
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriazole-based cross-linkers in radical polymerization processes: tuning mechanical properties of poly(acrylamide) and poly(N,N-dimethylacrylamide) hydrogels†
Tobias Götz‡
a,
Nicole Schädel‡b,
Nadja Petriab,
Manuel Kirchhofb,
Ursula Bilitewskic,
Günter E. M. Tovar*ad,
Sabine Laschat *b and
Alexander Southan*a
*b and
Alexander Southan*a
aInstitute of Interfacial Process Engineering and Plasma Technology IGVP, University of Stuttgart, Nobelstraße 12, 70569 Stuttgart, Germany. E-mail: alexander.southan@igvp.uni-stuttgart.de; guenter.tovar@igvp.uni-stuttgart.de; Tel: +49 711 68568162
bInstitute of Organic Chemistry IOC, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: sabine.laschat@oc.uni-stuttgart.de
cAG Compound Profiling and Screening, Helmholtz Centre for Infection Research (HZI), Inhoffenstr. 7, 38124 Braunschweig, Germany
dFraunhofer Institute for Interfacial Engineering and Biotechnology IGB, Nobelstraße 12, 70569 Stuttgart, Germany. E-mail: Guenter.Tovar@igb.fraunhofer.de
First published on 10th October 2018
Abstract
Triazole-based cross-linkers with different spacer lengths and different functional end groups (acrylamides, methacrylamides, maleimides and vinylsulfonamides) were synthesized, investigated for cytotoxic and antibacterial activity, and incorporated into poly(acrylamide) (PAAm) and poly(N,N-dimethylacrylamide) (PDMAAm) hydrogels by free-radical polymerization. Hydrogels prepared with different cross-linkers and cross-linker contents between 0.2% and 1.0% were compared by gel yields, equilibrium degrees of swelling (S) and storage moduli (G′). Generally with increasing cross-linker content, G′ values of the hydrogels increased, while S values decreased. The different polymerizable cross-linker end groups resulted in a decrease of G′ in the following order for cross-linkers with C4 spacers: acrylamide > maleimide > methacrylamide > vinylsulfonamide. Longer cross-linker alkyl spacer lengths caused an increase in G′ and a decrease in S. Independent of the cross-linker used, a universal correlation between G′ and equilibrium polymer volume fraction ϕ was found. For PAAm hydrogels, G′ ranged between 4 kPa and 23 kPa and ϕ between 0.07 and 0.14. For PDMAAm hydrogels, G′ ranged between 0.1 kPa and 4.9 kPa and ϕ between 0.02 and 0.06. The collected data were used to establish an empirical model to predict G′ depending on ϕ. G′ of PAAm and PDMAAm hydrogels is given by G′ = 4034 kPa ϕ2.66 and G′ = 4297 kPa ϕ2.46, respectively.
Introduction
In biological systems the structure and function of large biomacromolecules is often controlled by small organic compounds acting e.g. as cofactors, signal molecules or as cross-linkers to maintain certain biological or mechanical properties. Prominent examples are the lysine-derived pyridinium amino acids desmosine and pyridinoline,1,2 which act as cross-linkers of the connective tissue proteins elastin and collagen and contribute to the hydrogel properties, water binding ability, elasticity as well as tensile strength and cell adhesion.3 Much work has been devoted to elastin-like peptides, due to their promising applications in tissue engineering, drug delivery and biology.4,5 Mostly, diaminodicarboxylic acids or small peptidomimics were used as cross-linkers for peptides by incorporation into the main polymer via peptide bonds,6 for example in the site-specific cross-linking of collagen peptides by lysyl advanced glycation end products.7 When transferring the design principles of biological hydrogels to synthetic hydrogels, the challenge is to identify structure–property relationships in order to obtain analogous rheological properties by a different molecular scaffold and cross-linking chemistry. To meet this challenge, we have previously developed desmosine-inspired cross-linkers based on 2,5-diacylpyridinium salts with terminal acrylamide units, which were used to cross-link partially thiolated hyaluronan via thiol-Michael reaction8,9 and piperazinyl-functionalized poly(ethylene glycol) via aza-Michael reaction.9,10In a previous effort to simplify and streamline the synthetic access, we designed cross-linkers Cn Mal and Me-Cn+ I− Mal with a neutral triazole or positively charged triazolium core and two terminal maleimides connected via alkyl spacers of various lengths (Fig. 1). Cross-linking with partially thiolated hyaluronan provided hydrogels, where both the presence of a charged core as well as the spacer length had an impact on E-moduli and equilibrium degrees of swelling.11,12 While these hydrogels were obtained by Michael additions, the question occurred to us whether such triazole derivatives would be suitable for the cross-linking of acrylamides via radical polymerization.
 | ||
| Fig. 1 Maleimide-functional cross-linkers Cn Mal and Me-Cn+ I− Mal used in a previous study. | ||
Poly(acrylamide) (PAAm) hydrogels are one of the most commonly used polymer networks. Nowadays, PAAm hydrogels are the standard materials for gel electrophoresis13,14 and have been used in semi-interpenetrating networks (IPN)15 as well as in double network hydrogels.16 Further future uses include sensing17 and actuation.18 Additionally, PAAm hydrogels find use in biological applications to study cell adhesion and response depending on hydrogel stiffness and network structure.19,20 Despite the numerous applications of PAAm hydrogels, a disadvantage of PAAm and its monomer acrylamide (AAm) is low solubility in many solvents other than water, preventing the homogeneous integration of hydrophobic building blocks.21 Therefore, other representatives of acrylic based monomers and polymers like N,N-dimethylacrylamide (DMAAm) and poly(N,N-dimethylacrylamide) (PDMAAm) respectively have been used for hydrogel formulations with additional solvents such as DMSO or without any water in the formulations.22 Due to the presence of hydrogen bonding and hydrophobic interactions in PDMAAm hydrogels,23 these polymer networks have been used in the preparation of hybrid hydrogels by addition of clay,24 silica25 or hydrophobic domains.26
Generally, PAAm and PDMAAm hydrogels can be obtained by the free-radical polymerization of acrylamide or N,N-dimethyl-acrylamide in the presence of cross-linkers to generate a three-dimensional network structure.27,28 Several studies have investigated the influence of the monomer and cross-linker concentration on the properties of hydrogels generated by free-radical polymerization.19,29 Systematic investigations of structure–property relationships regarding cross-linkers are less common and these studies often suffer from limited comparability, as they try to correlate cross-linkers with either completely different polarities, e.g. divinylbenzene (DVB) and poly(ethylene glycol)diacrylate (PEGDA),30 or large differences in the cross-linker size,31 different number of functionalities such as bis-, tris- or tetrakis(acrylate) cross-linkers32 or different reactive groups such as acrylamides and methacrylates.33,34 Those mismatched comparisons are due to limited commercial availability of cross-linkers. Therefore, new cross-linkers have been synthesized to expand the possibilities of applications and to customize cross-linkers for special monomer and hydrogel requirements.35 Other cross-linkers have been designed to contain thermo-labile,36 acid-labile,37 base-labile38 or photocleavable39 groups to allow for degradation of the polymer network or to contain additional functionalities40 such as carboxylic acid groups for pH-responsive behavior due to protonation and deprotonation.
In the current manuscript a systematic study on the influence of the cross-linker structure on hydrogel properties is presented (Scheme 1). We synthesized and investigated cross-linkers containing four different polymerizable groups (maleimide, acrylamide, methacrylamide and vinylsulfonamide) tethered to a central triazole or triazolium core with hydrophobic alkyl spacers of different lengths. With respect to future applications the cytotoxicity and antimicrobial activity of the cross-linkers was evaluated. This dedicated compound library was used to cross-link acrylamide (AAm) and N,N-dimethylacrylamide (DMAAm) by free-radical polymerization and the material properties of the resulting hydrogels were examined.
 | ||
| Scheme 1 Outline of this study: monomers acrylamide (AAm) and N,N-dimethylacrylamide (DMAAm) were cross-linked to hydrogels with cross-linkers carrying different functional groups and spacer lengths and the hydrogel properties were assessed. | ||
Results and discussion
Chemical synthesis
For this systematic study on the influence of the cross-linker structure on PAAm and PDMAAm hydrogels, a library of cross-linkers with different spacer lengths and various end groups was synthesized (Scheme 2). They all contain a neutral triazole or a positively charged triazolium core, which is connected via alkyl spacers of different lengths ((CH2)n with n = 4, 6, 8 and 10) to various reactive units. As terminal groups, different Michael acceptors such as acrylamides, methacrylamides and vinylsulfonamides were employed to vary the reactivity in the thio-Michael addition41 or the copolymerisation parameters in radical reactions.42,43 All cross-linkers were synthesized from the previously reported diamines Cn NH2,11 which are accessible from the alkynes 1-Cn and azides 2-Cn in two steps (Scheme 2). The maleimide cross-linker C4 Mal was synthesized following the previously published procedure.11 The acrylamide and methacrylamide cross-linkers Cn AAm and Cn MeAAm were obtained by reaction of the diamines Cn NH2 with acryloyl chloride or methacryloyl chloride and NEt3 as base in 30–50% and 31–72% yield, respectively.44 The vinylsulfonamide cross-linkers Cn VSAm were synthesized from the diamines Cn NH2 with 2-chloroethane sulfonyl chloride in 18–50% yield.45 The positively charged triazolium cross-linkers Me-Cn+ I− AAm, Me-Cn+ I− MeAAm and Me-Cn+ I− VSAm were obtained by methylation with methyl iodide in acetonitrile at 30 °C.11 Detailed descriptions of the synthetic procedure together with characterization data can be found in the ESI.†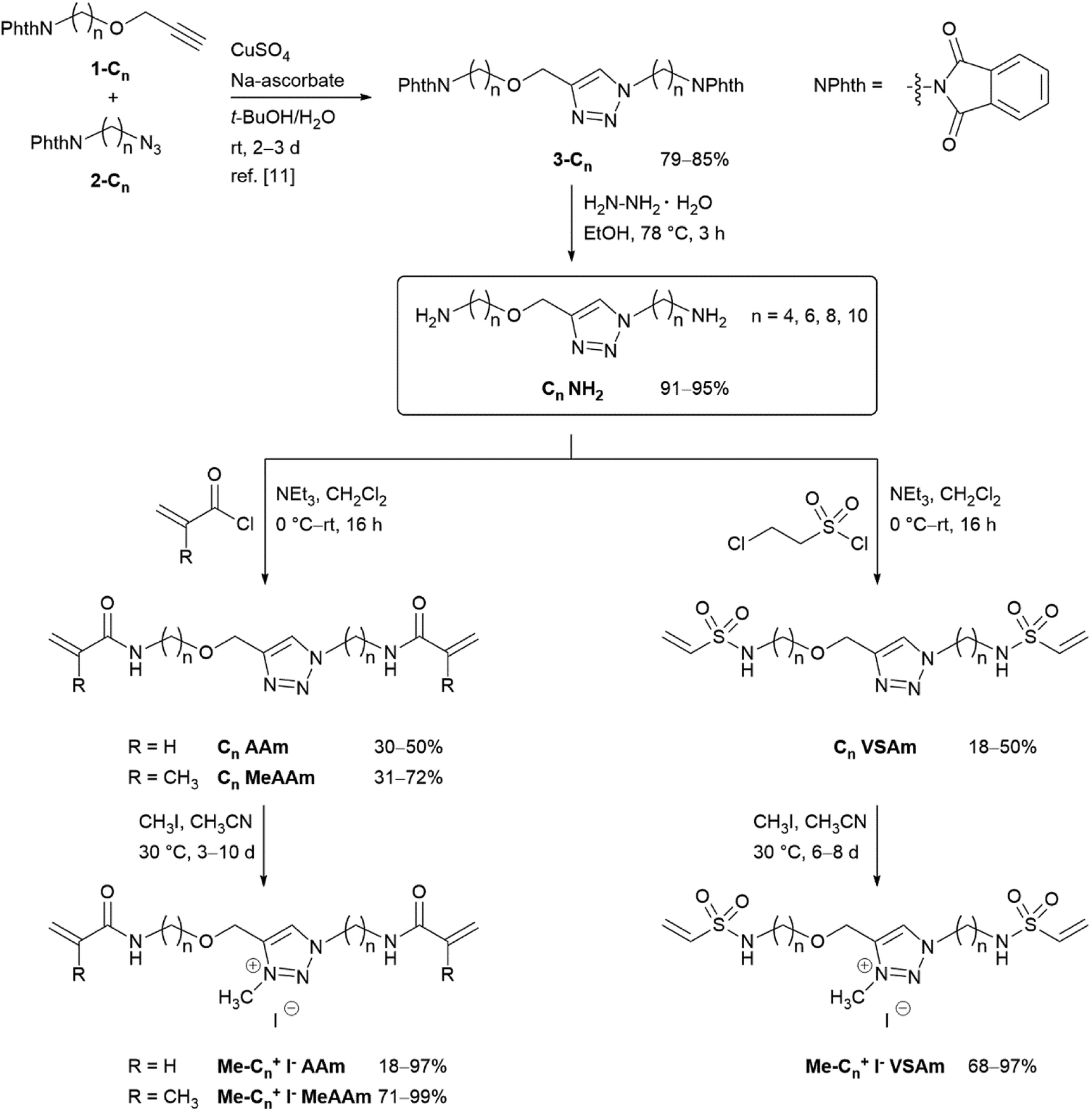 | ||
| Scheme 2 Synthesis of triazole- and triazolium-based cross-linkers with terminal acrylamides, methacrylamides and vinylsulfonamides. | ||
Biological studies of cross-linkers
PAAm and PDAAm hydrogels were extensively used in biomedical applications due to their low cytotoxicity and high biocompatibility.46,47 Thus, we surmised that unreacted cross-linkers carrying free Michael acceptor units entrapped in the hydrogel network are the most relevant species regarding any undesired biological activities such as cytotoxicity. Therefore, mouse fibroblast cells L929 were incubated for 3 days and cell viability was evaluated using the Alamar Blue assay with the test compounds at a final concentration of 50 μM.48,49 Both cross-linkers carrying a neutral triazole core and a cationic triazolium core were investigated (for details see ESI†). The results revealed that cross-linkers with terminal acrylamide or methacrylamide groups were mostly non-cytotoxic, i.e. they showed high cell viabilities irrespective of the spacer length or charge of the cross-linker. Only charged C10 derivatives showed some cytotoxicity (IC50 = 15.1 ± 0.3 μM for Me-C10+ I− AAm and IC50 = 20.4 ± 0.5 μM for Me-C10+ I MeAAm). The maleimides and vinylsulfonamides were cytotoxic, in particular the neutral maleimides showed cytotoxicity in the μM range (IC50 = 6–8 μM C4–C8 Mal). The vinylsulfonamides showed an increasing influence on cell viability with increasing chain lengths. For example, the vinylsulfonamide C4 VSAm only slightly reduced the cell viability to 87.8%, whereas the analogous cross-linker with C10 spacer C10 VSAm drastically reduced it to 0.9%. Introduction of a positive charge on the cross-linker core did not significantly change the cytotoxicity of the cross-linkers, except for the vinylsulfonamides, where cell viability was higher for the triazolium cross-linkers Me-C6+ I− VSAm (49.5%) and Me-C8+ I− VSAm (37.0%) than for their neutral counterparts C6 VSAm (21.1%) and C8 VSAm (1.2%). Overall, the cytotoxic activity increased with the chain length and was dependent on the reactivity of the terminal unit. Presumably, the observed increased cytotoxicity of cross-linkers carrying maleimides is due to their well-known high propensity to undergo Michael addition reactions with proteins, which is used in protein immobilization.50Antimicrobial activities were evaluated by monitoring growth of the Gram positive bacterium Staphylococcus aureus, and the Gram negative bacteria Escherichia coli K12 and its deletion mutant Escherichia coli ΔTolC in liquid culture via the turbidity of the bacterial suspension (optical density at 600 nm) after 24 h incubation of the bacteria with the cross-linkers. The ΔTolC deletion mutant indicates whether the inactivity of a compound on E. coli K12 is due to the efficient compound export from the cell, as TolC is part of an efflux pump system and forms a channel through the outer membrane. The results are summarized in the ESI.† Most compounds did not inhibit growth of the wild-type strain E. coli K12, whereas growth of the deletion mutant E. coli ΔTolC and of S. aureus was inhibited particularly by charged triazolium cross-linkers with C10 spacers. For the E. coli ΔTolC mutant this effect was almost independent on the functional group (IC50 – values from 4.7 μM to 13.7 μM for the ΔTolC mutant), whereas growth of S. aureus was mainly inhibited by the vinylsulfonamide (IC50 = 8.3 μM for Me-C10+ I− VSAm compared to IC50 = 21.5 μM for Me-C10+ I− MeAm).
Summarizing, the relatively low cytotoxic and antimicrobial activities of the cross-linkers should allow their use in hydrogel formulations also for biomedical applications as the other components in the formulations, i.e. AAm and PDAAm in this study, are known to result in non-toxic materials.
Hydrogel preparation and characterization
The cross-linkers available for hydrogel preparation in this study differed in their polymerizable groups (acrylamide, methacrylamide, vinylsulfonamide, maleimide), in their spacer lengths (n = 4, 6, 8 or 10) as well as in the charge of the central heterocycle core unit (triazole or triazolium). In order to study hydrogel formation depending on the cross-linker structure, the cross-linkers should be copolymerized with suitable acrylic monomers via free-radical polymerization under homogenous conditions. However, cross-linkers with the longer spacer lengths (C6, C8 and C10) were almost insoluble in water. Therefore, an ethanol-water mixture was employed. Because both DMAAm and PDMAAm proved to be soluble in this mixture, DMAAm was chosen as a monomer for comparing the different cross-linker spacer lengths. For comparing the reactivities of the different polymerizable groups, only the C4 cross-linkers were used which all showed sufficient water solubility. Therefore, cross-linkers with the different polymerizable groups were copolymerized with AAm in water. In all cases, N,N′-methylenebisacrylamide (MBA) was used as reference cross-linker. The cross-linker amount in the precursor solution was adjusted to 0.2%, 0.5%, 0.7% or 1.0% molar ratio of cross-linker (X) as part of a 15 wt% monomer content in the formulation to assess a possible tailoring of the hydrogel properties. Hydrogel formation was investigated by measuring the equilibrium degree of swelling (S), the gel yield (Y), and the storage moduli (G′) of the swollen gels in the linear viscoelastic range.51 From these macroscopic properties, the network structure on a molecular level was characterized by calculating the mesh size of each hydrogel.52Effect of the cross-linker charge
Concerning the charge of the central heterocyclic core, both neutral Cn Mal as well as charged Me-Cn+ I− Mal (Fig. 1) with values for n between 4 and 10 gave hydrogels by thiol-Michael reaction with thiolated hyaluronan in a previous study.11,12 However, by free-radical cross-linking no hydrogels were obtained with the charged cross-linkers. This behaviour was observed independent of the polymerizable terminal groups and the spacer length of the cross-linkers for both AAm and DMAAm. This effect can be ascribed to the presence of the iodide counter ions present in the charged cross-linkers rather than to the charge. This assumption is supported by the evolution of a yellow colour of the formulations with the charged cross-linkers during hydrogel preparation attempts. In contact with amylose, the yellow colour turned into a deep blue, indicating the presence of iodine in the solution53 (ESI†). Obviously, the iodide ions were oxidized to iodine by the radical initiator used,54 thus inhibiting the radical polymerization and making the formation of a polymer network impossible. Due to the non-radical reaction mechanism, no similar effect was observed during network formation with the thiol-Michael reaction. It can be concluded that for the formation of hydrogels with the charged cross-linkers using free-radical polymerization, the iodide counter ion has to be replaced with a less reactive ion such as chloride, either by ion exchange after synthesis or by using a different electrophile in the reaction step introducing the charge. Therefore, in this study hydrogels could only be prepared with the neutral cross-linkers.Effect of polymerizable group
To analyse the influence of the polymerizable group in the cross-linker, PAAm gels with all C4 cross-linkers and MBA as a reference were prepared at different cross-linker concentrations. In all cases, hydrogel preparation was possible in aqueous solution in a homogenous reaction, evidenced by the change of the liquid precursor solutions to solids upon curing and an optically clear appearance without any noticeable opacity either in the freshly cured or the swollen state. Physical characterization data of the resulting hydrogels are shown in Fig. 2.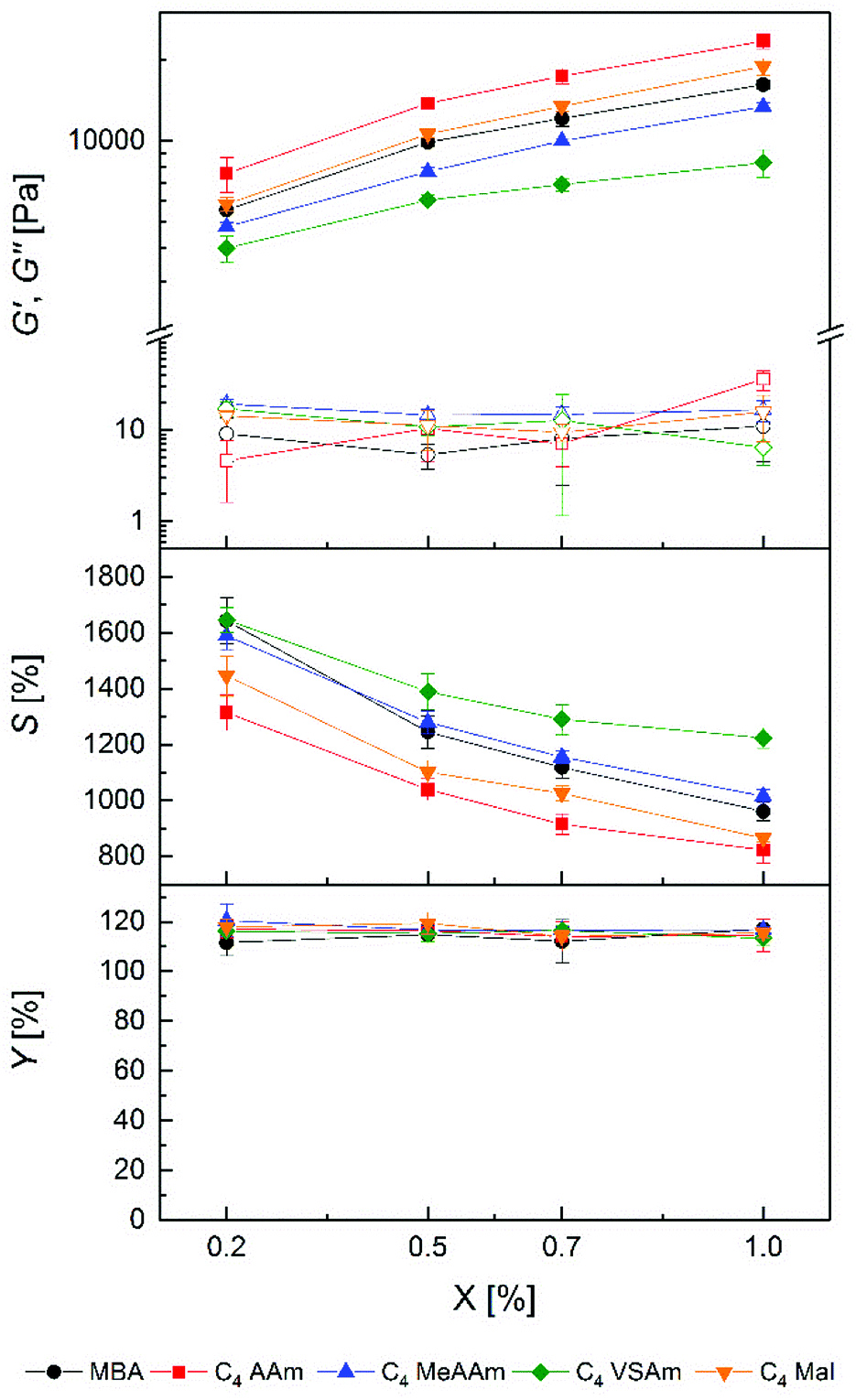 | ||
| Fig. 2 Storage modulus G′ (filled symbols), loss modulus G′′ (open symbols), equilibrium degree of swelling S and gel yield Y of prepared PAAm hydrogels with different ratios of C4 cross-linker (X). Standard deviation is given as error bars (n = 9). | ||
As a general trend for all cross-linkers, G′ values increased with increasing cross-linker concentration whereas S values decreased. These observations are in accordance with the expectation that a higher cross-linker concentration in the precursor solutions should lead to a higher cross-link density in the hydrogels, as also described by numerous other literature reports.55,56 This is further supported by common models linking macroscopic hydrogel properties with the molecular level.57 Therefore, it can be concluded that all cross-linkers were incorporated covalently into the hydrogel polymer networks. Thus, copolymerization of AAm with all cross-linkers can be considered as successful, as also shown by the high gel yields which are slightly above 100% but independent of the hydrogel composition. Since the gel yields were determined gravimetrically, the values above 100% probably resulted from remaining water in the hydrogels after drying because of the strong hydrogen bonding with the amide groups in the polymer network.58
Regarding the polymerizable groups, the order of achievable G′ values of hydrogels was C4 AAm > C4 Mal > MBA > C4 MeAAm > C4 VSAm at each cross-linker concentration. The corresponding loss moduli of the hydrogels stayed at a constant and very low level. Concerning the equilibrium degrees of swelling, the order was reversed as expected. Hydrogels with G′ values between 4.0 ± 0.4 kPa and 23.4 ± 1.4 kPa as well as S values between 823 ± 46% and 1646 ± 45% could be prepared which was not possible using only commercially available MBA at the given total monomer concentration in the precursor solution. Thus, the different cross-linkers allow tailoring the hydrogel properties over a wider range than with standard cross-linkers alone. This finding is relevant in various possible applications of hydrogels, for example in tissue engineering, where cells react both to the rigidity as well as the water content of the hydrogel.19,20 Also the diffusion of small molecules through the hydrogel network is highly dependent on the hydrogel water content.59 The possibility to tailor the water content over a broader range therefore has implications e.g. for tissue engineering (diffusion of cell nutrients) as well as for separation techniques like SDS-PAGE.14
Interestingly, even the two cross-linkers containing acrylamide groups (MBA and C4 AAm) resulted in significantly different hydrogel properties, with the MBA containing hydrogels showing lower stiffness and higher swelling. The observations can be explained either by a more efficient copolymerization of the triazole-based cross-linker with AAm compared to MBA or by additional forces like hydrophobic interactions or hydrogen bonds between the cross-linker core and the PAAm backbone. Another explanation could be a reduced cyclization reaction of C4 AAm compared to MBA57 or the longer chain length of C4 AAm.60 In both cases a more effective formation of cross-links during gelation would be expected.
A similar reasoning can be used to explain the differences using the other cross-linkers. However, whereas bisacrylamides and bismethacrylamides are widely used as cross-linkers in hydrogel preparation via radical polymerization,34 the use of bismaleimides and bisvinylsulfonamides is less frequent. In a copolymerization study of AAm and sodium N-(4-sulfophenyl)maleimide by Hocking, an increase of the maleimide content resulted in lower yields and lower molecular weights of the resulting polymer.61 Otherwise in a comparative study by Dušek, the gelation of polystyrene with p-maleimido benzoic anhydride as a cross-linker did not lead to different results compared to DVB, except for a higher gel point conversion.62 In our study the maleimide cross-linker C4 Mal led to slightly higher storage modulus and smaller equilibrium degree of swelling compared to MBA.
The C4 VSAm gels showed the highest equilibrium degrees of swelling and lowest storage moduli. To our knowledge nothing is known about vinylsulfonamides as cross-linkers in radical polymerization processes. The radical polymerization of N-propylvinylsulfonamide was conducted neat with a low yield of 18%.63 Copolymerization of AAm was only done with sodium vinylsulfonate, but again showed lower polymer yields with increasing amount of vinylsulfonate.64 Our data suggest that copolymerization of vinylsulfonamide cross-linkers proceeds less effective than with the other cross-linkers, especially with increasing amount of cross-linker. Summarizing, it was possible to prepare PAAm hydrogels with all uncharged C4 cross-linkers and acrylamide with high gel yields and with various tunable equilibrium degrees of swelling and storage moduli.
Effect of spacer length
To investigate the effect of spacer length, hydrogels with DMAAm in an ethanol-water mixture were prepared. As cross-linkers, C4 MeAAm–C10 MeAAm, C4 AAm–C10 AAm and MBA were used. The same amounts of cross-linker (X) were used as in the PAAm hydrogels described above. In contrast to the PAAm hydrogels, PDMAAm hydrogels with the lowest amount of cross-linker (0.2%) were not stable enough to be removed from the mold without breaking. Therefore, these hydrogels could not be characterized. At higher cross-linker concentrations, hydrogels were stable enough to be handled carefully. Most of the hydrogels were optically clear after preparation. Only hydrogels containing C10 MeAAm (1.0%) and C10 AAm (0.7% and 1.0%) were slightly opaque in the swollen state, probably due to inhomogeneous swelling caused by limited water affinity of the long spacers.In Fig. 3 and 4 the physical characteristics of PDMAAm hydrogels prepared with C4 MeAAm–C10 MeAAm and C4 AAm–C10 AAm, respectively, are shown. Compared to the gel yields of PAAm hydrogels, the gel yields of the PDMAAm hydrogels were generally lower and increased from about 80% at 0.5 mol% to about 90% at 1.0 mol%. An exception was the low gel yield of the hydrogels containing 0.5% MBA. This shows that at low cross-linker concentrations the polymer network formation occurred more efficiently with cross-linkers containing a spacer. Possible explanations of this observation are already given in the previous section.
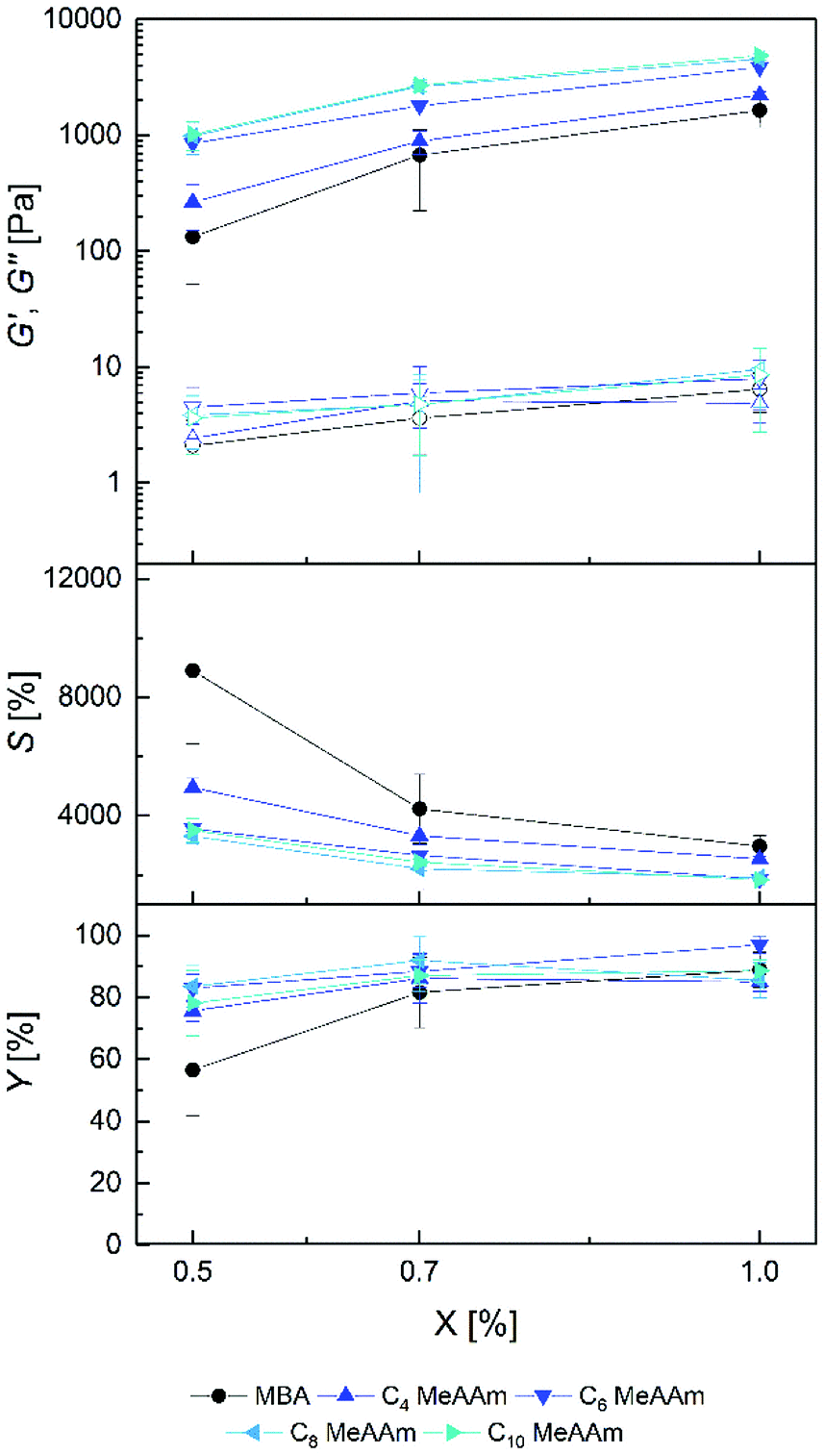 | ||
| Fig. 3 Storage modulus G′ (filled symbols), loss modulus G′′ (open symbols), equilibrium degree of swelling S and gel yield Y of prepared PDMAAm hydrogels with different ratios of C4–C10 MeAAm cross-linker (X). Standard deviation is given as error bars (n = 9). | ||
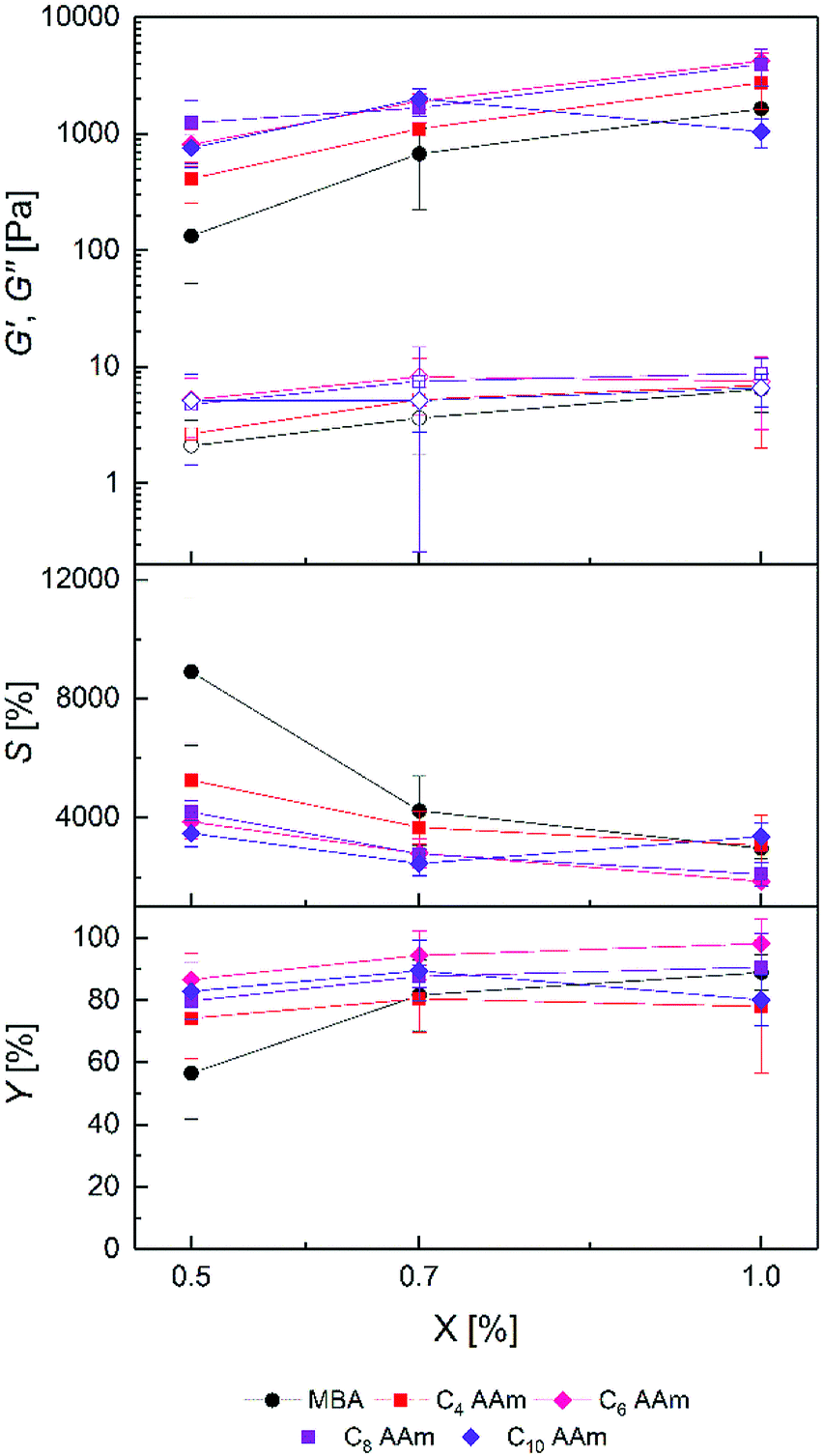 | ||
| Fig. 4 Storage modulus G′ (filled symbols), loss modulus G′′ (open symbols), equilibrium degree of swelling S and gel yield Y of prepared PDMAAm hydrogels with different ratios of C4–C10 AAm cross-linker (X). Standard deviation is given as error bars (n = 9). | ||
The G′ values increased with increasing cross-linker ratio X for both C4 MeAAm–C10 MeAAm and C4 AAm–C10 AAm, whereas the equilibrium degrees of swelling decreased. Like for the PAAm hydrogels, this indicates that cross-link density in the hydrogels was controllable by the cross-linker concentration in the precursor solution. In general, compared to the PAAm hydrogels the PDMAAm hydrogels showed lower storage moduli and higher equilibrium degrees of swelling. G′ values between 133 ± 81 Pa and 4.9 ± 0.4 kPa as well as S values between 1858 ± 183% and 8899 ± 2464% were achieved. Obviously cross-links were formed to a much lower extent in the copolymerization of DMAAm with the cross-linkers compared to AAm. Apart from differences in copolymerization parameters, which could also be reflected in the lower gel yields, other factors might play a role here. Since ethanol was present as co-solvent during the preparation of the gels, the polymerization rate was presumably reduced compared to the polymerization in pure water.65 Additionally, the tertiary amides in the PDMAAm networks are not able to form hydrogen bonds as in the PAAm networks. Those missing hydrogen bonds reduce the intermolecular forces and raise the degree of swelling.58 Such influences on the hydrogel properties are obviously more important than the difference between acrylamide and methacrylamide cross-linker end groups because the differences between the data from Fig. 3 and 4 is rather small.
The influence of spacer length in the PDMAAm hydrogels is clearly seen in Fig. 3. An extension of spacer length caused an increase of storage modulus and a decrease in equilibrium degree of swelling. This effect is mainly seen from MBA to C4 MeAAm to C6 MeAAm to C8 MeAAm and less pronounced from C8 MeAAm to C10 MeAAm. The behavior of the equilibrium degrees of swelling is vice versa. Here, the effect is most pronounced going from MBA to C4 MeAAm to C6 MeAAm. For the acrylamide cross-linkers, similar observations were made (Fig. 4). This observation could be explained by hydrophobic interactions which cause additional interactions in the polymer network.66,67 However, we tend to ascribe this effect rather to differences during the copolymerization of cross-linkers with different spacer lengths with DMAAm as will be discussed in the following section.
An exception to the trends described above is the low gel yield of the C10 AAm 1.0% hydrogels (Fig. 3) which is also reflected in their high equilibrium degrees of swelling and their low storage moduli. In contrast to C10 MeAAm, C10 AAm was not soluble in the mixture of DMAAm, water and ethanol at room temperature. Therefore, the formulations were preheated to 40 °C to dissolve the cross-linker and transferred into a preheated mold. With 1.0% the gels were opaque right after the 2.5 h reaction time which is a sign of network inhomogeneity. The C10 spacer can therefore be regarded as the upper limit of reasonable spacer lengths in this context. Another peculiarity occurred with the C8 AAm cross-linker which yielded stable hydrogels only in a single experiment with all amounts of cross-linker (X in %). Although the generated data matches into the dataset, in this case no reproducibility is given. Beside these two exceptions, hydrogel properties could be tuned over a wide range by the choice of the cross-linker spacer length.
Hydrogel network structure
On the molecular level, an increasing cross-linker concentration should result in a larger cross-link density, in smaller mesh sizes and smaller molecular weights between cross-links Mc. According to eqn (4) and (5) (Experimental part), concomitantly the stiffness should increase and the equilibrium degree of swelling should decrease. This was indeed observed for every individual cross-linker. Consequently, the trends in the swelling and stiffness data discussed above fit well with the expectations and generally allow tailoring of hydrogel properties. From the swelling and stiffness data, the Mc values for all hydrogels were calculated by using the Flory–Rehner theory for phantom networks68 and the rubber elasticity theory, respectively (ESI†).69,70 In both cases, the hydrogels were assumed to behave like swollen phantom networks which is generally preferred to the affine network model for highly swollen systems.71,72 The Mc calculated from the swelling data was in the range between 1.3 kg mol−1 and 7.0 kg mol−1 for PAAm hydrogels and 9.0 kg mol−1 and 93 kg mol−1 for PDMAAm hydrogels, which is generally in a similar range as reported before.33,73 The Mc values from G′ were generally greater than the Mc values from the swelling. A similar observation was made recently by Ammar, who ascribed the differences to weighing errors occurring during the determination of the equilibrium degree of swelling.74 On the other hand, Mahmudi pointed out that a small error for the Flory–Huggins interaction parameter χ has a large effect on calculated Mc values.69 Trends of Mc were generally the same as discussed above, showing the influence of the cross-linker end groups also on the molecular level.However, in order to gain a more comprehensive understanding of the effects determining the hydrogel properties, the G′ and swelling data in terms of the equilibrium polymer volume fraction ϕ of all hydrogels were collected in one graph (Fig. 5).
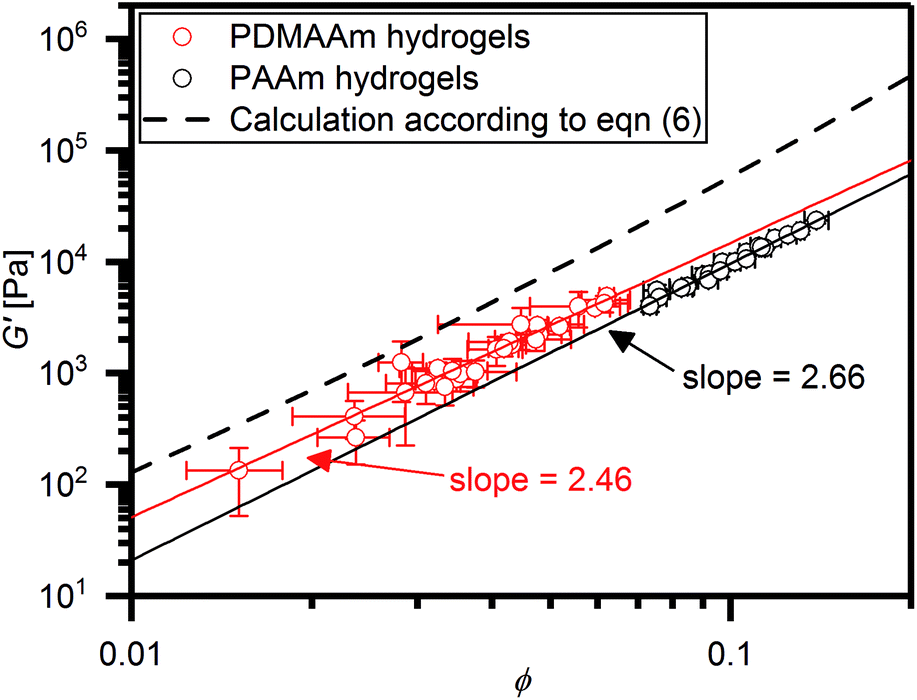 | ||
| Fig. 5 Double logarithmic plot of all G′ values collected in this study versus the corresponding equilibrium polymer volume fractions ϕ. In this plot, the data can be fitted with a linear function (straight lines) with slopes indicated in the figure. The dashed line represent the prediction of G′ with ϕ according to eqn (6) with a Flory–Huggins interaction parameter of 0.494. | ||
It is evident that in the double logarithmic plot the data for PAAm hydrogels fall on a straight line with a slope of 2.66, similarly to the data of the PDMAAm hydrogels with a slope of 2.46. The values for the slopes fit well with theoretical models which predict G′ to be proportional to 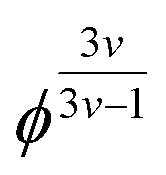 with v as the exponent of the excluded volume effect.75,76 From the slopes, values for v of 0.53 for PAAm hydrogels and 0.56 for PDAAm hydrogel are obtained, which is in the range of a good solvent for the polymers in accordance with the Flory–Huggins interaction parameters (see Experimental section). Because the slopes from Fig. 5 are expected to decrease with increasing v together with a decrease of χ, the slightly smaller slope of the PDMAAm hydrogels compared to the PAAm hydrogels reflects the smaller χ values of the PDMAAm hydrogels. This correlation between theoretically expected trends and experimental values shows that all cross-linkers tested effectively form hydrogels of a similar composition independent of the spacer length and polymerizable group due to the low cross-linker amounts used. Differences in the G′ and S data therefore can be explained by differences in copolymerization of the different cross-linkers with AAm and DMAAm, respectively, and probably not by the presence of a specific cross-link structure caused by the different cross-linkers.
with v as the exponent of the excluded volume effect.75,76 From the slopes, values for v of 0.53 for PAAm hydrogels and 0.56 for PDAAm hydrogel are obtained, which is in the range of a good solvent for the polymers in accordance with the Flory–Huggins interaction parameters (see Experimental section). Because the slopes from Fig. 5 are expected to decrease with increasing v together with a decrease of χ, the slightly smaller slope of the PDMAAm hydrogels compared to the PAAm hydrogels reflects the smaller χ values of the PDMAAm hydrogels. This correlation between theoretically expected trends and experimental values shows that all cross-linkers tested effectively form hydrogels of a similar composition independent of the spacer length and polymerizable group due to the low cross-linker amounts used. Differences in the G′ and S data therefore can be explained by differences in copolymerization of the different cross-linkers with AAm and DMAAm, respectively, and probably not by the presence of a specific cross-link structure caused by the different cross-linkers.
Comparing the absolute G′ values with values predicted by eqn (6), it is evident that the experimentally determined G′ values were roughly one order of magnitude smaller than the theoretical values. This can most likely be explained with inhomogeneity and defects like loops in the polymer network structure caused by the network formation mechanism. This effect is well known for PAAm hydrogels cross-linked with MBA.57,77 The solid lines depicted in Fig. 5 can be regarded as empirical models to predict hydrogel stiffness of PAAm and PDMAAm hydrogels with ϕ, thus yielding for PAAm hydrogels: G′ = 4034 kPa ϕ2.66 and for PDMAAm hydrogels: G′ = 4297 kPa ϕ2.46. Because all tested cross-linkers fall on the same line in Fig. 5, similar mechanisms of gel formation can be assumed for all hydrogels. Therefore, hydrogel properties are mainly determined by the polymer volume fraction ϕ in the equilibrium swollen state, which is a result of the combination of monomer, cross-linker amount and cross-linker structure (spacer length, polymerizable group).
Conclusions
Our results revealed that triazole-based cross-linkers with different polymerizable groups, i.e. acrylamide, methacrylamide, vinylsulfonamide and maleimide units, are suitable for chemical cross-linking of PAAm and PDMAAm hydrogels. Cross-linkers with acrylamide or methacrylamide end groups were mostly found to be non-cytotoxic. The cell viability depends on the terminal functional groups and increases with decreasing chain lengths and the presence of the cationic core. Acrylamides with C8 spacers show antibacterial but no cytotoxic activity, which might be potentially interesting for antibiotic development.Copolymerization of different cross-linker end groups with the monomers cause changes in the equilibrium degrees of swelling and hydrogel stiffness. Also the spacer lengths of the cross-linkers have a significant influence on hydrogel properties. The reason for these observations can be found in different copolymerization behaviour of the different cross-linkers with the monomers AAm and DMAAm. The cross-linkers thus influence the process leading to covalent cross-links rather than the properties of a hydrogel with a given cross-link density, e.g. by the introduction of non-covalent interactions. Due to the low concentrations of cross-linker necessary for hydrogel formation, non-covalent interactions in the hydrogels are still dominated by the monomer used for network formation. Thus, by measuring the equilibrium degree of swelling, mechanical properties can be predicted using an empirical relationship independent of the cross-linker used.
However, because hydrogel properties are controlled both by covalent and non-covalent interactions, using cross-linkers with different polymerizable groups offers another possibility to fine-tune covalent cross-link density and with this the hydrogel physical properties apart from the usual approaches based on changes in the monomer and cross-linker content and ratio. This might be useful especially in cases where certain hydrogel properties are needed, but for example solubility issues do not allow using the necessary concentrations of a monomer and a standard cross-linker.
Experimental
Hydrogel preparation and characterization
For adding APS and TEMED to the precursor solution 5% (w/w) aqueous stock solutions of both were freshly prepared.
To illustrate the synthetic procedure, a short example of the preparation of a DMAAm hydrogel with C4 AAm as cross-linker (X = 0.2%) is given. For 1 g precursor solution 149 mg DMAAm and 1 mg C4 AAm were dissolved in ethanol. Water, APS and then TEMED stock solution were added to yield 1 g in sum. After TEMED was added the solution was vortexed for 5 seconds and then transferred into aluminium molds of 15 mm in diameter and 1 mm in depth. The filled molds were covered with a glass slide and inserted into a Petri dish filled with wet tissues to guarantee a water saturated atmosphere during the curing of the solution at 40 °C for 2.5 hours in an oven. Subsequently the formed hydrogel discs were weighed and washed with water.
With every X of cross-linker 1 g of hydrogel precursor solution was prepared which yielded in three gels. This was repeated two times to attain triplicates.
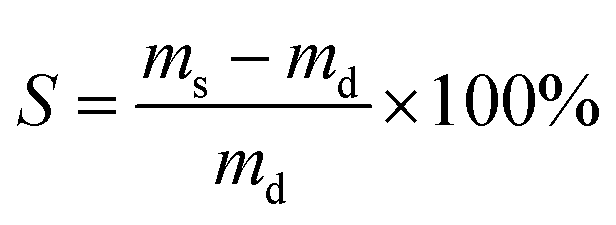 | (1) |
For gel yield the hydrogel mass m0 directly after curing was multiplied by the total mass concetration of monomers (0.15) to calculate the theoretical dry mass mt of the gel. The gel yield (Y) is the ratio between experimentally determined and theoretical dry mass of the gel.
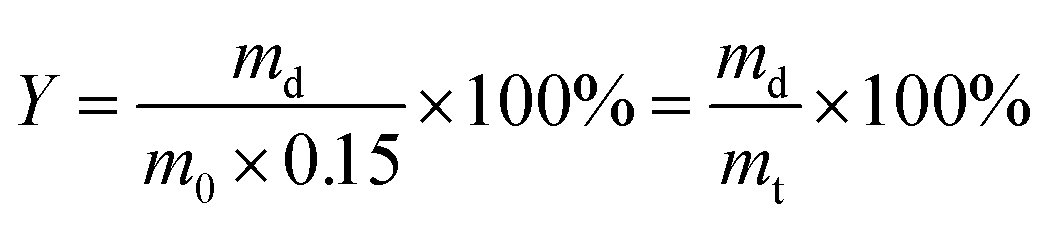 | (2) |
The equilibrium polymer volume fraction ϕ in the hydrogels was calculated, assuming additivity of volumes, by
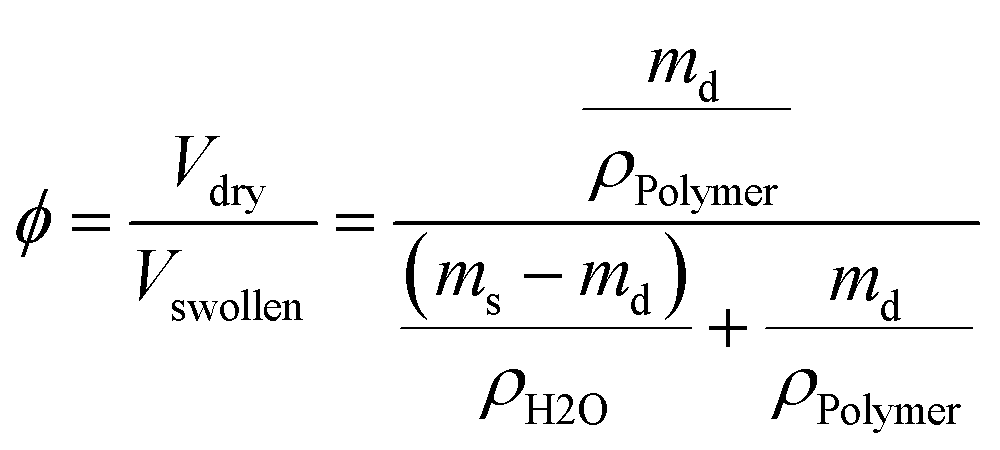 | (3) |
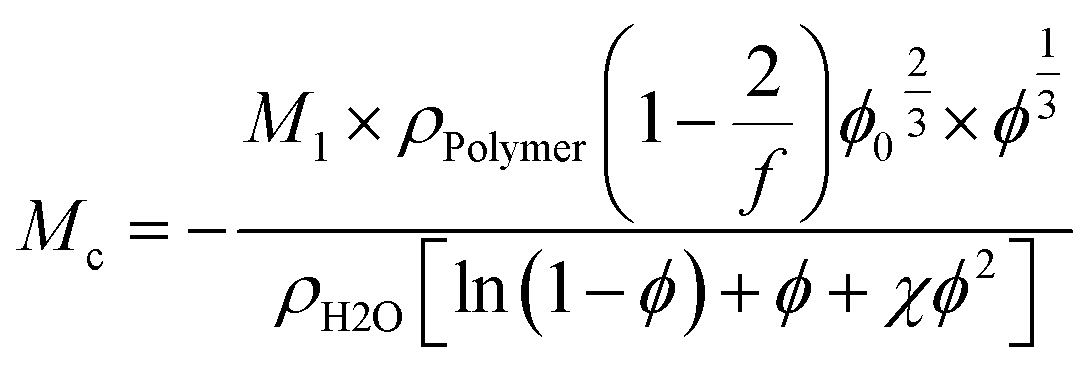 | (4) |
From rubber elasticity theory, Mc can be calculated for phantom networks according to:69
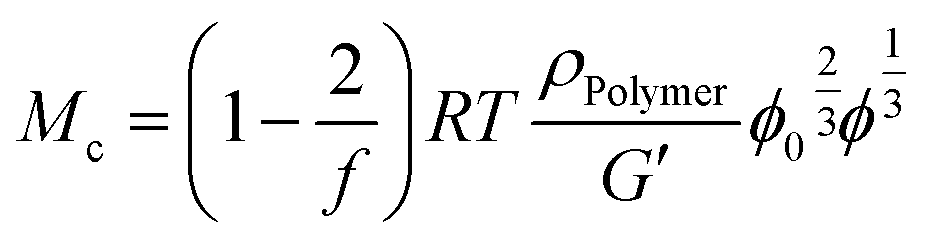 | (5) |
Combining eqn (4) and (5) yields a relationship between G′ and ϕ:
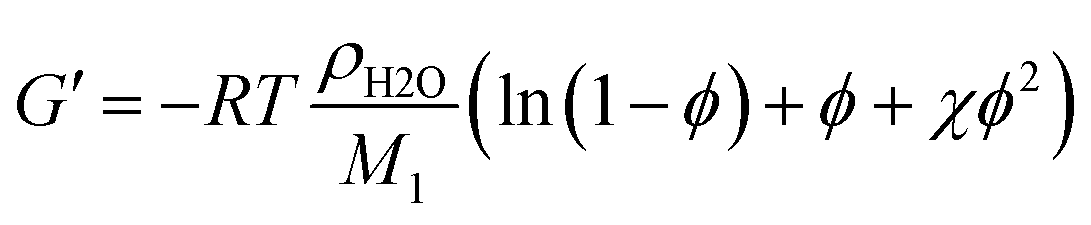 | (6) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous financial support by the Baden-Württemberg Stiftung (BiogelPlus, BioMatS11), the Ministerium für Wissenschaft, Forschung und Kunst des Landes Baden-Württemberg and the Fonds der Chemischen Industrie (fellowship for N. S.) is gratefully acknowledged. The biological assays significantly benefitted from the excellent technical assistance of Isabell Ramming, Brigitte Pawletta, Heike Overwin and Susanne Daenicke (COPS, HZI).Notes and references
- M. I. Tokareva, M. N. Ivantsova and M. A. Mironov, Chem. Heterocycl. Compd., 2017, 53, 21–35 CrossRef CAS.
- L. Anastasia, P. Rota, M. Anastasia and P. Allevi, Org. Biomol. Chem., 2013, 11, 5747–5771 RSC.
- D. Miranda-Nieves and E. L. Chaikof, ACS Biomater. Sci. Eng., 2017, 3, 694–711 CrossRef CAS.
- Y. Navon and R. Bitton, Isr. J. Chem., 2016, 56, 581–589 CrossRef CAS.
- J. C. Rodríguez-Cabello, F. J. Arias, M. A. Rodrigo and A. Girotti, Adv. Drug Delivery Rev., 2016, 97, 85–100 CrossRef PubMed.
- M. Kamalov, H. Kaur and M. A. Brimble, Chem.–Eur. J., 2016, 22, 3622–3631 CrossRef CAS PubMed.
- M. Kamalov, P. W. R. Harris, G. J. S. Cooper and M. A. Brimble, Chem. Commun., 2014, 50, 4944–4946 RSC.
- V. Hagel, M. Mateescu, A. Southan, S. V. Wegner, I. Nuss, T. Haraszti, C. Kleinhans, C. Schuh, J. P. Spatz, P. J. Kluger, M. Bach, S. Tussetschläger, G. E. M. Tovar, S. Laschat and H. Boehm, Sci. Rep., 2013, 3, 2043 CrossRef PubMed.
- M. Mateescu, I. Nuss, A. Southan, H. Messenger, S. V. Wegner, J. Kupka, M. Bach, G. E. M. Tovar, H. Boehm and S. Laschat, Synthesis, 2014, 46, 1243–1253 CrossRef.
- A. Southan, M. Mateescu, V. Hagel, M. Bach, C. Schuh, C. Kleinhans, P. J. Kluger, S. Tussetschläger, I. Nuss, T. Haraszti, S. V. Wegner, J. P. Spatz, H. Boehm, S. Laschat and G. E. M. Tovar, Macromol. Chem. Phys., 2013, 214, 1865–1873 CrossRef CAS.
- M. Martini, P. Hegger, N. Schädel, B. Minsky, M. Kirchhof, S. Scholl, A. Southan, G. Tovar, H. Boehm and S. Laschat, Materials, 2016, 9, 810 CrossRef PubMed.
- P. S. Hegger, J. Kupka, B. B. Minsky, N. Schädel, N. Petri, S. Laschat and H. Boehm, ChemistrySelect, 2017, 2, 7701–7705 CrossRef CAS.
- M. Fried and D. M. Crothers, Nucleic Acids Res., 1981, 9, 6505–6525 CrossRef CAS PubMed.
- H. Schägger and G. v. Jagow, Anal. Biochem., 1987, 166, 368–379 CrossRef.
- E. C. Muniz and G. Geuskens, Macromolecules, 2001, 34, 4480–4484 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- N. Dave, M. Y. Chan, P.-J. J. Huang, B. D. Smith and J. Liu, J. Am. Chem. Soc., 2010, 132, 12668–12673 CrossRef CAS PubMed.
- M. Bassil, J. Davenas and M. EL Tahchi, Sens. Actuators, B, 2008, 134, 496–501 CrossRef CAS.
- A. K. Denisin and B. L. Pruitt, ACS Appl. Mater. Interfaces, 2016, 8, 21893–21902 CrossRef CAS PubMed.
- B. Trappmann, J. E. Gautrot, J. T. Connelly, D. G. T. Strange, Y. Li, M. L. Oyen, M. A. Cohen Stuart, H. Boehm, B. Li, V. Vogel, J. P. Spatz, F. M. Watt and W. T. S. Huck, Nat. Mater., 2012, 11, 642–649 CrossRef CAS PubMed.
- S. Wu and R. A. Shanks, J. Appl. Polym. Sci., 2004, 93, 1493–1499 CrossRef CAS.
- G. Caria, V. Alzari, O. Monticelli, D. Nuvoli, J. M. Kenny and A. Mariani, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1422–1428 CrossRef CAS.
- M. P. Algi and O. Okay, Eur. Polym. J., 2014, 59, 113–121 CrossRef CAS.
- K. Haraguchi, R. Farnworth, A. Ohbayashi and T. Takehisa, Macromolecules, 2003, 36, 5732–5741 CrossRef CAS.
- R. Tamaki, K. Naka and Y. Chujo, Polym. J., 1998, 30, 60 CrossRef CAS.
- J. Hao and R. A. Weiss, Macromolecules, 2011, 44, 9390–9398 CrossRef CAS.
- N. Orakdogen and O. Okay, Polymer, 2006, 47, 561–568 CrossRef CAS.
- J. P. Baker, H. W. Blanch and J. M. Prausnitz, Polymer, 1995, 36, 1061–1069 CrossRef CAS.
- N. Gundogan, O. Okay and W. Oppermann, Macromol. Chem. Phys., 2004, 205, 814–823 CrossRef CAS.
- J. Xie, X. Liu, J. Liang and Y. Luo, J. Appl. Polym. Sci., 2009, 112, 602–608 CrossRef CAS.
- I. Iturralde, M. Paulis and J. R. Leiza, Eur. Polym. J., 2014, 53, 282–291 CrossRef CAS.
- T. Sharma and G. Madras, Bull. Mater. Sci., 2016, 39, 613–626 CrossRef CAS.
- D. Saraydın, E. Karadağ, Y. Işıkver, N. Şahiner and O. Güven, J. Macromol. Sci., Pure Appl. Chem., 2004, 41, 419–431 CrossRef.
- S. Abdurrahmanoglu and O. Okay, J. Macromol. Sci., Pure Appl. Chem., 2008, 45, 769–775 CrossRef CAS.
- P. Kasák, Z. Kroneková, I. Krupa and I. Lacík, Polymer, 2011, 52, 3011–3020 CrossRef.
- C. Gorsche, C. Schnoell, T. Koch, N. Moszner and R. Liska, Macromolecules, 2018, 51, 660–669 CrossRef CAS.
- V. Bulmus, Y. Chan, Q. Nguyen and H. L. Tran, Macromol. Biosci., 2007, 7, 446–455 CrossRef CAS PubMed.
- N. Metz and P. Theato, Macromolecules, 2009, 42, 37–39 CrossRef CAS.
- M. Kim and H. Chung, Polym. Chem., 2017, 8, 6300–6308 RSC.
- N. Zhou, Z. Cao and B. Xu, Chem.–Eur. J., 2017, 23, 15844–15851 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- Y. V. Bune, A. I. Barabanova, Y. S. Bogachev and V. F. Gromov, Eur. Polym. J., 1997, 33, 1313–1323 CrossRef CAS.
- V. F. Kurenkov and V. A. Myagchenkov, Eur. Polym. J., 1980, 16, 1229–1239 CrossRef CAS.
- A. F. Tominey, J. Liese, S. Wei, K. Kowski, T. Schrader and A. Kraft, Beilstein J. Org. Chem., 2010, 6, 66 Search PubMed.
- G. Martinez, J. Arumugam, H. K. Jacobs and A. S. Gopalan, Tetrahedron Lett., 2013, 54, 630–634 CrossRef CAS PubMed.
- K. Pafiti, Z. Cui, D. Adlam, J. Hoyland, A. J. Freemont and B. R. Saunders, Biomacromolecules, 2016, 17, 2448–2458 CrossRef CAS PubMed.
- T. F. Xi, C. X. Fan, X. M. Feng, Z. Y. Wan, C. R. Wang and L. L. Chou, J. Biomed. Mater. Res., 2006, 78A, 283–290 CrossRef CAS PubMed.
- S. N. Rampersad, Sensors, 2012, 12, 12347 CrossRef CAS PubMed.
- M. V. Lancaster and R. D. Fields, US Pat. US5501959A, 1996.
- A. Watzke, M. Gutierrez-Rodriguez, M. Köhn, R. Wacker, H. Schroeder, R. Breinbauer, J. Kuhlmann, K. Alexandrov, C. M. Niemeyer, R. S. Goody and H. Waldmann, Bioorg. Med. Chem., 2006, 14, 6288–6306 CrossRef CAS PubMed.
- K. S. Anseth, C. N. Bowman and L. Brannon-Peppas, Biomaterials, 1996, 17, 1647–1657 CrossRef CAS PubMed.
- T. Canal and N. A. Peppas, J. Biomed. Mater. Res., 1989, 23, 1183–1193 CrossRef CAS PubMed.
- W. Saenger, Naturwissenschaften, 1984, 71, 31–36 CrossRef CAS.
- P. C. Moews and R. H. Petrucci, J. Chem. Educ., 1964, 41, 549 CrossRef CAS.
- T. Ikeda-Fukazawa, N. Ikeda, M. Tabata, M. Hattori, M. Aizawa, S. Yunoki and Y. Sekine, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1017–1027 CrossRef CAS.
- N. Orakdogen and O. Okay, Eur. Polym. J., 2006, 42, 955–960 CrossRef CAS.
- H. J. Naghash and O. Okay, J. Appl. Polym. Sci., 1996, 60, 971–979 CrossRef CAS.
- Y. Zhang, Y. Li and W. Liu, Adv. Funct. Mater., 2015, 25, 471–480 CrossRef CAS.
- G. Majer and A. Southan, J. Chem. Phys., 2017, 146, 225101 CrossRef PubMed.
- S. Abdurrahmanoglu and O. Okay, Macromolecules, 2008, 41, 7759–7761 CrossRef CAS.
- M. B. Hocking, D. T. Syme, D. E. Axelson and K. H. Michaelian, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 2949–2968 CrossRef CAS.
- K. Dušek, L. Matějka, P. Špaček and H. Winter, Polymer, 1996, 37, 2233–2242 CrossRef.
- T. Fukuhara, Y. Shibasaki, S. Ando, S. Kishimura, M. Endo, M. Sasago and M. Ueda, Macromolecules, 2005, 38, 3041–3043 CrossRef CAS.
- J. Cao, Y. Tan, Y. Che and Q. Ma, J. Polym. Res., 2011, 18, 171–178 CrossRef CAS.
- A. Valdebenito and M. V. Encinas, Polym. Int., 2010, 59, 1246–1251 CrossRef CAS.
- S. Abdurrahmanoglu, V. Can and O. Okay, Polymer, 2009, 50, 5449–5455 CrossRef CAS.
- M. P. Mullarney, T. A. P. Seery and R. A. Weiss, Polymer, 2006, 47, 3845–3855 CrossRef CAS.
- J. C. Bray and E. W. Merrill, J. Appl. Polym. Sci., 1973, 17, 3779–3794 CrossRef.
- N. Mahmudi, M. Şen, S. Rendevski and O. Güven, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 265, 375–378 CrossRef CAS.
- J. E. Mark and B. Erman, Rubberlike elasticity: A molecular primer, Wiley, New York, 1988 Search PubMed.
- Y. Akagi, J. P. Gong, U.-i. Chung and T. Sakai, Macromolecules, 2013, 46, 1035–1040 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1977, 66, 5720–5729 CrossRef CAS.
- T. Caykara, S. Küçüktepe and E. Turan, Polym. Int., 2007, 56, 532–537 CrossRef CAS.
- N. E. Ben Ammar, T. Saied, M. Barbouche, F. Hosni, A. H. Hamzaoui and M. Şen, Polym. Bull., 2018, 75, 3825–3841 CrossRef CAS.
- T. Katashima, M. Asai, K. Urayama, U.-i. Chung and T. Sakai, J. Chem. Phys., 2014, 140, 074902 CrossRef PubMed.
- K. Urayama, T. Kawamura and S. Kohjiya, J. Chem. Phys., 1996, 105, 4833–4840 CrossRef CAS.
- M. Y. Kizilay and O. Okay, Polymer, 2003, 44, 5239–5250 CrossRef CAS.
- M. Şen, N. Pekel and O. Güven, Die Angewandte makromolekulare Chemie, 1998, 257, 1–6 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures for the cross-linkers, 1H and 13C spectra of the cross-linkers, rheological characterization of the hydrogels, mesh size calculations of the hydrogels, cytotoxicity data on the cross-linkers. See DOI: 10.1039/c8ra07145d |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |