DOI:
10.1039/C8RA04511A
(Paper)
RSC Adv., 2018,
8, 29301-29308
A novel and practical synthesis of CAT3: a phenanthroindolizidine alkaloid with potential in treating glioblastoma†
Received
29th May 2018
, Accepted 13th July 2018
First published on 17th August 2018
Abstract
CAT3, one of the (+)-deoxytylophorinine-based phenanthroindolizidine alkaloids, is a promising therapeutic agent for the treatment of hedgehog (Hh)-driven glioblastoma and is currently being evaluated in preclinical studies. In this paper, a novel and practical synthetic route for CAT3 was firstly demonstrated with 10% overall yield in 11 steps and has been successfully validated for pilot-plant scale preparation. Investigation of the substitution at the 3-position of phenanthrene revealed that the electron-donating functionality can well preserve the S configuration. In particular, the excellent enantiomeric excess of CAT3 (≥99% ee) was achieved by introducing the strongly electron-donating tert-butyldimethylsilyl (TBS) group.
Introduction
Glioblastoma (GBM) is one of the most common and malignant brain tumors in the human central nervous system along with high invasion, high mortality and poor prognoses,1,2 which ends with an average survival of 15–17 months even with the best available treatment.3 Temozolomide (TMZ), the principal first-line chemotherapeutic agent, is widely used for the treatment of recurrent and high-grade glioma and is proven to be an efficient inhibitor of GBM proliferation.4,5 However, due to the emergence of intrinsic and acquired TMZ resistance in GBM cells, its use is seriously limited.6,7 Therefore, there is an urgent need to develop new therapeutic agents for GBM disease.
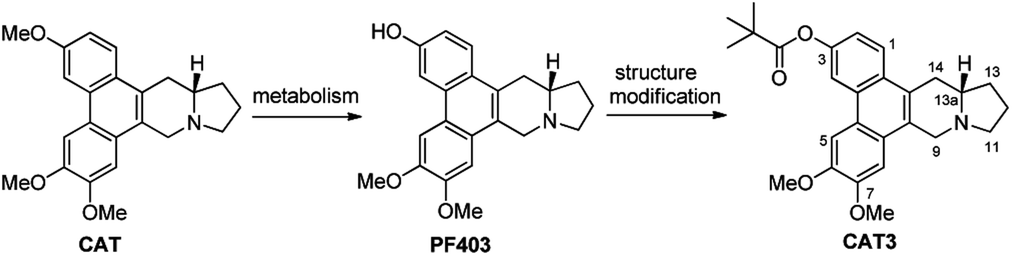
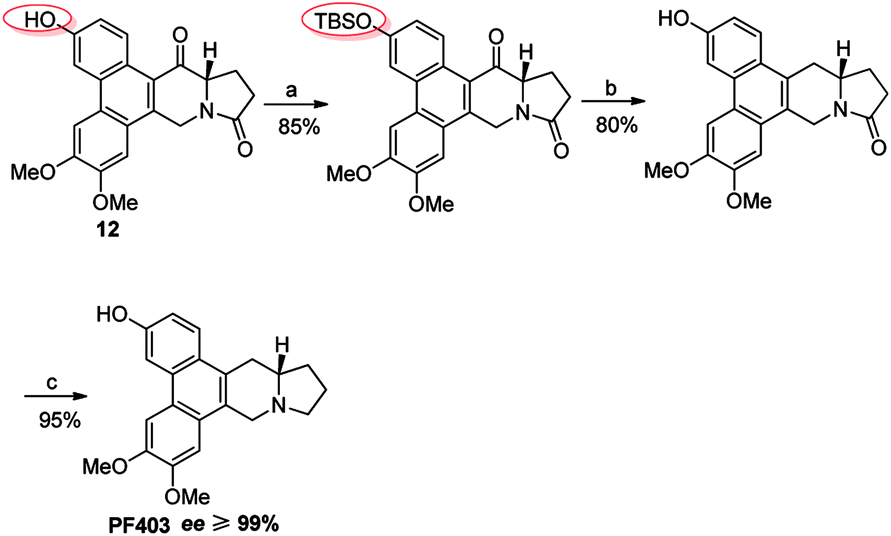
(+)-Deoxytylophorinine (CAT) was isolated from the roots of Tylophora atrofolliculata and Tylophora ovata and showed significant anticancer properties in vivo with its ability to well penetrate the blood brain barrier,8–11 while serious damage on gastrointestine was also observed. In the study of its metabolic products in vivo, (13aS)-3-hydroxyl-6,7-dimethoxyphenanthro[9,10-b]indolizidine (PF403, Fig. 1) was found to possess excellent antiproliferative activity with the IC50 value less than 0.01 nM in multiple cancer cell lines in vitro.10–12 However, the excellent data in vitro cannot be reflected to its data in vivo probably due to its poor pharmacokinetic properties. To address this issue, the prodrug strategy was employed herein and (13aS)-3-pivaloyloxyl-6,7-dimethoxyphenanthro[9,10-b]indolizidine (CAT3) was designed and synthesized as the prodrug of PF403 (Fig. 1). A series of preclinical studies has proven that CAT3 is a promising therapeutic agent against hedgehog (Hh)-driven GBM.10,12 Although the structure of CAT3 only differs slightly from that of CAT, the synthesis of which has been accomplished in several total syntheses,13–15 the mutigram synthesis of CAT3 is still challenging. A main problem lies in the completely different starting materials due to the methoxy and pivaloyoxy group for CAT and CAT3, respectively, used in the synthesis. When the same synthetic strategy of CAT was used to synthesize CAT3,13–15 it did not work after attempting multiple times in the past. This challenge, together with its excellent potency for GBM, stimulated us to investigate a novel and efficient synthetic route for CAT3.
 |
| | Fig. 1 The development process of the preclinical drug CAT3. | |
Results and discussion
Strategically, CAT3 can be easily synthesized from acylation of PF403. Therefore, the efficient synthesis of PF403 is a key question. Takashi and coworkers disclosed the synthesis of PF403 starting from 4-benzyloxybenzaldehyde and 2-(3,4-dimethoxyphenyl)acetic acid with 10.8% yield in 13 steps,16,17 which was the first meaningful synthetic route. However, photo-induced electron cyclization reaction was used to construct the phenanthrene ring, and it was not efficient for large-scale preparation.18 PF403 was also successfully prepared in 16 steps in our previous work, but the route was very tedious.15 Moreover, it has been demonstrated that it is only applicable for small-amount synthesis. When we tried multigram synthesis, the yield decreased to as low as 2%, making it unacceptable for the large-scale preparation of CAT3.
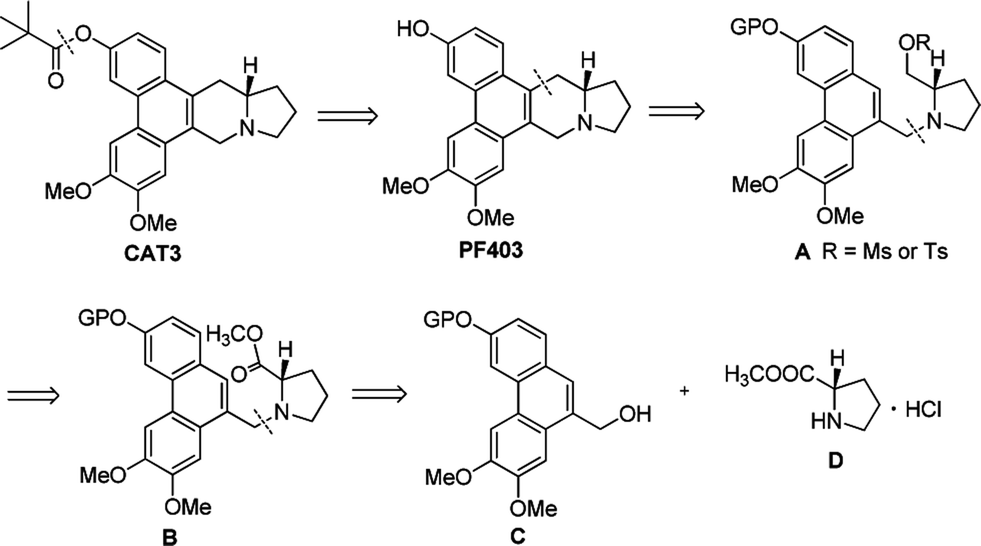
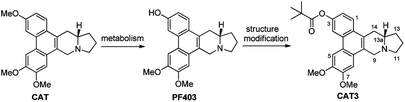
Thus it is necessary to develop a novel synthesis of PF403. The retrosynthetic analysis was illustrated in Scheme 1. We envisioned that PF403 could be accessible from the intermediate A, which could be readily made by reduction and protection from B. It was envisaged that B could be assembled from compound C and D by a substitution reaction.
 |
| | Scheme 1 The retrosynthetic analysis of CAT3. | |
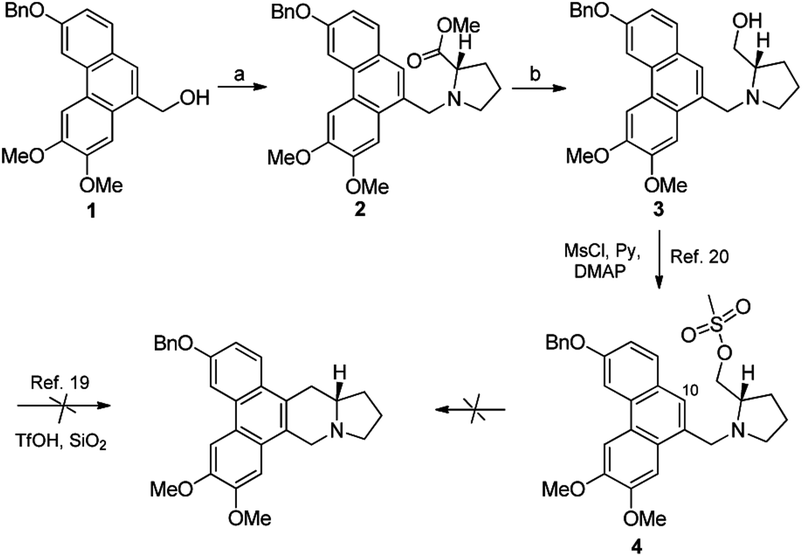
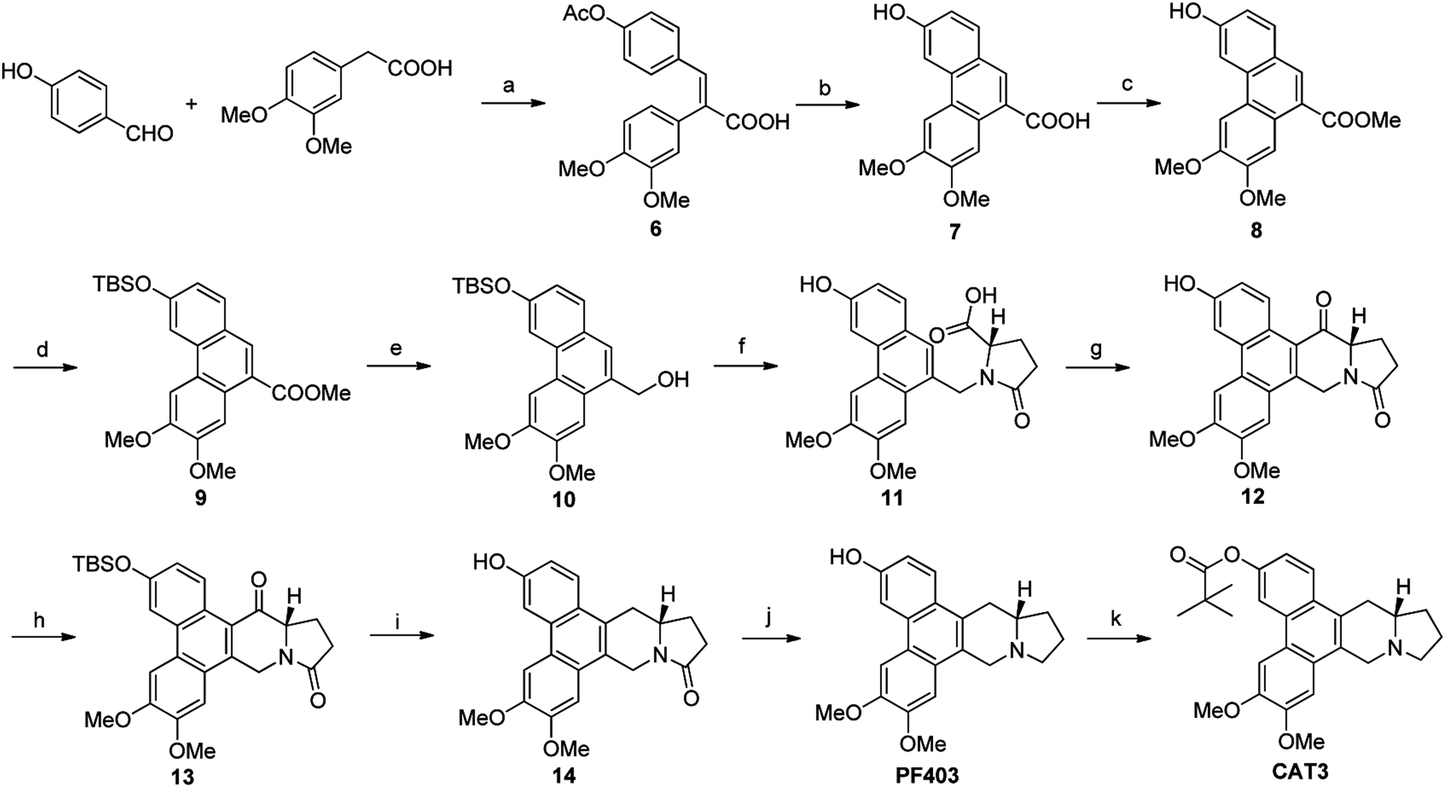
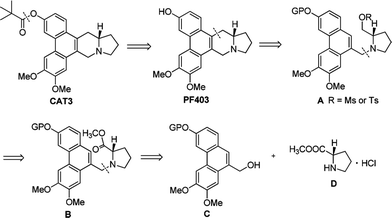
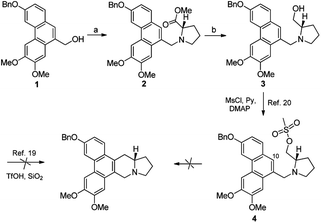
Based on our retrosynthetic analysis, the synthesis of PF403 commenced with the known compound 1, which was obtained using our reported method in 6 steps.15 The ester 2 was easily prepared from the condensation of 1 with commercially available methyl L-prolinate hydrochloride. Compound 3, which was expected to produce phenanthroindolizidine by eliminating one molecule of H2O, was easily obtained by reducing 2 with diisobutylaluminum hydride.19 However, multiple attempts to construct the indolizidine ring from 3 in one step failed according to literature,20,21 even if we tried to convert the hydroxy group to a better leaving group such as MsO–, TfO– or TsO–. Except compound 1, other different substitutions on 3-position was tried, such as TBSO– and HO–, but both result were disappointed. Thus the preparation of PF403 via this route proved probably impracticable (Scheme 2).
 |
| | Scheme 2 A simple synthetic route firstly designed for PF403. Reagents and conditions: (a) PPh3, imidazole, I2, DCM, r.t., then methyl L-prolinate hydrochloride, imidazole, r.t.; (b) DIBAL-H, DCM. 0 °C; 40% for two steps. | |
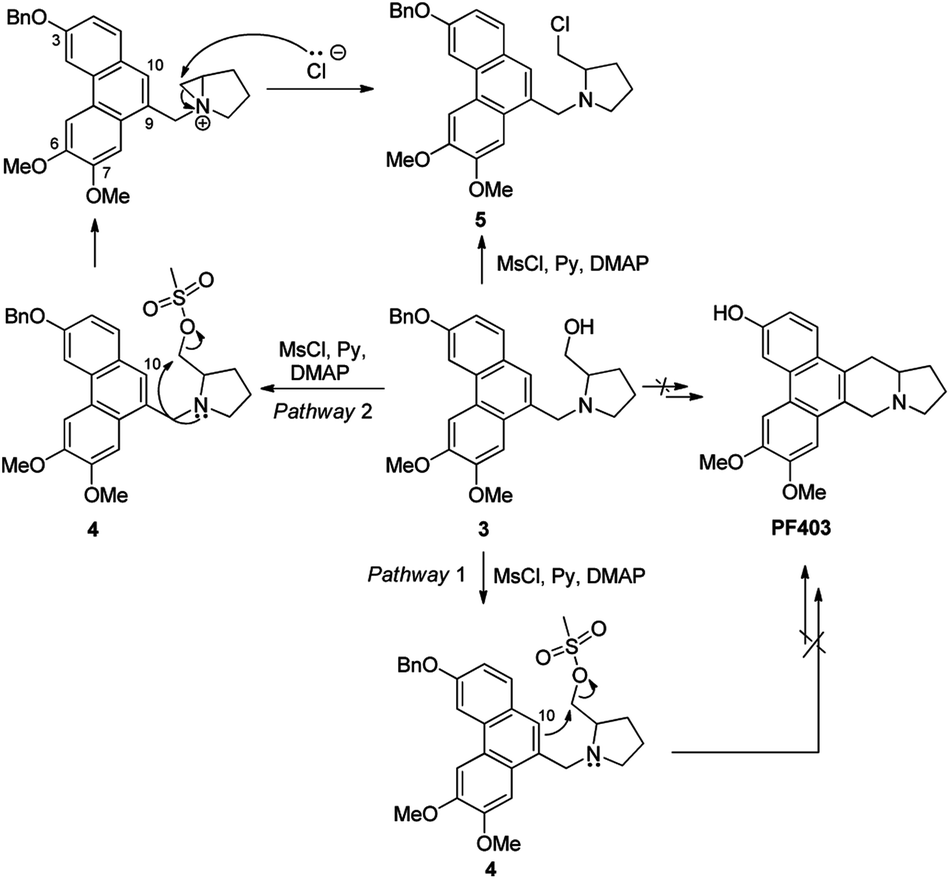
We deduce that there are two probable pathways starting from 3 under the acid condition to construct phenanthroindolizidine. Theoretically, the route should work well if the aromatic electron at 10-position of phenanthrene ring attacks carbocation to generate phenanthroindolizidine via pathway 1. However, identification of the chlorinated compound 5 suggests that the carbocation, which probably arised from transition state 4 with the aid of the nitrogen lone-pair electrons, appears to favor the formation of the nitrogen-containing three-membered ring structure as a transition state via pathway 2 (Scheme 3).
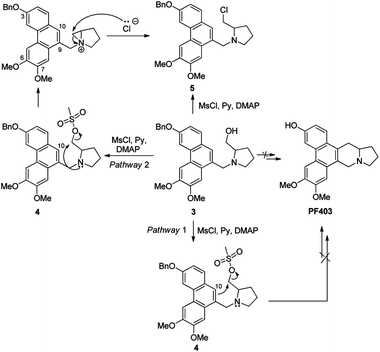 |
| | Scheme 3 A plausible mechanism explaining the failure in constructing the phenanthroindolizidine. | |
After multiple attempts, we eventually found a novel and practical synthetic route to produce CAT3 in 11 steps (Scheme 4). The condensation between 2-(3,4-dimethoxyphenyl)acetic acid and 4-hydroxybenzaldehyde gave compound 6 in almost quantitative yield, which was confirmed by difference NOE spectrum as cis-configuration (see ESI†). Compound 9 was prepared in 50% overall yield in three steps, including the scroll coupling in the presence of FeCl3, esterification in methanol with con. H2SO4, substitution of 3-OH with tert-butyldimethylsilyl chloride (TBSCl). The key intermediate 10 was achieved in 95% yield by the reduction of 9 with diisobutylaluminum hydride, which was transformed to 11 in 50% yield through a reaction sequence including iodine substitution, nucleophilic substitution with L-glutamic acid dimethyl ester hydrochloride, and subsequent hydrolysis. After formation of 12 in 80% yield by Friedel–Crafts reaction from 11, its 3-position hydroxyl group was again protected by TBS to give 13 in 85% yield. The 14-position carbonyl of 13 was then reduced to compound 14 in 80% yield, in which the lactam carbonyl group was further reduced to methylene in 95% yield, and PF403 was successfully synthesized till now. After PF403 in hand, we firstly tried to prepare CAT3 by acylation of PF403 using pivaloyl chloride. However, a main by-product chlorinated substitution on 4-position of CAT3 was produced, which was difficult to be separated because of its polarity almost the same as CAT3. At last, pivaloyl chloride was replaced by pivalic anhydride and CAT3 was smoothly achieved with a yield as high as 70% avoiding the by-product. In general, there are several aspects of advantages for this route as follows: (1) the conventional design for the synthesis of 6 is broken so that no protection step is required for 4-hydroxybenzaldehyde, thus leading to a shorter synthesis; (2) the operation and post treatment of the reactions in this route are easy and convenient, which also result in a high overall yield (10%), and no traditional column chromatography is used; (3) pilot-scale preparation has been achieved for this route with cheap reagents and materials used, indicating that it is probably an economical route for large-scale synthesis; (4) all the chiral intermediates and products are prepared in high optical purity (≥99% ee), which is vital for a drug candidate.
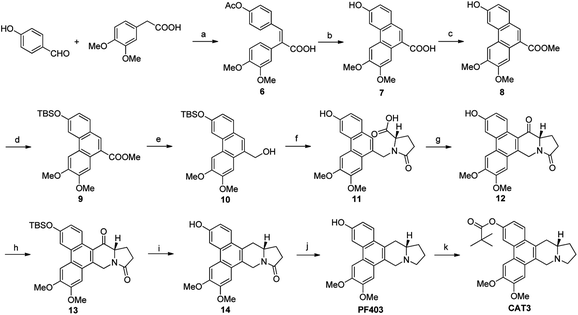 |
| | Scheme 4 Reagents and conditions for the large-scale preparation route of CAT3: (a) Ac2O, Et3N, reflux; (b) FeCl3, m-CPBA, TFA, r.t.; (c) MeOH, H2SO4, reflux; (d) TBSCl, imidazole, DCM, r.t.; 50% in four steps; (e) DIBAL-H, DCM. 0 °C to r.t., 95%; (f) i: NaI, TMSCl, then L-dimethyl glutamate, K2CO3, r.t.; ii: 5% HAc, r.t.; iii: NaOH, CH3OH, r.t., then HCl; 55%; (g) TFAA, r.t., then BF3–Et2O, reflux, 85%; (h) TBSCl, imidazole, DMF, r.t., 85%; (i) NaBH4, DCM-MeOH, then Et3SiH, BF3–Et2O, r.t., 80%; (j) Red-Al, THF, r.t., 95%; (k) pivalic anhydride, Et3N, DMAP, DCM, r.t., 70%. | |
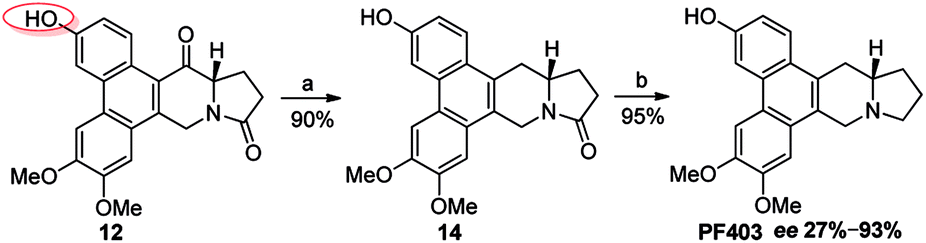
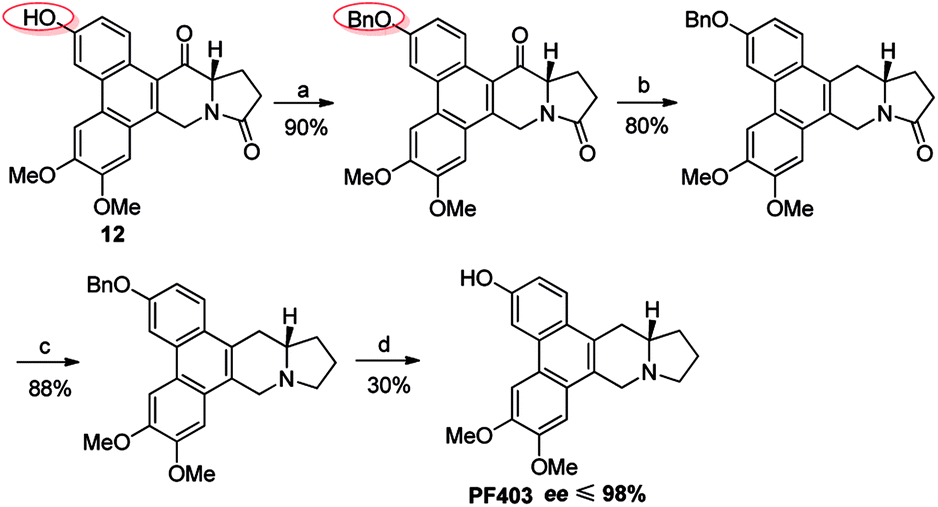
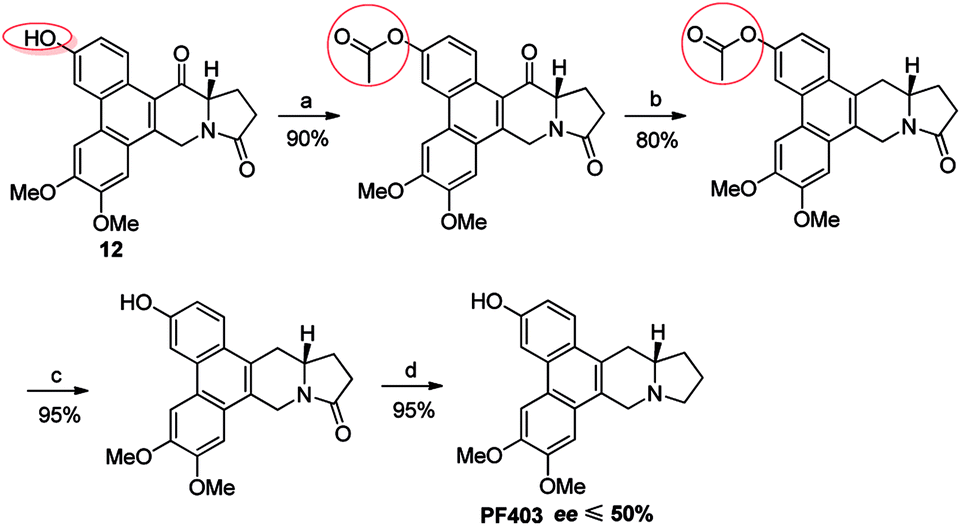
It should be noted that compound 14 can be synthesized in one step from 12 with high yield (Scheme 5). However, the optical purity of CAT3 from this approach was not satisfactory. We studied all the chiral intermediates and noticed that the optical purity obviously decreased when the 14-position ketone of compound 12 was reduced to the hydroxyl group. In order to figure it out, a batch of experiments were performed. As a common sense, we first focused our efforts on the solvent and temperature used in this step. Different kinds of solvents or/and their combination were tested, such as diethylene glycol dimethyl, tetrahydrofuran, methanol, ethanol, dichloromethane and so on. Meanwhile, we tried the reaction at different temperatures, including −20 °C, 0 °C and 20 °C. Unfortunately, the results were unsatisfactory (27–93% ee, Scheme 5) in all these cases. Then we turned to modify the 3-position of compound 12. Three different substitutes were selected and compared, including the free hydroxyl group, and the hydroxyl protected with the electron-donating benzyl and electron-withdrawing acetyl group. The best enantiomeric excess was obtained when 3-position hydroxyl was protected as benzyl ether (95–98% ee, Scheme 6) while the worst result obtained when protected as acetyl ester (10–50% ee, Scheme 7). Thus it can be preliminarily inferred that substitution on 3-position is related with the enantiomeric excess of the final product, and stronger electron-donating ability may further increase the optical purity of the product. As we expect, excellent enantiomeric excess was achieved when the 3-position hydroxyl was protected by the strong electron-donating TBS group (≥99% ee, Scheme 8). We deduce that the electron-donating group at 3-position, which appears far from the ketone at C-14 in the plane structure, has a remote control on the stereochemistry of the phenanthroindolizidine moiety in favor of the formation of the S-configured center. The yields and ee values generated from different substitutions were illustrated in Schemes 5–8 and Table 1.
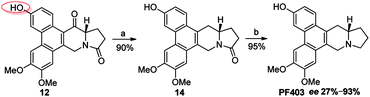 |
| | Scheme 5 Reagents and conditions for the synthesis of PF403 starting from 12: (a) NaBH4, then Et3SiH, BF3–Et2O, r.t.; (b) Red-Al, THF, r.t. | |
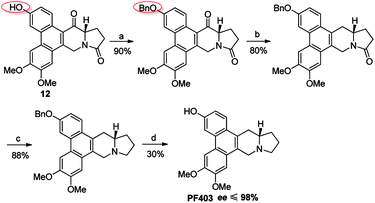 |
| | Scheme 6 Reagents and conditions for the synthesis of PF403 starting from 12 with benzyl substitution at 3-position: (a) BnBr, K2CO3, acetone, reflux; (b) NaBH4, then Et3SiH, BF3–Et2O, r.t.; (c) Red-Al, THF, r.t.; (d) Pd–C, H2, MeOH, r.t. | |
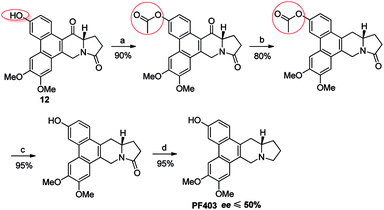 |
| | Scheme 7 Reagents and conditions for the synthesis of PF403 starting from 12 with acetyl substitution at 3-position: (a) Ac2O, TEA, DMAP, DCM, r.t.; (b) NaBH4, then Et3SiH, BF3–Et2O, r.t.; (c) NaOH, CH3OH, r.t., then HCl; (d) Red-Al, THF, r.t. | |
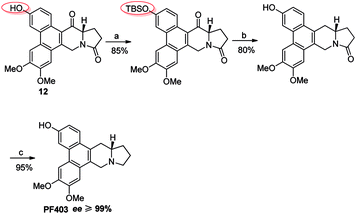 |
| | Scheme 8 Reagents and conditions for the synthesis of PF403 starting from 12 with TBS substitution at 3-position: (a) TBSCl, imidazole, DMF, r.t.; (b) NaBH4, then Et3SiH, BF3–Et2O, r.t.; (c) Red-Al, THF, r.t. | |
Table 1 The ee and yield of the final product from the different synthetic route
| |
Substitution at 3-position of compound 12 |
| HO |
BnO |
TBSO |
AcO |
| ee |
27–93% |
95–98% |
≥99% |
10–50% |
| Yield |
13.2% |
2.9% |
10.0% |
10.1% |
Experimental
General methods
All the reactions sensitive to air or moisture were carried out under argon atmosphere in anhydrous solvents. The purchased extra dry Tetrahydrofuran (THF) with the resealable package was used directly. Thin layer chromatography (TLC) was performed using glass precoated silica gel GF254 plates. Spots were visualized under 254 nm or 365 nm UV light or colored by 1% KMnO4 in 5% NaCO3 aqueous solution. NMR spectra were obtained on Bruker-400 MHz and Bruker-500 MHz spectrometers. Chemical shifts are reported in δ (ppm) relative to solvent peaks. High-resolution electrospray ionization mass spectra (HRESIMS) data were obtained on micromass Autospec-Ultima ETOF spectrometer. High performance liquid chromatography (HPLC) was performed the HPLC system was equipped with a UV-vis detector (G1315C/D), an Agilent 1200 series G1322A IV pump and software for process control and data handling (Agilent ChemStation for LC 3D Systems) equipping an GL Intersil ODS column (250 × 10 mm, 5 μm). UV spectra were measured on a JASCO V650 spectrophotometer. IR spectra were recorded on a Nicolet 5700 FT-IR microscope instrument (FT-IR microscope transmission).
(S)-(1-((3-(Benzyloxy)-6,7-dimethoxyphenanthren-9-yl)methyl)pyrrolidin-2-yl)methanol (3)
To a stirring solution of imidazole (90 mg, 1.32 mmol) and triphenylphosphine (267 mg, 1.02 mmol) in DCM (15 ml) was added iodine (214 mg, 0.84 mmol). After stirring for 10 min at room temperature, compound 1 (224 mg, 0.6 mmol) was added. After completion of the substitution, imidazole (122 mg, 1.8 mmol) and methyl L-prolinate hydrochloride (198 mg, 1.2 mmol) was added to the stirring reaction in sequences. After completion of the reaction, it was quenched by the addition of the aqueous solution of saturated NH4Cl (20 ml). The reaction mixture was extracted into dichloromethane CH2Cl2 (3 × 20 ml). The combined organic layer was washed with brine (2 × 30 ml), dried with anhydrous Na2SO4, and concentrated under vacuum to furnish the residue as crude product (2). To the cooled (−10 °C) solution of the crude product in dry CH2Cl2 (30 ml) was added DIBALH (0.8 ml, 1.2 mmol, 1.5 M in toluene) dropwise under argon, and the reaction mixture was allowed to stir at room temperature for 12 h. Then the reaction mixture was quenched with the solution of seignette salt along with stirring for additional 12 h. The reaction mixture was separated as two layers and the water layer was extracted with dichloromethane CH2Cl2 (3 × 20 ml). The combined organic layer was washed with brine (2 × 30 ml), dried with anhydrous Na2SO4, and concentrated under vacuum to furnish the crude product, which was purified by PTLC (DCM/MeOH = 10/1) to provide 3 (109 mg, 0.24 mmol) as yellow oil with 40% yield in two steps. 1H NMR (500 MHz, DMSO-d6) δ 8.15 (s, 1H), 8.03 (s, 1H), 7.93 (s, 1H), 7.81 (d, J = 8.7 Hz, 1H), 7.59–7.53 (m, 3H), 7.43 (d, J = 7.5 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H),7.36–7.34 (m, 1H), 7.26 (dd, J = 8.7, 1.7 Hz, 1H), 5.36 (s, 2H), 4.70–4.67 (m, 1H), 4.62–4.59 (m, 1H), 4.03 (s, 3H), 3.93 (s, 3H), 3.66–3.61 (m, 1H), 3.51–3.45 (m, 2H), 2.66–2.59 (m, 2H), 2.25–2.22 (m, 1H), 1.91–1.87 (m, 1H), 1.61–1.50 (m, 3H). 13C NMR (125 MHz, DMSO-d6) δ 156.9, 148.8, 148.7, 137.3, 131.5, 130.6, 129.8, 128.8, 128.5, 128.1, 127.9, 126.5, 125.3, 124.3, 116.1, 106.4, 105.4, 104.2, 69.7, 65.3, 64.9, 59.0, 55.9, 55.3, 54.5, 28.0, 22.5. HRESIMS, m/z calculated for C29H32NO4 [M + H]+: 458.2331. Found: 458.2338.
(S)-1-((3-(Benzyloxy)-6,7-dimethoxyphenanthren-9-yl)methyl)-2-(chloromethyl)pyrrolidine (5)
To the cooled (0 °C) solution of 3 (55 mg, 0.12 mmol) in dry CH2Cl2 (10 ml) was added pyridine (21 mg, 0.26 mmol), DMAP (1.2 mg, 0.01 mmol) and methanesulfonyl chloride. After stirring for 12 h at room temperature, the reaction was quenched by the addition of the saturated sodium bicarbonate aqueous. The reaction mixture was separated as two layers and the water layer was extracted with dichloromethane CH2Cl2 (2 × 20 ml). The combined organic layer was washed with brine (2 × 20 ml), dried with anhydrous Na2SO4, and concentrated under vacuum to furnish the crude product, which was purified by PTLC (ethyl acetate/petroleum ether = 1/1) to provide 5 (33 mg, 0.07 mmol) as light yellow oil with 60% yield. 1H NMR (500 MHz, CDCl3) δ 7.93 (d, J = 2.2 Hz, 1H), 7.92 (s, 1H), 7.83 (s, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.56 (d, J = 2.2, 1H), 7.54 (s, 1H), 7.48 (s, 1H), 7.43 (d, J = 7.5 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H),7.38–7.33 (m, 1H), 7.25 (dd, J = 8.7, 2.2 Hz, 1H), 5.29 (s, 2H), 4.10 (s, 3H), 4.05 (s, 3H), 3.98–3.92 (m, 1H), 3.85–3.80 (m, 1H), 3.07–3.02 (m, 1H), 2.70–2.65 (m, 1H), 2.42–2.39 (m, 1H), 2.26–2.21 (m, 1H), 2.12–2.08 (m, 1H), 1.82–1.77 (m, 1H), 1.70–1.53 (m, 2H), 1.26–1.23 (m, 1H). HRESIMS, m/z calculated for C29H31ClNO3 [M + H]+: 476.1992. Found: 476.1996.
(E)-3-(4-Acetoxyphenyl)-2-(3,4-dimethoxyphenyl)acrylic acid (6)
To a stirring solution of 3, 4-dimethoxyphenyl acetic acid (196 g, 1 mol) and 4-hydroxybenzaldehyde (123 g, 1 mol) in acetic anhydride (400 ml) was added triethylamine (200 ml) dropwise and the mixture was refluxed overnight under argon. After completion of the reaction, it was cooled to room temperature and 800 ml water was added. The reaction was stirred for 1 h and furnished 6 (341 g, 1 mol) as yellow solid after a sequence of filtration, water washing and drying. 1H NMR (500 MHz, CD3OD) δ 7.78 (s, 1H), 7.08 (d, J = 8.7 Hz, 2H), 6.93 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.7 Hz, 2H), 6.73 (s, 1H), 6.72 (dd, J = 8.2, 2.0 Hz, 1H), 3.82 (s, 3H), 3.69 (s, 3H), 2.20 (s, 3H). 13C NMR (125 MHz, CD3OD) δ 171.1, 170.8, 152.6, 150.6, 150.3, 140.1, 134.2, 133.9, 132.6, 129.9, 123.6, 122.6, 114.7, 112.97, 56.4, 56.3, 49.5, 49.3, 49.2, 49.0, 48.8, 48.7, 48.5, 20.9. IR (KBr) νmax = 3072, 2992, 2838, 2630, 1768, 1680, 1612, 1514, 1419, 1456, 1369, 1021, 912, 848 cm−1. HRESIMS, m/z calculated for C19H18O6Na [M + Na]+: 365.0996. Found: 365.0986.
Methyl 3-((tert-butyldimethylsilyl)oxy)-6,7-dimethoxy phenanthrene-9-carboxylate (9)
To a solution of 6 (341 g, 1 mol) and ferric chloride in trifluoroacetic acid (1400 ml) was added dropwise a solution of 3-chloroperbenzoic acid (189.8 g, 1.1 mol) in trifluoroacetic acid (1700 ml) below 0 °C and then stirred for 12 h at room temperature. After completion of the reaction, a solution of sodium thiosulfate pentahydrate (20 g in 100 ml water) was added and then stirred for 30 min. The reaction was evaporated under reduced pressure to afford crude product 7 (800 g), which was used for the next step without further purification. To the solution of 7 (800 g) in methanol (2500 ml) was added concentrated sulfuric acid (125 ml), and the reaction was refluxed for 12 h. After completion of the reaction, it was quenched by the addition of imidazole (306 g) and was stirred for another 30 min. Then the mixture was concentrated under vacuum to furnish the residue as crude product (8). To the stirring solution of the crude product and imidazole (238 g, 3.5 mol) in dichloromethane (2000 ml) was added TBSCl (530 g, 3.5 mol). After stirring for 12 h, the produced solid was filtered and the filtrate was washed by brine (2 × 2000 ml), dried with anhydrous Na2SO4, and concentrated under vacuum to furnish the residue, which was purified by fast silica column chromatography (PE/EtOAc, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 9 (213 g, 0.5 mol) as yellow solid with 50% yield in four steps. Mp 144–146 °C; 1H NMR (500 MHz, CDCl3) δ 8.65 (s, 1H), 8.45 (s, 1H), 7.86 (s, 1H), 7.86 (d, J = 2.2 Hz, 1H), 7.82 (d, J = 8.6 Hz, 1H), 7.13 (dd, J = 8.6, 2.2 Hz, 1H), 4.12 (s, 3H), 4.09 (s, 3H), 4.02 (s, 3H), 1.06 (s, 9H), 0.32 (s, 6H), 0.07 (s, 4H). 13C NMR (125 MHz, CDCl3) δ 168.3, 156.6, 149.9, 148.9, 133.6, 131.9, 131.4, 125.0, 124.9, 124.7, 121.9, 120.8, 111.3, 106.9, 103.2, 55.95, 55.90, 52.1, 25.9, 18.5, −4.0. IR (KBr) νmax = 3120, 3004, 2951, 2857, 1708, 1617, 1520, 1469, 1421, 1258, 1208, 1113, 1033, 992, 938, 910, 832, 778 cm−1. HRESIMS, m/z calculated for C24H30O5Si [M + H]+: 427.1938. Found: 427.1946.
:1) to give 9 (213 g, 0.5 mol) as yellow solid with 50% yield in four steps. Mp 144–146 °C; 1H NMR (500 MHz, CDCl3) δ 8.65 (s, 1H), 8.45 (s, 1H), 7.86 (s, 1H), 7.86 (d, J = 2.2 Hz, 1H), 7.82 (d, J = 8.6 Hz, 1H), 7.13 (dd, J = 8.6, 2.2 Hz, 1H), 4.12 (s, 3H), 4.09 (s, 3H), 4.02 (s, 3H), 1.06 (s, 9H), 0.32 (s, 6H), 0.07 (s, 4H). 13C NMR (125 MHz, CDCl3) δ 168.3, 156.6, 149.9, 148.9, 133.6, 131.9, 131.4, 125.0, 124.9, 124.7, 121.9, 120.8, 111.3, 106.9, 103.2, 55.95, 55.90, 52.1, 25.9, 18.5, −4.0. IR (KBr) νmax = 3120, 3004, 2951, 2857, 1708, 1617, 1520, 1469, 1421, 1258, 1208, 1113, 1033, 992, 938, 910, 832, 778 cm−1. HRESIMS, m/z calculated for C24H30O5Si [M + H]+: 427.1938. Found: 427.1946.
(3-((tert-Butyldimethylsilyl)oxy)-6,7-dimethoxyphenanthren-9-yl)methanol (10)
To a cooled (0 °C) solution of 9 (110 g, 0.26 mol) in dry CH2Cl2 (1000 ml) was added DIBALH (378 ml, 0.57 mol, 1.5 M in toluene) dropwise under argon, and the reaction mixture was allowed to stir at room temperature for 12 h. Then the reaction mixture was quenched with 3 M HCl (550 ml) and extracted with CH2Cl2 (2 × 500 ml). The combined organic layer was washed with brine (1000 ml), dried with anhydrous Na2SO4, and concentrated under vacuum to furnish the product 10 (98 g, 0.25 mol) with 95% yield, which was pure enough for the next step. 1H NMR (500 MHz, CDCl3) δ 7.84 (s, 1H), 7.82 (d, J = 2.2 Hz, 1H), 7.67 (d, J = 8.6 Hz, 1H), 7.53 (s, 1H), 7.47 (s, 1H), 7.08 (dd, J = 8.6, 2.2 Hz, 1H), 5.03 (s, 2H), 4.10 (s, 3H), 4.02 (s, 3H), 1.06 (s, 9H), 0.30 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 154.5, 149.4, 148.9, 131.7, 131.4, 130.1, 125.9, 125.7, 124.9, 124.6, 120.4, 111.4, 104.8, 103.8, 64.8, 56.0, 25.9, 18.5, −4.1. IR (KBr) νmax = 3368, 2954, 2932, 2896, 2857, 1606, 1524, 1508, 1423, 1390, 1361, 1268, 1204, 1159, 1112, 1078, 1033, 982, 921, 893, 835, 780 cm−1. HRESIMS, m/z calculated for C23H30O4SiNa [M + Na]+: 421.1806. Found: 421.1823.
(S)-1-((3-Hydroxy-6,7-dimethoxyphenanthren-9-yl)methyl)-5-oxopyrrolidine-2-carboxylic acid (11)
To the stirring mixture of alcohol 10 (98.0 g, 0.246 mmol), NaI (8.14 g, 53.3 mmol), 1,4-dioxane (400 ml), and MeCN (600 ml) was added TMSCl (46.8 ml, 0.387 mmol) dropwise below 20 °C. After stirring 1 h at room temperature, anhydrous K2CO3 and dimethyl L-glutamate hydrochloride was added. When the reaction finished, it was filtered and acetic acid was added to the filtration. After completion of the reaction, the reaction was evaporated under vacuum to furnish the residue. The obtained residue was dissolved in methanol (600 ml) and dichloromethane (100 ml), and 4 M NaOH (240 ml) was added to the solution dropwise until the pH was stable around 11–12. After completion of the hydrolysis, most methanol was evaporated under reduced pressure. The obtained crude residue was mixed with 500 ml water and then extracted with dichloromethane (3 × 500 ml). The aqueous phase was carefully acidified with 4 M HCl to pH 2 at 0–5 °C and off-white solid precipitated out of the reaction mixture. Simple filtration and baking of the solid afforded 11 (53.5 g, 0.135 mol) with 55% yield. Mp 190–192 °C; 1H NMR (500 MHz, CD3OD) δ 7.92 (s, 1H), 7.83 (d, J = 2.1 Hz, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.48 (s, 1H), 7.44 (s, 1H), 7.08 (dd, J = 8.6, 2.1 Hz, 1H), 5.49 (d, J = 14.6 Hz, 1H), 4.30 (d, J = 14.6 Hz, 1H), 4.03 (s, 3H), 3.95 (s, 3H), 3.78 (dd, J = 9.4, 3.3 Hz, 1H), 2.62–2.55 (m, 1H), 2.46–2.40 (m, 1H), 2.19–2.13 (m, 1H), 2.04–1.98 (m, 1H). 13C NMR (125 MHz, CD3OD) δ 177.5, 175.0, 157.8, 150.9, 150.2, 133.2, 131.2, 129.0, 127.0, 126.5, 126.1, 125.9, 117.5, 106.7, 106.1, 105.3, 60.0, 56.5, 56.3, 45.6, 30.95, 23.8. IR (KBr) νmax = 3240, 2994, 2966, 2838, 1742, 1664, 1634, 1612, 1581, 1532, 1511, 1443, 1391, 1359, 1320, 1159, 1122, 1064, 1030, 984, 880, 859, 841, 796 cm−1. HRESIMS, m/z calculated for C22H20NO6 [M − H]−: 394.1296. Found: 394.1308.
(S)-3-Hydroxy-6,7-dimethoxy-13,13a-dihydrodibenzo[f,h]pyrrolo[1,2-b]isoquinoline-11,14(9H,12H)-dione (12)
To the solution of 11 (52.0 g, 0.132 mol) in trifluoroacetic anhydride (300 ml) was added boron fluoride ethyl ether (420 ml) and the reaction was stirred for 30 min at 45 °C. After completion of the reaction, it was poured to ice water (1500 ml) and stirred for 1 h with light green precipitating out. Simple filtration and baking of the solid afforded 12 (43.7 g, 0.116 mol) with 85% yield. Mp 300 °C (decomposed); 1H NMR (500 MHz, DMSO-d6) δ 9.92 (s, 1H), 9.13 (d, J = 9.2 Hz, 1H), 8.02 (d, J = 2.5 Hz, 1H), 8.00 (s, 1H), 7.48 (s, 1H), 7.16 (dd, J = 9.2, 2.5 Hz, 1H), 5.53 (d, J = 18.1 Hz, 1H), 4.80 (d, J = 18.1 Hz, 1H), 4.57 (t, J = 6.7 Hz, 1H), 4.07 (s, 3H), 4.03 (s, 3H), 2.48–2.29 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ 196.2, 173.1, 156.2, 151.5, 149.7, 137.4, 131.1, 128.6, 126.9, 122.8, 122.3, 120.8, 117.5, 106.5, 105.2, 104.0, 60.8, 55.84, 55.79, 29.5, 20.4. IR (KBr) νmax = 3167, 3000, 2925, 2835, 1708, 1663, 1613, 1519, 1457, 1401, 1353, 1245, 1207, 1178, 1117, 1029, 940, 924, 844, 806, 760 cm−1. HRESIMS, m/z calculated for C22H20NO5 [M + H]+: 378.1336. Found: 378.1335.
(S)-3-((tert-Butyldimethylsilyl)oxy)-6,7-dimethoxy-13,13a-dihydrodibenzo[f,h]pyrrolo[1,2-b]isoquinoline-11,14(9H,12H)-dione (13)
To the stirring solution of 12 (43.7 g, 0.116 mol) and N,N-diisopropylethylamine (45.0 g, 0.348 mol) in DMF (300 ml) was added TBSCl (34.9 g, 0.232 mol). After completion of the reaction, water (800 ml) was carefully added to the reaction dropwise. After it was stirred for additional 1 h at ice water bath, yellow solid precipitated. Simple filtration, washing by methanol and baking of the solid afforded 13 (48.2 g, 0.098 mol) with 85% yield. Mp 212–214 °C; 1H NMR (500 MHz, CDCl3) δ 9.27 (d, J = 9.2 Hz, 1H), 7.86 (d, J = 2.5 Hz, 1H), 7.85 (s, 1H), 7.29 (s, 1H), 7.20 (dd, J = 9.2, 2.5 Hz, 1H), 5.70 (d, J = 17.8 Hz, 1H), 4.65 (d, J = 17.8 Hz, 1H), 4.44–4.41 (m, 1H), 4.15 (s, 3H), 4.09 (s, 3H), 2.63–2.51 (m, 4H), 1.05 (s, 9H), 0.30 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 195.5, 174.2, 154.6, 151.7, 149.9, 137.4, 131.4, 129.1, 127.7, 123.1, 122.8, 122.6, 121.6, 111.4, 104.3, 103.6, 61.2, 56.3, 56.1, 40.7, 30.2, 25.9, 21.0, 18.5, −4.01, −4.02. IR (KBr) νmax = 3106, 2953, 2934, 2856, 1691, 1612, 1582, 1521, 1462, 1419, 1391, 1260, 1209, 1177, 1120, 1025, 953, 886, 825, 785 cm−1. HRESIMS, m/z calculated for C25H34NO9 [M + H]+: 492.2228. Found: 492.2241. HPLC (Daicel Chiralpak OD-H, 4.6 mm × 250 mml, 10 μm, hexane/iPrOH = 50:50, flow rate = 1 ml min−1, λ = 264, 355 nm): tR = 25.9 min; 99.5% ee.
(S)-3-Hydroxy-6,7-dimethoxy-12,13,13a,14-tetrahydrodibenzo[f,h]pyrrolo[1,2-b]isoquinolin-11(9H)-one (14)
To the cooled solution of 13 (48.2 g, 0.098 mol) in dichloromethane (200 ml) and methanol (200 ml) was slowly added NaBH4 (11.1 g, 0.294 mol). When the reaction finished, 1 M HCl was added to the reaction to make sure pH 7–8. After stirring for additional 1 h, the reaction mixture was extracted into CH2Cl2 (3 × 200 ml). The combined organic layer was concentrated under vacuum to furnish the intermediate. To a solution of the intermediate was added triethylsilane (120 ml) and boron fluoride ethyl ether (120 ml). After completion of the reaction, lots of off-white solid precipitated. Simple filtration, washing by dichloromethane and baking of the solid afforded 14 (28.3 g, 0.078 mol) with 80% yield. Mp 257–259 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.97 (d, J = 2.4 Hz, 1H), 7.95 (s, 1H), 7.88 (d, J = 9.0 Hz, 1H), 7.20 (s, 1H), 7.12 (dd, J = 9.0, 2.4 Hz, 1H), 5.09 (d, J = 17.2 Hz, 1H), 4.48 (d, J = 17.2 Hz, 1H), 4.00 (s, 3H), 3.97 (s, 3H), 3.94–3.85 (m, 1H), 3.45 (dd, J = 16.2, 4.1 Hz, 1H), 2.80 (dd, J = 16.2, 10.9 Hz, 1H), 2.48–2.35 (m, 3H), 1.99–1.85 (m, 1H). 13C NMR (125 MHz, DMSO-d6) δ 173.2, 155.8, 149.3, 148.4, 130.5, 125.7, 125.2, 124.2, 123.4, 122.7, 121.4, 116.4, 106.6, 104.1, 103.2, 55.53, 55.51, 52.4, 32.2, 29.4, 24.5. IR (KBr) νmax = 3446, 2946, 2835, 1660, 1618, 1519, 1470, 1441, 1358, 1272, 1202, 1169, 941, 864, 670 cm−1. HRESIMS, m/z calculated for C22H22NO4 [M + H]+: 364.1543. Found: 364.1543. HPLC (Daicel Chiralpak OD-H, 4.6 mm × 250 mml, 10 μm, hexane/iPrOH = 50:50, flow rate = 1 ml min−1, λ = 258, 286 nm): tR = 11.9 min; 99.5% ee.
(S)-6,7-Dimethoxy-9,11,12,13,13a,14-hexahydrodibenzo[f,h]pyrrolo[1,2-b]isoquinolin-3-ol (PF403)
To the cooled solution of 14 (28.3 g, 0.078 mol) in THF (200 ml) was added a solution of Red-Al (78 ml, 3.5 M in toluene) in toluene under argon, and the reaction was stirred at room temperature until it finished. Then the reaction was cooled to 0 °C and the solution of seignette salt (78 g in 500 ml water) was added along with stirring for additional 12–24 h. The precipitation was filtered and baked to afford PF403 (25.8 g, 0.074 mol) as white solid with 95% yield. Mp 176–178 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.72 (s, 1H), 7.95 (d, J = 2.2 Hz, 1H), 7.92 (s, 1H), 7.84 (d, J = 8.9 Hz, 1H), 7.18 (s, 1H), 7.10 (dd, J = 8.9, 2.2 Hz, 1H), 4.53 (d, J = 15.0 Hz, 1H), 3.99 (s, 3H), 3.93 (s, 3H), 3.64–3.57 (m, 1H), 3.50 (d, J = 15.0 Hz, 1H), 2.75 (dd, J = 15.6, 10.6 Hz, 1H), 2.36–2.31 (m, 2H), 2.22–2.09 (m, 1H), 1.91–1.79 (m, 2H), 1.78–1.73 (m, 1H), 1.68–1.56 (m, 1H). 13C NMR (125 MHz, DMSO-d6) δ 155.6, 149.1, 148.0, 130.3, 126.6, 125.0, 124.8, 124.5, 123.8, 122.6, 116.3, 106.4, 104.0, 103.4, 59.9, 55.5, 55.4, 54.6, 53.4, 33.1, 30.9, 21.3. IR (KBr) νmax = 3565, 3092, 2965, 2825, 1626, 1585, 1535, 1511, 1481, 1461, 1437, 1414, 1386, 1359, 1323, 1256, 1237, 1150, 1033, 1009, 920, 844, 788 cm−1. HRESIMS, m/z calculated for C22H24NO3 [M + H]+: 350.1751. Found: 350.1760. HPLC (Daicel Chiralpak AD-H, 4.0 mm × 10 mml, 5 μm, hexane (with 0.2% TEA)/iPrOH = 82:18, flow rate = 1 ml min−1, λ = 258, 287 nm): tR = 14.6 min; 99.6% ee.
(13aS)-3-Pivaloyloxyl-6,7-dimethoxyphenanthro[9,10-b]-indolizidine (CAT3)
To the cooled solution of PF403 (25.8 g, 0.077 mol), triethylamine (34.5 ml, 0.23 mol) and DMAP (0.94 g, 7.7 mmol) in dichloromethane (300 ml) was added trimethylacetic anhydride (28.1 g, 0.15 mol). After completion of the reaction, it was quenched by water. The reaction mixture was extracted into dichloromethane CH2Cl2 (3 × 200 ml). The combined organic layer was washed with brine (3 × 200 ml), dried with anhydrous Na2SO4, and concentrated under vacuum to furnish the residue, which was recrystallized by anhydrous ethanol (400 ml) to afford CAT3 (22.4 g, 0.052 mol) as off-white crystal with 70% yield. Mp 200.4–200.8 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.41 (s, 1H), 8.07 (s, 1H), 8.01 (d, J = 8.9 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.22 (s, 1H), 4.58 (d, J = 15.3 Hz, 1H), 4.02 (s, 3H), 3.94 (s, 3H), 3.53 (d, J = 15.3 Hz, 1H), 3.38–3.32 (m, 2H), 2.87–2.73 (m, 1H), 2.37–2.31 (m, 2H), 2.16–2.13 (m, 1H), 1.85–1.81 (m, 2H), 1.68–1.59 (m, 1H), 1.40 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 176.9, 149.5, 148.8, 148.6, 129.6, 128.0, 127.8, 126.4, 124.9, 124.8, 122.9, 120.5, 114.9, 104.6, 103.4, 59.8, 55.9, 55.5, 54.5, 53.4, 38.7, 33.1, 30.8, 27.0, 21.3. IR (KBr) νmax = 3097, 2965, 2830, 1740, 1619, 1530, 1510, 1468, 1427, 1415, 1367, 1339, 1256, 1205, 1164, 1135, 1034, 899, 841, 821, 786 cm−1. HRESIMS, m/z calculated for C27H32NO4 [M + H]+: 434.2331. Found: 434.2332. HPLC [Daicel Chiralpak OZ-H, 4.6 mm × 250 mml, 5 μm, hexane (with 0.2% TEA)/iPrOH = 90:10, flow rate = 1 ml min−1, λ = 259, 281 nm]: tR = 11.8 min; 99.6% ee.
Conclusions
In summary, a novel and facile synthesis for CAT3 was achieved in 11 steps with 10% overall yield after multiple attempts. This route represents a very economical and practical synthesis, in which all the reactions and post treatments are easy to handle. Importantly, pilot-scale preparation has been achieved following this route, and it would be suitable for large-scale preparation. Based on preliminary studies of different substitutions at 3-position of phenanthrene, the relationship between the electronic effect of the 3-position substitute group and the enantiomeric excess of chiral intermediates and the final product has been preliminarily elucidated. The introduction of electron-donating group significantly retains the configuration of phenanthroindolizidine as S at its 13a position. By virtue of the strong electron-donating TBS group, the excellent enantiomeric excess was achieved for CAT3 (≥99% ee) which is critical for its further preclinical investigations. This interesting finding not only represents a big breakthrough in the synthesis of CAT3, but also greatly inspires the construction and studies of chiral phenanthroindolizidine or other similar structures which is highly important for researchers in diverse fields, including natural product synthesis, organic chemistry, and medicinal chemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to the Department of Instrumental Analysis of our institute for the acquisition of UV, IR, NMR, and MS spectra. This work was supported by grants from National Mega-project for Innovative Drugs (No. 2014ZX09304306-001), from CAMS Innovation Fund for Medical Sciences (No. 2016-I2M-1-010) and from the Natural Science Foundation of China (No. 81602996).
References
- P. M. Zeltzer, J. M. Boyett, J. L. Finlay, A. L. Albright, L. B. Rorke, J. M. Milstein, K. R. Stevens, P. Stanley, H. Li, J. H. Wisoff, J. R. Geyer, P. McGuire-Cullen, J. A. Stehbens, S. B. Shurin and R. J. Packer, J. Clin. Oncol., 1999, 17, 832 CrossRef PubMed.
- J. M. Maris, M. D. Hogarty, R. Bagatell and S. L. Cohn, Lancet, 2007, 369, 2106 CrossRef.
- M. R. Gilbert, E. P. Sulman and M. P. Mehta, N. Engl. J. Med., 2014, 370, 2048 CrossRef PubMed.
- R. Bei, L. Marzocchella and M. Turriziani, Recent Pat. Anti-Cancer Drug Discovery, 2010, 5, 172 CrossRef PubMed.
- V. A. Trinh, S. P. Patel and W. J. Hwu, Expert Opin. Drug Saf., 2009, 8, 493 CrossRef PubMed.
- T. C. Johannessen and R. Bjerkvig, Expert Rev. Anticancer Ther., 2012, 12, 635 CrossRef PubMed.
- J. Zhang, M. F. Stevens and T. D. Bradshaw, Curr. Mol. Pharmacol., 2012, 5, 102 CrossRef PubMed.
- F. F. Yu, H. N. Lv, W. J. Dong, J. Ye, H. Z. Hao, S. G. Ma, S. S. Yu and Y. L. Liu, J. Pharm. Biomed. Anal., 2015, 107, 223 CrossRef PubMed.
- Z. J. Liu, H. N. Lv, H. Y. Li, Y. Zhang, H. J. Zhang, F. Q. Su, S. Xu, Y. Li, Y. K. Si, S. S. Yu and X. G. Chen, Chemotherapy, 2011, 57, 310 CrossRef PubMed.
- J. Chen, H. N. Lv, J. P. Hu, M. Ji, N. N. Xue, C. Li, S. G. Ma, Q. Zhou, B. Lin, Y. Li, S. S. Yu and X. G. Chen, Cancer Lett., 2016, 381, 391 CrossRef PubMed.
- C. Li, Y. Li, H. Lv, S. W. Li, K. Tang, W. Q. Zhou, S. S. Yu and X. G. Chen, Internet J. Oncol., 2015, 47, 179 CrossRef PubMed.
- M. Ji, L. Y. Wang, J. Chen, N. N. Xue, C. Y. Wang, F. F. Lai, R. B. Wang, S. S. Yu, J. Jin and X. G. Chen, OncoTargets Ther., 2018, 11, 3671 CrossRef PubMed.
- H. N. Lv, J. H. Ren, S. G. Ma, S. Xu, J. Qu, Z. J. Liu, Q. Zhou, X. G. Chen and S. S. Yu, PLoS One, 2012, 7, e30342 CrossRef PubMed.
- H. N. Lv, S. Xu, S. G. Ma, Y. B. Liu, Y. Li, J. Qu, Z. J. Liu and S. S. Yu, Synthesis, 2015, 47, 1399 CrossRef.
- P. F. Yu, H. N. Lv, C. Li, J. H. Ren, S. G. Ma, S. Xu, X. G. Chen and S. S. Yu, Synthesis, 2012, 44, 3757 CrossRef.
- I. Takashi, Y. Takashi, M. Takeshi, Y. Ryuta, H. Syusuke and S. Seigo, Bioorg. Med. Chem. Lett., 2011, 21, 5978 CrossRef PubMed.
- I. Takashi, S. Seigo, Y. Takashi, M. Takeshi, H. Shusuke and Y. Ryuta, PCT Int. Appl., 2010, 047127 Search PubMed.
- L. B. Liu, B. W. Yang, T. J. Katz and M. K. Poindexter, J. Org. Chem., 1991, 56, 3769 CrossRef.
- R. B. Wang, S. S. Zhou, H. D. G. L. Jiang, X. G. Zheng, W. Zhou and S. S. Li, Eur. J. Org. Chem., 2012, 2012, 1373 CrossRef.
- P. J. Kropp, G. W. Breton, S. L. Craig, S. D. Crawford, W. F. Durland, J. E. Jones and J. S. Raleigh, J. Org. Chem., 1995, 60, 4146 CrossRef.
- S. Chandrasekhar, P. K. Mohanty, K. Harikishan and P. K. Sasmal, Org. Lett., 1999, 1, 877 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04511a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Hai-Ning Lv,
Shan-Shan Zhu,
Xiao-Dong Ren
,
Hai-Ning Lv,
Shan-Shan Zhu,
Xiao-Dong Ren