes are limited.7 Notably, the catalytic efficiency of NHCE metal complexes is significantly remarkable as compared to NHC–metal complexes (Chart 2).7 For example, in copper chemistry, NHCE supported copper(I) complexes {[(IMS)2CuCl]; IMS = 1,3-dimethylimidazoline-2-thione, and [(IMesSe)2Cu][BF4]; IMesSe = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-selone} were found to be more regioselective in the hydroborylation of alkynes over NHC–Cu.7d,e Recently, we investigated the efficiency of [(Bptp/Bpsp)12Cu8][PF6]8 (Bptp = 2,6-bis(1-isopropylimidazole-2-thione)pyridine and Bpsp = 2,6-bis(1-isopropylimidazole-2-selone)pyridine) in click catalysis as well as in the hydro-amination of alkynes, and they were found to be as good as the NHC–Cu catalyst.8 However, to the best of our knowledge the catalytic comparisons of phosphine–copper, NHC–copper and NHCE–copper have never been investigated.
 | ||
| Chart 2 Known catalytic comparisons between NHCE–metal and NHC–metal-supported Cu(I) complexes. | ||
Herein, we present the first comparative study of phosphine–copper, NHC–copper and NHCE–copper. The coordination properties of ligands, structural features of copper(I) complexes and catalytic efficiency of new copper(I) complexes are reported in detail. The mononuclear copper(I) complexes (IMesE)CuX [E = S, Se, IMesE 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-thone (1–3) and 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-selone (4–5)], (NHCE)2CuX, NHCE; IMesSe, X = CuCl2 (7), NHCE; IMesSe, X = PF6 (9) and NHCE = IPrSe, X = PF6 (11) [IPrSe, 1,3-bis(2,6-diisopropylphenyl)imidazoline-2-selone], and polynuclear copper(I) complexes [(Ebis)CuI]n (13) [where Ebis = 1,2-bis(3-methyl-4-imidazolin-2-selone)ethane], [(Ebpis)1.5CuBF4]n (14) [Ebpis = 1,2-bis(3-isopropyl-4-imidazolin-2-selone)ethane] were isolated. Interestingly, 7, 9 and 11 can also be isolated from [(IMes)CuCl] (6) by the ligand transfer method. Besides, the complexes 13 and 14 were synthesized from the tetranuclear copper(I) complex, [(PPh3)4Cu4I4] (12). Complexes 1–14 were characterized by CHN analysis, FT-IR, multinuclear NMR and single crystal X-ray diffraction techniques.
Experimental
Materials and methods
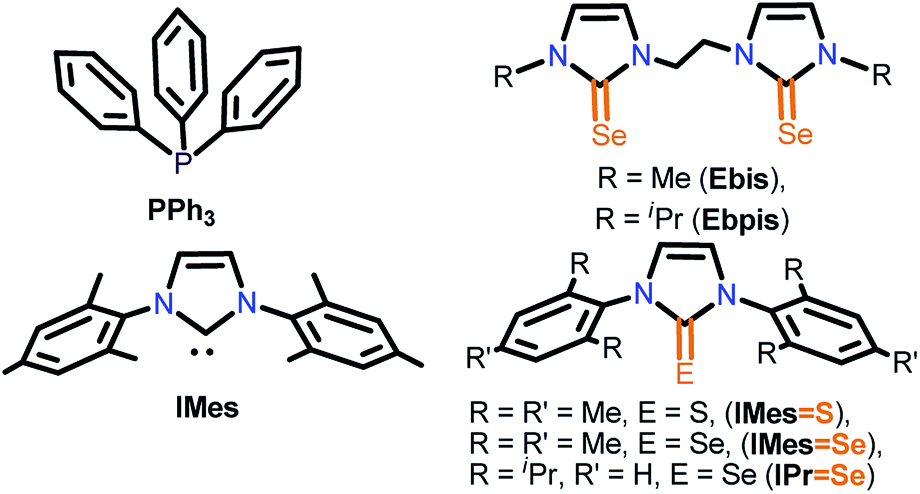
The necessary manipulations were carried out under an argon atmosphere in a glove box or using standard Schlenk techniques. The solvents were purchased from commercial sources and purified according to standard procedures and freshly distilled under argon atmosphere prior to use.9 IMes·HCl (1,3-dimesityl-1H-imidazol-3-ium chloride), IMesS (1,3-dimesityl-1H-imidazole-2(3H)-thione), IMesSe (1,3-dimesityl-1H-imidazole-2(3H)-selenone), IPrSe (1,3-bis(2,6-diisopropylphenyl)-1H-imidazole-2(3H)-selenone), Ebis (3,3′-(ethane-1,2-diyl)bis(1-methyl-1H-imidazole-2(3H)-selenone)) and Ebpis (3,3′-(ethane-1,2-diyl)bis(1-isopropyl-1H-imidazole-2(3H)-selenone)) were prepared as previously reported (Chart 3).7k,l Unless otherwise stated, the chemicals were purchased from commercial sources. CuCl, CuBr, CuI, [Cu(CH3CN)4]PF6, KPF6 and NH4BF4 were purchased from Sigma Aldrich and used as received.
 | ||
| Chart 3 Types of ligands studied for copper(I)-mediated 1,3-dipolar cycloaddition of alkynes with azides. | ||
FT-IR measurement (neat) was carried out on a Bruker Alpha-P Fourier transform spectrometer. The UV-vis spectra were obtained on a T90+ UV-visible spectrophotometer. NMR spectra were recorded on a Bruker Ultrashield-400 spectrometer at 25 °C unless otherwise stated. Chemical shifts are given relative to TMS and were referenced to the solvent resonances as internal standards. Elemental analyses were performed by the Euro EA-300 elemental analyzer. The crystal structures of 1–5, 7, 9, 11, 13 and 14 were obtained on an Oxford Supernova diffractometer. Single crystals of complexes suitable for the single crystal X-ray analysis were mounted on a Goniometer KM4/Xcalibur equipped with a Sapphire 2 (large Be window) detector (CuKα radiation source, λ = 1.5418 Å) at ambient temperature (298 K) in inert oil. Using Olex2,10 the structure was solved with the ShelXS11 structure solution program using Direct Methods and refined with the Olex2 refine refinement package using Gauss–Newton minimization. Absorption corrections were performed on the basis of multi-scans. Non-hydrogen atoms were anisotropically refined. H atoms were included in the refinement in calculated positions riding on their carrier atoms. No restraint was made for any of the compounds. Due to the severely disordered (positional disorder) imidazole ring with Ueq > 0.2, molecule 14 gave “B” level alerts and atoms are not shown in the ellipsoid view for clarity.
Synthesis of 1
A mixture of IMesS (0.100 g, 0.297 mmol) and CuCl (0.035 g, 0.356 mmol) in methanol (5 mL) was refluxed at 80 °C for 12 h. The clear solution was then brought to ambient temperature to form colorless crystals of 1. Yield: 78% (based on CuCl). Mp: 207–209 °C (decomposed). Elemental analysis calcd (%) for C21H24ClCuN2S (435.50): C, 57.92; H, 5.55; N, 6.43; found: C, 57.84; H, 5.57; N, 6.39. 1H NMR (400 MHz, DMSO-d6): δ = 7.66 (s, 2H, ImH), 7.07 (s, 4H, CHmeta), 2.26 (s, 6H, CH3para), 1.95 (s, 12H, CH3ortho). 13C NMR (100 MHz, DMSO-d6): δ = 155.66 (CS), 140.04, 134.65, 131.85, 129.61, 121.67 (ArC), 20.69 (p-CH3), 17.11 (o-CH3). FT-IR (neat): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3148(w), 3112(w), 3082(w), 2917(m), 1605(m), 1558(w), 1482(s), 1449(m), 1382(s), 1290(m), 1235(s), 1167(w) (CS), 1140(w), 1034(m), 925(w), 853(s), 750(s), 693(s), 666(w), 606(m), 572(m), 520(m) cm−1.
= 3148(w), 3112(w), 3082(w), 2917(m), 1605(m), 1558(w), 1482(s), 1449(m), 1382(s), 1290(m), 1235(s), 1167(w) (CS), 1140(w), 1034(m), 925(w), 853(s), 750(s), 693(s), 666(w), 606(m), 572(m), 520(m) cm−1.
Synthesis of 2
Complex 2 was prepared in the same manner as described for 1 using IMesS (0.100 g, 0.297 mmol) and CuBr (0.051 g, 0.356 mmol) in methanol (5 mL). Yield: 84% (based on CuBr). Mp: 220–222 °C (decomposed). Elemental analysis calcd (%) for C21H24BrCuN2S (479.93): C, 52.55; H, 5.04; N, 5.84; found: C, 52.54; H, 5.07; N, 5.79. 1H NMR (400 MHz, DMSO-d6): δ = 7.31 (s, 2H, ImH), 6.97 (s, 4H, CHmeta), 2.22 (s, 6H, CH3para), 1.93 (s, 12H, CH3ortho). 13C NMR (100 MHz, DMSO-d6): δ = 159.87 (CS), 138.59, 135.02, 133.21, 128.87, 119.70 (ArC), 20.62 (p-CH3), 17.32 (o-CH3). FT-IR (neat): = 3147(w), 3113(w), 3084(w), 2916(m), 1604(m), 1556(w), 1481(s), 1449(m), 1382(s), 1345(w), 1289(m), 1234(s), 1167(w) (CS), 1141(w), 1033(m), 924(w), 885(s), 851(m), 748(s), 693(s), 644(w), 605(m), 573(m), 519(m) cm−1.
Synthesis of 3
A mixture of IMesS (0.100 g, 0.297 mmol) and CuI (0.068 g, 0.356 mmol) in methanol (5 mL) was refluxed at 80 °C for 12 h. The resulting white precipitate was dissolved in hot acetonitrile and allowed to crystallize under ambient conditions over 2 days. Yield: 71% (based on CuI). Mp: 232–234 °C (melting). Elemental analysis calcd (%) for C21H24CuIN2S (526.95): C, 47.87; H, 4.59; N, 5.32; found: C, 47.84; H, 4.57; N, 5.29. 1H NMR (400 MHz, DMSO-d6): δ = 7.61 (s, 2H, ImH), 7.08 (s, 4H, CHmeta), 2.28 (s, 6H, CH3para), 1.98 (s, 12H, CH3ortho). 13C NMR (100 MHz, DMSO-d6): δ = 156.55 (CS), 139.76, 134.78, 132.17, 129.50, 121.27 (ArC), 20.69 (p-CH3), 17.23 (o-CH3). FT-IR (neat): = 3145(w), 3111(w), 3084(w), 2913(m), 1597(m), 1556(w), 1478(s), 1445(m), 1380(s), 1286(m), 1230(s), 1165(w) (CS), 1138(w), 1028(m), 921(w), 848(s), 743(s), 690(s), 604(m), 570(m) cm−1.
Synthesis of 4
Complex 4 was prepared in the same manner as described for 1 using IMesSe (0.100 g, 0.260 mmol) and CuBr (0.048 g, 0.312 mmol) in methanol (5 mL). Yield: 87% (based on CuBr). Mp: 227–229 °C (melting). Elemental analysis calcd (%) for C21H24BrCuN2Se (526.85): C, 47.88; H, 4.59; N, 5.32; found: C, 47.91; H, 4.57; N, 5.29. 1H NMR (400 MHz, DMSO-d6): δ = 7.73 (s, 2H, ImH), 7.02 (s, 4H, CHmeta), 2.25 (s, 6H, CH3para), 1.95 (s, 12H, CH3ortho). 13C NMR (100 MHz, DMSO-d6): δ = 148.22 (CSe), 139.24, 134.58, 133.32, 129.13, 123.30 (ArC), 20.68 (p-CH3), 17.57 (o-CH3). FT-IR (neat): = 3145(w), 3108(w), 3078(w), 2916(m), 1605(m), 1553(w), 1480(s), 1447(m), 1371(s), 1337(w), 1291(m), 1232(s), 1166(w) (CSe), 1125(w), 1032(m), 925(w), 851(s), 752(s), 688(s), 594(w), 568(s), 520(m) cm−1.
Synthesis of 5
Complex 5 was prepared in the same manner as described for 3 using IMesSe (0.100 g, 0.260 mmol) and CuI (0.060 g, 0.312 mmol) in methanol (5 mL). Yield: 77% (based on CuI). Mp: 224–226 °C (melting). Elemental analysis calcd (%) for C21H24CuIN2Se (573.85): C, 43.95; H, 4.22; N, 4.88; found: C, 43.91; H, 4.17; N, 4.89. 1H NMR (400 MHz, DMSO-d6): δ = 7.79 (s, 2H, ImH), 7.08 (s, 4H, CHmeta), 2.30 (s, 6H, CH3para), 1.97 (s, 12H, CH3ortho). 13C NMR (100 MHz, DMSO-d6): δ = 149.14 (CSe), 139.85, 134.57, 132.94, 129.53, 123.31 (ArC), 20.77 (p-CH3), 17.38 (o-CH3). FT-IR (neat): = 3143(w), 3107(w), 3079(w), 2913(m), 1599(m), 1551(m), 1477(s), 1443(m), 1369(s), 1334(w), 1289(m), 1228(s), 1163(w) (CSe), 1123(m), 1026(m), 922(w), 849(s), 746(s), 684(s), 599(w), 566(m) cm−1.
Synthesis of 7
Se (0.100 g, 0.260 mmol) and CuCl (0.031 g, 0.312 mmol) in methanol (5 mL). Yield: 75% (based on CuCl).Se (0.100 g, 0.260 mmol) was treated with excess [(IMes)CuCl] (0.16 g, 0.426 mmol) in acetone under refluxing conditions overnight to yield 7. Yield: 65% (based on IMesCuCl). Mp: 218–220 °C (melting). Elemental analysis calcd (%) for C42H48Cl2Cu2N4Se2 (964.79): C, 52.29; H, 5.01; N, 5.81; found: C, 52.30; H, 5.07; N, 5.84. 1H NMR (400 MHz, DMSO-d6): δ = 7.81 (s, 2H, ImH), 7.07 (s, 4H, CHmeta), 2.28 (s, 6H, CH3para), 1.94 (s, 12H, CH3ortho). 13C NMR (100 MHz, DMSO-d6): δ = 148.59 (CSe), 140.00, 134.46, 132.76, 129.58, 123.45 (ArC), 20.74 (p-CH3), 17.21 (o-CH3). FT-IR (neat): = 3146(w), 3106(w), 3076(w), 2915(m), 1607(m), 1553(w), 1482(s), 1448(m), 1371(s), 1338(m), 1292(m), 1233(s), 1165(w) (CSe), 1125(w), 1033(s), 925(w), 852(s), 754(s), 688(s), 595(s), 569(s) cm−1.Synthesis of 8
Complex 8 can be prepared as a by-product during the synthesis of 7 in method 2. Yield: 30% based on [(IMes)CuCl]. Mp: 277–279 °C (decomposed). Elemental analysis calcd (%) for C42H48ClCuN4 (707.85): C, 71.26; H, 6.83; N, 7.91; found: C, 71.30; H, 6.87; N, 7.94. 1H NMR (400 MHz, CDCl3): δ = 7.00 (s, 4H, ImH), 6.89 (s, 8H, CHmeta), 2.41 (s, 12H, CH3para), 1.66 (s, 24H, CH3ortho). 13C NMR (100 MHz, CDCl3): δ = 177.35 (C–Cu), 139.39, 134.53, 134.45, 129.16, 122.78 (ArC), 21.18 (p-CH3), 16.95 (o-CH3). FT-IR (neat): = 2912(m), 1604(m), 1542(w), 1483(s), 1400(m), 1266(s), 1230(s), 1163(m), 1069(m), 1036(m), 929(m), 857(s), 733(s), 641(m), 573(m) cm−1.
Synthesis of 9
Se (0.100 g, 0.260 mmol) and [Cu(CH3CN)4]PF6 (0.097 g, 0.260 mmol) in methanol (5 mL). Yield: 80% (based on [Cu(CH3CN)4]PF6).Se (0.100 g, 0.260 mmol) was treated with [(IMes)CuCl] (0.208 g, 0.520 mmol) and an excess of KPF6 (0.239 g, 1.300 mmol) in acetone under refluxing conditions overnight to yield the desired product 9. Yield: 68% (based on [(IMes)CuCl]. Mp: 259 °C (decomposed). Elemental analysis calcd (%) for C42H48CuF6N4PSe2 (975.28): C, 51.72; H, 4.96; N, 5.74; found: C, 51.73; H, 4.97; N, 5.80. 1H NMR (400 MHz, CDCl3): δ = 7.36 (s, 4H, ImH), 7.04 (s, 8H, CHmeta), 2.37 (s, 12H, CH3para), 2.10 (s, 24H, CH3ortho). 13C NMR (100 MHz, CDCl3): δ = 142.28 (CSe), 140.96, 134.70, 132.53, 129.88, 124.58 (ArC), 21.35 (p-CH3), 18.47 (o-CH3). 31P NMR (CDCl3, 161 MHz): −157.59 to −131.22 (sept, PF6). 19F NMR (CDCl3, 376 MHz): −74.72 to −72.83 (d, PF6). FT-IR (neat): = 1484(m), 1435(m), 1264(s), 1185(m) (CSe), 1121(m), 834(s) (P–Fstretching), 731(s), 697(s), 549(s) cm−1.Synthesis of 10
Complex 10 can be prepared as a by-product during the synthesis of 9 and also 11 by method 2. Yield: 28% along with 9 and 25% along with 11 based on [(IMes)CuCl]. Mp: 238–240 °C (melting). Elemental analysis calcd (%) for C42H48Cu2F6N4P (817.36): C, 61.72; H, 5.92; N, 6.85; found: C, 61.73; H, 5.97; N, 6.84. 1H NMR (400 MHz, CDCl3): δ = 7.01 (s, 4H, ImH), 6.89 (s, 8H, CHmeta), 2.41 (s, 12H, CH3para), 1.66 (s, 24H, CH3ortho). 13C NMR (100 MHz, CDCl3): δ = 177.35 (C–Cu), 139.40, 134.52, 134.45, 129.16, 122.78 (ArC), 21.18 (p-CH3), 16.96 (o-CH3). 31P NMR (CDCl3, 161 MHz): −157.59 to −131.21 (sept, PF6). 19F NMR (CDCl3, 376 MHz): −74.73 to −72.84 (d, PF6). FT-IR (neat): = 2920(w), 1608(w), 1482(s), 1454(m), 1372(m), 1341(w), 1265(s), 1234(m), 1035(m), 840(s) (P–Fstretching), 734(s), 700(m), 557(s) cm−1.
Synthesis of 11
Se (0.10 g, 0.213 mmol) and [Cu(CH3CN)4]PF6 (0.040 g, 0.106 mmol) in methanol (5 mL). Yield: 78% (based on [Cu(CH3CN)4]PF6).Se (0.100 g, 0.213 mmol) was treated with IMesCuCl (0.160 g, 0.426 mmol) and an excess of KPF6 (0.19 g, 1.065 mmol) in acetone under refluxing conditions overnight to yield the desired product 11. Yield: 70% based on [(IMes)CuCl]. Mp: 268–270 °C (decomposed). Elemental analysis calcd (%) for C54H72CuF6N4PSe2 (1143.60): C, 56.71; H, 6.35; N, 4.90; found: C, 56.70; H, 6.37; N, 4.89. 1H NMR (400 MHz, CDCl3): δ = 7.43–7.39 (t, 2H, CHpara), 7.23–7.22 (d, 4H, CHmeta), 7.20 (s, 2H, ImH), 2.34–2.28 (sept, 4H, iPrCH), 1.20–1.19, 1.12–1.10 (d, 24H, CH3). 13C NMR (100 MHz, CDCl3): δ = 155.76 (CSe), 145.79 (ImC), 133.11, 131.11, 124.61, 123.37 (ArC), 29.22 (iPrCH), 24.95, 23.44 (CH3). 31P NMR (CDCl3, 161 MHz): −157.59 to −131.21 (sept, PF6). 19F NMR (CDCl3, 376 MHz): −74.82 to −72.82 (d, PF6). FT-IR (neat): = 2962(m), 2867(m), 1558(w), 1463(s), 1420(m), 1345(s), 1265(m), 1212(w), 1181(m) (C–Se), 1120(w), 1060(m), 937(m), 839(s) (P–Fstretching), 803(w), 737(s), 555(s) cm−1.Synthesis of 12
CuI (0.50 g, 0.263 mmol) and excess of PPh3 (1.38 g, 0.526 mmol) were mixed together in acetonitrile (20 mL) and allowed to stir at 85 °C overnight to yield a colourless clear solution, which upon cooling to room temperature gave, 12. Yield: 82% (based on CuI). Mp: 221–223 °C (melting). Elemental analysis calcd (%) for C72H60Cu4I4P4 (1810.94): C, 47.75; H, 3.34; found: C, 47.80; H, 3.37. 1H NMR (400 MHz, CDCl3): δ = 7.30–7.25 (m, 3H, ArH), 7.17–7.13 (m, 2H, CHortho). 13C NMR (100 MHz, CDCl3): δ = 134.09, 133.94, 133.79, 133.58, 129.46, 128.47, 128.38 (ArC). 31P NMR (CDCl3, 161 MHz): −4.96 (s, PPh3). FT-IR (neat): = 1677(w), 1562(w), 1475(m), 1429(s), 1212(w), 1178(m), 1093(m), 1025(w), 995(w), 744(s), 691(s), 521(m) cm−1.
Synthesis of 13
Se), 119.95, 119.75 (ImC), 47.22 (CH2–CH2), 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 091 (CH3). FT-IR (neat): = 2932(m), 1679(s), 1561(m), 1479(m), 1436(m), 1405(m), 1244(m), 1180(m) (CSe), 1115(s), 1046(m), 924(w), 808(m), 751(w), 717(s), 690(s), 533(s) cm−1.
091 (CH3). FT-IR (neat): = 2932(m), 1679(s), 1561(m), 1479(m), 1436(m), 1405(m), 1244(m), 1180(m) (CSe), 1115(s), 1046(m), 924(w), 808(m), 751(w), 717(s), 690(s), 533(s) cm−1.Synthesis of 14
Se), 120.38, 114.77, 50.96, 46.71, 21.95 (CH3). 11B{1H} NMR (128.4 MHz, CDCl3): δ = −0.99. 19F{1H} NMR (376.4 MHz, CDCl3): δ = −154.02. FT-IR (neat): = 3170(w), 3141(w), 2977(w), 1567(m), 1453(m), 1418(s), 1325(s), 1218(m), 1175(m) (CSe), 1135(m), 1036(s) (B–Fstretching), 741(s), 684(s), 640(m), 518(m) cm−1.General synthetic procedure for the [3+2] cycloaddition of azides and terminal alkynes
The azide (1.0 mmol), alkyne (1.2 mmol), catalyst (1 mol%) and water (1 mL) were loaded. The solution was stirred at room temperature for 1 h and the conversion was followed by TLC to ensure the completion of the reaction. After the complete conversion water (5 mL) was added, followed by the addition of ethyl acetate (5 mL). The reaction mixture was allowed to stir at room temperature for a further 5–10 minutes, after which the ethyl acetate layer was collected and the volatiles were evaporated to obtain the solid. The acquired solid was further washed with n-hexane and dried under vacuo to yield the desired product. The isolated 1,2,3-triazole products were characterized by 1H NMR and 13C NMR spectroscopy to confirm the product purity.General procedure for the synthesis of C–Si bonding
The catalyst (1 mol%) was placed in an oven-dried Schlenk flask that was evacuated for 5 minutes then refilled with an argon gas. CH3CN (1 mL) was added to the flask under argon, followed by terminal alkyne (1.0 mmol), hydrosilane (1.2 mmol) and pyridine (0.2 mmol). The resulting mixture was allowed to stir at 100 °C for 12 h. After completion of the reaction, a saturated aqueous NH4Cl solution (10 mL) was added to the mixture and the aqueous phase was extracted with ethylacetate (5 mL × 3). The combined organic layer was washed with brine (10 mL) and then dried over anhydrous sodium sulfate. Filtration and evaporation of the solvent followed by column chromatography on silica gel gave the corresponding product.Results and discussion
The N-heterocyclic thione and selone ligands such as IMesS, IMesSe, IPrSe, Ebis and Ebpis were synthesized in fairly good yields from their corresponding imidazolium salts using elemental chalcogen powders in the presence of potassium carbonate.7k,l These organochalcogen ligands were demonstrated as promising ligands for investigating the coordination abilities with copper metal. The copper(I) complexes isolated herein can be organized in three different categories such as neutral monomeric copper(I) complexes, cationic monomeric copper(I) complexes, neutral 2D copper(I) sheets and cationic 2D copper(I) sheets.
Neutral mononuclear copper(I) complexes 1–5 were synthesized by treating copper(I) halides with one equivalent of IMesE in methanol (Scheme 1). The monomeric copper(I) complexes 1–5 were isolated in excellent yield. The crystalline solids 1–5 are only soluble in hot acetonitrile and in DMSO. The formation of these compounds was established by elemental analysis, FT-IR, multi nuclear NMR spectroscopy, UV-vis and single crystal X-ray diffraction techniques. In 1H NMR, the aryl hydrogens were slightly downfield shifted upon complexation, while the imidazole hydrogens were largely downfield shifted due to hydrogen bonding interactions. The CE signal in 13C NMR was shifted up-field by 5–10 ppm due to the decrease in the π-acceptance nature of the carbene carbon upon complexation. The FT-IR spectra of molecules 1–5 show the existence of the CE stretching frequency at 1163–1167 cm−1, which is in agreement with the complex formation.
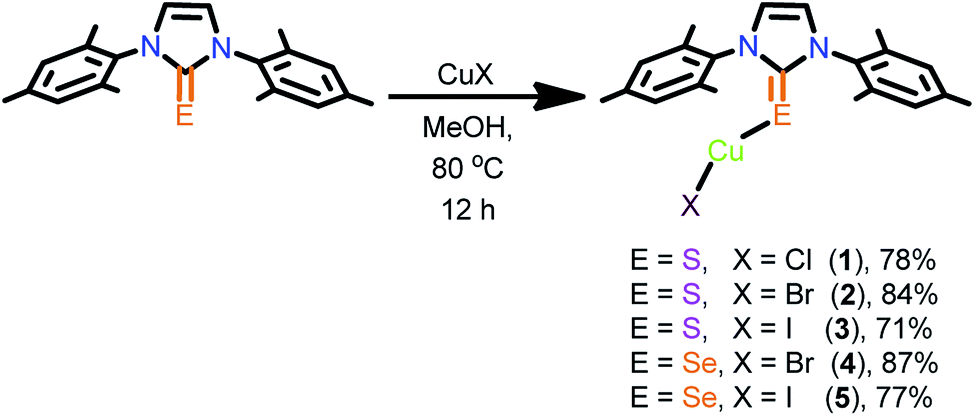 | ||
| Scheme 1 Synthesis of 1–5. | ||
The solid state structures of 1–5 were unambiguously determined by single crystal X-ray diffraction (Fig. 1). Complexes 1–5 crystallized in the monoclinic space group, P21/c. The crystallographic data for 1–5 are listed in table S1 (see ESI-1, table S1†) and the important bond parameters are listed in the figure caption. The molecular structures of 1–5 are isostructural and are neutral monomeric copper(I) chalcogenone complexes with a copper:chalcogen ratio of 1:1. The copper(I) centre in 1–5 is two-coordinate with one imidazole thione/selone and one halogen atom. Interestingly, the molecular structures of 1–5 are comparable with NHC analogues of [(IMes)CuX].12,13
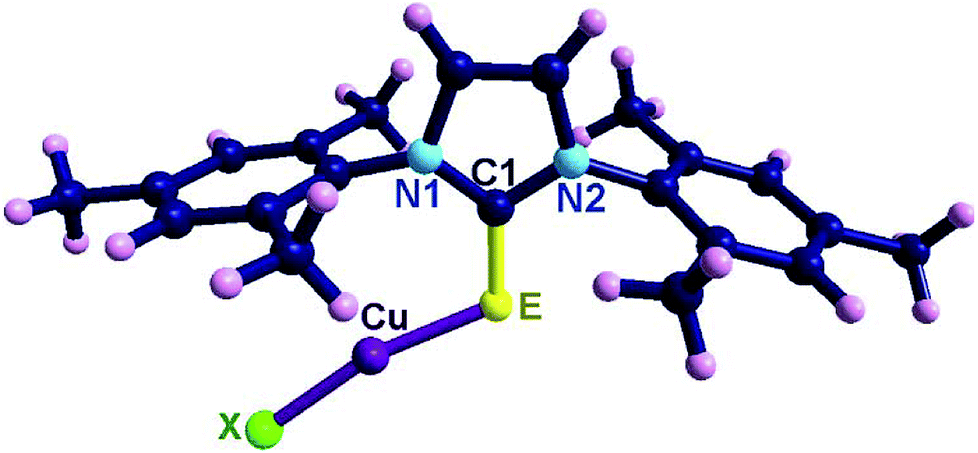 | ||
| Fig. 1 Selected bond lengths (Å) and angles (°) for 1 (E = S, X = Cl): C(1)–S(1), 1.699(3), S(1)–Cu(1), 2.129(11), Cu(1)–Cl, 2.098(13), C(1)–S(1)–Cu(1), 109.13(11), N(1)–C(1)–N(2), 109.2(3), N(1)–C(1)–S(1), 129.6(3), N(2)–C(1)–S(1), 123.7(2), S(1)–Cu(1)–Cl, 165.94(6); for 2 (E = S, X = Br): C(1)–S(1), 1.699(3), S(1)–Cu(1), 2.135(9), Cu(1)–Br, 2.222(6), C(1)–S(1)–Cu(1), 108.61(10), N(1)–C(1)–N(2), 106.4(2), N(1)–C(1)–S(1), 129.6(2), N(2)–C(1)–S(1), 124.1(2), S(1)–Cu(1)–Br, 163.97(4); for 3 (E = S, X = I): C(1)–S(1), 1.699(8), S(1)–Cu(1), 2.142(3), Cu(1)–I, 2.385(16), C(1)–S(1)–Cu(1), 108.0(3), N(1)–C(1)–N(2), 105.4(7), N(1)–C(1)–S(1), 129.3(6), N(2)–C(1)–S(1), 125.3(6), S(1)–Cu(1)–I, 160.72(10); for 4 (E = Se, X = Br): C(1)–Se(1), 1.855(5), Se(1)–Cu(1), 2.241(11), Cu(1)–Br, 2.222(12), C(1)–Se(1)–Cu(1), 105.84(15), N(1)–C(1)–N(2), 106.6(5), N(1)–C(1)–Se(1), 129.3(4), N(2)–C(1)–Se(1), 124.1(4), Se(1)–Cu(1)–Br, 163.34(6); for 5 (E = Se, X = I): C(1)–Se(1), 1.842(4), Se(1)–Cu(1), 2.252(9), Cu(1)–I, 2.389(8), C(1)–Se(1)–Cu(1), 104.86(13), N(1)–C(1)–N(2), 105.9(4), N(1)–C(1)–Se(1), 129.5(3), N(2)–C(1)–Se(1), 124.6(3), Se(1)–Cu(1)–I, 159.60(4). | ||
Upon coordination, the CS bond lengths (1.699(3) Å for 1, 1.699(3) Å for 2, 1.699(8) Å for 3) and CSe bond lengths (1.855(5) Å for 4, 1.842(9) Å for 5) were marginally increased related to their corresponding ligands (IMesS (1.675(18) Å) and IMesSe (1.830(6) Å)).5 The E–Cu–X bond angle in molecules 1–5 lies between 159.60(4)–165.94(6)°, suggesting the quasi-linear arrangement around the metal centre.
Molecules 1–5 have quite strong C–H⋯Cl hydrogen bonds, and also moderately strong C–H⋯S and C–H⋯Se hydrogen bonding interactions in their solid state structures as evidenced by single crystal X-ray diffraction analysis (see ESI-1, Fig. S16,† Chart 4). Surprisingly, the molecular packing of 1 is not comparable with 2–5. The hydrogen bonded polymeric chain through C(3)–H(3)⋯Cl (2.811(2) Å, 164.54(3)°) interactions are observed in 1, and are marginally stronger than the C–H⋯Cl interactions reported for [(IPrS)BiCl3]·CHCl3 (C(2)–H(2)⋯Cl(1); 2.871 Å, 145.32°).7k The solid state structure elucidated from the single crystal X-ray diffraction technique revealed the oppositely arranged molecular layers in molecule 1, while all the other molecules (2–5) were arranged as AA′AA′AA′AA′AA′ (see ESI-1, Fig. S16 and S17†) in their solid states structures. In addition to this, an unusual C–H⋯S bonding was noticed in 2 (3.101(1) Å, 134.84(1)°) and 3 (3.044(3) Å, 136.51(7)°), while the very rare C–H⋯Se bonding was observed in 4 (3.158(1) Å, 132.74(3)°) and 5 (3.1241(1) Å, 132.134(3)°).7k,14 The observed C–H⋯S interactions in 2 and 3 are quite weak compared to the interactions reported for [(IPrS)BiCl3]·CHCl3 (H⋯S; 2.797 Å, 175.23°)7k and are stronger than the interactions reported for N,N′-dimethylthioformamide (H⋯S; 3.781(7) Å, 175.4(7)°).15 Besides, the imidazole protons signal appeared to be shifted downfield (about 0.3 to 0.7 ppm) for molecules 1–5 due to the existence of hydrogen bonding (see ESI-1, S18†). Moreover, the C(1)–E(1)–Cu(1) bond angles are almost comparable. The existing E⋯H bond distances (3.044(3)–3.157(1) Å) and bond angles (132.13(3)–136.50(7)°) additionally support the moderately strong hydrogen bonding (Chart 4).7k,16
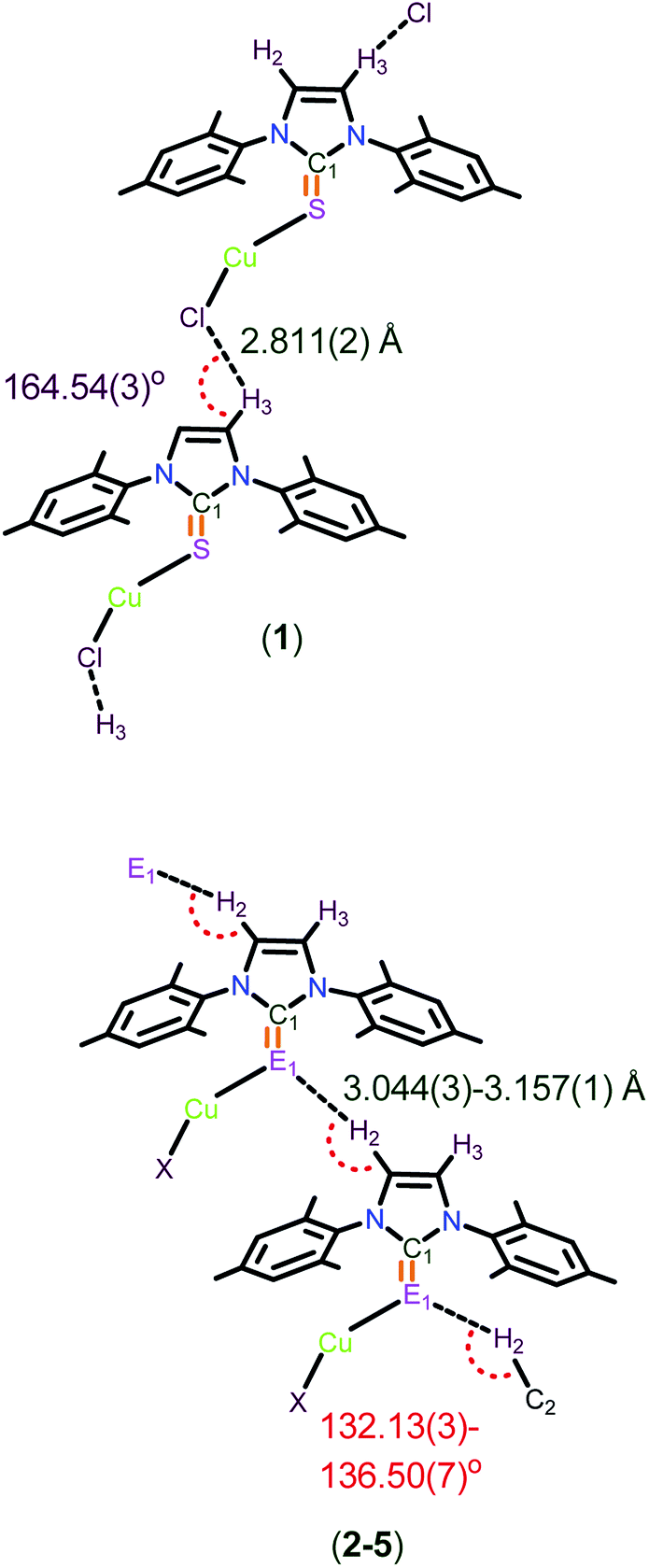 | ||
| Chart 4 The representation of H(3)⋯Cl(1) bond distances in 1 and H(2)⋯E(1) bond distances and C(2)–H(2)–E(1) bond angles in molecules 2–5. | ||
The cationic copper(I) complexes (7–11) were isolated in very good yields by treating IMesSe (for 7 and 9) and IPrSe (for 11) with [Cu(CH3CN)4]PF6 in methanol (Scheme 2 and 3). Interestingly, these complexes can also be isolated in very good yield along with cationic NHC-copper(I) complexes 8 and 10 by the ligand transfer method from [(IMes)CuCl] (6).17 The ligand exchange reaction signifies the better orbital overlap between the softer Lewis donor (NHCSe) and the soft Cu(I) metal center.18 Compounds 7, 9 and 11 were crystallized at an ambient temperature. The compounds 8 and 10 were separated by handpicking them from the mixture and were purified by recrystallization from saturated dichloromethane solutions. The formation of 7, 9 and 11 was established by elemental analysis, FT-IR, multinuclear NMR spectroscopy and single crystal X-ray diffraction techniques.
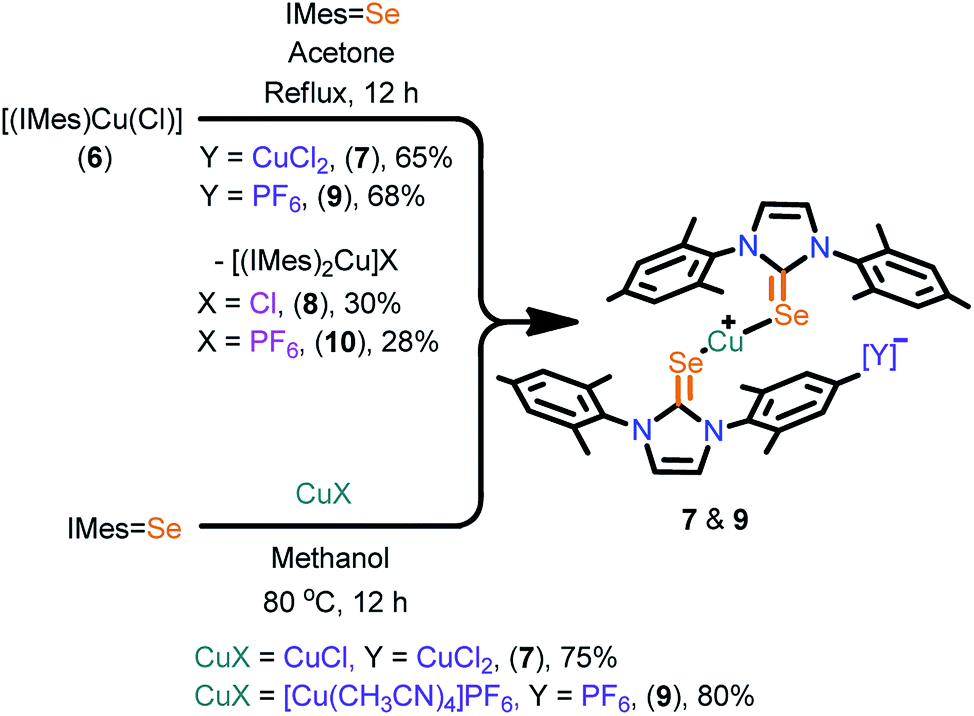 | ||
| Scheme 2 Synthesis of 7 and 9. | ||
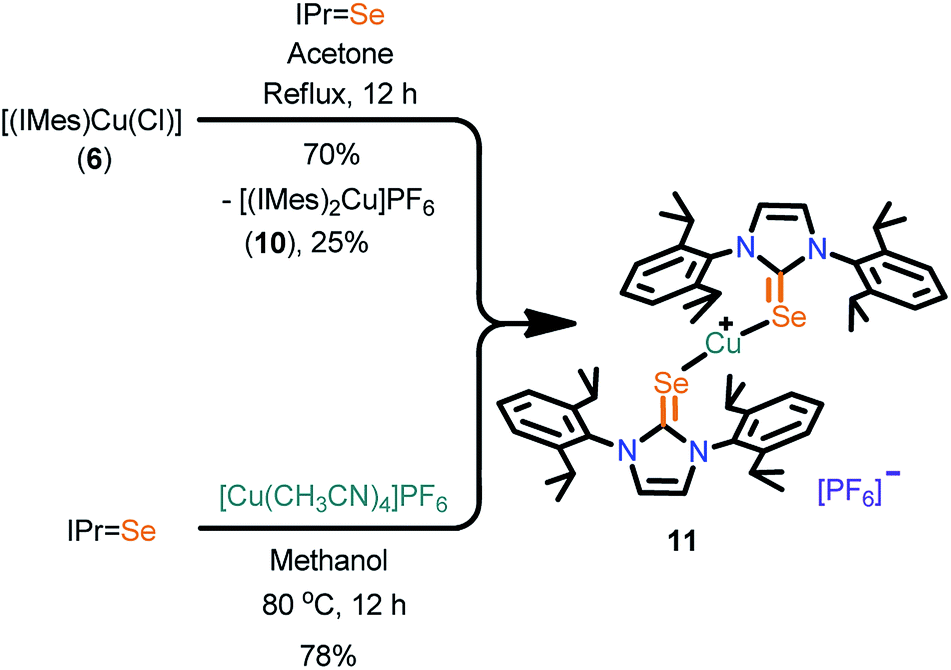 | ||
| Scheme 3 Synthesis of 11. | ||
The PF6 counter anion in molecules, 9 (834 cm−1), 10 (840 cm−1) and 11 (839 cm−1) were confirmed by IR spectroscopy. The 31P NMR displayed a septet for the presence of PF6 (−131.21 to −157.59 ppm) ion and the 19F NMR showed a doublet for the PF6 (−72.82 to −74.82 ppm) ion. The CSe was shifted upfield for 6 (178.98 ppm), for 7 (148.59 ppm), for 9 (142.28 ppm) and for 11 (154.21 ppm). Molecules 7 and 9 displayed (10–15 ppm) upfield shift after complexation, compared to their corresponding ligand (IMesSe, 157.49 ppm); 11 showed an upfield shift around 8 ppm, compared to its ligand (IPrSe, 162.14 ppm), suggesting the strong σ-donor nature of NHCE over NHC.7d,k,l
The solid-state structures of 7, 9 and 11 were confirmed by single crystal X-ray diffraction (Fig. 2). Complexes 7, 9 and 11 crystallized in the monoclinic space group, P21/n (for 7 and 9), C2/c (for 11). The crystallographic data for 7, 9 and 11 are given in Table S2 (see ESI†) and the important bond parameters are listed in the figure caption. The molecular structures of 7, 9 and 11 are cationic homoleptic mononuclear copper(I) selones. The copper(I) centre in 7, 9 and 11 are two coordinate with two imidazoline-2-selones and its valence satisfied with one counter anion, particularly, [CuCl2]− for 7, [PF6]− ion for 9 and 11. The cationic salt 7 represent the first copper–NHCE salt with loosely bound [CuCl2]− salt.
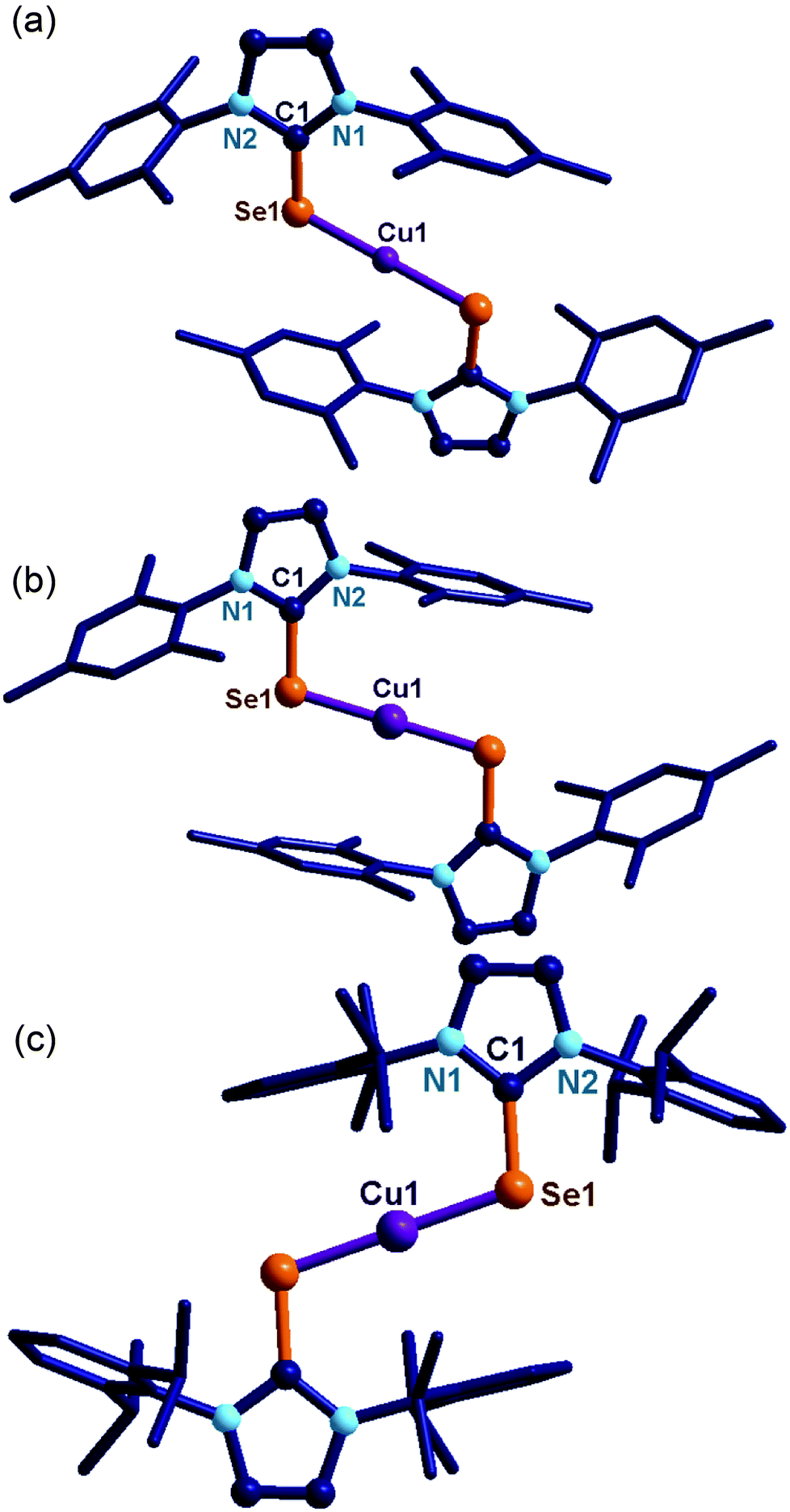 | ||
| Fig. 2 (a) Molecular structure of 7. Hydrogen atoms and dichlorocuprate counter ions have been omitted for clarity. Selected bond lengths (Å) and angles (°): C(1)–Se(1), 1.856(3), Se(1)–Cu(1), 2.253(3), Cu(2)–Cl(1), 2.090(2), C(1)–Se(1)–Cu(1), 105.24(9), N(1)–C(1)–N(2), 106.1(3), N(1)–C(1)–Se(1), 131.1(2), N(2)–C(1)–Se(1), 122.7(2), Se(1)–Cu(1)–Se(1′), 180.0. (b) The molecular structure of 9. Hydrogen atoms and hexafluoro phosphate counter anions have been omitted for clarity. Selected bond lengths (Å) and angles (°): C(1)–Se(1), 1.853(3), Se(1)–Cu(1), 2.252(3), C(1)–Se(1)–Cu(1), 104.61(10), N(1)–C(1)–N(2), 106.0(3), N(1)–C(1)–Se(1), 123.0(2), N(2)–C(1)–Se(1), 130.9(2), Se(1)–Cu(1)–Se(2), 180.0. (c) The molecular structure of 11. Hydrogen atoms and hexafluoro phosphate counter anions have been omitted for clarity. Selected bond lengths (Å) and angles (°): C(1)–Se(1), 1.849(3), Se(1)–Cu(1), 2.267(3), C(1)–Se(1)–Cu(1), 105.89(9), N(1)–C(1)–N(2), 105.7(2), N(1)–C(1)–Se(1), 122.7(2), N(2)–C(1)–Se(1), 131.6(2), Se(1)–Cu(1)–Se(2), 180.0. | ||
The CSe bond lengths in 7 (1.856(3) Å), 9 (1.853(3) Å) and 11 (1.849(3) Å) are considerably longer than in the corresponding ligands [IMesSe (1.830(6) Å) and IPrSe (1.822(4) Å)].7d,k,l The Se–Cu bond distances are similar and the Se–Cu–Se bond angles found in 7, 9 and 11 are perfectly linear as reported by our group for linear copper(I) chalcogenones.7d Molecules 8 and 10 produced poor quality crystals but the bonding modes of NHC to the copper(I) metal centre were clearly established (see ESI†) by single crystal X-ray measurement.
Interestingly, the presence of the dichlorocuprate counter ion in 7 is responsible for the expected existence of the mononuclear complex as shown in Scheme 4. The NMR studies (1H, 13C, HMBC and HSQC) on molecule 7 and its spectral changes as compared with 1 suggest the existence of a mononuclear complex without any counter ion. To the best of our knowledge, this is one of the rare examples of metal–NHCE that shows the dynamic equilibrium between homoleptic and heteroleptic species.
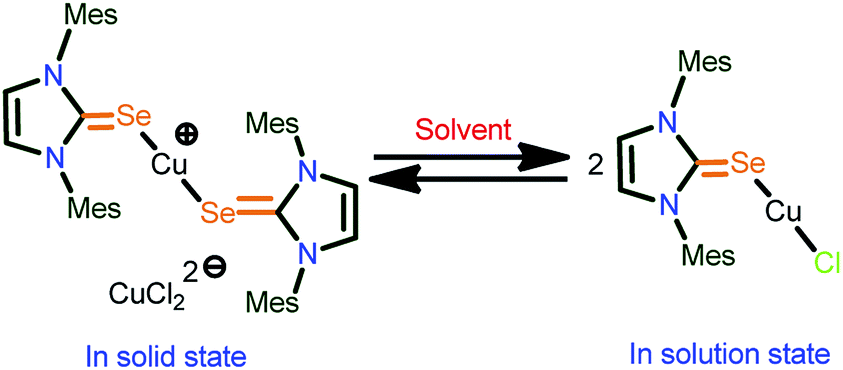 | ||
| Scheme 4 Expected solution-state structure of 7 as suggested by NMR studies. | ||
The 2D copper(I) layer 13 was isolated in good yield by treating [(PPh3)3Cu4I4] with Ebis. 13 can also be isolated from the direct reaction between CuI, PPh3 and Ebis (Scheme 5). The two-dimensional ionic coordination polymer of 14 was isolated from the one-pot reaction between CuI, PPh3, Ebpis and NH4BF4 or by treating 12 with Ebpis and NH4BF4 (Scheme 6). However, 12 was isolated in very good yield by treating CuI with an excess of PPh3 in acetonitrile.
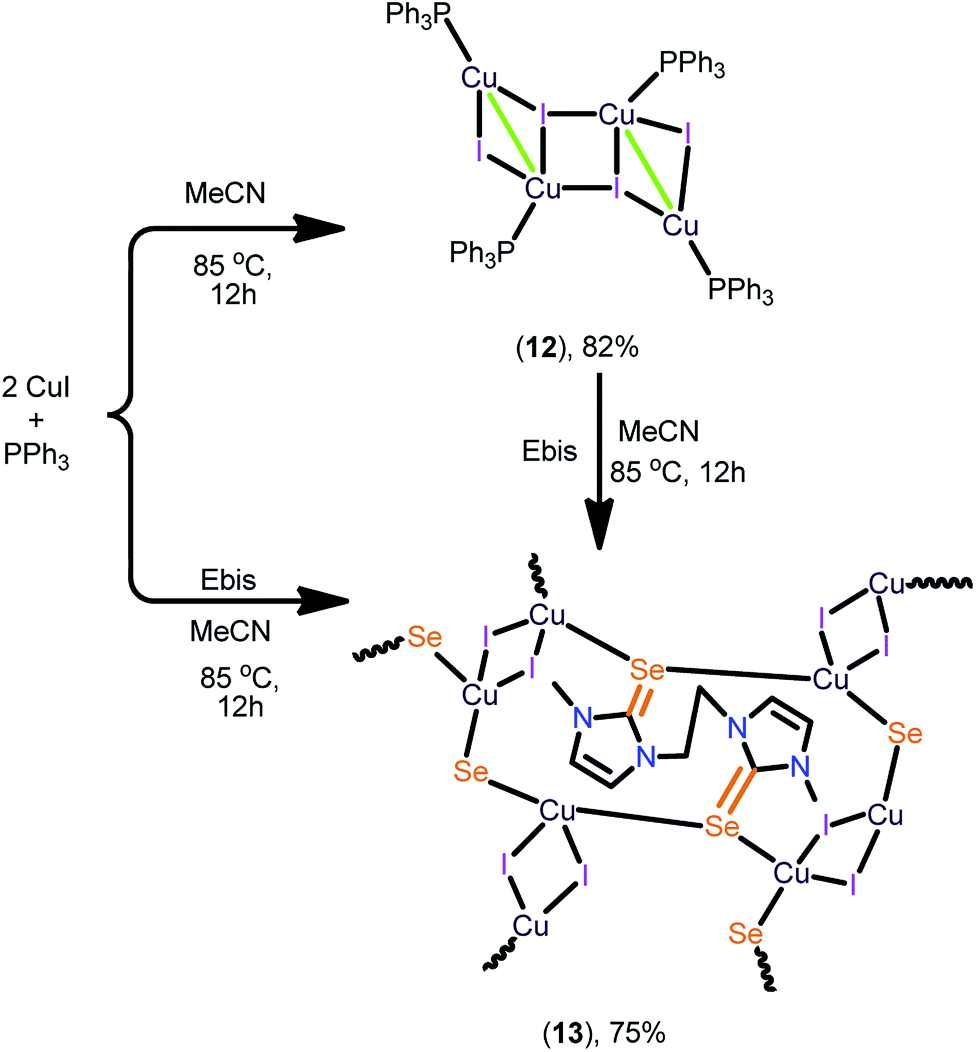 | ||
| Scheme 5 Synthesis of 12 and 13. | ||
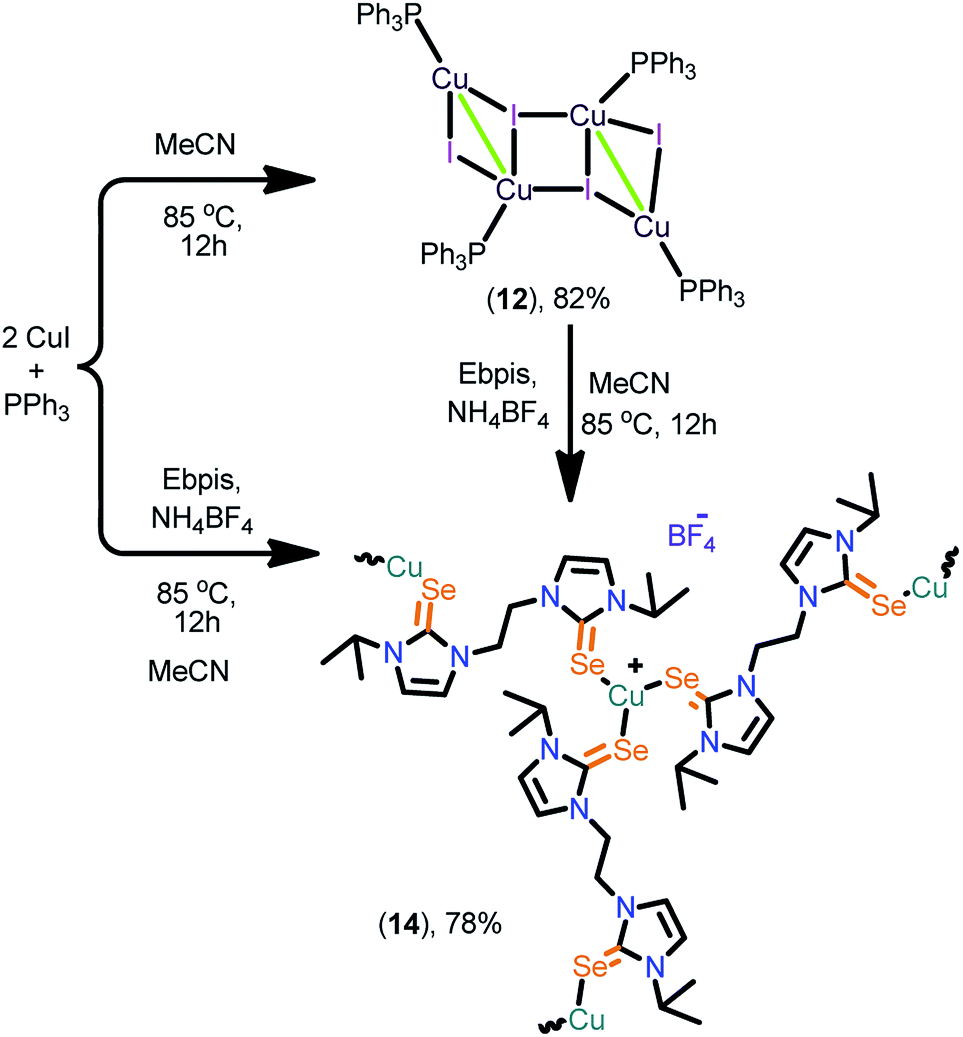 | ||
| Scheme 6 Synthesis of 14. | ||
These experiments signify the greater σ-donor strength of NHCSe compared to PPh3.16 The formation of 13 and 14 was established by elemental analysis, FT-IR, multinuclear NMR spectroscopy and single crystal X-ray diffraction techniques. The FT-IR spectrum of 14 indicates the existence of the BF4 counter ion (B–Fstrectching, = 1036 cm−1), and was also approved by a signal at −0.99 ppm in 11B NMR and a signal at −154.02 ppm in 19F NMR. The carbene carbon signal in 13 (155.73 ppm) and 14 (154.13 ppm) was up-field shifted as expected after coordination to copper.7d
The solid-state structures of 12–14 were determined by the single crystal X-ray diffraction technique (Fig. 3 and 4). Molecule 12 crystallized in the monoclinic space group, C2/c, while 13 crystallized in the monoclinic space group, P21/c and 14 crystallized in the tetragonal space group, P41212. The crystallographic data for 12, 13 and 14 are listed in Table S3 (see ESI†) and the important bond parameters are listed as figure captions.
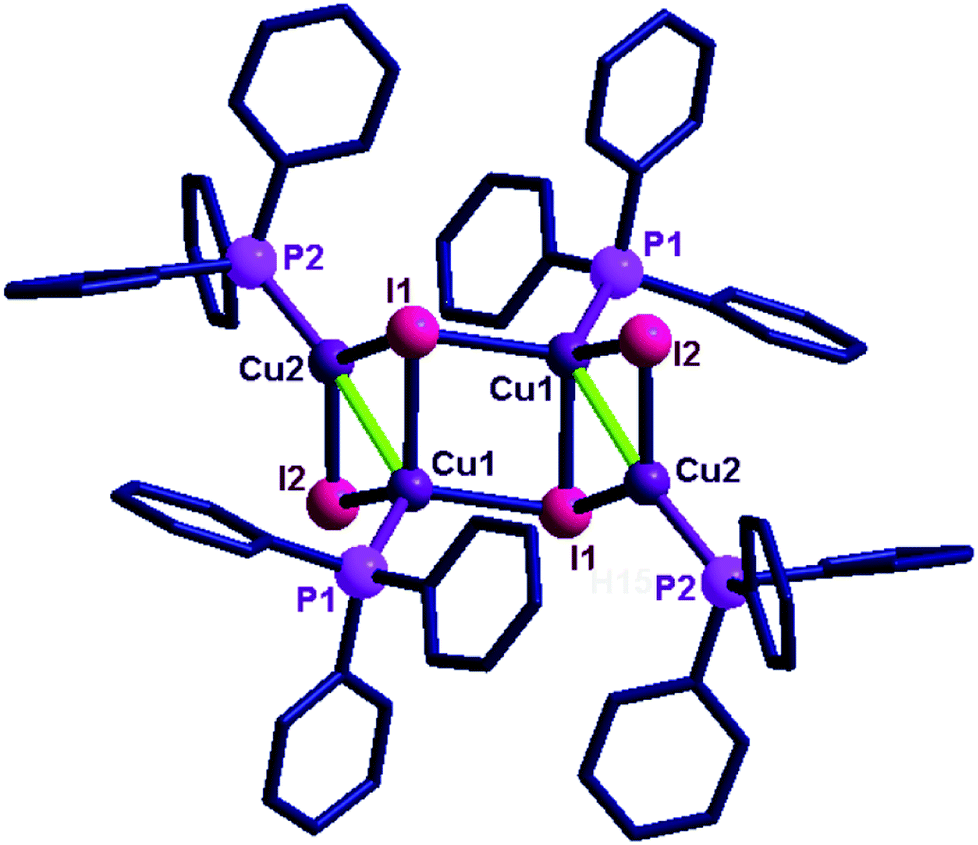 | ||
| Fig. 3 Solid state structure of 12. Selected bond lengths (Å) and angles (°): Cu(1)–P(1), 2.248(2), Cu(2)–P(2), 2.227(2), Cu(1)–Cu(2), 2.843(19), Cu(1)–I(1), 2.713(14), Cu(1)–I(2), 2.645(14), Cu(2′)–I(1), 2.590(14), Cu(2)–I(2), 2.533(14), Cu(1)–I(1)–Cu(2′), 105.86(4), Cu(1)–I(2)–Cu(2), 66.57(4), Cu(1′)–I(2)–Cu(2′), 64.72(2). | ||
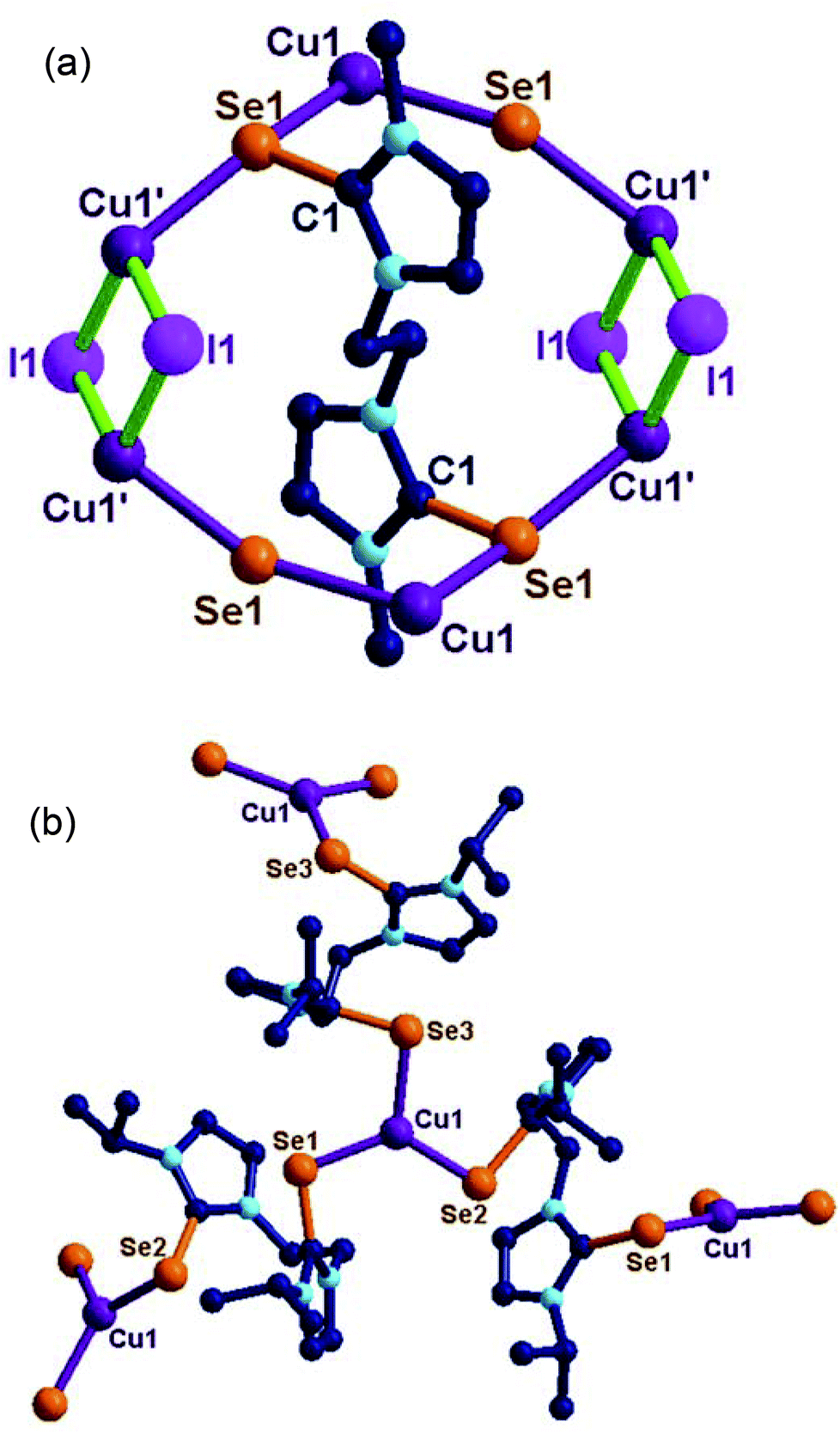 | ||
| Fig. 4 (a) Solid-state structure of 13. Selected bond lengths (Å) and angles (°): C(1)–Se(1), 1.867(7), Se(1)–Cu(1), 2.553(14), Cu(1)–I, 2.650(12), Cu(1′)–I, 2.660(13), C(1)–Se(1)–Cu(1), 95.3(2), C(1)–Se(1)–Cu(1′), 103.4(2), N(1)–C(1)–N(2), 106.8(6), N(1)–C(1)–Se(1), 127.3(5), N(2)–C(1)–Se(1), 125.9(5), Cu(1)–Se(1)–Cu(1′), 160.72(10). (b) Solid state structure of 14. Selected bond lengths (Å) and angles (°): C(1)–Se(1), 1.859(5), Se(1)–Cu(1), 2.336(9), C(8)–Se(2), 1.874(5), Se(2)–Cu(1), 2.323(10), C(15)–Se(3), 1.842(10), Se(3)–Cu(1), 2.352(10), C(1)–Se(1)–Cu(1), 102.35(15), C(8)–Se(2)–Cu(1), 99.85(16), C(15)–Se(3)–Cu(1), 98.6(3), Se(1)–Cu(1)–Se(2), 126.86(4), Se(1)–Cu(1)–Se(3), 114.60(4), Se(2)–Cu(1)–Se(3), 118.49(4), N(1)–C(1)–N(2), 106.0(5), N(1)–C(1)–Se(1), 125.6(4), N(2)–C(1)–Se(1), 128.3(4), N(3)–C(8)–N(4), 106.0(5), N(3)–C(8)–Se(2), 127.1(4), N(4)–C(8)–Se(2), 126.9(4), N(5)–C(15)–N(6), 108.7(12), N(5)–C(15)–Se(3), 125.1(10), N(6)–C(15)–Se(3), 126.2(11). | ||
The copper(I) centre in 12 shows two types of coordination environments, namely, tetra and penta with μ2 and μ3 bridging iodides. The coordination environment of the penta coordinated copper in 12 is fulfilled by three iodine atoms, one phosphine and one copper atom. The coordination environment of the tetra coordinated copper is fulfilled by two iodine atoms, one phosphorus and one copper atom. The μ3 bridged iodine and copper distances are longer than the distance noted between the μ2 bridged iodine with copper centres. The μ3 bonded Cu–I distances (2.713(14) Å) are slightly longer than the μ2 bonded Cu–I distances (2.645(14) Å). The Cu–P bond lengths [Cu(1)–P(1), 2.248(2) Å, Cu(2)–P(2), 2.227(2) Å] are comparable. The Cu–Cu distance in molecule 12 is 2.843(19) Å, which is comparable with the sum of the van der Waals radii for copper (2.8 Å).17
Molecule 13 is a two-dimensional sheet consisting of a Cu2I2 core. Each Cu2I2 core is further connected by Ebis ligands to form an interesting 2D layer of 13 (see ESI-1, S47†). The copper(I) centre in 13 adopts a tetrahedral geometry (106.384(0)–113.20(1)°) with two selone units and with two iodides. The bridging Cu–I distance (2.650(12) Å to 2.660(13) Å) are in the expected range. The Se–Cu distance is (2.553(14) Å) considerably longer than that of 4, 5, 7, 9, 10 and 14. This is rare structural evidence for the μ2 bridging mode of bis-imidazolin-2-chalcogenone ligands.6d,e,17b
Molecule 14 exists as a two-dimensional sheet through tri-coordinated homoleptic copper selenide (114.60(4)° to 126.86(4)°). The geometry of the copper(I) centre in 14 can be described as trigonal planar. The coordination environment around copper(I) is satisfied by μ3 bridging Ebpis ligands. The Se–Cu distances in 14 are slightly elongated (2.323(10)–2.336(9) Å) due to the formation of extended coordination (see ESI-1, S48†).
The solution-state UV-vis absorption spectra of 1–14 were measured in CH3CN at 25 °C (Fig. 5(a)–(f)). As shown in Fig. 5, the solution-state UV-vis absorption spectra of 1–14 were measured in CH3CN at 25 °C (Fig. 5(a)–(f)). The solid-state UV-visible spectra of 1–14 are broad compared to their solution-state UV-visible spectra, mainly due to the molecular association in the solid state. The selone derivatives (4 and 5) of IMesE show a slight bathochromic shift compared to the thione derivatives (1–3). Besides, all the complexes had absorption at higher wavelengths than the corresponding ligands IMesS (243, 273 nm), IMesSe (246 and 290 nm).7k The solution-state spectra are almost identical except for 5, which shows a slight red shift. Molecules 8 and 10 show slight red shifts for n–π* transitions, while in the solid-state π–π* and n–π* transitions are merged together to show a broad absorption range to support the molecular association. Upon coordination, 7, 9 and 11 showed shifts by about 50 nm for n–π* transitions due to the influence of the selone moiety. The solid-state UV-visible spectra of complexes 12–14 display broad signals in the range of 200–400 nm, while the same appeared to be distinct signals for π–π*and n–π* transitions in the solution-state UV-visible study. The absorption bands at 274 nm (for 13) and 278 nm (for 14) can be assigned to ligand–metal charge transfer.
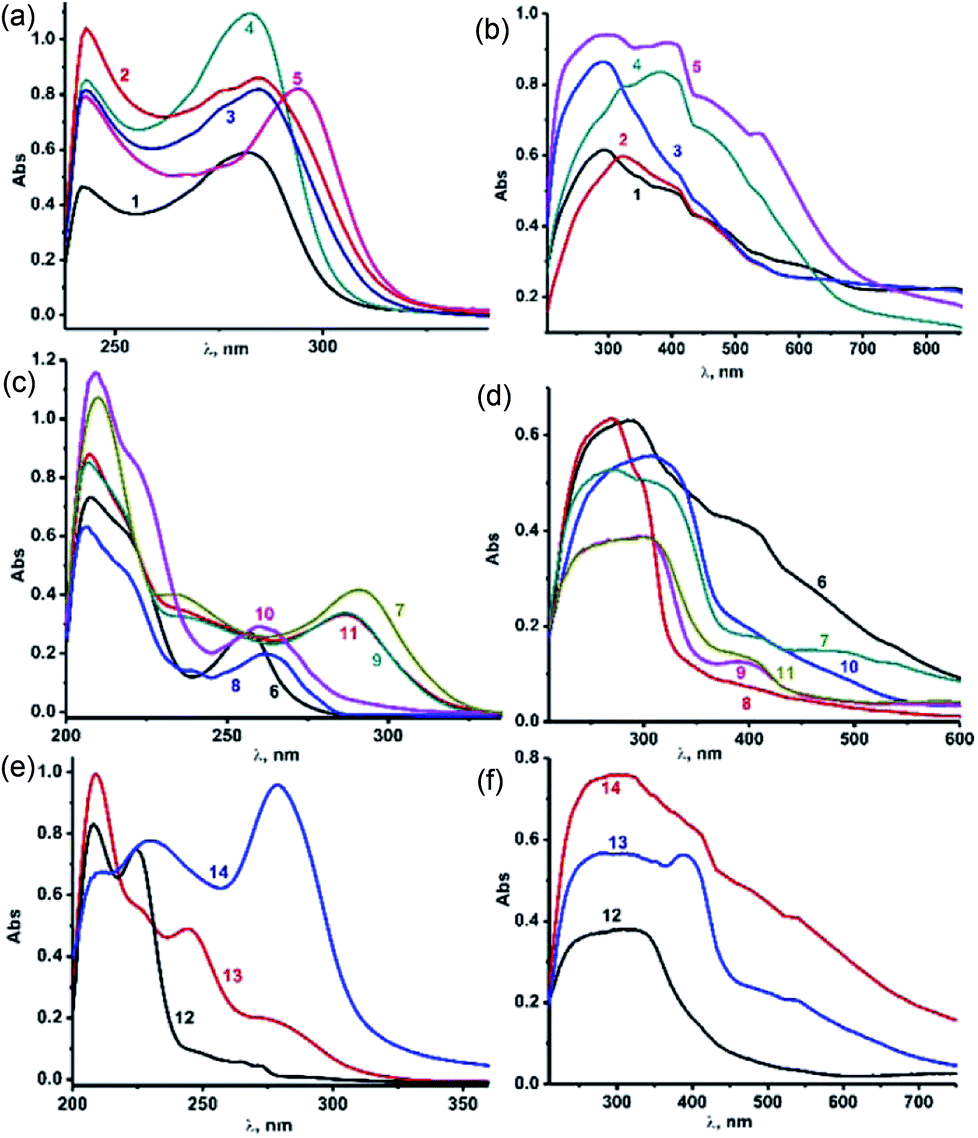 | ||
| Fig. 5 (a) Solution UV-vis spectra of complexes 1–5 in acetonitrile at 298 K with 1.2 × 10−5 M solutions. (b) Solid-state UV-vis spectra of complexes 1–5 at 298 K. (c) Solution UV-vis spectra of complexes 6–11 in acetonitrile at 298 K with 1.2 × 10−5 M solutions. (d) Solid state UV-vis spectra of complexes 6–11 at 298 K. (e) Solution UV-vis spectra of complexes 12–14 in acetonitrile at 298 K with 1.2 × 10−5 M solutions. (f) Solid state UV-vis spectra of complexes 12–14 at 298 K. | ||
The applications of the newly isolated catalysts (1–5, 7, 9 and 11–14) were investigated for the click reaction of azides with terminal alkynes. The well-known catalysts, such as [(IMes)CuCl] (6) together with the homoleptic NHC derivatives such as [(IMes)2Cu][Cl] (8) and [(IMes)2Cu][PF6] (10) were also tested for comparison.18 Although various bis-NHC, abnormal-NHC and few chalcogenone based ligands were also employed in CuAAC reactions, a detailed study comparing the catalytic efficiency of the NHCE supported copper(I) complexes with NHC or phosphine supported Cu(I) has not been conducted.7,20,21 Therefore, we have demonstrated the click catalysis using the NHCE supported copper(I) complexes and compared the catalytic efficiency with NHC and phosphine supported copper(I) complexes for the first time. To the best of our knowledge, the significance of ancillary ligands such as PPh3, NHC, NHCS and NHCSe have never been compared in any catalysis.7d,e,8,20
The catalytic reactions were carried out under neat conditions at room temperature (Scheme 7) (Fig. 6). Notably, the catalysts 1–5, 6, 8 and 10 (Entries 1–5) gave very good conversion (70–92%) within 1 h;19 the linear copper(I) chalcogenones (7, 9 and 11) gave moderate yields (68–76%, Entries 7, 9 and 11).
 | ||
| Scheme 7 [3+2] cycloaddition of benzylazide with phenyl acetylene. | ||
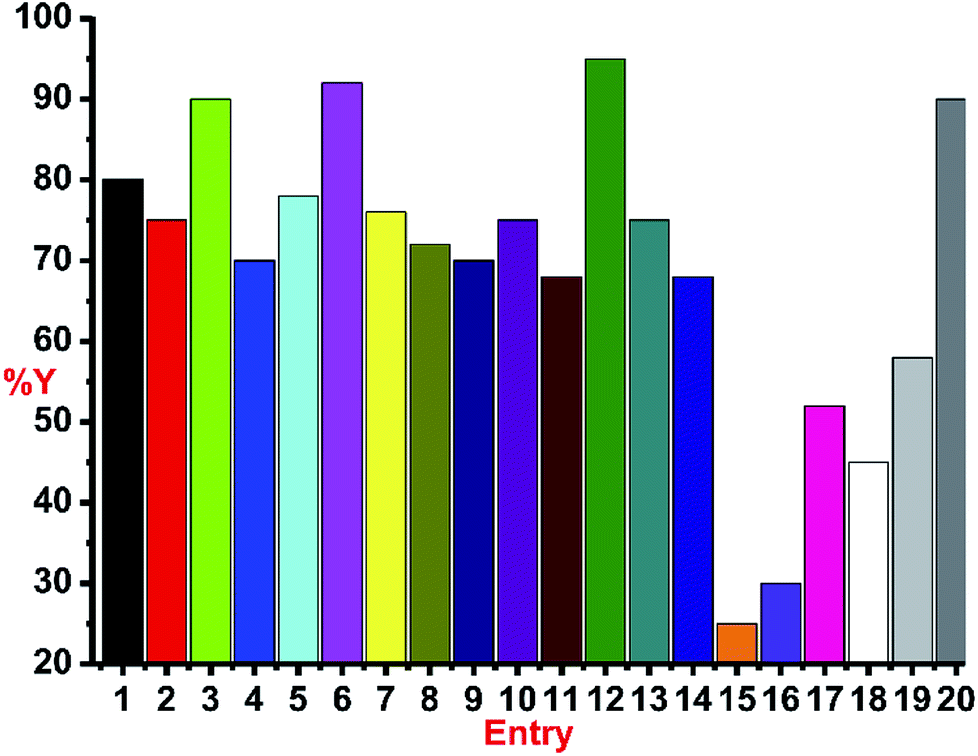 | ||
| Fig. 6 The screening of catalysts 1–14 in click catalysis. Reaction conditions: phenyl acetylene (1.2 mmol), benzyl azide (1.0 mmol), catalyst (1 mol%) and neat conditions at RT. Entries: 15, CuCl only; 16, only CuI; 17, IMesS and CuCl; 18, IMes.HCl and CuCl; 19, PPh3 and CuI; 20, 0.5 mol% 12 for 4 h; %Y, %isolated yield by column chromatography. | ||
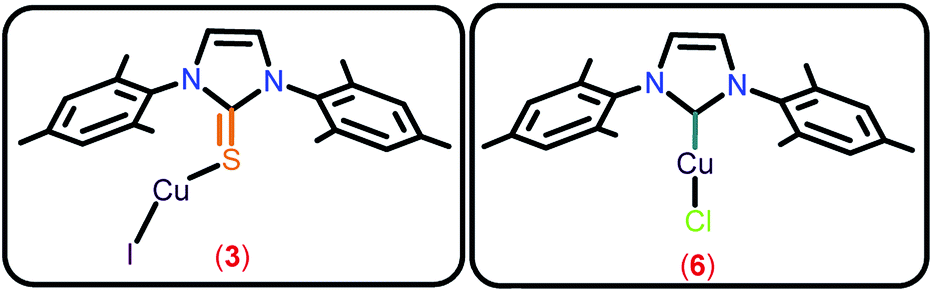
The coordination polymers (13 and 14) (68–75%, Entries 13–14) were found to be as active as linear chalcogenones in this catalysis. The phosphine-based copper(I) iodide (12) was found to be the most efficient (95%, Entry 12) among all the catalysts isolated herein. Catalysts 3, 6 and 12 were found to be the most efficient in this catalysis (90–95%, Entries 3, 6 and 12) among all the isolated catalysts 1–14 (Chart 5). The effect of the ligand for this transformation was investigated by carrying out the experiments with only copper(I) chloride (Entry 15) or copper(I) iodide (Entry 16) and a poor yield was obtained. The in situ generated catalysts gave considerable yields (52–58%, Entries 17 and 19). The IMes.HCl addition to CuCl also produced a decent yield (45%, Entry 18). Entries 15–19 show the significance of the presence of ligand for this reaction and indicate the prominence of a well-defined catalyst. The decrease in the quantity of catalyst 12 to 0.5 mol% led to the isolation of 90% yield but after 4 h (Entry 20). Thus, catalysts 3, 6 and 12 seem to be the efficient catalysts.
 | ||
| Chart 5 Catalysts used for the substrate scope in click catalysis. | ||
In order to investigate the effect of solvent in this reaction, the better catalysts (3, 6 and 12) were subjected to click catalysis in various polar solvents as described in Fig. 7. Virtually identical output was perceived for all three catalysts in different solvents, but water was found to be a better choice (Entry 1).
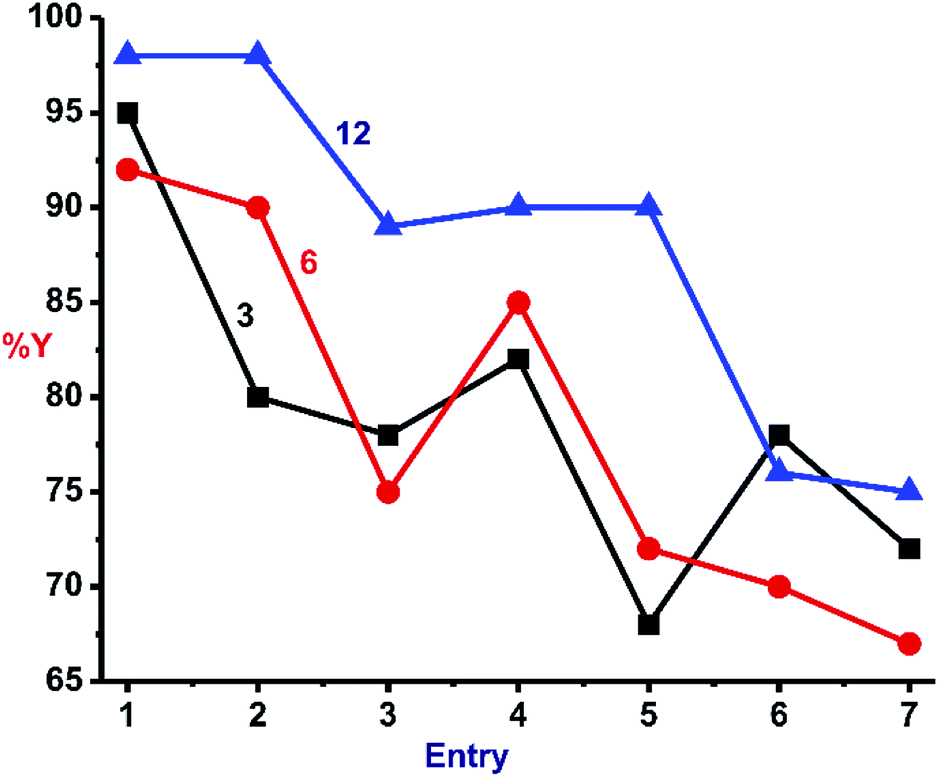 | ||
| Fig. 7 Solvent screening in various solvents using catalysts 3 (black), 6 (red) and 12 (green); reaction conditions: phenylacetylene (1.2 mmol), benzylazide (1.0 mmol), catalyst (1 mol%) and solvent at RT. Entries: 1, in water; 2, in DMSO:water; 3, in THF:water; 4, in tBuOH:water; 5, in tBuOH; 6, in DMSO; and 7 in THF. % Y, % isolated yield by column chromatography. | ||
All three catalysts 3, 6 and 12 were examined for the substrate scope in water (Scheme 8, Chart 6). Interestingly, the substantial discrepancies in reactivity were not observed. The catalysts 3, 6 and 12 were remarkably efficient to produce 18 different heterocyclic compounds.
 | ||
| Scheme 8 [3+2] cycloaddition of arylazides with terminal alkynes. | ||
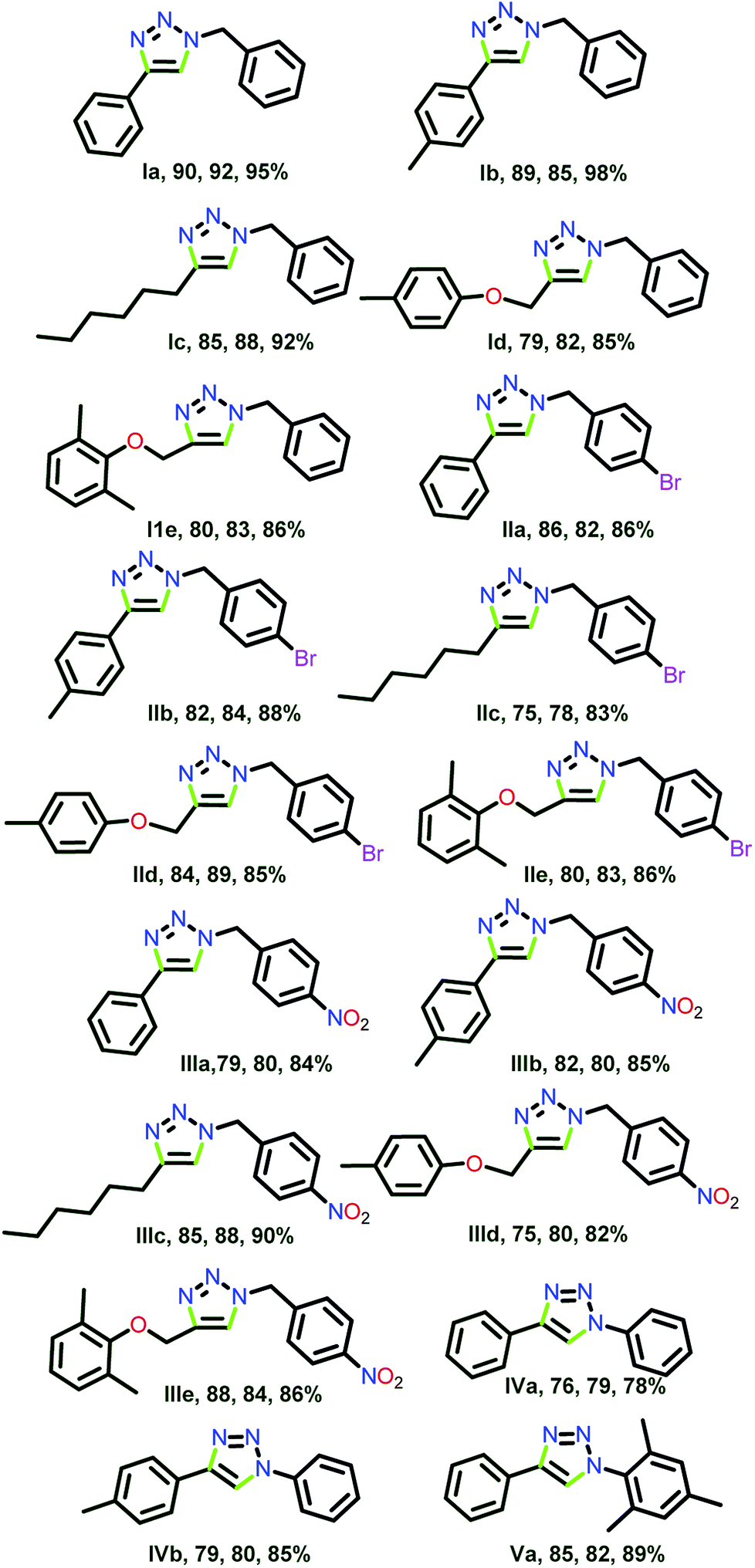 | ||
| Chart 6 1,2,3-Triazoles isolated by click catalysis by 3, 6 and 12 in water (see ESI-2, Table S2-1†). | ||
The plausible mechanistic pathway through which the thione/selone supported copper(I) complex proceeds, the click catalysis, has been displayed in chart 722
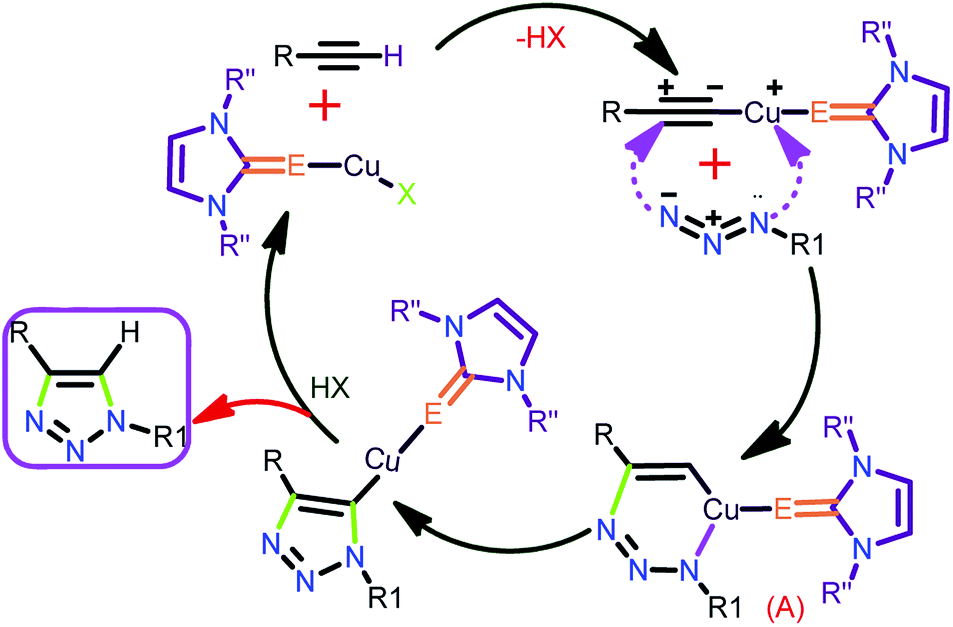 | ||
| Chart 7 Plausible mechanisms for the Huisgen coupling reaction by catalyst 3.22 | ||
The copper(I) catalyst is expected to form an intermediate A by coordinating with both terminal alkyne and azide (Chart 7). Followed by the successive reproduction of catalyst to yield the desired 1,2,3-triazoles.
It is worth stating that the steric hindrance in molecules 7–11 and 13–14 has a major influence in constructing the intermediate A, which disfavours the product formation compared to catalysts 3 and 6. The reasonably less steric hindrance exists at the metal centre in 3 due to the localization of the metal centre away from NHC via the thione, favouring the formation of the intermediate A in addition to the easy-leaving iodine attached to the metal (Chart 8). The greater efficiency of 12 was anticipated based on the presence of a greater number of electrophilic metal centers and also the more π-accepting (PPh3) ligand, while 3 and 6 displayed relatively less activity because of the presence of the weakly π-accepting ligands attached to the metal.
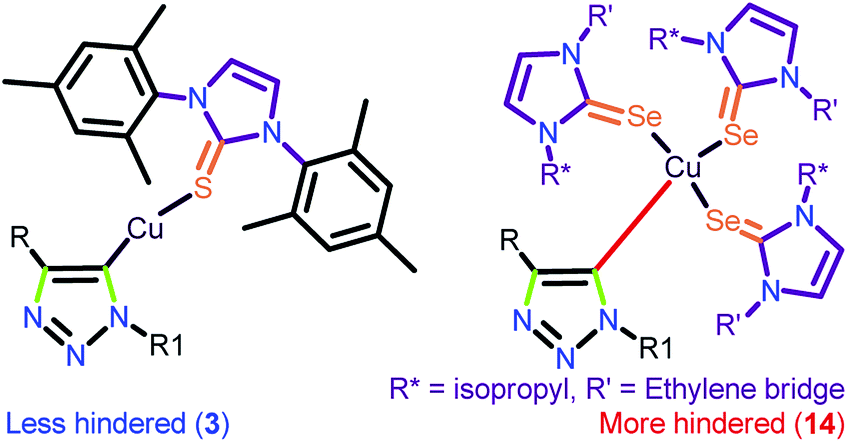 | ||
| Chart 8 Expected steric hindrance at the metal centre in 3 and 14. | ||
Alkynylsilane derivatives are a noticeable class of structural motifs in organic synthesis as Si-masked synthetic intermediates, particularly for C–C and C–X (X = heteroatom) bond formation reactions.23 The cross-dehydrogenative coupling of terminal alkynes and hydrosilanes has been studied with various metal salts such as H2PtCl6/I2, CuCl/TMEDA (TMEDA = N,N,N′,N′-tetraethylenediamine), LiAlH4, Zn(SO2CF3)2/pyridine, MgO, and KNH2/Al2O3.24 However, the metal complexes-mediated cross-dehydrogenative coupling is limited, and the only example known so far is M(η2-Ph2CNPh)(hmpa)3 (M = Yb or Ca, hmpa = hexamethylphosphoramide) (Chart 9).25
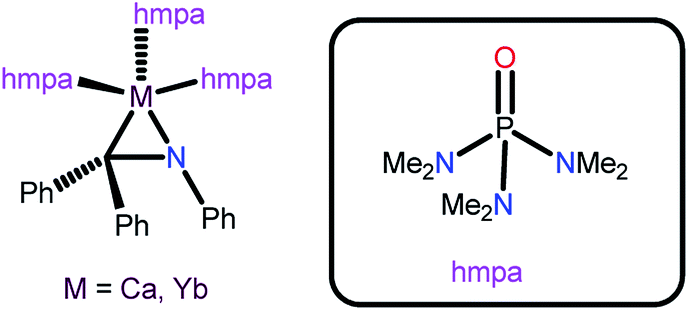 | ||
| Chart 9 Catalysts used for the cross-dehydrogenative coupling of alkynes with hydrosilanes. | ||
Catalysts 1–14 were screened for the cross-dehydrogenative coupling of alkynes with hydrosilanes (Scheme 9 and Table 1). Initially, the dehydrogenative silylation was examined for suitable reaction conditions using phenylacetylene and triethylsilane (1:1.2 equivalents) in acetonitrile at 100 °C for 12 h without base (Entry 1) with 1 mol% catalyst 3 to produce 65% of the desired product. Interestingly, the addition of catalytic amounts (20 mol%) of organic base such as pyridine produced a quantitative yield (Entry 2). Nevertheless, catalysts 6 and 12 also gave the desired product in very good yield (Entries 13 and 14).
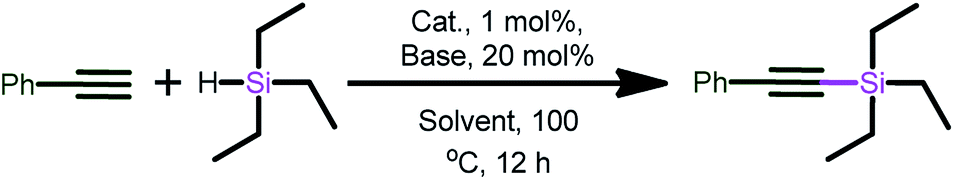 | ||
| Scheme 9 Cross-dehydrogenative coupling of terminal alkynes with hydrosilanes. | ||
| E | Solvent | Base | SMCb |
|---|---|---|---|
| a Reaction conditions: phenylacetylene (0.40 mmol), triethylsilane (0.60 mmol), catalyst (1 mol%), base (20 mol%), solvent (1.0 mL); SMC: starting material conversion.b Percentage of conversion is based on GC (the given GC conversion values are the average of at least two independent measures).c Without catalyst.d Reaction at room temperature.e With catalyst 6.f With catalyst 12. | |||
| 1 | CH3CN | — | 65% |
| 2 | CH3CN | Pyridine | 98% |
| 3c | CH3CN | Pyridine | 10% |
| 4 | CH3CN | K2CO3 | 57% |
| 5 | CH3CN | KOtBu | 78% |
| 6 | CH3CN | NEt3 | 55% |
| 7 | CH3CN | KOH | 68% |
| 8 | CH3OH | Pyridine | 12% |
| 9 | THF | Pyridine | 19% |
| 10 | 1,4-Dioxane | Pyridine | 14% |
| 11 | Toluene | Pyridine | 30% |
| 12d | CH3CN | Pyridine | 62% |
| 13e | CH3CN | Pyridine | 95% |
| 14f | CH3CN | Pyridine | 92% |
Catalyst 3 was utilized for this transformation to determine suitable conditions. The effect of base was investigated by employing the reaction with K2CO3 (Entry 4), KOtBu (Entry 5), NEt3 (Entry 6) and KOH (Entry 7), but none of them produced quantitative yields (55–78% only). The effect of solvent on this reaction was studied by using CH3OH (Entry 8), THF (entry 9), 1,4-dioxane (Entry 10) and toluene (Entry 11). The change in solvent did not favor the formation of the desired product. Subsequently, the influence of temperature on the reaction rate was investigated (Entry 12) by performing the reaction at room temperature. Even after extending the time for 24 h, the desired product formed was not satisfactory. Similarly, the significance of catalyst in this transformation was reviewed by performing this experiment without catalyst, and the yields were found to be inadequate (Entry 3). The plausible mechanism for the reaction is shown in Chart 10. The initial step may necessitate the nucleophilic attack to a copper center by alkyne to result transition state A followed by the nucleophilic attack to a silane to give the hypervalent silicon hydride via four membered transition-state B with a hydrogen bond, followed by the elimination of hydrogen to give the silane coupled product and regeneration of the catalyst.
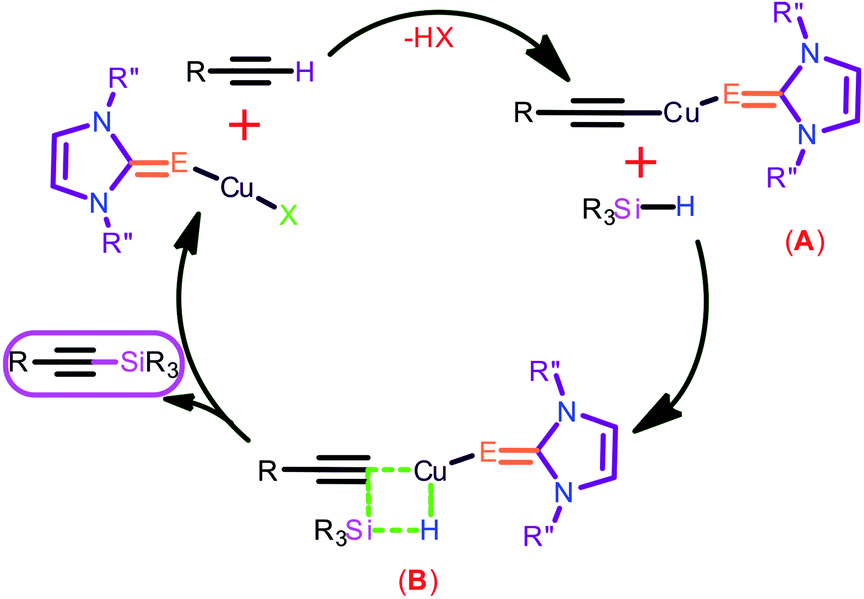 | ||
| Chart 10 Possible mechanisms for the dehydrogenative coupling of silanes by catalyst 3. | ||
As presented in Chart 11 phenyl acetylene and 1-octyne were treated with triethylsilane, dimethylphenylsilane and over 90% yields were achieved (compounds VI–IX). Similarly, the double dehydrogenative coupling was carried out using diphenylsilane to afford the silicon-tethered diyene building blocks (X–XI) with very good yield. It is worth mentioning that the dehydrogenative coupling of alkynes with triphenyl silane led to the recovery of starting materials, which supports the unfavorable condition of the steric bulk of silane in this transformation.
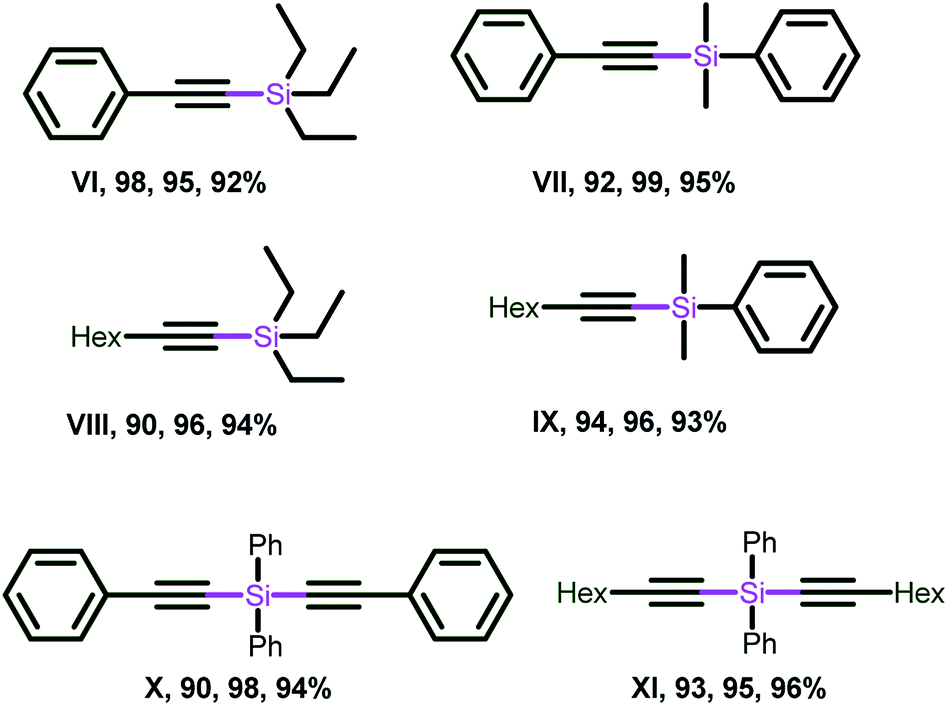 | ||
| Chart 11 Alkynylsilanes isolated by 3, 6 and 12. Reaction conditions for X and XI: phenylacetylene (0.80 mmol), diphenylsilane (0.40 mmol), catalyst (1 mol%), base (20 mol%), solvent (1.0 mL). (See ESI-2, Table S2-2†). | ||
Conclusions
Copper(I) complexes supported by NHC (for 6, 8 and 10), NHCE (for 1–5, 7, 9, 11, 13 and 14) and PPh3 (for 12) were synthesized and structurally characterized. The molecules 1–5 were isolated as rare mononuclear NHCE supported neutral copper(I) chalcogenones. The molecules 7, 9 and 11 were isolated by ligand exchange reaction between [(IMes)CuCl] and 6, 8 and 10. The synthetic methodology for 6, 8 and 10 represents the first synthetic strategy to isolate copper chalcogenones from copper carbene derivatives. Complexes 7–11 display a perfectly linear geometry around the copper(I) center. Molecules 13 and 14 were isolated from the copper(I) phosphine iodide complex 12. These newly isolated molecules 1–14 were used as catalysts for the [3+2] cycloaddition of azides with terminal alkynes. The catalysts 3, 6 and 12 were relatively more active for cycloaddition reactions. The cationic 3, 6 and 12 were found to be efficient in the C–Si bond formation reaction. (i) The ligand exchange experiment signifies the higher σ-donor abilities of NHCE. (ii) The PPh3 based catalyst (12) is effective in click catalysis over NHCE and NHC based catalysts. (iii) Less steric hindrance and more Lewis acidic metal centres facilitate the reaction. (iv) The efficiencies derived in this work are 12> 3> 6> 1= 2= 4= 5> 7= 8= 9= 10= 11= 13= 14. (v) The softer Lewis donor (NHCE) seems to have better orbital overlap with the soft Cu(I) metal center than NHC or PPh3. Investigations are currently in progress in our laboratory comparing NHC and NHC–analogous metal complexes in terms of stability and reactivity in organic transformations in order to reduce the reaction times and produce quantitative yields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the DST-SERB (EMR/2017/001211) for financial support. KS thank UGC for the fellowship.Notes and references
- (a) S. V. C. Vummaleti, D. J. Nelson, A. Poater, A. G. –Suárez, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 6, 1895–1904 RSC; (b) M. K. Barman, A. K. Sinha and S. Nembenna, Green Chem., 2016, 18, 2534–2541 RSC; (c) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef PubMed; (d) D. J. Nelson, F. Nahra, S. R. Patrick, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2014, 33, 3640–3645 CrossRef.
- For the catalytic reactions studied with both NHC–metal and NHCE–metal:
(a) M. M. Kimani, C. A. Bayse, B. S. Stadelman and J. L. Brumaghim, Inorg. Chem., 2013, 52, 11685–11687 CrossRef PubMed;
(b) E. E. Battin, M. T. Zimmerman, R. R. Ramoutar, C. E. Quarles and J. L. Brumaghim, Metallomics, 2011, 3, 503–512 RSC.
- J. Choi, N. Kang, H. Y. Yang, H. J. Kim and S. U. Son, Chem. Mater., 2010, 22, 3586–3588 CrossRef.
- (a) D. J. D. Wilson, S. A. Couchman and J. L. Dutton, Inorg. Chem., 2012, 51, 7657–7668 CrossRef PubMed; (b) K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 14, 2416–2425 CrossRef; (c) D. J. Nelson, A. Collado, S. Manzini, S. Meiries, A. M. Z. Slawin, D. B. Cordes and S. P. Nolan, Organometallics, 2014, 33, 2048–2058 CrossRef; (d) A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef.
- (a) Y. Rong, A. A. Harbi, B. Kriegel and G. Parkin, Inorg. Chem., 2013, 52, 7172–7182 CrossRef PubMed; (b) V. Rani, H. B. Singh and R. J. Butcher, Eur. J. Inorg. Chem., 2017, 31, 3720–3728 CrossRef.
- (a) C. A. Bayse and J. L. Brumaghim, Biochalcogen Chemistry: The Biological Chemistry of Sulphur, Selenium and Tellurium, ACS Symposium series 1152, American Chemical Society, Washington, DC, 2013, pp. 57–64 Search PubMed; (b) M. M. Kimani, J. L. Brumaghim and D. VanDerveer, Inorg. Chem., 2010, 49, 9200–9211 CrossRef PubMed; (c) M. M. Kimani, H. C. Wang and J. L. Brumaghim, Dalton Trans., 2012, 41, 5248–5259 RSC; (d) M. M. Kimani, C. A. Bayse and J. L. Brumaghim, Dalton Trans., 2011, 40, 3711–3723 RSC; (e) M. M. Kimani, D. Watts, L. A. Graham, D. Rabinovich, G. P. A. Yap and J. L. Brumaghim, Dalton Trans., 2015, 44, 16313–16324 RSC; (f) B. S. Stadelman, M. M. Kimani, C. A. Bayse, C. D. McMillen and J. L. Brumaghim, Dalton Trans., 2016, 45, 4697–4711 RSC.
- (a) E. Alvarado, A. C. Badaj, T. G. Larocque and G. G. Lavoie, Chem.–Eur. J., 2012, 18, 12112–12121 CrossRef PubMed; (b) J. Jin, H. –W. Shin, J. H. Park, J. H. Park, E. Kim, T. K. Ahn, D. H. Ryu and S. U. Son, Organometallics, 2013, 32, 3954–3959 CrossRef; (c) N. Ghavale, S. T. Manjare, H. B. Singh and R. J. Butcher, Dalton Trans., 2015, 44, 11893–11900 RSC; (d) K. Srinivas, C. N. Babu and G. Prabusankar, Dalton Trans., 2015, 44, 15636–15644 RSC; (e) H. R. Kim, G. Jung II, K. Yoo, K. Jang, E. S. Lee, J. Yun and S. U. Son, Chem. Commun., 2010, 46, 758–760 RSC; (f) C. N. Babu, K. Srinivas and G. Prabusankar, Dalton Trans., 2016, 45, 6456–6465 RSC; (g) W. G. Jia, Y. B. Huang, Y. J. Lin and G. X. Jin, Dalton Trans., 2008, 5612–5620 RSC; (h) Y. B. Huang, W. G. Jia and G. X. Jin, J. Organomet. Chem., 2009, 694, 86–90 CrossRef; (i) A. K. Sharma, H. Joshi, R. Bhaskar and A. K. Singh, Dalton Trans., 2017, 46, 2228–2237 RSC; (j) O. Prakash, K. N. Sharma, H. Joshi, P. L. Gupta and A. K. Singh, Organometallics, 2014, 33, 2535–2543 CrossRef; (k) K. Srinivas, P. Suresh, C. N. Babu, A. Sathyanarayana and G. Prabusankar, RSC Adv., 2015, 5, 15579–15590 RSC; (l) K. Srinivas, A. Sathyanarayana, C. N. Babu and G. Prabusankar, Dalton Trans., 2016, 45, 5196–5209 RSC; (m) L. M. Zhang, H. Y. Li, H. X. Li, D. J. Young, Y. Wang and J. P. Lang, Inorg. Chem., 2017, 56, 11230–11243 CrossRef PubMed.
- K. Srinivas and G. Prabusankar, Dalton Trans., 2017, 46, 16615–16622 RSC.
- D. D. Perrin and W. L. F. Armarego, Purification of laboratory chemicals, Pergamon Press, London, 3rd edn, 1988 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef; (b) G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, Universität Göttingen, Göttingen, 1997 Search PubMed.
- N. Parvin, S. Pal, S. Khan, S. Das, S. K. Pati and H. W. Roesky, Inorg. Chem., 2017, 56, 1706–1712 CrossRef PubMed.
- W. Xie, J. H. Yoon and S. Chang, J. Am. Chem. Soc., 2016, 138, 12605–12614 CrossRef PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef.
- H. Borrmann, I. Persson, M. Sandström and C. M. V. Stålhandske, J. Chem. Soc., Perkin Trans. 1, 2000, 1, 393–402 RSC.
- (a) M. R. L. Furst and C. S. J. Cazin, Chem. Commun., 2010, 46, 6924–6925 RSC; (b) N. M. Scott and S. P. Nolan, Eur. J. Inorg. Chem., 2005, 1815–1828 CrossRef; (c) G. A. Blake, J. P. Moerdyk and C. W. Bielawski, Organometallics, 2012, 31, 3373–3378 CrossRef.
- (a) V. Charra, P. d. Frémont and P. Braunstein, Coord. Chem. Rev., 2017, 293, 48–79 Search PubMed; (b) M. Slivarichova, R. C. d. Costa, J. Nunn, R. Ahmad, M. F. Haddow, H. A. Sparkes, T. Gray and G. R. Owen, J. Organomet. Chem., 2017, 847, 224–233 CrossRef.
- (a) T. Nakamura, T. Terashima, K. Ogata and S. i. Fukuzawa, Org. Lett., 2011, 13, 620–623 CrossRef PubMed; (b) S. D. González, A. Correa, L. Cavallo and S. P. Nolan, Chem.–Eur. J., 2006, 12, 7558–7564 CrossRef PubMed.
- S. D. González, E. C. E. Adán, J. B. Buchholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC.
- (a) R. Berg, J. Straub, E. Schreiner, S. Mader, F. Rominger and B. F. Straub, Adv. Synth. Catal., 2012, 354, 3445–3450 CrossRef; (b) S. C. Sau, S. R. Roy, T. K. Sen, D. Mullangi and S. K. Mandal, Adv. Synth. Catal., 2013, 355, 2982–2991 CrossRef; (c) S. Hohloch, C. Y. Su and B. Sarkar, Eur. J. Inorg. Chem., 2011, 3067–3075 CrossRef; (d) F. Wang, H. Fu, Y. Jiang and Y. Zhao, Green Chem., 2008, 10, 452–456 RSC; (e) V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef PubMed; (f) S. I. Presolski, V. Hong, S.-H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef PubMed; (g) S. Q. Bai, L. L. Koh and T. S. A. Hor, Inorg. Chem., 2009, 48, 1207–1213 CrossRef PubMed.
- (a) F. Lazreg and C. S. J. Cazin, Organometallics, 2018, 37, 679–683 CrossRef; (b) F. Lazreg, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7969–7975 CrossRef.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (b) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef PubMed.
- (a) Y. R. Leroux, H. Fei, J.-M. Noël, C. Roux and P. Hapiot, J. Am. Chem. Soc., 2010, 132, 14039–14041 CrossRef PubMed; (b) R. Gleiter and D. B. Werz, Chem. Rev., 2010, 110, 4447–4488 CrossRef PubMed; (c) J. P. Brand, D. F. Gonzàlez, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102–115 RSC; (d) B. M. Trost, M. T. Rudd, M. G. Costa, P. I. Lee and A. E. Pomerantz, Org. Lett., 2004, 6, 4235–4238 CrossRef PubMed.
- (a) M. G. Voronkov, N. I. Ushakova, I. I. Tsykhanskaya and V. B. Pukhnarevich, J. Organomet. Chem., 1984, 264, 39 CrossRef; (b) H. Q. Liu and J. F. Harrod, Can. J. Chem., 1990, 68, 1100 CrossRef; (c) M. Itoh, M. Kobayashi and J. Ishikawa, Organometallics, 1997, 16, 3068 CrossRef; (d) T. Tsuchimoto, M. Fujii, Y. Iketani and M. Sekine, Adv. Synth. Catal., 2012, 354, 2959 CrossRef; (e) M. Itoh, M. Mitsuzuka, T. Utsumi, K. Iwata and K. Inoue, J. Organomet. Chem., 1994, 476, c30 CrossRef; (f) T. Baba, A. Kato, H. Yuasa, F. Toriyama, H. Handa and Y. Ono, Catal. Today, 1998, 44, 271 CrossRef.
- (a) K. Takaki, M. Kurioka, T. Kamata, K. Takehira, Y. Makioka and Y. Fujiwara, J. Org. Chem., 1998, 63, 9265 CrossRef; (b) F. Buch and S. Harder, Organometallics, 2007, 26, 5132 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, NMR, X-ray data, figures and GC data. 1H and 13C NMR spectra of the synthesized complex. Catalysis protocols and characterization data of catalytic products. CCDC 1855114–1855124. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06057f |
| This journal is © The Royal Society of Chemistry 2018 |