
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSplit-anion solvent extraction of light rare earths from concentrated chloride aqueous solutions to nitrate organic ionic liquids†
Mercedes Regadío a,
Tom Vander Hoogerstraetea,
Dipanjan Banerjeeb and
Koen Binnemans*a
a,
Tom Vander Hoogerstraetea,
Dipanjan Banerjeeb and
Koen Binnemans*a
aKU Leuven – University of Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. Box 2404, 3001 Heverlee, Belgium. E-mail: koen.binnemans@kuleuven.be
bDutch-Belgian Beamline (DUBBLE), ESRF – The European Synchrotron, CS 40220, F-38043 Grenoble Cedex 9, France
First published on 10th October 2018
Abstract
Despite its benefits, the extraction of rare earths (REEs) from chloride solutions with neutral or basic extractants is not efficient, so that separation is currently carried out by using acidic extractants. This work aims to improve this process by replacing the conventional molecular diluents in the organic phase by ionic liquids (ILs) which contain coordinating anions. The extraction of La(III), Ce(III) and Pr(III) from concentrated chloride solutions was tested with a quaternary ammonium and a phosphonium nitrate IL extractant. Dissolution of a trialkylphosphine oxide neutral extractant (Cyanex 923) in the nitrate ILs changed the preference of the organic phase from lighter to heavier REE and increased the overall extraction efficiency and the loading capacity of the organic phase. An increase of the CaCl2 concentration in the feed solution resulted in higher extraction efficiencies, due to a lower activity of water and hence to a poorer hydration of the REE ions. In that respect, chloride ions were not coordinating to the REE ion after extraction from concentrated chloride solutions. To achieve selectivity, one should fine-tune the loading by varying the CaCl2 and/or Cyanex 923 concentrations. Adjustment of the CaCl2 concentration in the feed and stripping solutions is essential for the separation of mixtures of REE. However, and unlike in the case of acidic extractants, no control of equilibrium pH is required. The split-anion extraction offers the possibility to separate mixtures of REEs in different groups without having to change the chloride feed solution. It leads to safer and environmentally friendlier extraction processes by (1) using solvents that are not volatile, not flammable and do no accumulate static electricity, (2) consuming no acids or alkali, (3) easy stripping with water and (4) avoidance to create nitrate-containing effluents.
1 Introduction
The group of rare-earth elements (REEs) comprises the 15 lanthanides together with scandium (Sc) and yttrium (Y). In nature, the REEs occur together in the same ores. Lanthanum (La) and especially cerium (Ce) are the most abundant REEs, but they are used in relatively few applications. On the contrary, the other REEs are scarcer, while their demand is constantly growing, particularly that of neodymium (Nd) because of its use in permanent magnets.1,2 Thus, the demand for the different REEs is not according to their availability. This contributes to the so-called balance problem, which is the challenge to keep the production and the demand of individual REEs in equilibrium over time.3 As a consequence, La and Ce are produced in relatively large excess, with, being a key step in the production of REE concentrates, their separation from the other REEs.The most important technique for the separation of mixtures of REEs in industry is solvent extraction.4 In solvent extraction, a REE-containing feed solution is intensively mixed with an immiscible organic phase. After phase disengagement, the organic phase selectively extracts the target metal ions, leaving the other metal ions in the raffinate. Co-extracted metal ions are subsequently removed from the loaded organic phase by a scrubbing stage, after which the target metal ions are stripped and thereafter the organic phase is regenerated. Full separation is achieved by many extraction, scrubbing and stripping stages. Conventional solvent extraction uses molecular organic solvents, which have a chance of igniting due to a spark after static electricity build-up. Furthermore, REE solvent extraction processes consume significant volumes of acid and alkali when using acidic extractants, or generates nitrate-containing waste water when extracting from nitrate aqueous feed solution with neutral and basic extractants. On the other hand, neutral and basic extractants have the advantages that no pH control is required, consumption of acid and alkali is low, stripping can easily be done by water and gel formation in the organic phase is largely prevented. However, the extraction efficiencies of REE from chloride feed solutions with neutral and basic extractants are typically quite low, thus requiring the use of nitrate feed solutions.
Recently, a novel solvent extraction approach called split-anion extraction, was introduced to allow efficient extraction of REEs from chloride medium with basic extractants, by replacing the molecular organic solvents by ionic liquids.5 Ionic liquids (ILs) are solvents that consist entirely of ions, in general a polyatomic organic cation and an inorganic or organic anion. ILs might offer an alternative to conventional organic solvents such as kerosene, because of their negligible vapour pressure (low volatility), very low flammability, little accumulation of static electricity and high chemical stability.6–10 Thus, using ILs leads to safer and greener extraction processes.11 In split-anion extraction, different anions are present in the aqueous and organic phases, and metal extraction occurs through ion complexation instead of (an)ion exchange. The anions of the IL in the organic phase form stable complexes with the REE and prefer to stay in the organic phase, while the anions in the aqueous phase are highly hydrated and have a strong affinity to stay in the aqueous phase.
During the processing of REE concentrates, it is common practice to obtain two different REE solutions: a high value solution containing Pr(III) and the heavier REE, and another one consisting of La(III) and Ce(III). The latter can be applied in car exhaust catalysts, fluid cracking catalysts (FCC) and nickel metal hydride (NiMH) batteries. Despite its importance, the separation of La(III) and Ce(III) from the rest of REE has often been neglected. The objective of this work is to optimize the separation of Pr(III) from La(III) + Ce(III), in chloride aqueous solutions by split-anion extraction. For that purpose, simulated leachates from a REE ore deposit and nitrate ionic liquids were used. The performance of two ILs, tricaprylmethylammonium nitrate (Aliquat 336 nitrate, abbreviated to [A336][NO3]) and trihexyl(tetradecyl)phosphonium nitrate (Cyphos IL 101 nitrate, abbreviated to [C101][NO3]) is evaluated, with and without the addition of the neutral extractants.
2 Experimental materials and methods
2.1 Chemicals and materials
Hydrochloric acid (HCl, 37% in water), ethylene glycol (99.9%), Triton™ X-100 (≥99.0%) and praseodymium(III) chloride hexahydrate (PrCl3·6H2O 99.9%) were purchased from Acros Organics (Geel, Belgium), nitric acid (HNO3, 67% in water) from Sigma-Aldrich (Diegem, Belgium), sodium hydroxide pellets (NaOH, ACS grade) and absolute ethanol from VWR International Chemicals (Leuven, Belgium), lanthanum(III) chloride heptahydrate (LaCl3·7H2O, 99.99%), cerium(III) chloride heptahydrate (CeCl3·7H2O, 99.9%) and tri-n-butylphosphate (TBP, 99%) from Alfa Aesar (Karlsruhe, Germany) and calcium chloride dihydrate (CaCl2·2H2O, >99.5%) and potassium nitrate (KNO3, 99%) were purchased from Chem-Lab (Zedelgem, Belgium). Cyphos IL 101 (97.7% trihexyl(tetradecyl)phosphonium chloride) and Cyanex 923 (93%, a mixture of four trialkylphosphine oxides, mainly dioctylhexylphosphine oxide (42%) and dihexyloctylphosphine oxide (31%)) were obtained from Cytec Industries (Ontario, Canada) and Aliquat 336 (88.2–90.6% quaternary compounds, mixture of methyltrioctyl, predominant and methyltridecylammonium chlorides, manufactured by BASF) was purchased from Sigma-Aldrich (Diegem, Belgium). All chemicals were used as received without any further purification. Single-element ICP aqueous standard solutions (1000 mg L−1 of neodymium and samarium in 2–5% HNO3) were obtained from Chem-Lab (Zedelgem, Belgium). Ultrapure water (deionized to a resistivity of 18.2 MΩ cm at 25 °C) was prepared with a Sartorius Arium Pro ultrapure water system.A CaCl2 stock solution (5 mol L−1) was made by dissolving CaCl2·2H2O in ultrapure water. Four aqueous REE stock solutions were also prepared in ultrapure water and acidified with a drop of 37 wt% HCl solution to avoid hydrolysis of the REE ions. The first REE solution (solution A) was made by dissolving a mixture of 0.3 mol L−1 (40 g L−1) of metal ions in total, specifically La(III), Ce(III) and Pr(III) chlorides in an ion mass ratio of 29, 63 and 8%, respectively. This composition was chosen to simulate a pregnant leach solution (PLS) obtained after HCl leaching of a REE concentrate produced from the eudialyte deposit of the Norra Kärr in Sweden.12,13 The aqueous feed solutions for the extraction tests were prepared by mixing the CaCl2 stock solution and solution A to achieve a final CaCl2 concentration between 1 and 4 mol L−1 and a total metal concentration between 0.05–0.3 mol L−1 (7 and 40 g L−1). In some cases, ethylene glycol (between 10 and 50 v%) was also added. The other three aqueous REE stock solutions contained 3.26 mol L−1 CaCl2 plus an individually dissolved REE chloride: 47.2 g L−1 La(III) (solution B), 50.8 g L−1 Ce(III) (solution C) and 47.0 g L−1 Pr(III) (solution D). These single-element REE–CaCl2 aqueous solutions were used as prepared to study the effect of the volume ratio of Cy923/[C101][NO3] on the loading of the organic phase and the nature of the REE complex extracted to the organic phase. The aqueous solutions used for the recovery of metals from the loaded organic phases (stripping agents), consisted of water with different concentrations of CaCl2 (from 0 to 5 mol L−1 of CaCl2).
The ionic liquids employed in the organic phases were the nitrate forms derived from tricaprylmethylammonium chloride (trade name: Aliquat 336, abbreviated to [A336][Cl]) and trihexyl(tetradecyl)phosphonium chloride (trade name: Cyphos IL 101, abbreviated to [C101][Cl]). Cyphos IL 101 and Aliquat 336 conveniently have safety and technical information, good characterization and established extraction mechanism.12–18 Aliquat 336 is a well-known basic extractant that is used diluted in molecular diluents and for REE extraction from nitrate and thiocyanate aqueous media.17 However, it is not often used as pure IL or as diluent, neither for extraction from chloride solutions, like in this work.15 Both nitrate ionic liquids [A336][NO3] and [C101][NO3] were synthesized by a metathesis reaction from the above chloride ILs. The metathesis was performed by three contacts with fresh 2.5 mol L−1 KNO3 solution at a phase volume ratio ionic liquid-to-aqueous of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (100 mL each phase). Excess ions were removed by washing three times with dilute HNO3 (pH 3–4) in a 1:1 phase ratio.5 The success of the metathesis reaction was checked by measuring the potassium and chloride content in the prepared ILs with TXRF. To decrease the viscosity, and to avoid phase volume changes in solvent extraction and REE hydrolysis; the nitrate ILs were pre-saturated with slightly acidic water (pH 3–4). ILs with cations containing several long alkyl chains have in general low toxicity due to the low solubility in water and difficult uptake through the cell membrane due to the absence of surfactant properties.19–22 The nitrate ion is known to have a low toxicity. The ILs considered in this study have no fluorinated anions, moderate viscosity, and no cation exchange with the aqueous phase during extraction. Their long alkyl chains provide high hydrophobicity, low losses to the aqueous solutions and fast disengagement time; all desirable characteristics for solvent extraction. Additionally, compared to other ILs, [C101][NO3] and [A336][NO3] are liquids at room temperature, hydrophobic, relatively cheap and good diluents for other extractants. As a result, these ILs are good candidates for replacing the organic diluents commonly used in solvent extraction processes.14,23–28
:1 (100 mL each phase). Excess ions were removed by washing three times with dilute HNO3 (pH 3–4) in a 1:1 phase ratio.5 The success of the metathesis reaction was checked by measuring the potassium and chloride content in the prepared ILs with TXRF. To decrease the viscosity, and to avoid phase volume changes in solvent extraction and REE hydrolysis; the nitrate ILs were pre-saturated with slightly acidic water (pH 3–4). ILs with cations containing several long alkyl chains have in general low toxicity due to the low solubility in water and difficult uptake through the cell membrane due to the absence of surfactant properties.19–22 The nitrate ion is known to have a low toxicity. The ILs considered in this study have no fluorinated anions, moderate viscosity, and no cation exchange with the aqueous phase during extraction. Their long alkyl chains provide high hydrophobicity, low losses to the aqueous solutions and fast disengagement time; all desirable characteristics for solvent extraction. Additionally, compared to other ILs, [C101][NO3] and [A336][NO3] are liquids at room temperature, hydrophobic, relatively cheap and good diluents for other extractants. As a result, these ILs are good candidates for replacing the organic diluents commonly used in solvent extraction processes.14,23–28
The solvent of the extractions tests consisted of the above mentioned ionic liquids, with or without a neutral extractant. Two neutral extractants were investigated: tri-n-butylphosphate (TBP) and a commercial mixture of trialkylphosphine oxides (trade name: Cyanex 923, abbreviated to Cy923). The working organic solutions used for the stripping tests (loaded organic phase), consisted of 20 v% Cy923 in [C101][NO3] loaded with 30 g L−1 of REE (1.8 g L−1 La(III), 9.1 g L−1 Ce(III) and 20.3 g L−1 Pr(III)).
2.2 Methods and analyses
The split-anion extraction study was carried out by varying different parameters: initial pH, water:ethylene glycol ratio, REE and CaCl2 concentrations in the aqueous feed solution; the type of IL, the type and amount of neutral extractant in the organic phase, and the temperature. The experiments were performed in 4 mL glass vials with 1 mL of each phase, unless otherwise stated. The samples were shaken horizontally in a Nemus Life TMS-200 turbo thermoshaker (50 °C, 2500 rpm, for 45 min, unless otherwise stated), followed by centrifuging in a Heraeus Labofuge 200 (3500 rpm, 5 min) to accelerate the phase disengagement. The bottom aqueous phase was separated from the upper IL phase by a needle syringe. Samples were taken from both the IL and aqueous phases for analysis of the concentration of metal ions with a benchtop TXRF spectrometer (Bruker S2 Picofox). The TXRF samples were prepared by dilution and addition of an optimum internal standard, for counteracting matrix effects.29 The dilution was performed with 5 v% TritonX-100 in water in the case of aqueous samples, and with ethanol absolute in the case of organic samples, until the analyte had an expected concentration of 50 mg L−1. One or two specific internal standards were added in a concentration close to the one expected for the analyte.30 Nd and Sm were chosen as preferred internal standard for La, Ce and Pr, as the energy of their X-ray fluorescence lines are close to the analyte lines.
The extraction data are displayed in terms of the distribution ratio (D), percentage extraction (E) and separation factor (α). The distribution ratio (D) is the ratio of the concentration of the element in the two different phases at equilibrium:
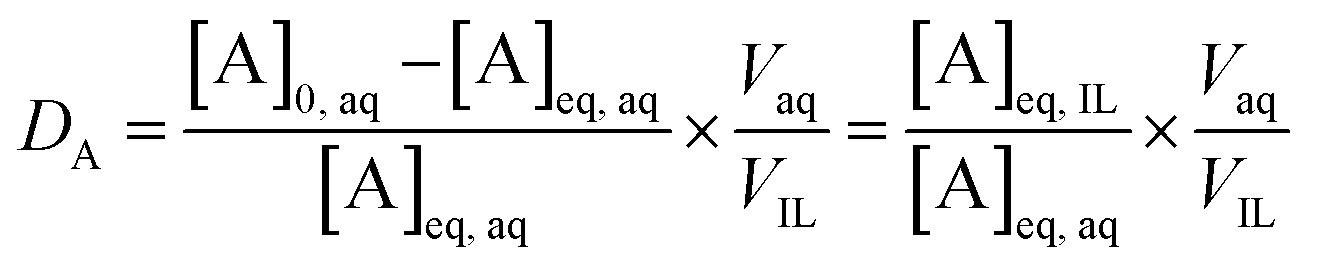 | (1) |
The percentage extraction (E) expresses the proportion of an element moving from one phase to the other:
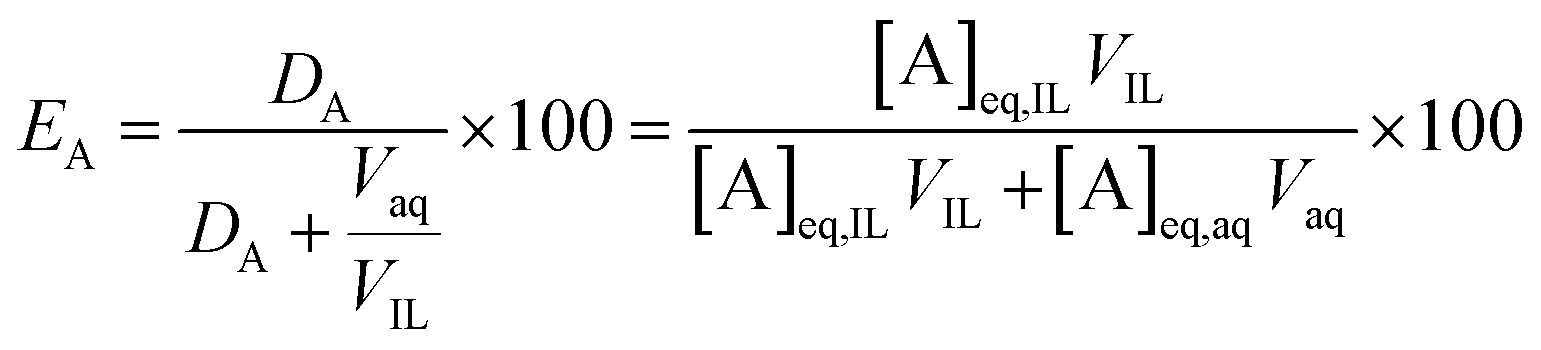 | (2) |
At a phase volume ratio (VIL/Vaq) of unity, eqn (2) can be simplified into:
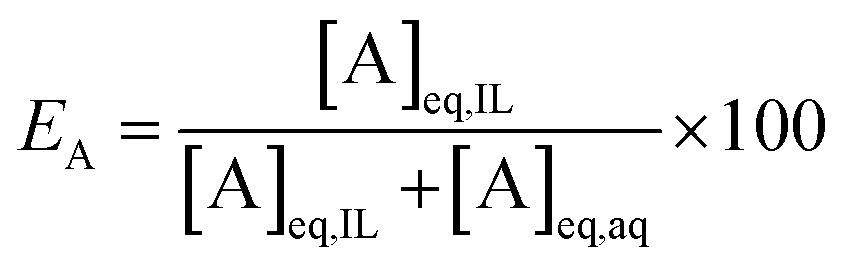 | (3) |
In this paper, the efficiency is preferably expressed in terms of E instead of D, since the former is less sensitive to changes in the extraction parameters. For example, at equal phase ratio, when the D value varies from 500 to 1000, the E changes only from 99.8% to 99.9% (eqn (2)).
The separation factor is used to compare the extractability between two elements (αAB) or group of elements (αA+BC), distributed from one to another phase. This factor α is the equilibrium constant of the exchange reaction of the elements between the two immiscible phases (eqn (4)).
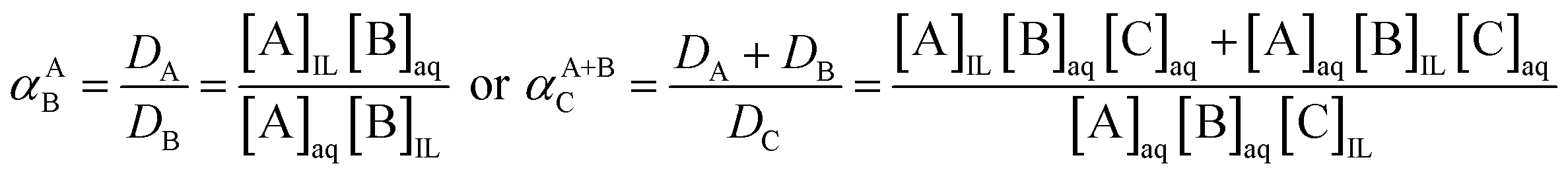 | (4) |
The percentage stripping (S) was used to describe the recovery of the elements from the loaded organic phase (eqn (5)).
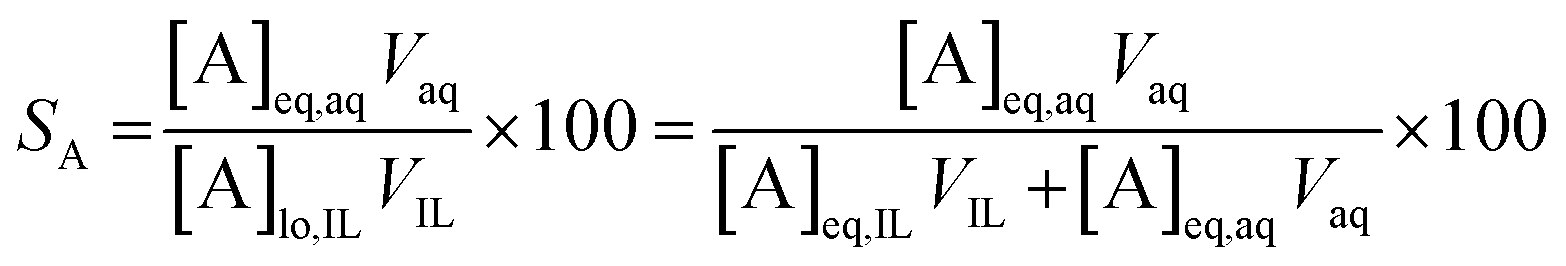 | (5) |
At a phase volume ratio (VIL/Vaq) of unity, eqn (5) can be simplified into eqn (6):
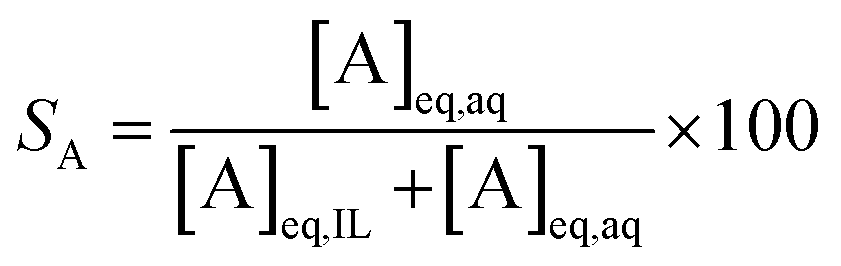 | (6) |
The loading of the organic phase as a function of the volume ratio of Cy923/[C101][NO3] was tested with the feed solution B, C and D (Section 2.1), in 15 mL centrifuge tubes. Four consecutive contact steps of the same organic phase with fresh new feed solution and at phase volume ratio organic-to-aqueous 1:1 (except the last one at 1:2.85) were performed in a Thermo Scientific MaxQ™ 2000 benchtop orbital shaker (8 h, RT, 300 rpm). The REE content in the samples after each contact was measured via TXRF for calculating the extension of the metal loading in the organic phases at each step (eqn (S1)†).
The speciation in the loaded organic phases was investigated using Extended X-ray Absorption Fine Structure (EXAFS) and X-ray Absorption Near Edge Structure (XANES), on the Dutch-Belgian Beamline (DUBBLE, BM26A) at the European Synchrotron Radiation Facility (ESRF) in Grenoble (France). Absorption spectra in the region of the lanthanum, cerium and praseodymium K-edges (38.925, 40.443, 41.991 keV, respectively) were collected. The full experimental description can be found in the ESI (Section SI 5†). The obtained data on the degeneracies and path lengths were used to elucidate the chemical speciation of the extracted REE complexes as a function of the volume ratio of Cy923/[C101][NO3].
The pH of aqueous samples was controlled by a Slimtrode (Hamilton) pH-electrode connected to a S220 SevenCompact™ pH/Ion meter (Mettler–Toledo). Viscosities and densities of selected IL phases before and after extraction tests were determined using a rolling-ball viscometer (Anton Paar Lovis 2000 M/ME) and a pycnometer or an oscillating U-tube densitometer (Anton Paar, DMA 4500 ME).
3 Results and discussion
3.1 Extraction tests using an organic phase composed entirely of an IL
Extraction tests from chloride feed solutions to a nitrate IL (named organic or IL phase) were performed. The feed solutions were prepared from the 5 mol L−1 solution of CaCl2 and the solution A and contained between 1 and 4 mol L−1 of CaCl2 and between 7 and 40 g L−1 of La(III) + Ce(III) + Pr(III) (Section 2.1). CaCl2 was used as salting-out agent to accelerate the phase separation after solvent extraction. The presence of ions promote the cohesion between hydrophobic particles due to van der Waals interactions, which would enhance the disengagement between the hydrophilic feed solution and the hydrophobic IL during the settling. Ca2+ and Cl− ions have a relatively high charge density, providing a high hydration mantle and therefore a high hydrophilicity (Hofmeister series and hard/soft acids/bases ranking).The REE were extracted well by both ionic liquids [C101][NO3] and [A336][NO3] (Fig. 1a). The extraction behaviour of La(III) was similar to that of Pr(III) and lower than the one of Ce(III), in agreement with results reported before.5 On the other hand, other studies showed that when [A336][NO3] was diluted in mixed xylenes (molecular diluent), the distribution of La(III) in the organic phase was higher than that of Ce(III).31,32 This might be due to the formation of different REE complexes as the concentration of the extractant [A336][NO3] differed.24,26 [C101][NO3] had a slightly higher extraction capacity, less solubility in water and lower separation factors than [A336][NO3] (Fig. 1). In the case of [A336][NO3], emulsions were formed, even at low loadings of REE in the IL (0.8 g L−1) and at high concentrations of the CaCl2 (4 mol L−1) in the feed solution; showing poor phase disengagement after reaching the extraction equilibrium. The shorter octyl chains on the quaternary ammonium centre of [A336][NO3] compared to the tetradecyl chain on the quaternary phosphonium centre of [C101][NO3] leaded to a higher miscibility with the aqueous feed solution and enhanced emulsion formation.33 With both ILs, the separation factors of Pr from La (αPrLa) and of Pr from Ce (αPrCe), were low (Fig. 1b). This is common for consecutive REE in the periodic table, with values typically ranging between 1 and 3.5,16,34–40
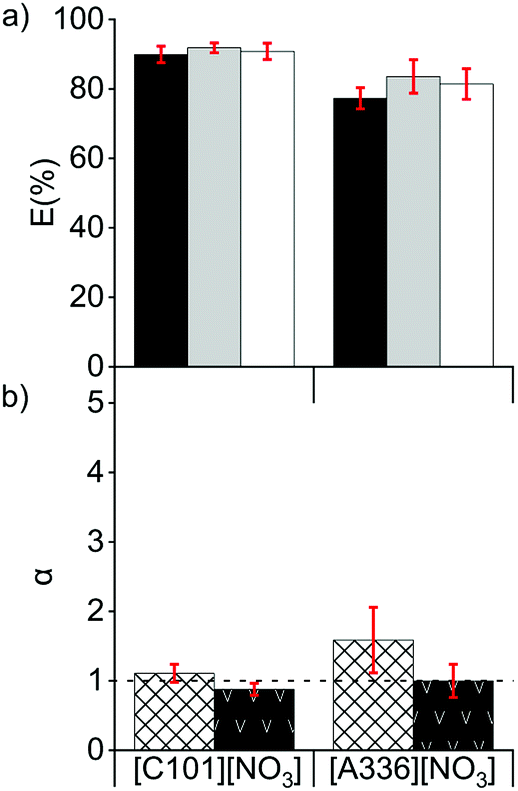 | ||
Fig. 1 (a) Extraction of La ( ), Ce ( ), Ce ( ) and Pr ( ) and Pr ( ), and (b) separation factors of αPrLa ( ), and (b) separation factors of αPrLa ( ) and αPrCe ( ) and αPrCe ( ), after one contact with [C101][NO3] and [A336][NO3] at 50 °C, 3000 rpm, O/A 1:1. Chloride feed solution: 3.4, 9.3 and 1.2 g L−1 of La(III), Ce(III) and Pr(III), 4 mol L−1 CaCl2. ), after one contact with [C101][NO3] and [A336][NO3] at 50 °C, 3000 rpm, O/A 1:1. Chloride feed solution: 3.4, 9.3 and 1.2 g L−1 of La(III), Ce(III) and Pr(III), 4 mol L−1 CaCl2. | ||
Secondly, the effect of the temperature, initial pH and chloride concentration were examined with [C101][NO3] as the organic phase. The salting-out agent CaCl2 was now also used for modifying the chloride concentration, because its 2:1 chloride/cation mole ratio and its high solubility constant. At 80 °C, the extraction of the REE remained similar as at 50 °C, although Pr(III) showed lower extraction, suggesting that the change in enthalpy is negative, and heat is released (Fig. 2). Previous studies reported that the extraction of mixed or single REE, with neutral organophosphorus extractants by nitrato complexes decreases when increasing the temperature, showing to be an exothermic reaction.36,41–45 However, as the metal-loaded ILs have higher viscosities than conventional loaded organic phases, temperatures above 35 °C are needed to lower the viscosity enough for having a fast enough mass transfer and kinetics of the extraction.
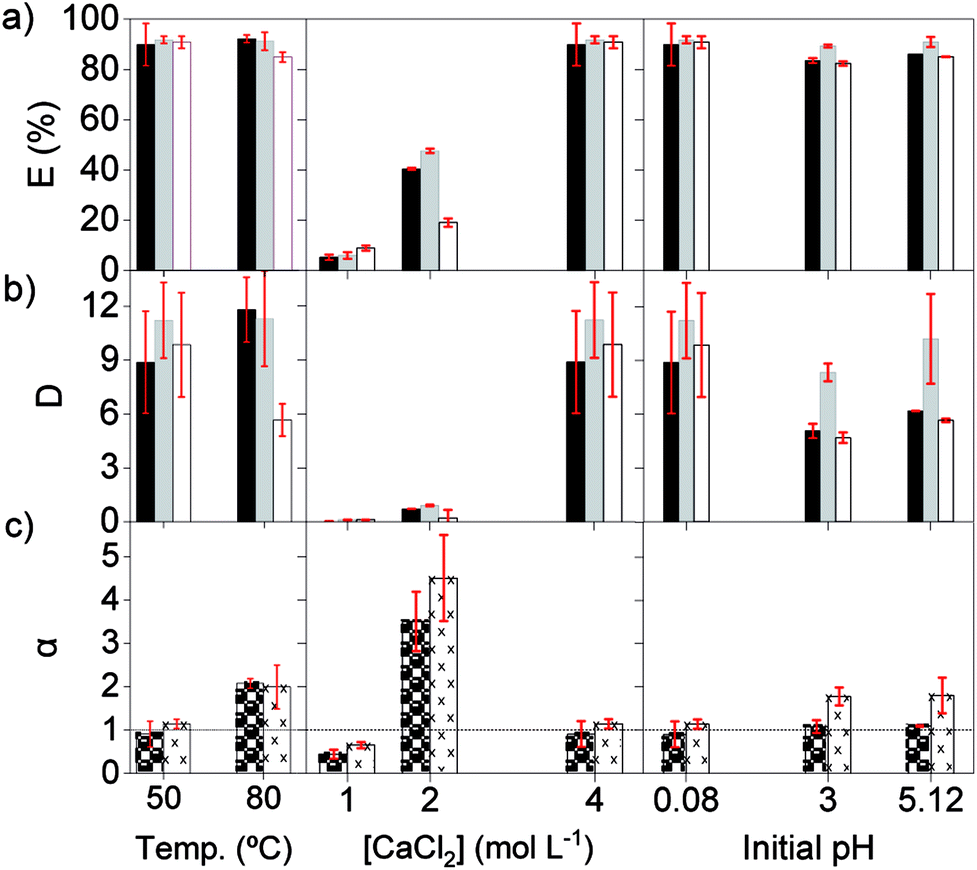 | ||
Fig. 2 Extraction (a) and distribution ratios (b) of La ( ), Ce ( ), Ce ( ) and Pr ( ) and Pr ( ), and separation factors of αLaPr ( ), and separation factors of αLaPr ( ) and αCePr ( ) and αCePr ( ) (c) after one contact with [C101][NO3], at 3000 rpm, volume O/A 1:1. Chloride feed solution: 3.4, 9.3 and 1.2 g L−1 of La(III), Ce(III) and Pr(III). Initial pH 0.08, 4 mol L−1 CaCl2 and 50 °C, except where noted. ) (c) after one contact with [C101][NO3], at 3000 rpm, volume O/A 1:1. Chloride feed solution: 3.4, 9.3 and 1.2 g L−1 of La(III), Ce(III) and Pr(III). Initial pH 0.08, 4 mol L−1 CaCl2 and 50 °C, except where noted. | ||
The initial pH of the feed solution did not show a clear effect on the extraction of REE at the studied concentrations. However, these ILs can extract mineral acids as molecules, which has a negative impact on the extraction of metal ions. Therefore, very acidic feed solutions should be avoided.5,46
The extraction of REEs was most affected by the concentration of chloride ions in the aqueous phase. After extraction, the amount of REE loaded in [C101][NO3] sharply increased from 0.8, to 6.0 and to 12.6 g L−1, when CaCl2 went from 1, to 2 and to 4 mol L−1 (Fig. 2). The increase in metal extraction by increasing the concentration of the chloride salting-out agent is well known in metal ions that can easily form chloro complexes (Zn(II), Fe(III), Co(II), Pd(II) and Au(III)).46,47 However, REE chloro complexes have very low stability constants in water.48 In these experiments, adding CaCl2 most likely decreases the activity of water and probably also decreases the number of water molecules coordinated to the REE ions. Thus, the stability of the hydrated REE aquo complexes in the aqueous phase decreases, which would in turn increase their availability for extraction to the organic phase, resulting in higher E (%).49 Regarding the selectivity, the adjustment of the concentration of CaCl2 in the feed solution is essential to improve the separation of mixtures of REEs. The extraction with pure [C101][NO3] showed the highest values of αLaPr and αCePr at intermediate E (%), which corresponds to an intermediate CaCl2 concentration (2 mol L−1).
Finally, a solution with a low concentration of REEs was compared to a solution containing a high concentration of REEs using [C101][NO3] as the organic phase. The feed solutions contained 7 and 35 g L−1 of REE and an intermediate CaCl2 concentration of 2.5 mol L−1. As expected, the percentage extraction of REE was lower with the high initial REE content than with 7 g L−1 REE. However, the separation factors were higher when the concentration of REEs in the feed solution was high (Fig. 3). Therefore, concentrated REE feed solutions are preferred and, to counteract the extraction decrease that brings with it, more CaCl2 could be added (Fig. 2a). To optimize the separation, it is advisable to work with feed solutions where the concentration of the target REE is close to the maximum loading of the IL phase and at medium CaCl2 concentrations, so that the overall REEs percentage extraction is not below 20% or above 90%.
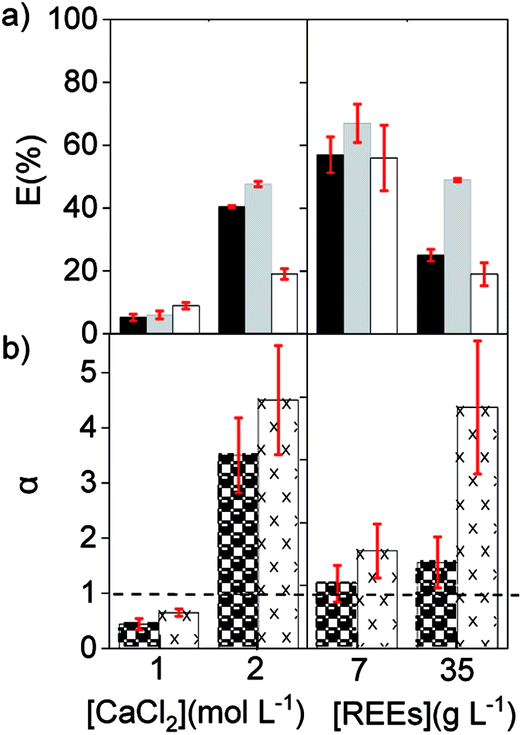 | ||
Fig. 3 (a) Extraction of La ( ), Ce ( ), Ce ( ) and Pr ( ) and Pr ( ), and (b) separation factors of αLaPr ( ), and (b) separation factors of αLaPr ( ) and αCePr ( ) and αCePr ( ), from different chloride feed solutions: 14 g L−1 REE (left) and 2.5 mol L−1 CaCl2 (right). Pure [C101][NO3], 3000 rpm, volume O/A 1:1. ), from different chloride feed solutions: 14 g L−1 REE (left) and 2.5 mol L−1 CaCl2 (right). Pure [C101][NO3], 3000 rpm, volume O/A 1:1. | ||
3.2 Extraction tests using an organic phase composed of mixtures of a neutral extractant and an IL
The effect of dissolving neutral extractants in the IL phase and ethylene glycol in the aqueous phase was studied. The aqueous feed solutions used here were the same used in Section 3.1. Tri-n-butyl phosphate (TBP) and Cyanex 923 (Cy923) were chosen because they are the most frequently used neutral extractants for REE extraction from nitrate media.12,13,15,36,50–53 However, in the present study, these extractants were applied for extraction from chloride aqueous media and dissolved in the ILs.23 The results showed that the extraction of REE from feed solutions of 7 g L−1 of REE and 2 mol L−1 of CaCl2 with [C101][NO3] and [A336][NO3] was not affected by the presence of TBP in both ILs. When increasing the TBP from 1 to 20 v%, the extraction remained constant ≈24%, and the separation factors αLaPr and αPrCe were low, close to 1 (Section SI 1 and Fig. S1†). On the other hand, addition of Cy923 to the ILs increased the extraction efficiencies considerably (Fig. 4a). This is due to the strong Lewis acid–base interaction between the REEs and Cy923. Cy923 has a greater Lewis basicity than TBP. Phosphine oxides are molecules with a high Gutmann donor number, so that they are hard Lewis bases, which strongly interact with the REEs.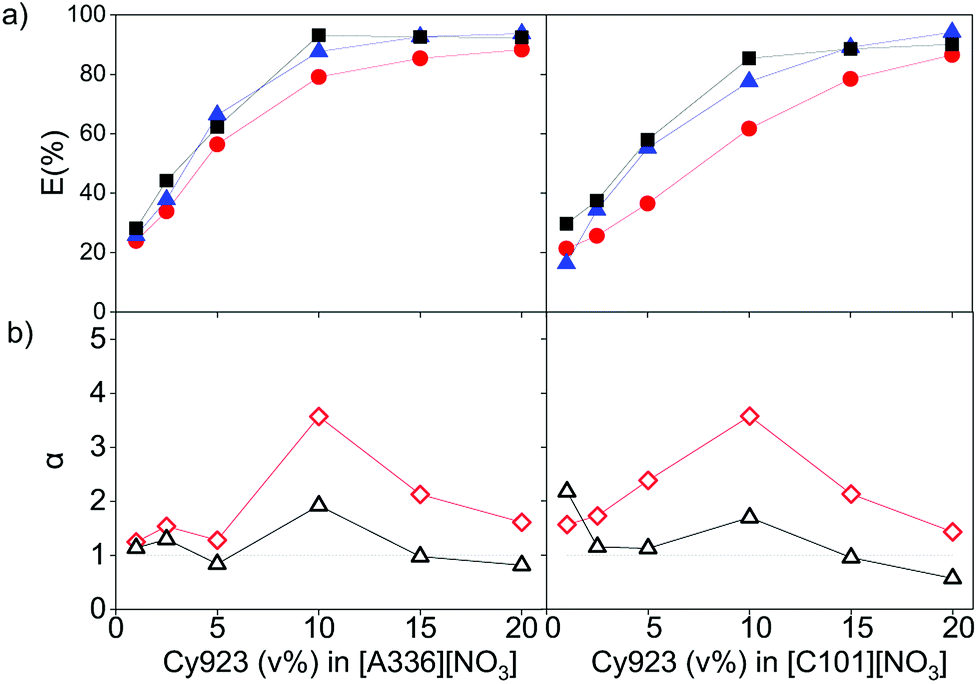 | ||
Fig. 4 (a) Extraction of La ( ), Ce ( ), Ce ( ) and Pr ( ) and Pr ( ), and (b) separation factors of αPrLa ( ), and (b) separation factors of αPrLa ( ) and αPrCe ( ) and αPrCe ( ) after one contact with different Cy923 concentrations in [A336][NO3] (left) and in [C101][NO3] (right), at 70–80 °C, 3000 rpm, volume O/A 1:1. Chloride feed solution: 2.0, 4.4 and 0.6 g L−1 of La(III), Ce(III) and Pr(III), 2 mol L−1 CaCl2. Distribution ratios from 0.3 till 15 and from 0.2 till 9 for [A336][NO3] and [C101][NO3], respectively. ) after one contact with different Cy923 concentrations in [A336][NO3] (left) and in [C101][NO3] (right), at 70–80 °C, 3000 rpm, volume O/A 1:1. Chloride feed solution: 2.0, 4.4 and 0.6 g L−1 of La(III), Ce(III) and Pr(III), 2 mol L−1 CaCl2. Distribution ratios from 0.3 till 15 and from 0.2 till 9 for [A336][NO3] and [C101][NO3], respectively. | ||
Regarding the selectivity, the separation trend of Pr(III) from La(III) + Ce(III) followed a bell-shaped curve with a maximum around 10 v% of Cy923 (0.25 mol L−1) in the ILs (1.53 mol L−1) (Fig. 4b). The content of Cy923 in the IL should be high enough to extract considerable amounts of REEs, until an optimum value. Beyond that value, the selectivity was lost due to lack of competition between the extractable REE complexes because of the excess of extractant. Thus, at equal phase volume ratios, if the REEs concentration increases, the concentration of Cy923 should increase to keep the selectivity towards the heaviest REE (Section S2, Fig. S2†).
A second observed effect of Cy923 is that the extraction sequence of the lanthanides follows the order La(III) < Ce(III) < Pr(III), which means that the E (%) increases with increase in atomic number. Cy923 forms more stable complexes with the heavier REE (higher atomic number), since these have greater Lewis acidity and charge density due to the small ionic radii, high oxidation state and hindered polarization. Such a positive extraction sequence is preferable when the objective is to extract heavier REEs. When no Cy923 molecules are present, the lighter REEs (lower atomic number and greater hydration) are preferably extracted.
A third favourable effect of Cy923 is the improvement of the physical properties of the IL phase by addition of Cy923. In that sense, Cy923 acted both as an extractant and a modifier. Cy923 decreased the viscosity of the organic phase (Fig. 5) and enhanced the disengagement of both phases. No emulsions were formed when [A336][NO3] was mixed with Cy923, as opposed to the extraction tests initially performed with pure [A336][NO3] (Section 3.1).
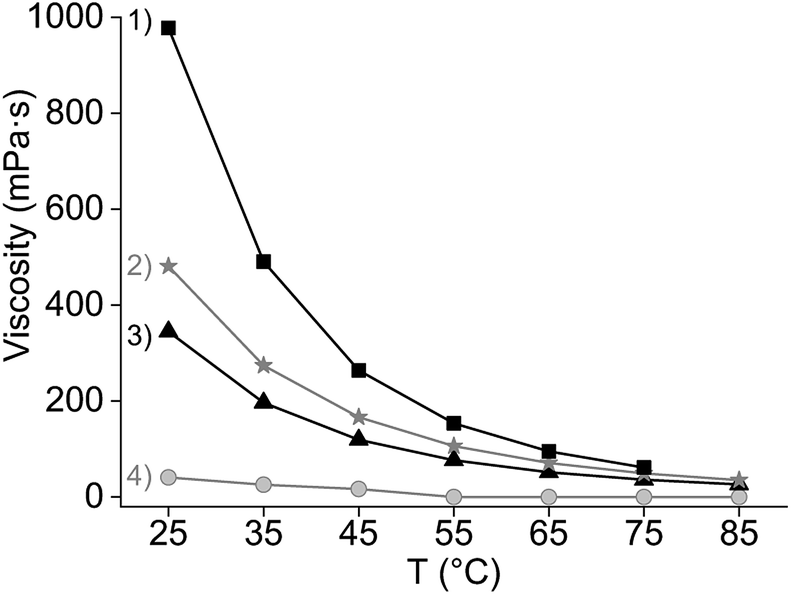 | ||
| Fig. 5 Viscosity as a function of the temperature and the organic phase composition: (1) pure Cy923, (2) 20 v% Cy923 in [C101][NO3], (3) pure [C101][NO3] and (4) after loading 39 g L−1 of REE in 20 v% Cy923 in [C101][NO3]. | ||
As discussed above, the extraction of REEs from aqueous solutions is facilitated by decreasing the water content, since REEs are highly hydrated in the aqueous phase. This was previously achieved by dissolving CaCl2 in the feed solution. Alternatively, an organic solvent immiscible with the IL phase could be used, for example ethylene glycol (EG). When part of the water in the feed solution is replaced by EG, the extraction efficiency increased. However, the effect is less pronounced that the effect of the addition of Cy923 to the IL phase or CaCl2 to the aqueous phase (Fig. 6, and S3†). This might be due to the smaller decrease in water activity by addition of neutral EG molecules than when increasing the ionic strength by addition of CaCl2. Furthermore, the use of EG decreased the selectivity, as the separation factors decreased with an increase in EG concentration (Fig. 6). Even when varying the concentrations of CaCl2, the extractability and selectivity of the system [EG-chloride-aqueous phase]/[Cyanex-nitrate-IL phase] did not substantially improve (Fig. S3 left and Table S1†). Thus, depending on the nature of the molecules substituting the water molecules, the solvation properties, attraction and association between REE and extractants molecules differ. The poorer selectivity with EG in the aqueous phase may be due to the lower difference in polarity between the aqueous and the organic phases.
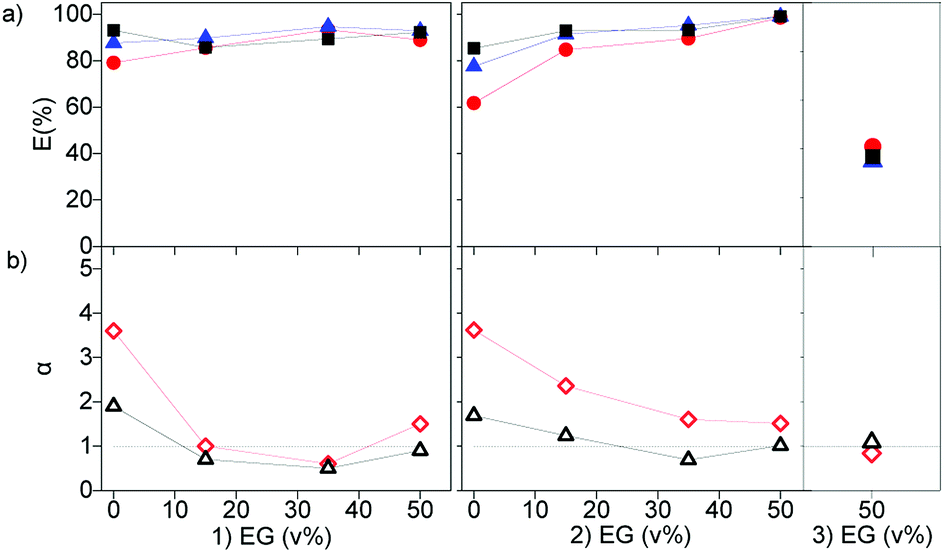 | ||
Fig. 6 (a) Extraction of La ( ), Ce ( ), Ce ( ) and Pr ( ) and Pr ( ), and (b) separation factors of αPrLa ( ), and (b) separation factors of αPrLa ( ) and αPrCe ( ) and αPrCe ( ) as a function of ethylene glycol (EG) in the feed solution. One contact with (1) 10 wt% Cy923 in [A336][NO3], (2) 10 wt% Cy923 in [C101][NO3] and (3) pure [C101][NO3]. At 80 °C, 3000 rpm, volume O/A 1:1. Chloride feed solution: 1.7, 4.0, 0.6 g L−1 of La(III), Ce(III) and Pr(III), 2 mol L−1 CaCl2. ) as a function of ethylene glycol (EG) in the feed solution. One contact with (1) 10 wt% Cy923 in [A336][NO3], (2) 10 wt% Cy923 in [C101][NO3] and (3) pure [C101][NO3]. At 80 °C, 3000 rpm, volume O/A 1:1. Chloride feed solution: 1.7, 4.0, 0.6 g L−1 of La(III), Ce(III) and Pr(III), 2 mol L−1 CaCl2. | ||
After extraction, the REE can easily be recovered from the loaded organic phase by water. In only one stripping stage; 93, 78 and 81% of La(III), Ce(III) and Pr(III) were stripped from a loaded organic phase composed of 20 v% Cy923/[C101][NO3], with concentrations in the strip liquor of 1.7, 7.1 and 16.5 g L−1, respectively.
3.3 Loading capacities of organic phases composed of [C101][NO3] and/or Cy923
As indicated before, an amount of the target REE in the feed-aqueous phase equivalent to the loading capacity of the IL-organic phase, is needed to enhance selectivity thanks to the competition between REE to form complexes. At equal phase volume ratios, this competitive complex formation depends on the CaCl2 and REE concentrations relative to the loading capacity of the IL phase. The CaCl2 concentration affects mostly the selectivity, the REE concentration influences the percentage extraction, and the composition of the organic phase (Cy923/IL volume ratio) determines the loading capacity of the IL phase. The loading was studied as a function of the Cy923 content in the IL phase, with three single REE solutions of La(III), Ce(III) and Pr(III), containing 3.26 mol L−1 CaCl2 (solution B, C and D) as directed in Section 2.2. The results show that the loading capacity increased with increasing the Cy923 content (Fig. 7). The maximum value was reached with 75 v% of Cy923, remaining constant at higher concentration of the extractant (Fig. 7). Loading with La, Ce and Pr was very similar, with slightly higher values for Ce and Pr, especially at higher Cy923 concentration (Fig. 7). The determination of the loading capacity of the organic phase is explained in the Section S4,† and can be calculated through eqn (S2) and Table S3.†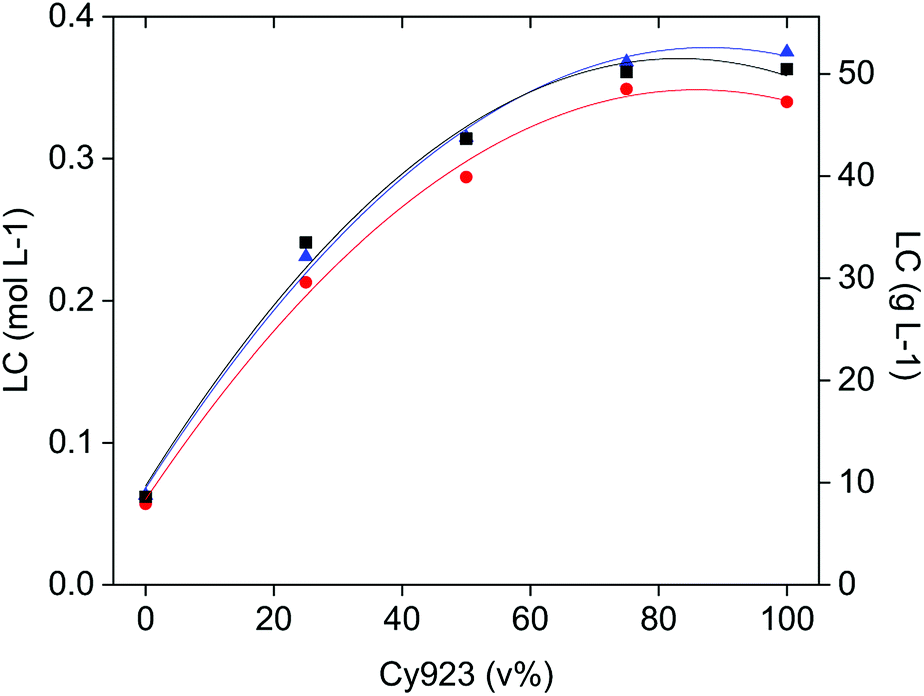 | ||
Fig. 7 Fitting polynomial curves to loading capacities (LC) data points of different mixtures of Cy923 and [C101][NO3] with La ( ), Ce ( ), Ce ( ) and Pr ( ) and Pr ( ) after the fourth contact with solutions B, C and D. Consecutive contacts at 1:1, 1:1, 1:1 and 1:2.85 O/A, 3.26 mol L−1 CaCl2, RT, 300 rpm, 8 h. ) after the fourth contact with solutions B, C and D. Consecutive contacts at 1:1, 1:1, 1:1 and 1:2.85 O/A, 3.26 mol L−1 CaCl2, RT, 300 rpm, 8 h. | ||
To maximise the selectivity, one should select the organic phase having a loading capacity similar to the amount of the easiest extractable REE in the feed. This affinity changes from light REE (LREE) preferentially extracted when Cy923 is absent, to heavy REE (HREE, Y and Sc) when Cy923 is present. The fact that the lowest loading capacity belongs to the system that preferentially extracts LREE and the highest loading capacity to the one that preferentially extract HREE is unfortunate, since LREE are generally present in ores in much greater quantity than HREE. For instance, typical PLSs from steenstrupine have 50–110 g L−1 of LREE versus 1–3 g L−1 of HREE, and typical PLSs from eudialyte have 20–40–110 g L−1 of LREE versus 3–5–7 g L−1 of HREE.54 This means that for extracting the LREE with the [C101][NO3] system, a lot of CaCl2 should be dissolved in the PLS (within the limitation of its solubility product) to avoid diluted stripping liquors and numerous flowing back cycles of raffinate to the extraction stage. In the case of extracting the HREE with the Cy923/[C101][NO3] system, working at low volume ratios of O/A phases is convenient to avoid the co-extraction of the far more abundant LREE.
Overall, the separation factors in Section 3.1 and 3.2 were between 1 and 4. In the case of pure [C101][NO3], the highest αLa+CePr was achieved at the minimum CaCl2 concentration where the maximum extraction was reached (2 mol L−1 CaCl2 in feed solutions of 14 g L−1 of REEs). In the case of mixtures of Cy923/[C101][NO3], the highest αPrLa+Ce was attained at the minimum Cy923 concentration where the maximum extraction was reached. Specifically, 10 v% Cy923 was the case for a feed solution of 7 g L−1 of REEs in 2 mol L−1 CaCl2, while 20 v% Cy923 was the case for a feed of 40 g L−1 of REEs in 2 mol L−1 CaCl2. These separation values are noteworthy since neighbouring REEs in the periodic table are the most difficult to separate, compared to more distant REEs, especially in chloride solutions with neutral extractants, due to similarities in chemical and complexation properties. Currently, in the case of nitrate solutions with neutral extractants, the selectivity between neighbouring REEs is not large enough to be of practical use for separations. In the case of acidic (alkylphosphoric) extractants, i.e., the extractants with the highest selectivity between the REE used in industry today, the mean separation factor is between 2.4–3.0.
3.4 Speciation of the REE in the [C101][NO3] and Cy923/[C101][NO3] organic phases
XANES and EXAFS studies were performed to know the nature of the complexes of La, Ce and Pr after split-anion extraction in the different organic phases (Table S4†). EXAFS was used to determine the structure of the REE complexes, thanks to the provided information on the degeneracies and half path lengths, regarding the position of neighbouring atoms. Giving complementary information, XANES was also used because it is very sensitive to coordination changes and helped to discriminate different species present. Unlike the path lengths which are determined very accurately with EXAFS, the degeneracies have a considerable error, which is why they were not taken as exact values. Because of the noisy data and multi-electron excitation, the data analysis was not straightforward (Section SI 6†).The samples studied consisted of mixtures of Cy923 and [C101][NO3] in volume proportions of 0/100, 25/75, 50/50, 75/25 and 100/0 (Table S4†). The results confirmed that different species were formed, depending on the composition of the organic phase (Section SI 6†). The type of organic phase influenced the absorbance, the bandwidth and the position of the absorption maximum (Fig. 8). In general, no isosbestic points appeared in the region of 50 eV after the edge, confirming that the REE were extracted in different predominant species, by each organic phase. Increments in Cy923/[C101][NO3] volume ratio shifted the edge to higher energy denoting different species, which are summarized in Table 1. The transition between different species (change in speciation), when increasing the ratio Cy923/[C101][NO3], is gradual. Thus, other minor complexes may accompany the main complex, coexisting together at that certain organic composition.
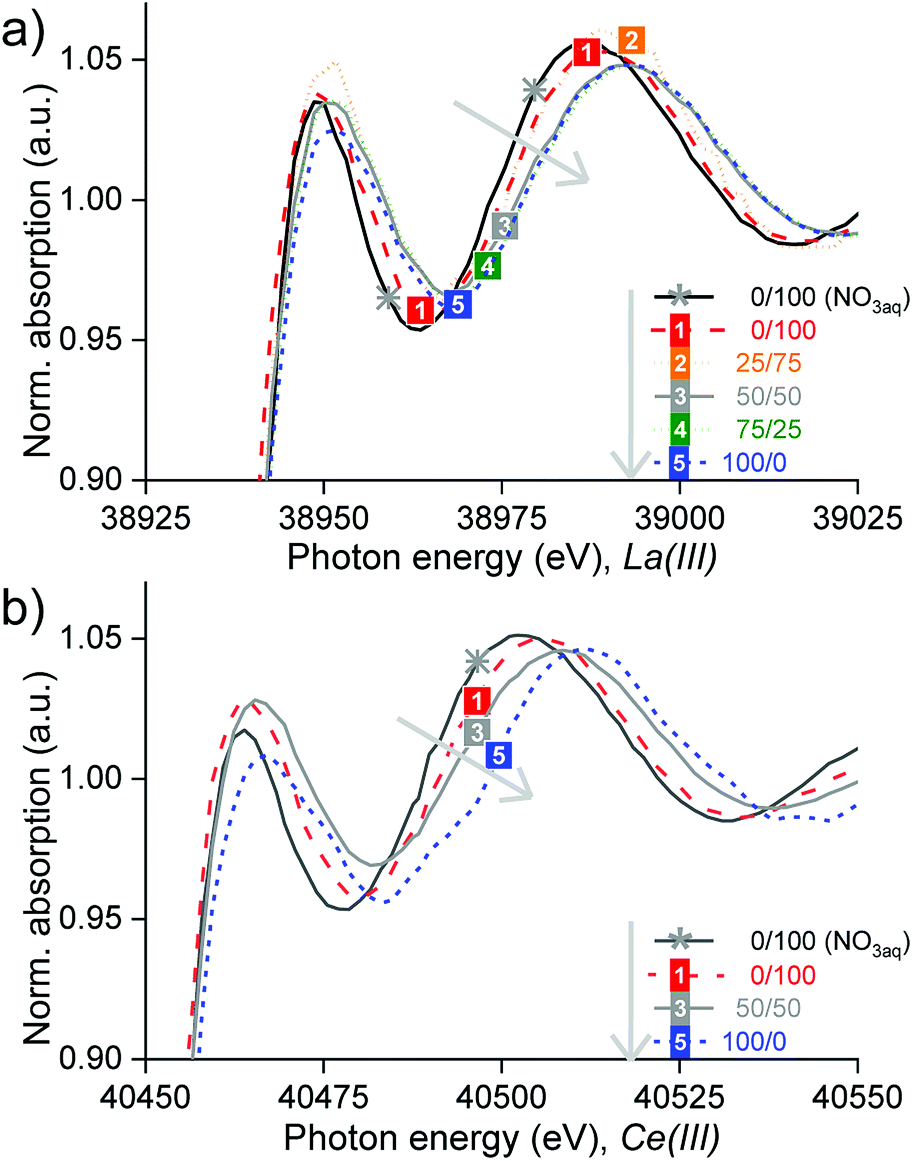 | ||
| Fig. 8 Normalized XANES spectra of the (a) lanthanum and (b) cerium K-edges at increasing Cy923/[C101][NO3] volume ratio from 0/100 till 100/0 (grey arrow). | ||
| Legend | v/v Cy923/[C101][NO3] | Major species | Minor species |
|---|---|---|---|
a  : organic sample obtained after extraction from concentrated nitrate aqueous solutions (NO3,aq). The rest were obtained from concentrated chloride aqueous solutions. Ln: La, Ce or Pr. x: probably smaller than 7–9, and possibly a lower figure for LREE than for HREE. y: figure of [NO3−] molecules in the outer sphere. : organic sample obtained after extraction from concentrated nitrate aqueous solutions (NO3,aq). The rest were obtained from concentrated chloride aqueous solutions. Ln: La, Ce or Pr. x: probably smaller than 7–9, and possibly a lower figure for LREE than for HREE. y: figure of [NO3−] molecules in the outer sphere. |
|||
 |
0/100 (NO3,aq) | [Ln(NO3)5]2− | |
 |
0/100 | [(Ln(H2O)x)–(NO3)y]3−y | |
 |
25/75 | [Ln(H2O)x]3+ | Ln(NO3)3·3Cy923 |
 |
50/50 | Ln(NO3)3·3Cy923 | [Ln (H2O)x]3+ |
 |
75/25 | Ln(NO3)3·3Cy923 | LnCl3·3Cy923 |
 |
100/0 | LnCl3·3Cy923 | |
Firstly, when [C101][NO3] comprised the total organic phase, it was found that La(III) and Ce(III) were extracted from concentrated chloride aqueous media as hydrated cationic complexes, i.e. [La(H2O)x]3+ and [Ce(H2O)x]3+ (Section S6.1, Fig. S6, Fig. S7 and Table S5†). These complexes are probably stabilized by nitrate anions in the outer coordination sphere: [(Ln(H2O)x)–(NO3)y]3−y. This could explain the long bond distances found between the La–O and Ce–O compared to the ones in water reported by Persson et al.55 Although the EXAFS analysis showed that 7 water molecules are coordinated to La(III) and 9 water molecules for Ce(III), these values are uncertain because they are strongly correlated to the data isolation and to different fitting parameters such as the Debye–Waller factor and the amplitude reduction factor (S0); what severely affects their resolution and accuracy.56 In general, the small lanthanide ions (Yb(III), Lu(III)) form 8-coordinate aqua complexes and the large ones (La(III), Ce(III)) form 9-coordinate complexes, with both coordination numbers possible in the middle of the lanthanide series.57 However, these coordination numbers of the aqua molecules may vary with the ionic strength and concentration of the solution. Therefore, the complexes in the IL solutions are denoted as [La(H2O)x]3+ and [Ce(H2O)x]3+ and may have a smaller × number than in diluted aqueous solutions (without CaCl2). No evidence of nitrato complexes of La(III) or Ce(III) was found by the EXAFS analyses. However, these complexes are highly stable and did appear after extraction from nitrate aqueous media (Fig. S8†) and after split-anion extraction of Sm or Eu from chloride aqueous media.58
Secondly, when Cy923 composed the whole organic phase, La(III) and Pr(III) were extracted from concentrated chloride aqueous media via neutral chloride complexes with Cy923, specifically LaCl3·3Cy923 and PrCl3·3Cy923 (Section S6.2, Fig. S9, Fig. S10 and Table S6†) similar to the RECl3·2.5Cy923 extracted species reported by Lee et al.37
Thirdly, when [C101][NO3] and Cy923 were mixed in equal volume proportion (1.61:1 molar ratio); Ce(III) was extracted from concentrated chloride aqueous media mainly via a neutral nitrate complex solvated by Cy923, represented as Ce(NO3)3·3Cy923 (Section S6.3, Fig. S12 and Table S7†). This species is the most commonly observed lanthanide complex extracted by trialkylphosphine oxides from nitrate solutions32,51 and similar to the RE(NO3)3·2Cy923 reported in other works.44,45,50 Thus, the IL not only acts as a diluent for the extractant but also as a nitrate source for the formation of a REE-nitrato complexes from a chloride, nitrate free, feed solution. The cerium-anion paths Ce–O, Ce–N and Ce–P, showed shorter bond distances than in the crystal structure of tris(nitrato)-tris(triethylphosphine oxide)–cerium(III)59 and relatively high Debye-Waller factors. Therefore, other minor species might also be present, such as [Ce(H2O)x]3+ (formed when extracting with [C101][NO3]) and CeCl3·3Cy923 (formed when extracting with Cy923), which were not possible to identify in the analysis because of their low concentration compared to the main complex formed.
Finally, the data from 25/75 and 75/25 v/v mixtures of Cy923/[C101][NO3] were of an insufficient quality to enable solving the EXAFS and therefore no complexes could be determined by curve fitting to a theoretical standard model with the software. However, their main species in the lanthanum-loaded organic phase, may be the same as in the case of 0/100 and 50/50, respectively, because of the largely linear superposition of the X-ray absorption spectra 0/100 over 25/75, and especially of 50/50 over 75/25 (Fig. 8a). In all cases, only the chloride ions required to maintain electrical neutrality were extracted to the organic phase. Here, the EXAFS measurements did not find chlorides coordinating to the REE ion in its first/inner coordination sphere. However, anionic REE-chloro complexes in extractants of the type of [A336][Cl], were previously reported, after extraction from concentrated chloride aqueous media.48,60,61
The separation of REE with neutral and basic extractants are based on the differences in the complex formation constants between the REE cations, the coordinating anion and the extractant.62 The more efficient extraction of La(III) and Ce(III) than Pr(III) by a solvent composed entirely of [C101][NO3] is related to the faster and easier formation of hydrated complexes by La(III) and Ce(III), compared to Pr(III). The preferential extraction of Pr(III) (lower ionic radius than La(III) and Ce(III)) by a mixture of Cy923 in [C101][NO3], go along with that the distribution ratios of REE increase with decreasing ionic radii for RE(NO3)3·3Cy923 complexes.45,51
4 Conclusions
Split-anion extraction allows to recover REE from acidic chloride leaching solutions which supplements the conventional solvent extraction procedures. This process uses ILs to replace the molecular diluent, and also provide nitrate anions for extracting the REE-(aquo or Cy923)-nitrato complexes from a chloride feed solution, free of nitrate ions. REE chlorides are not extracted because they are weak inner sphere ligands and cannot compensate for the high hydration energy of the REE ions. As opposed to the extraction with TBP in the ILs, Cy923 in the ILs affects the extraction greatly. On the one hand, the extraction preference changed from lighter to heavier REE. On the other hand, it increased the overall extraction efficiency and the loading capacity of the organic phase. The extraction of light REE by the IL was through ion pairs of the hydrated REE cation stabilized by nitrates in the outer sphere. The extraction of light REE by 50 v% Cy923 in the IL was through REE nitrate salts solvated by Cy923 (1:3 stoichiometry). The transfer of the cation or neutral REE complexes to the (IL) organic phase is boosted by high CaCl2 concentrations in the feed solution, as the high charge density of these ions decreases the hydration of the REE. Replacing part of the water by EG showed minor increments on the extraction efficiencies and decreased the separation factors. The separation factors of La and Ce, from Pr (IL system) and of Pr, from La and Ce (Cy923/IL system) were between 1 and 4.5. Higher values are difficult to achieve due to the similarities between the extractable REE complexes. Nevertheless, the results are well positioned with regards to the mean separation factor of the currently used REE extractants (2.4–3). Split-anion extraction has the advantages of no need for pH control, lower acid/alkali consumption, cheaper operating costs and easier stripping and waste water treatment than acidic extractants. Conversely, split-anion extraction is dependent on the REEs concentration in the aqueous phase and the loading of the organic phase. Thus, it requires optimization of CaCl2 and REEs concentrations in the feed solution related to the loading capacity of the organic phase, in order to attain the separation of mixtures of REEs, based on competitive complex formation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received funding from the European Commission's FP7 Framework Programme (2007-13) under grant agreement number 309373 (EURARE). This publication reflects only the authors' view, exempting the Commission from any liability.References
- J. Hykaway, The Rare Earths- Pick your spots carefully, Byron Capital Markets, London, 2010 Search PubMed.
- D. Schüler, M. Buchert, R. Liu, S. Dittrich and C. Merz, Study on Rare Earths and Their Recycling, Öko-Institut e.V, 2011, p. 162 Search PubMed.
- K. Binnemans and P. T. Jones, J. Sust. Metall., 2015, 1, 29–38 CrossRef.
- R. D. Abreu and C. A. Morais, Miner. Eng., 2014, 61, 82–87 CrossRef CAS.
- K. Larsson and K. Binnemans, Hydrometallurgy, 2015, 156, 206–214 CrossRef CAS.
- R. D. Rogers, K. Richard and S. Volkov, Green Industrial Applications of Ionic Liquids, Springer Science & Business Media, Dordrecht, 2012 Search PubMed.
- P. A. Thomas and B. B. Marvey, Molecules, 2016, 21, 16 CrossRef PubMed.
- A. E. Visser, R. P. Swatloski, W. M. Reichert, H. D. Willauer, J. G. Huddleston and R. D. Rogers, in Green Industrial Applications of Ionic Liquids, ed. R. D. Rogers, K. R. Seddon and S. Volkov, Springer Netherlands, Dordrecht, 2002, vol. 92, pp. 137–156 Search PubMed.
- F. Kerton and R. Marriott, Alternative Solvents for Green Chemistry 2nd edition, RSC Green Chemistry No. 20, Cambridge, 2013 Search PubMed.
- R. D. Rogers and K. R. Seddon, Ionic Liquids as Green Solvents, American Chemical Society, Washington, 2003 Search PubMed.
- J. N. Chubb, P. Lagos and J. Lienlaf, J. Electrost., 2005, 63, 119–127 CrossRef CAS.
- E. Balomenos, P. Davris, E. Deady, J. Yang, D. Panias, B. Friedrich, K. Binnemans, G. Seisenbaeva, C. Dittrich, P. Kalvig and I. Paspaliaris, Johnson Matthey Technol. Rev., 2017, 61, 142–153 CrossRef.
- A. S. L. Sjoqvist, D. H. Cornell, T. Andersen, M. Erambert, M. Ek and M. Leijd, Minerals, 2013, 3, 94–120 CrossRef.
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2012, 14, 1657–1665 RSC.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- K. L. Nash, Solvent Extr. Ion Exch., 1993, 11(4), 729–768 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 96–100 CrossRef CAS PubMed.
- T. Wang, C. B. Gu, S. X. Wang, P. Kou, J. Jiao and Y. J. Fu, J. Cleaner Prod., 2018, 172, 827–836 CrossRef CAS.
- T. P. Thuy Pham, C.-W. Cho and Y.-S. Yun, Water Res., 2010, 44, 352–372 CrossRef PubMed.
- A. Cieniecka-Rosłonkiewicz, J. Pernak, J. Kubis-Feder, A. Ramani, A. J. Robertson and K. R. Seddon, Green Chem., 2005, 7, 855–862 RSC.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- S. P. M. Ventura, C. S. Marques, A. A. Rosatella, C. A. M. Afonso, F. Gonçalves and J. A. P. Coutinho, Ecotoxicol. Environ. Saf., 2012, 76, 162–168 CrossRef CAS PubMed.
- K. Larsson and K. Binnemans, Green Chem., 2014, 16, 4595–4603 RSC.
- S. Riaño and K. Binnemans, Green Chem., 2015, 17, 2931–2942 RSC.
- A. Rout and K. Binnemans, Dalton Trans., 2014, 43, 1862–1872 RSC.
- T. Vander Hoogerstraete and K. Binnemans, Green Chem., 2014, 16, 1594–1606 RSC.
- S. Wellens, R. Goovaerts, C. Moeller, J. Luyten, B. Thijs and K. Binnemans, Green Chem., 2013, 15, 3160–3164 RSC.
- S. Wellens, T. Vander Hoogerstraete, C. Moller, B. Thijs, J. Luyten and K. Binnemans, Hydrometallurgy, 2014, 144, 27–33 CrossRef.
- M. Regadío, S. Riaño, K. Binnemans and T. Vander Hoogerstraete, Anal. Chem., 2017, 89, 4595–4603 CrossRef PubMed.
- S. Riaño, M. Regadío, K. Binnemans and T. Vander Hoogerstraete, Spectrochim. Acta, Part B, 2016, 124, 109–115 CrossRef.
- I. Komasawa, K. Hisada and M. Miyamura, J. Chem. Eng. Jpn., 1990, 23, 308–315 CrossRef CAS.
- Y. Marcus and I. Abrahamer, J. Inorg. Nucl. Chem., 1961, 22, 141–150 CrossRef CAS.
- A. Rout and K. Binnemans, Phys. Chem. Chem. Phys., 2016, 18, 16039–16045 RSC.
- T. Sato, Hydrometallurgy, 1989, 22, 121–140 CrossRef CAS.
- M. Anitha, M. K. Kotekar, D. K. Singh, R. Vijayalakshmi and H. Singh, Hydrometallurgy, 2014, 146, 128–132 CrossRef CAS.
- C. Deqing, M. Genxiang and L. Deqian, Presented in part at the ISEC’99: Int. Solvent Extract. Conf. Solvent Extract. for 21th Century, Barcelona, 11–16 July, 1999 Search PubMed.
- G. S. Lee, M. Uchikoshi, K. Mimura and M. Isshiki, Sep. Purif. Technol., 2009, 67, 79–85 CrossRef CAS.
- J. N. Mathur, Solvent Extr. Ion Exch., 1983, 1, 349–412 CrossRef CAS.
- J. Starý, Talanta, 1966, 13, 421–437 CrossRef.
- E. P. Horwitz, A. C. Muscatello, D. G. Kalina and L. Kaplan, Sep. Sci. Technol., 1981, 16, 417–437 CrossRef CAS.
- H. Yang, W. Wang, H. Cui, D. Zhang, Y. Liu and J. Chen, J. Chem. Technol. Biotechnol., 2012, 87, 198–205 CrossRef CAS.
- F. Habashi, Text book of Hydrometallurgy, Laval University, Quebec, Canada, 1999 Search PubMed.
- E. Jorjani and M. Shahbazi, Arabian J. Chem., 2016, 9, S1532–S1539 CrossRef CAS.
- Y. A. El-Nadi, N. E. El-Hefny and J. A. Daoud, Solvent Extr. Ion Exch., 2007, 25, 225–240 CrossRef CAS.
- J. A. Daoud, Arab J.Nucl.Sci.Appl., 2016, 94, 187–205 Search PubMed.
- J. E. Quinn, M. D. Ogden and K. Soldenhoff, Solvent Extr. Ion Exch., 2013, 31, 538–549 CrossRef CAS.
- M. Regel-Rosocka, A. M. Sastre and J. Szymanowski, Environ. Sci. Technol., 2001, 35, 630–635 CrossRef.
- M. Sohsah, J. Krejzler and S. Siekierski, Solvent Extr. Ion Exch., 1990, 8, 875–892 CrossRef CAS.
- F. David, V. Vokhmin and G. Ionova, J. Mol. Liq., 2001, 90, 45–62 CrossRef CAS.
- B. Gupta, P. Malik and A. Deep, Solvent Extr. Ion Exch., 2003, 21, 239–258 CrossRef CAS.
- M. L. P. Reddy, R. L. Varma, T. R. Ramamohan, S. K. Sahu and V. Chakravortty, Solvent Extr. Ion Exch., 1998, 16, 795–812 CrossRef CAS.
- C. Tunsu, C. Ekberg, M. Foreman and T. Retegan, Solvent Extr. Ion Exch., 2014, 32, 650–668 CrossRef CAS.
- C. K. Gupta and N. Krishnamurthy, Int. Mater. Rev., 1992, 37, 197–248 CrossRef CAS.
- V. Innocenzi, N. M. Ippolito, L. Pietrelli, M. Centofanti, L. Piga and F. Veglio, J. Cleaner Prod., 2018, 172, 2840–2852 CrossRef CAS.
- I. Persson, P. D'Angelo, S. De Panfilis, M. Sandstrom and L. Eriksson, Chem. - Eur. J., 2008, 14, 3056–3066 CrossRef CAS PubMed.
- S. Calvin, XAFS for Everyone, CRC Press, Boca Raton, 2013 Search PubMed.
- E. N. Rizkalla and G. R. Choppin, in Handbook on the Physics and Chemistry of Rare Earths, vol. 26, ed. J. K. A. Gschneidner and L. Eyring, 1991, vol. 15, ch. 103 Search PubMed.
- S. Sobekova-Foltova, K. Binnemans and T. Vander Hoogerstraete, ERES 2017, 2nd conference on European Rare Earth Resources, Santorini, 2017 Search PubMed.
- A. Bowden, K. Singh and A. W. G. Platt, Polyhedron, 2012, 42, 30–35 CrossRef CAS.
- A. I. Mikhailichenko, E. G. Goryacheva, N. P. Sokolova, N. M. Aksenova, L. V. Vdovina and A. P. Emelyanov, Radiochemistry, 1984, 26, 25–29 Search PubMed.
- P. K. Khopkar and J. N. Mathur, J. Inorg. Nucl. Chem., 1981, 43, 1035–1040 CrossRef CAS.
- S. Nishihama, T. Hirai and I. Komasawa, J. Solid State Chem., 2003, 171, 101–108 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Effect of TBP in the IL, competitive complex formation effect, extraction from 10 v% ethylene glycol solutions at different chloride concentrations, polynomial fitting of the loading capacities of Cy923/[C101][NO3] (3.26 mol L−1 CaCl2), methodology and results interpretation of EXAFS and XANES. See DOI: 10.1039/c8ra06055j |
| This journal is © The Royal Society of Chemistry 2018 |