DOI:
10.1039/C8RA05885G
(Paper)
RSC Adv., 2018,
8, 31037-31047
Development of an Fe3O4@Cu silicate based sensing platform for the electrochemical sensing of dopamine†
Received
10th July 2018
, Accepted 13th August 2018
First published on 4th September 2018
Abstract
Abnormal levels of dopamine (DA) in body fluids is an indication of serious health issues, hence development of highly sensitive platforms for the precise detection of DA is highly essential. Herein, we demonstrate an Fe3O4@Cu silicate based electrochemical sensing platform for the detection of DA. Morphology and BET analysis shows the formation of ∼320 nm sized sea urchin-like Fe3O4@Cu silicate core–shell nanostructures with a 174.5 m2 g−1 surface area. Compared to Fe3O4 and Fe3O4@SiO2, the Fe3O4@Cu silicate urchins delivered enhanced performance towards the electrochemical sensing of DA in neutral pH. The Fe3O4@Cu silicate sensor has a 1.37 μA μM−1 cm−2 sensitivity, 100–700 μM linear range and 3.2 μM limit of detection (LOD). In addition, the proposed Fe3O4@Cu silicate DA sensor also has good stability, selectivity, reproducibility and repeatability. The presence of Cu in Fe3O4@Cu silicate and the negatively charged surface of the Cu silicate shell play a vital role in achieving high selectivity and sensitivity during DA sensing. The current investigation not only represents the development of a highly selective DA sensor but also directs towards the possibility for the fabrication of other Cu silicate based core–shell nanostructures for the precise detection of DA.
1 Introduction
The development of various sensors plays a vital role in our day to day life.1–16 Among them, the development of dopamine (DA) sensors has drawn much attention, since DA, a vital neurotransmitter existing in the central nervous system of mammals, plays a significant role in physiological process regulation.17 Typically, either a deficiency or large excess of DA in human serum is associated with several diseases,18–20 which urges the development of highly sensitive methods for the detection of DA. So far, a variety of methods, including chromatography,21 fluorometry,22 chemiluminescence analysis,23 colorimetry,24 and electrochemical assays25 etc. have been used for the detection of DA. Among them, the electrochemical method offers many advantages such as high sensitivity, fast response, cheap instruments, simple operation, and so on.26 However, DA sensing over conventional electrodes suffers from poor performance in terms of response or sensitivity. Additionally, other biological compounds i.e. uric acid (UA) and ascorbic acid (AA) also undergo oxidation at nearly the same potential thereby interfering with the DA signal leading to poor selectivity because of the fouling of the electrode surface by the DA oxidation product.27 Hence, enhancement in both sensitivity and selectivity of the DA sensor is vital for precise detection of DA.
In this regard, several modified electrodes have been developed and used in the detection of DA.28 For example, carbon nanotubes,29 graphene,30 CNT-graphene,31 graphene-conducting polymer,32 N-doped graphene,33 gold nanoparticles,34 metal oxides,35 CNT-metal nanoparticles,36 CNT-metal oxide,37 graphene metal nanoparticles,38 graphene metal oxide nanocomposite,39 metal organic framework,40 metal phthalocyanine,41 and g-C3N4-metal oxide nanocomposites42 based electrochemical interface etc. have been used in the sensitive detection of DA. Besides the aforementioned electrocatalysts, in the recent past, Fe2O3 and Fe3O4 nanostructures have proven their potential in the area of electrochemical sensing of DA43,44 owing to their nontoxicity, biological and chemical inertness.45,46 For example, Tai et al. reported the synthesis of porous α-Fe2O3 nanoparticles on carbon cloth and their application in the sensitive detection of DA.43 In another study, Fang et al. demonstrated the Fe3O4 nanoparticle based electrochemical sensing platform for the voltammetric sensing of dopamine.44 Conversely, these materials have poor conductivity and undergo aggregation leading to a poor electrochemical sensing performances. Hence, either functionalization of their surface or synthesis of Fe2O3/Fe3O4 based nanocomposites could be an effective approach to achieve high DA sensing performance.
Therefore, synthesis of Fe based nanocomposites have gained worldwide attention and in the recent past, several Fe nanocomposites based sensing platforms have been developed for the high sensitive detection of DA.47–59 For example, Zhang et al. demonstrated that Fe2O3-graphene-polyimide aerogel nanocomposites could be used in the detection of DA.47 In another study, Adekunle et al. synthesized SWCNT-Fe2O3 nanocomposites and used them in the detection of DA.48 Salamon et al. developed Fe3O4 nanorod-graphene nanocomposites for the high sensitive electrochemical sensing of DA.49 Recently, Fayemi et al. synthesized polyaniline/NiO, ZnO, and Fe3O4 nanocomposites, compared to polyaniline-NiO and polyaniline-ZnO nanocomposites, the polyaniline-Fe3O4 nanocomposite delivered much enhanced activity towards DA sensing.50 Rani et al. demonstrated that RGO-Fe3O4 flower nanocomposite is a potential material for the highly sensitive and selective electrochemical detection of DA.51 Sulaiman et al., Bagheri et al. and Teymourian et al. have synthesized Fe3O4-RGO nanocomposites and used these nanocomposites in the electrochemical sensing of DA.52–54 In another study, Wu et al. demonstrated the synthesis of mesoporous Fe3O4 decorated over the graphene sheets which delivered much better DA sensing performance.55 In addition, Fe3O4@C/GNS nanocomposites, Fe3O4–SnO2-Gr ternary nanocomposite, Fe3O4 nanoparticles N-doped carbon nanotube nanocomposite and Pd–Fe3O4 nanocomposites etc. have also shown their potentials towards the selective and sensitive determination of DA.56–59 In spite of these reports on the synthesis of iron oxide based nanocomposites and their application in the high sensitive detection of DA, still there are plenty of opportunities are existing into which no researcher in this field have paid attention and needs to be explored.
Considering this fact, in this paper, we report Fe3O4@Cu silicate based highly sensitive platform for the electrochemical detection of DA. The Fe3O4@Cu silicate based sensor displayed better performance than virgin Fe3O4 and Fe3O4@SiO2. The Fe3O4@Cu silicate based sensor showed a linier response for DA from 100–700 μM, sensitivity of 1.37 μA μM−1 cm−2 and 3.2 μM LOD. The current Fe3O4@Cu silicate based sensor showed high selectivity towards DA in the coexistence of AA and UA glucose. To the best of our knowledge, this is the first report on the electrochemical sensing of DA using sea urchin like Fe3O4@Cu silicate nanostructure.
2 Experimental section
2.1 Materials and methods
All chemical FeCl3·6H2O, Cu(NO3)2·3H2O sodium acetate(NaAc), polyvinylpyrrolidone (PVP), urea, tetraethyl orthosilicate (TEOS), dopamine, uric acid and ascorbic acid were purchased from sigma Aldrich and used as received. Ethanol, ethylene glycol (EG), ammonia solution, H3PO4, NaH2PO4 and Na2HPO4 were purchased from samchun chemicals Korea. All solutions were prepared using Millipore water (Milli-Q system).
2.2 Synthesis of Fe3O4 nanospheres, Fe3O4@SiO2 core–shell nanospheres and sea urchin like Fe3O4@Cu silicate core–shell nanocomposites
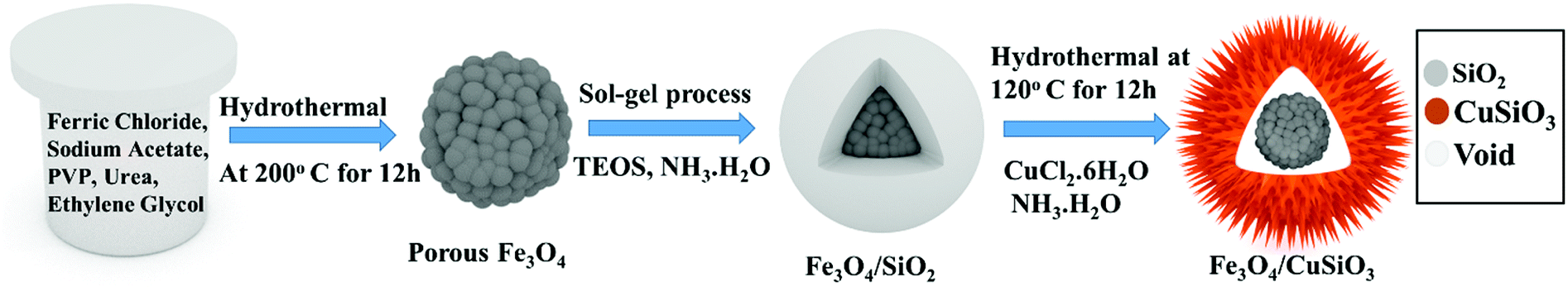
Synthesis of Fe3O4 nanospheres, Fe3O4@SiO2 core–shell nanospheres and sea urchin like porous Fe3O4@Cu silicate core–shell nanocomposite is schematically displayed in Scheme 1. The Fe3O4 nanospheres were first prepared by a hydrothermal method as described previously.60 Briefly, 5 mM of FeCl3·6H2O, 24.4 mM of NaAc were first dissolved in EG (30 mL) and stirred well. Afterward, 1 g of PVP, and 1 g of urea were added and then the mixture was stirred to ensure the mixing of all reagents. The mixed solution was transferred into a Teflon-lined stainless-steel autoclave with a capacity of 45 mL. The autoclave was heated at 200 °C for 12 h. Then, the black colored product was washed with water and ethanol by a magnetic decantation for three times, followed by vacuum dry at 60 °C for 12 h to achieve Fe3O4 nanospheres. Using this Fe3O4 nanospheres, Fe3O4@SiO2 nanospheres were synthesized by a modified Stober method. Initially, as-prepared Fe3O4 nanospheres (0.05 g) were dispersed in the mixed solvent (ethanol/DI water (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2)) and then 2 mL of ammonia solution was added. To this dispersion, 0.6 mL of TEOS was added drop wise under stirring and after TEOS addition the stirring was continued for additional 10 h. The Fe3O4@SiO2 nanospheres were collected from the solution and washed three times with ethanol via magnetic decantation and then dried in the vacuum oven. These Fe3O4@SiO2 nanospheres were used as substrate for the hydrothermal growth of Fe3O4@Cu silicate sea urchins like core–shell nanocomposite. Typically, 0.03 g of Fe3O4@SiO2 nanosphere was dispersed in 30 mL of DI water by ultrasonication for 20 min. To this dispersion, 1.5 mL of ammonia solution and 1.5 mL of 0.1 M Cu(NO3)2 solution were then added under stirring. This reaction mixture was then transferred into an autoclave and hydrothermally treated for 12 h at 120 °C. The product was washed through magnetic decantation with ethanol and DI water for three times and then dried in the vacuum oven at 60 °C for 12 h to achieve Fe3O4@Cu silicate sea urchins.
:2)) and then 2 mL of ammonia solution was added. To this dispersion, 0.6 mL of TEOS was added drop wise under stirring and after TEOS addition the stirring was continued for additional 10 h. The Fe3O4@SiO2 nanospheres were collected from the solution and washed three times with ethanol via magnetic decantation and then dried in the vacuum oven. These Fe3O4@SiO2 nanospheres were used as substrate for the hydrothermal growth of Fe3O4@Cu silicate sea urchins like core–shell nanocomposite. Typically, 0.03 g of Fe3O4@SiO2 nanosphere was dispersed in 30 mL of DI water by ultrasonication for 20 min. To this dispersion, 1.5 mL of ammonia solution and 1.5 mL of 0.1 M Cu(NO3)2 solution were then added under stirring. This reaction mixture was then transferred into an autoclave and hydrothermally treated for 12 h at 120 °C. The product was washed through magnetic decantation with ethanol and DI water for three times and then dried in the vacuum oven at 60 °C for 12 h to achieve Fe3O4@Cu silicate sea urchins.
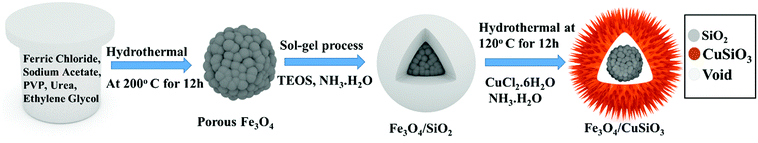 |
| | Scheme 1 Schematic illustration of the synthesis of Fe3O4@Cu silicate core–shell urchins. | |
2.3 Characterization
The phase and crystal structure was investigated by X-ray diffraction (XRD; Bruker AXS D8) analysis. The morphology, size and composition was investigated by field-emission scanning electron microscopic (FE-SEM, S-4800 Hitachi), transmission electron microscopic (TEM; Tecnai G2 F30, FEI) and energy dispersive X-ray spectroscopic (EDS, FE–SEM) measurements. The sample for TEM analysis was prepared by dispersing the samples (0.1 mg mL−1) in ethanol with the help of ultrasonication and from this dispersion 5 μL was drop-casted on the TEM grid. Brunauer–Emmett–Teller (BET) surface area analysis was performed using Micromeritics Tristar 3000 analyzer. For electrochemical sensing application, a Zive-SP1 electrochemical workstation (Wonatech, Korea) was used and the study was performed in a three-electrode mode, where carbon paper (CP) containing active materials, Pt wire, and Ag/AgCl (3 M NaCl) were used as the working, counter, and reference electrodes, respectively. Electrochemical dopamine sensing was carried out in 0.1 M PBS supporting electrolyte. For real sample analysis, human urine samples were collected from three healthy persons. Before sample collection, we had informed all human subjects and they had given their consent to perform the experiments using the sample collected from them.
3 Results and discussion
The phase and crystal structure of the synthesized Fe3O4, Fe3O4@SiO2 and Fe3O4@Cu silicate core–shell urchins were examined by XRD analysis and is represented in Fig. 1. All the diffraction peaks of Fe3O4 are well matched with the cubic spinel structure phase of magnetite (JCPDS card no. 79-0419).61 Identical diffraction peaks without any other characteristic peaks similar to the Fe3O4 were observed in the case of Fe3O4@SiO2 spectrum, indicating that amorphous SiO2 layer has been coated on the porous Fe3O4 surface. However, the porous core–shell Fe3O4@Cu silicate showed the presence of new characteristic peaks (marked with •) along with the peaks corresponding to the cubic spinel Fe3O4 (marked with *). These new characteristic peaks (marked with •) in the core–shell Fe3O4@Cu silicate well matched with the tetragonal rutile phase of CuSiO3 (JCPDS card no. 32-346).62 The XRD result reveals the presence of Fe3O4 and CuSiO4 in Fe3O4@Cu silicate core–shell urchins.
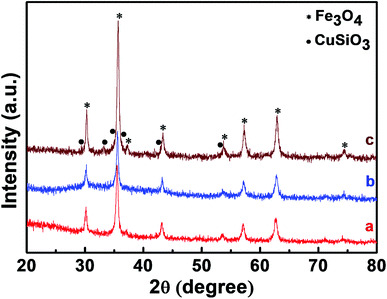 |
| | Fig. 1 XRD patterns of Fe3O4 nanospheres (a), Fe3O4@SiO2 nanospheres (b), and Fe3O4@Cu silicate core–shell urchins (c). | |
The surface area and porosity of Fe3O4, Fe3O4@SiO2 and Fe3O4@Cu silicate core–shell urchins were further investigated by Brunauer–Emmett–Teller (BET) and N2 adsorption–desorption measurements and is represented in Fig. 2. All the samples have type IV isotherm and hysteresis loop in the P/P0 range of 0–1.0 (Fig. 2(a)), indicating that the samples are mesoporous. The BET surface area of Fe3O4, Fe3O4@SiO2 and Fe3O4@Cu silicate core–shell urchins were estimated to be 33.78, 6.17, and 174.5 m2 g−1, respectively. Barrett–Joyner–Halenda (BJH) pore size distribution (Fig. 2(b)) profile shows that the average pore diameter and pore volume are 15.19, 1.5 and 7.76 nm and 0.094, 0.002, and 0.33 cm3 g−1 for Fe3O4, Fe3O4@SiO2 and Fe3O4@Cu silicate core–shell urchins, respectively. These results indicate that the needle-like structure of Cu silicate shell account for the increase in surface area and porosity of Fe3O4@Cu silicate core–shell urchins.
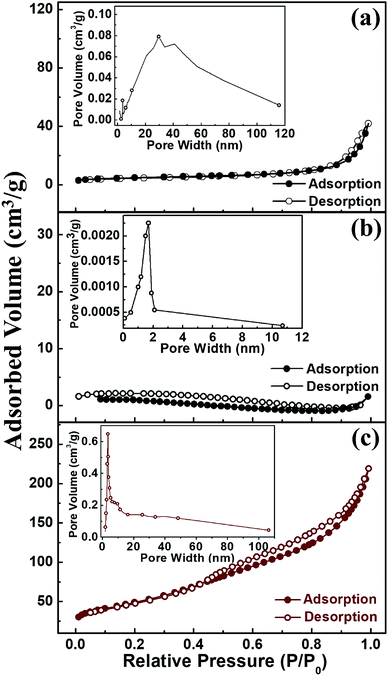 |
| | Fig. 2 N2 adsorption–desorption isotherms of Fe3O4 (a), Fe3O4/SiO2 (b), and Fe3O4@Cu silicate core–shell urchins (c). Inset shows the corresponding pore width distribution. | |
Fig. 3 represents the FE-SEM images of Fe3O4, Fe3O4@SiO2 and Fe3O4@Cu silicate core–shell urchins. Fe3O4 has porous sphere like morphology [Fig. 3(a)] with a mean diameter of ∼280 nm made up of many quasi-spherical nanoparticles. The Fe3O4@SiO2 nanocomposite shows the formation of core–shell sphere like morphology [Fig. 3(b)] with a smooth surface than Fe3O4 nanosphere, due to uniformly coated SiO2 layer. The FE-SEM image of Fe3O4@Cu silicate core–shell urchins shows the formation of core–shell nanostructure with sea urchin like morphology [Fig. 3(c)]. High magnified FE-SEM image [Fig. 3(d)] exhibits that the outer shell of Fe3O4@Cu silicate core–shell urchins has needle-like structures. The SiO2 shell layer in the Fe3O4@SiO2 acted as a sacrificial template for the hydrothermal growth of Cu silicate needle-like structures. The elemental mapping and energy dispersive X-ray spectroscopic (EDS) investigations (Fig. 4) were performed for Fe3O4@Cu silicate core–shell urchins to examine the composition. The elemental mapping study shows that the core–shell Fe3O4@Cu silicate core–shell urchins is made up of Fe, Cu, Si and O elements [Fig. 4(c, d, e and f)]. In the EDS spectrum [Fig. 4(b)], except from the Fe, Cu, O and Si element peaks, two additional peaks for Pt and C were also observed. The peaks for C and Pt were observed due to the use of carbon tape for sample pasting and Pt sputtering before FE-SEM analysis. These results suggests that sea urchin like Fe3O4@Cu silicate core–shell urchins is highly pure and made up of Fe, Cu, Si and O elements. Additionally, the TEM study was performed (Fig. 5) to investigate the morphology of Fe3O4, Fe3O4@SiO2 and Fe3O4@Cu silicate core–shell urchins. In the case of virgin Fe3O4, ∼280 nm sized porous sphere like morphology [Fig. 5(a)] composed of quasi-spherical nanoparticles was observed. The Fe3O4@SiO2 core–shell urchins has core–shell nanosphere morphology [Fig. 5(b)], with Fe3O4 core and SiO2 shell. In contrast, the TEM [Fig. 5(c)] and high magnified TEM image [Fig. 5(d)] of Fe3O4@Cu silicate depicts the growth of ∼320 nm sized sea urchin like core–shell morphology containing many Cu silicate spikes in the shell and with uniform void space in between the core and shell. High resolution TEM image [Fig. 5(e)] demonstrates that the Cu silicate shell thickness in Fe3O4@Cu silicate urchin like core–shell nanostructure is ∼30 nm. The diffraction rings in the selected area electron diffraction (SAED) pattern [Fig. 5(f)] demonstrates the polycrystalline nature of Fe3O4@Cu silicate urchins.
 |
| | Fig. 3 SEM images of Fe3O4 nanospheres (a), Fe3O4@SiO2 nanospheres (b), and Fe3O4@Cu silicate core–shell urchins (c and d). | |
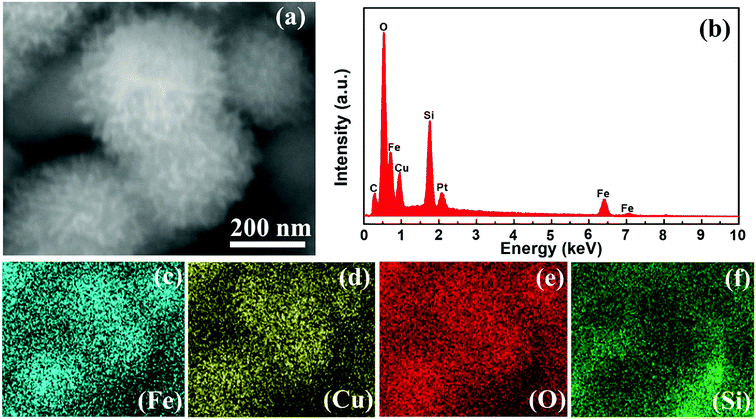 |
| | Fig. 4 SEM image (a), EDS profile of Fe3O4@Cu silicate core–shell urchins (b) and corresponding elemental mapping of Fe, Cu, O and Si (c–f). | |
 |
| | Fig. 5 TEM images of Fe3O4 nanospheres (a), Fe3O4@SiO2 nanospheres (b), and Fe3O4@Cu silicate core–shell urchins (c and d). HRTEM image (e) and SAED pattern taken from the shell of the Fe3O4@Cu silicate core–shell structure (f). | |
The redox probe Fe(CN)64− gives an overall idea about the characteristic of electrode surface and the electron transfer behavior at the electrode interface. Hence, the electrochemical activity of the CP, CP-Fe3O4, CP-Fe3O4@SiO2 and CP-Fe3O4@Cu silicate electrodes were first investigated by CV and electrochemical impedance spectroscopic (EIS) measurements in 0.1 M KCl containing 1 mM Fe(CN)64− redox probe and the response is shown in Fig. 6. The voltammetric profiles [Fig. 6(A)] of the aforementioned electrodes at 50 mV s−1 scan rate in 0.1 M KCl containing 1 mM Fe(CN)64− redox probe. The CP electrode shows an irreversible broad peak for the redox probe. On the other hand, the CP-Fe3O4 electrode shows a well-defined redox peak having a peak-to-peak separation (ΔEp) of ∼140 mV with a much higher anodic peak current (Ipa) than the CP electrode. In the case of CP-Fe3O4@SiO2 electrode an irreversible broad peak with a lower Ipa than CP-Fe3O4 electrode was observed. The achievement in an irreversible broad peak with low Ipa value could be attributed to the electrostatic repulsion between negatively charged Fe(CN)64− redox probe63 and negatively charged Fe3O4@SiO2 (ref. 64) there by hindering the diffusion of Fe(CN)64− redox probe to the electrode surface. It has been well established that, coating of silica layer over the Fe3O4 surface leads to the formation of Fe3O4@SiO2 nanocomposite with a negatively charged surface.64 The CP-Fe3O4@Cu silicate electrode has lowest ΔEp value of ∼85 mV with a higher Ipa value than the CP-Fe3O4@SiO2 electrode but it has lower Ipa value than CP-Fe3O4 electrode. Comparison of the Ipa and ΔEp value obtained on the CP, CP-Fe3O4, CP-Fe3O4@SiO2 and CP-Fe3O4@Cu silicate electrodes demonstrate that, CP-Fe3O4 and CP-Fe3O4@Cu silicate electrode have highest Ipa and lowest ΔEp value for Fe(CN)64− redox probe, respectively. Due to the highest BET surface area of Fe3O4@Cu silicate, a superior performance in terms of both highest Ipa and lowest ΔEp value was expected because it could provide a large number of active sites to interact with the redox probe. However, the discrepancy in the electrochemical behavior of CP-Fe3O4@Cu silicate electrode towards the redox probe i.e. achievement in low Ipa value than CP-Fe3O4 electrode could be attributed to the occurrence of same repulsion phenomenon as in the case of CP-Fe3O4@SiO2 electrode. Now one question arises, if both Fe3O4@SiO2 and Fe3O4@Cu silicate electrodes are negatively charged, then, in principle, both the electrodes should have shown similar electrochemical responses towards the negatively charged redox probe Fe(CN)64−. Interestingly, the achievement in a higher Ipa and lower ΔEp value on the CP-Fe3O4@Cu silicate electrode than the CP-Fe3O4@SiO2 electrode could be ascribed to the significant role of both Cu and highest surface area of Fe3O4@Cu silicate urchins in the electrochemical reaction. Additionally, these CP, CP-Fe3O4, CP-Fe3O4@SiO2 and CP-Fe3O4@Cu silicate electrodes were further characterized by EIS study and their responses are shown in Fig. 6(B). Examination of the Nyquist plot shows that CP electrode has largest semicircle suggesting high charge transfer resistance. Compared to the CP electrode, the diameter of the semicircle is smaller in the case of CP-Fe3O4 electrode suggesting that the presence of Fe3O4 facilitates the rate of electron transfer. However, in the case of CP-Fe3O4@SiO2 and CP-Fe3O4@Cu silicate electrode the diameter of the semicircle is almost similar and larger than the CP-Fe3O4 electrode. This result signifies the occurrence of same repulsion phenomena between the negatively charged CP-Fe3O4@SiO2 and Fe3O4@Cu silicate surface with the negatively charged redox probe as observed during the CV study. The BET surface area, CV and EIS results indicate that the Fe3O4@Cu silicate has highest surface area and is negatively charged, hence, it could interact well with the positively charged DA molecule than the CP, CP-Fe3O4, CP-Fe3O4@SiO2 electrodes resulting in a much enhanced electrochemical performance towards the sensing of DA.
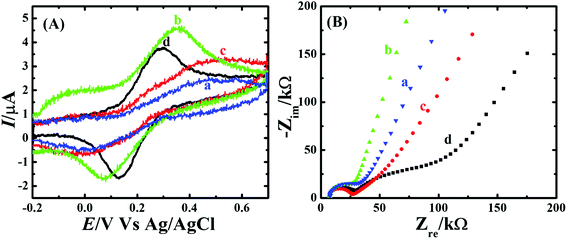 |
| | Fig. 6 (A) Cyclic voltammograms of (a) CP, (b) CP-Fe3O4, (c) CP-Fe3O4@SiO2 and (d) CP-Fe3O4@Cu silicate core–shell urchins in 0.1 M KCl containing 1 mM Fe(CN)64− at 50 mV s−1 scan rate and (B) Nyquist plot. | |
The performance of the CP, CP-Fe3O4, CP-Fe3O4@SiO2 and CP-Fe3O4@Cu silicate electrodes towards the DA sensing was first evaluated by CV study. Fig. 7 represents the CV responses of CP, CP-Fe3O4, CP-Fe3O4@SiO2 and CP-Fe3O4@Cu silicate electrodes in 0.1 M PBS containing 1.8 mM DA at 50 mV s−1 scan rate in the potential range from −0.2 and +0.7 V. As shown in Fig. 7(a), CP electrode shows a broad peak for the oxidation of DA. On the other hand, CP-Fe3O4, CP-Fe3O4@SiO2 and CP-Fe3O4@Cu silicate electrodes exhibit well-defined irreversible redox peaks [Fig. 7(a–c)] corresponding to the oxidation of DA to dopaminequinone (prominent peak) in forward scan and the reduction of electrogenerated dopaminequinone to DA (small hump) in reverse scan.65 On the CP-Fe3O4 electrode, the DA oxidation was noticed at 0.38 V and reduction of electrogenerated dopaminequinone was noticed at 0.05 V. The CP-Fe3O4@SiO2 electrode catalyzed DA electrooxidation with a higher catalytic current at 0.34 V, which is 40 mV less than that observed on the CP-Fe3O4 electrode. The achievement in higher catalytic current and 40 mV less oxidation potential on CP-Fe3O4@SiO2 electrode than CP-Fe3O4 electrode could be ascribed to the negatively charged surface of Fe3O4@SiO2 nanostructures. As a result, a high electrostatic interaction between negatively charged Fe3O4@SiO2 and positively charged DA molecule is anticipated facilitating the diffusion of positively charged DA molecule to the electrode surface, resulting in higher catalytic current than the CP-Fe3O4 electrode. In the previous section, with the help of negatively charged Fe(CN)64− redox probe, we have also probed that the surface of Fe3O4@SiO2 and Fe3O4@Cu silicate are negatively charged and is in well accordance with the previously reported literature.64 As mentioned in the previous section, the catalytic activity of the Fe3O4@SiO2 and Fe3O4@Cu silicate should have been almost similar, since both are negatively charged and DA is positively charged. Interestingly, on the CP-Fe3O4@Cu silicate electrode, DA oxidation occurred at 0.27 V, which is 80 mV lesser than the Fe3O4@SiO2 and the catalytic current was also much higher than the CP-Fe3O4@SiO2 electrode. The comparison of the voltammetric result obtained on those four electrodes clearly reveals that, compared to the CP, CP-Fe3O4 and CP-Fe3O4@SiO2 electrodes, the oxidation of DA occurred at a lowest potential with a highest peak current on the CP-Fe3O4@Cu silicate electrode, suggesting the excellent electrocatalytic activity of Fe3O4@Cu silicate towards DA oxidation. The achievement in excellent performance on CP-Fe3O4@Cu silicate electrode over CP, CP-Fe3O4 and CP-Fe3O4@SiO2 electrode is believed to be originated from the combined effect of urchin like morphology, negatively charged surface of the nanostructure, highest surface area and significant role of Cu during the electrochemical reaction with positively charged DA, thereby facilitating the rate of electron transfer66 resulting in a much better response towards the DA sensing than other electrodes.
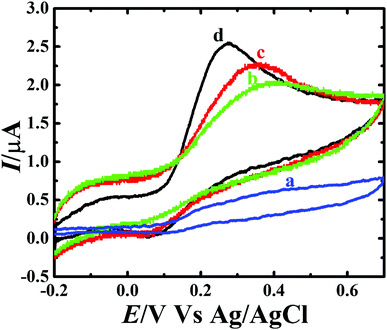 |
| | Fig. 7 Cyclic voltammograms of (a) CP, (b) CP-Fe3O4, (c) CP-Fe3O4@SiO2 and (d) CP-Fe3O4@Cu silicate core–shell urchins electrodes in 0.1 M PBS containing 1.8 mM DA at 50 mV s−1 scan rate. | |
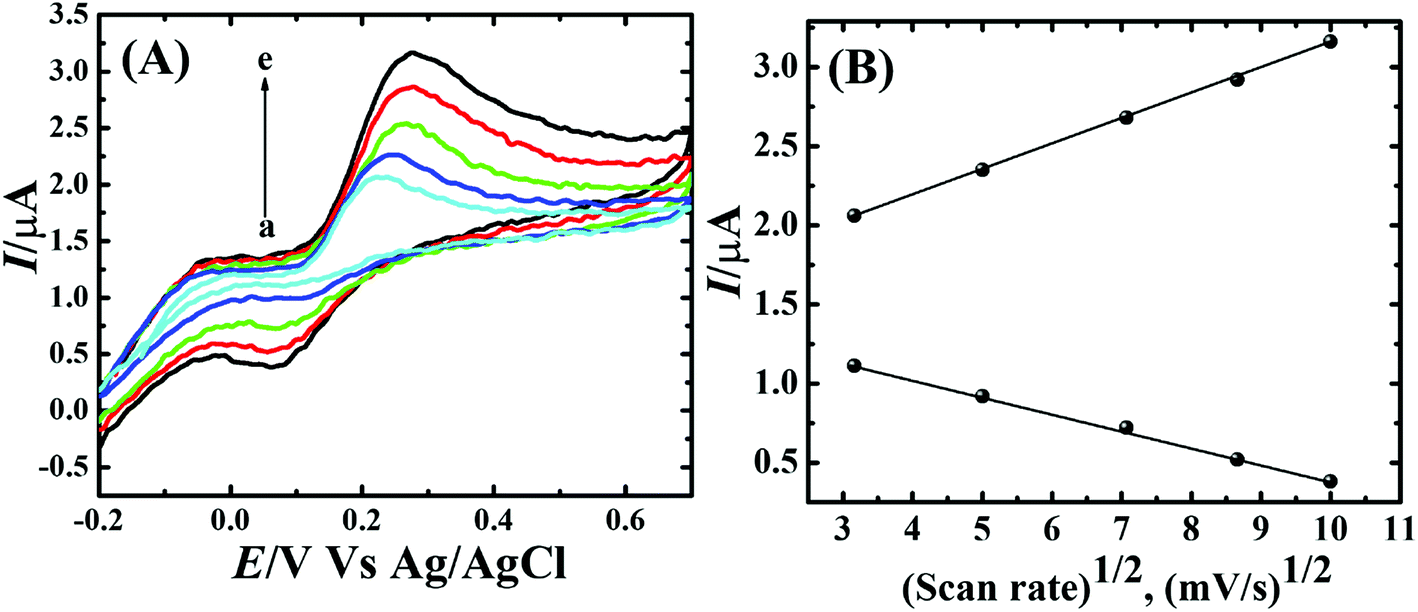
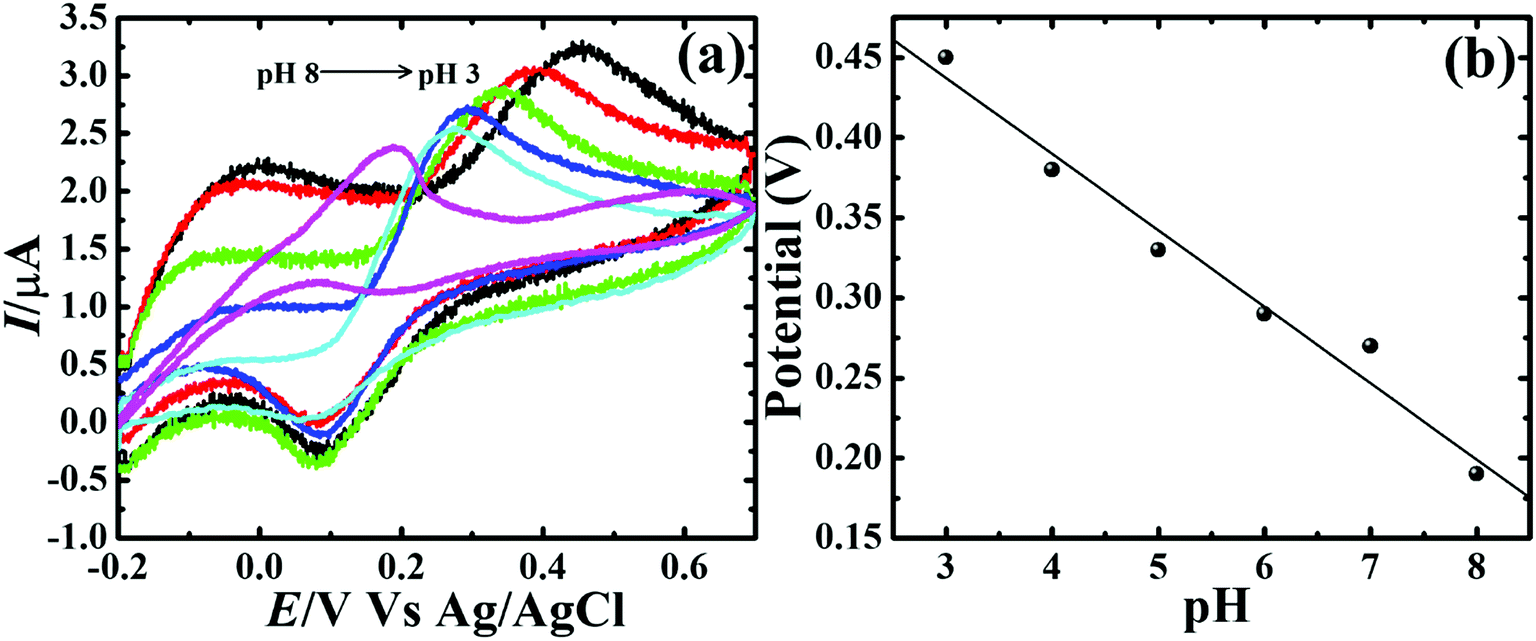
To assess the kinetics of DA electrooxidation, the effect of scan rate on the DA oxidation over CP-Fe3O4@Cu silicate electrode was investigated and the response is shown in Fig. 8. As revealed in Fig. 8(a), upon enhancing the scan rate from 10 to 100 mV a respective positive and negative shift in both oxidation and reduction peak potentials as well as the enhancement in anodic and cathodic peak currents were noticed. The plot of anodic and cathodic peak currents vs. square root of scan rate [Fig. 8(b)] shows a linear relationship with the correlation coefficient of 0.9883 and 0.9947, respectively, signifying that the DA oxidation is governed by diffusion controlled process.67 Further, the voltammetric profiles for the gradual addition of different concentration of DA were recorded at a fixed scan rate of 50 mV s−1 and the response is displayed in Fig. 9. The increase in the DA concentration from 0.2 to 1.8 mM in the 0.1 M PBS supporting electrolyte results in an enhancement in both anodic and cathodic peak currents [Fig. 9(a)]. The CV result of DA gradual addition indicates the excellent electro-oxidation behavior of DA at CP-Fe3O4@Cu silicate electrode. The calibration plot made from the anodic peak currents and DA concentrations shows a linear relationship [Fig. 9(b)] with a correlation coefficient (R2) of 0.9936. The good linear relationship between anodic peak currents and DA concentrations suggests that precise DA sensing could be achieved by CP-Fe3O4@Cu silicate electrode. In addition, the effect of electrolyte pH values on the electrochemical responses of CP-Fe3O4@Cu silicate electrode towards DA oxidation were also investigated at 50 mV s−1 scan rate in 0.1 M PBS of different pH values containing 1.8 mM DA and the response is shown in Fig. 10. The anodic peak current diminished and the oxidation potential shifted towards negative side as the electrolyte pH value was increased from 3 to 8 [Fig. 10(a)]. Though a highest anodic peak current and lowest oxidation potential was noticed in pH 3 and 8, respectively, yet, the electrolyte pH 7 was selected for rest of the investigations taking account the pH of human fluid is 7. Fig. 10(b), shows a linear relationship with a slope value of 56 mV pH−1 comparable with the Nernst equation theoretical slope value (59 mV pH−1) suggesting the involvement of equal number of protons and electrons during the DA oxidation.68 The analytical performance of Fe3O4@Cu silicate based DA sensor was evaluated by amperometric method as it is a suitable analytical method for the low concentration detection of analytes.37 Fig. S1(a)† represents the amperometric response of DA on Fe3O4@Cu silicate based sensor. As can be seen in Fig. S1(a),† the sequential addition of 100 μM DA to 0.1 M PBS electrolyte at a regular time interval resulted in the significant enhancement in the response current. The calibration plot (inset in Fig. S1(a)†) for current response vs. DA concentration clearly shows a linear relationship in the concentration range from 100–700 μM, while for a high concentration (800–1300 μM) of DA the calibration plot deviated from linearity (inset in Fig. S1(a)†). Hence, we had considered the concentration range from 100–700 μM for the sensitivity and limit of detection (LOD) calculation (Fig. S1(b)† and its inset). The sensitivity and limit of detection (LOD) of the Fe3O4@Cu silicate based DA sensor were calculated to be 1.37 μA μM−1 cm−2 and 3.2 μM, respectively. Based on these significant electrochemical performances, it was confirmed that the proposed Fe3O4@Cu silicate based electrochemical sensing platform is an excellent candidate for DA detection (Table 1).
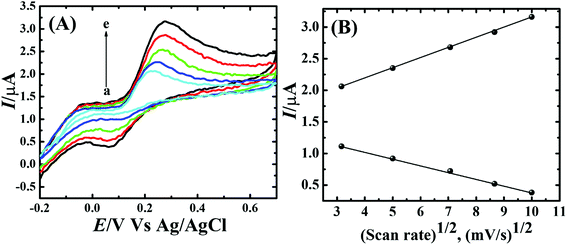 |
| | Fig. 8 (A) Cyclic voltammograms obtained on CP-Fe3O4@Cu silicate core–shell urchin electrode in 0.1 M PBS containing 1.8 mM DA at different scan rates (10–100 mV s−1) and (B) plot of anodic and cathodic peak currents vs. square root of scan rate. | |
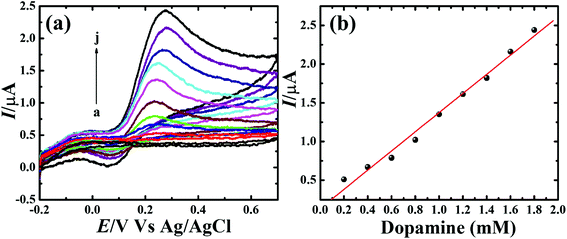 |
| | Fig. 9 (a) Cyclic voltammograms obtained for the different concentration of DA (0.2–1.8 mM) on CP-Fe3O4@Cu silicate core–shell urchin electrode in 0.1 M PBS at a scan rate of 50 mV s−1 and (b) corresponding calibration plot. | |
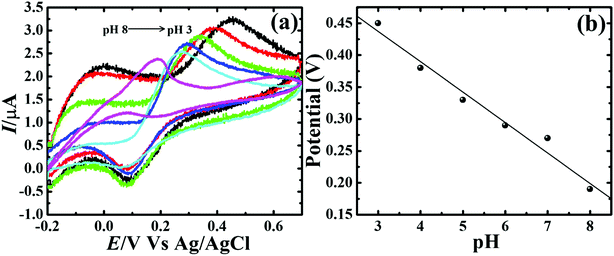 |
| | Fig. 10 (a) Cyclic voltammograms obtained on CP-Fe3O4@Cu silicate core–shell urchin electrode at 50 mV s−1 scan rate in 0.1 M PBS of different pH (3–8) containing 1.8 mM DA. (b) Calibration plot for pH vs. oxidation potential. | |
Table 1 Comparison of the performance of current sensor with earlier reported DA sensors
| Sensors |
Method |
Linear range (μM) |
Detection limit (μM) |
References |
| RGO@Pd-NC |
Amperometry |
20–220 |
7.02 |
69 |
| Graphene/chitosan/GCE |
DPV |
5–200 |
5 |
70 |
| Activated carbon tyrosinase/Nafion®-modified GCE |
Amperometry |
50–1000 |
50 |
71 |
| Fe3O4@Cu silicate sea urchins |
Amperometry |
100–700 |
3.2 |
This work |
Selectivity of the sensor is a vital factor since during the electrochemical sensing other electroactive species could interfere with the signal of main analyte. Therefore, interference of AA, UA and metal ions e.g. Cu2+, Pb2+, Cd2+, Hg2+, Zn2+ and Ni2+ during the amperometric sensing of DA was investigated. The sensor delivered a well-defined amperometric signal only for the 0.1 mM DA addition (Fig. S2†). The addition of AA, UA, Cu2+, Pb2+, Cd2+, Hg2+, Zn2+ and Ni2+ did not show obvious change in the amperometric response of DA, indicating that the sensor is highly selective towards the DA only. The reason for the high selectivity of the sensor only towards the DA molecule could be ascribed to the negatively charged surface of Fe3O4@Cu silicate and difference in the reduction potentials of the aforementioned metal ions with the DA potential. Due to the negatively charged surface of Fe3O4@Cu silicate, a high repulsion between Fe3O4@Cu silicate and the analytes AA and UA is anticipated, since the ascorbate and urate anions are negatively charged in neutral pH.72,73 At the same time, due to the potential difference, the sensor only detected DA, though the aforementioned metal ions undergo diffusion to the negatively charged electrode surface through the electrostatic interaction. The stability of the Fe3O4@Cu silicate based DA sensor was evaluated by CV measurement at a scan rate of 50 mV s−1 for 50 cycles in 0.1 M PBS containing 1.8 mM DA and the response is represented in Fig. 11. Irrespective of the cycle number the position of oxidation peak potential remained almost same with only small decrease in the catalytic current. For clear visualization of the relation between catalytic current and cycle number, voltammetric plot was made using the data of 1st, 10th, 20th, 30th, 40th and 50th CV cycles [Fig. 11(a)]. The sensor successfully retained 84% of catalytic current even after 50th cycle [Fig. 11(b)], signifying the excellent stability of Fe3O4@Cu silicate based DA sensor. In addition, the storage stability of the sensor was evaluated from the CV responses of the sensor recorded for one week. The sensor showed 19.7% degradation in the catalytic current after one week of storage (Fig. S3†), signifying that the sensor has outstanding storage stability. To further understand the reliability of DA sensor, the reproducibility of the sensor was investigated by fabricating three different sensors at the same experimental conditions and recording their CV responses towards the 1.8 mM of DA. In all cases, DA oxidation potential and catalytic current is almost similar, suggesting the excellent reproducibility of the sensor (Fig. S4†). Additionally, the repeatability of the Fe3O4@Cu silicate based DA sensor was evaluated using the same sensor in five different measurements, where the supporting electrolyte contains same concentration (1.8 mM) of DA. In all measurements, the sensor catalyzed DA oxidation at nearly same potential with virtually comparable catalytic current (Fig. S5†), signifying excellent repeatability of the sensor.
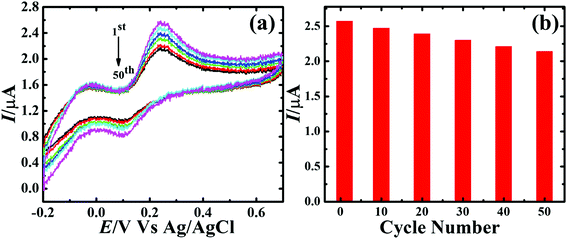 |
| | Fig. 11 (a) Voltammetric response of CP-Fe3O4@Cu silicate core–shell urchin electrode in 0.1 M PBS containing 1.8 mM DA at 50 mV s−1 scan rate after 1st, 10th, 20th, 30th, 40th and 50th cycles. (b) Plot of catalytic current vs. cycle number. | |
The utility of the current sensor in the detection of DA in human urine sample was evaluated by collecting the urine samples from three healthy persons. First the urine samples were diluted to 100 times using 0.1 M PBS and then subjected for the DA analysis, which shows to be free from DA. Therefore, to estimate the recovery rate and relative standard deviation (RSD) of DA in the real sample, known quantity of DA was spiked to these real samples. Then the concentration of DA was measured in these spiked samples by standard addition method and the results are represented in the Table S1.† The sensor exhibited recovery rates in the range from 96–104% with 1.4–2.1% RSD range for 5–15 μM spiking of DA to the real samples. These RSD and recovery rate values are reasonably acceptable, which clearly suggests the viability of the sensor in the analysis of real sample.
4 Conclusions
In summary, for the first time we have developed a Fe3O4@Cu silicate based DA sensor for the selective detection of DA. Owing to the negatively charged surface of Fe3O4@Cu silicate and presence of Cu in the Fe3O4@Cu silicate composite, the developed DA sensor showed excellent performance towards DA with high selectivity in the presence of UA and AA. Moreover, the Fe3O4@Cu silicate based DA sensor has high stability, reproducibility and repeatability. The sensor delivered excellent DA sensing performance with a sensitivity of 1.37 μA μM−1 cm−2, linear range from 100–700 μM and LOD of 3.2 μM. Most importantly, the present study excavate a new path for the development of various Cu silicate based core–shell nanostructures for the high sensitive, selective and precise detection of DA.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by KRF grant no 2017H1D3A1A01052414. This work is supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (No. 2016R1A2B4012847, 2013R1A4A1069528, 2012K2A1A2033057). This work supported by the DGIST R&D Program of the MSIP (18-BT-02).
References
- V. K. Gupta, S. Kumar, R. Singh, L. P. Singh, S. K. Shoora and B. Sethi, J. Mol. Liq., 2014, 195, 65–68 CrossRef.
- M. H. Dehghani, D. Sanaei, I. Ali and A. Bhatnagar, J. Mol. Liq., 2016, 215, 671–679 CrossRef.
- S. K. Srivastava, V. K. Gupta, M. K. Dwivedi and S. Jain, Anal. Proc., 1995, 32, 21–23 RSC.
- S. K. Srivastava, V. K. Gupta and S. Jain, Anal. Chem., 1996, 68, 1272–1275 CrossRef PubMed.
- S. K. Srivastava, V. K. Gupta and S. Jain, Analyst, 1995, 120, 495–498 RSC.
- M. L. Yola, V. K. Gupta, T. Eren, A. E. Şen and N. Atar, Electrochim. Acta, 2014, 120, 204–211 CrossRef.
- V. K. Gupta, H. Karimi-Maleh and R. Sadegh, Int. J. Electrochem. Sci., 2015, 10, 303–316 Search PubMed.
- V. K. Gupta, B. Sethi, R. A. Sharma, S. Agarwal and A. Bharti, J. Mol. Liq., 2013, 177, 114–118 CrossRef.
- V. K. Gupta, L. P. Singh, R. Singh, N. Upadhyay, S. P. Kaur and B. Sethi, J. Mol. Liq., 2012, 174, 11–16 CrossRef.
- V. K. Gupta, A. Nayak, S. Agarwal and B. Singhal, Comb. Chem. High Throughput Screening, 2011, 14, 284–302 CrossRef PubMed.
- S. Karthikeyan, V. K. Gupta, R. Boopathy, A. Titus and G. Sekaran, J. Mol. Liq., 2012, 173, 153–163 CrossRef.
- V. K. Gupta, N. Mergu, L. K. Kumawat and A. K. Singh, Talanta, 2015, 144, 80–89 CrossRef PubMed.
- V. K. Gupta, A. K. Singh and L. K. Kumawat, Sens. Actuators, B, 2014, 195, 98–108 CrossRef.
- V. K. Gupta, N. Mergu, L. K. Kumawat and A. K. Singh, Sens. Actuators, B, 2015, 207, 216–223 CrossRef.
- H. K. Maleh, F. T. Javazmi, N. Atar, M. L. Yola, V. K. Gupta and A. A. Ensafi, Ind. Eng. Chem. Res., 2015, 54, 3634–3639 CrossRef.
- V. K. Gupta, M. R. Ganjali, P. Norouzi, H. Khani, A. Nayak and S. Agarwal, Crit. Rev. Anal. Chem., 2011, 41, 282–313 CrossRef PubMed.
- T. Vijayaraghavan, R. Sivasubramanian, S. Hussain and A. Ashok, ChemistrySelect, 2017, 2, 5570–5577 CrossRef.
- S. Reyes, Y. Fu, K. L. Double, V. Cottam, L. H. Thompson, D. Kirik, G. Paxinos, C. Watson, H. M. Cooper and G. M. Halliday, Neurobiol. Aging, 2013, 34, 873–886 CrossRef PubMed.
- A. A. Grace, Neuropharmacology, 2013, 62, 1342–1348 CrossRef PubMed.
- D. Diaz-Diestra, B. Thapa, J. Beltran-Huarac, B. R. Weiner and G. Morell, Biosens. Bioelectron., 2017, 87, 693–700 CrossRef PubMed.
- M. Kim, J. Lee, C. H. Yang and S. Lee, Anal. Chim. Acta, 2016, 923, 55–65 CrossRef PubMed.
- Y. Suzuki, Sens. Actuators, B, 2017, 239, 383–389 CrossRef.
- W. Gao, L. Qi, Z. Liu, S. Majeed, S. A. Kitte and G. Xu, Sens. Actuators, B, 2017, 238, 468–472 CrossRef.
- D. Wen, W. Liu, A. Herrmann, D. Haubold, M. Holzschuh, F. Simon and A. Eychmüller, Small, 2016, 12, 2439–2442 CrossRef PubMed.
- D. O. Matos and W. A. Alves, ACS Appl. Mater. Interfaces, 2011, 3, 4437–4443 CrossRef PubMed.
- S. Reddy, B. E. Kumara Swamy and H. Jayadevappa, Electrochim. Acta, 2012, 61, 78–86 CrossRef.
- S. Liu, X. Xing, J. Yu, W. Lian, J. Li, M. Cui and J. Huang, Biosens. Bioelectron., 2012, 36, 186–191 CrossRef PubMed.
- M. Sajid, M. K. Nazal, M. Mansha, A. Alsharaa, S. M. S. Jillani and C. Basheer, TrAC, Trends Anal. Chem., 2016, 76, 15–29 CrossRef.
- J. Zhao, W. Zhang, P. Sherrell, J. M. Razal, X. F. Huang, A. I. Minett and J. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 44–48 CrossRef PubMed.
- Y. R. Kim, S. Bong, Y. J. Kang, Y. Yang, R. K. Mahajan, J. S. Kim and H. Kim, Biosens. Bioelectron., 2010, 25, 2366–2369 CrossRef PubMed.
- X. Dong, Y. Ma, G. Zhu, Y. Huang, J. Wang, M. B. Chan-Park, L. Wang, W. Huang and P. Chen, J. Mater. Chem., 2012, 22, 17044–17048 RSC.
- T. Qian, C. Yu, S. Wu and J. Shen, Biosens. Bioelectron., 2013, 50, 157–160 CrossRef PubMed.
- P. Wiench, Z. González, R. Menéndez, B. Grzyb and G. Gryglewicz, Sens. Actuators, B, 2018, 257, 143–153 CrossRef.
- N. Yusoff, A. Pandikumar, R. Ramaraj, H. N. Lim and N. M. Huang, Microchim. Acta, 2015, 182, 2091–2114 CrossRef.
- A. C. Anithaa, N. Lavanya, K. Asokan and C. Sekar, Electrochim. Acta, 2015, 167, 294–302 CrossRef.
- F. R. Caetano, L. B. Felippe, A. J. G. Zarbin, M. F. Bergamini and L. H. Marcolino-Junior, Sens. Actuators, B, 2017, 243, 43–50 CrossRef.
- A. Numan, M. M. Shahid, F. S. Omar, S. Rafique, S. Bashir, K. Ramesh and S. Ramesh, Microchim. Acta, 2017, 184, 2739–2748 CrossRef.
- S. J. Li, D. H. Deng, Q. Shi and S. R. Liu, Microchim. Acta, 2012, 177, 325–331 CrossRef.
- A. Numan, M. M. Shahid, F. S. Omar, K. Ramesh and S. Ramesh, Sens. Actuators, B, 2017, 238, 1043–1051 CrossRef.
- J. J. Fang, N. N. Yang and E. Q. Gao, Electrochem. Commun., 2018, 89, 32–37 CrossRef.
- N. C. de Lucena, C. M. Miyazaki, F. M. Shimizu, C. J. L. Constantino and M. Ferreira, Appl. Surf. Sci., 2018, 436, 957–966 CrossRef.
- J. Zou, S. Wu, Y. Liu, Y. Sun, Y. Cao, J. P. Hsu, A. T. Shen Wee and J. Jiang, Carbon, 2018, 130, 652–663 CrossRef.
- K. P. O. Mahesh, I. Shown, L.-C. Chen, K.-H. Chen and Y. Tai, Appl. Surf. Sci., 2018, 427, 387–395 CrossRef.
- B. Fang, G. Wang, W. Zhang, M. Li and X. Kan, Electroanalysis, 2005, 17, 744–748 CrossRef.
- D. Peng, R. P. Liang, H. Huang and J. D. Qiu, J. Electroanal. Chem., 2016, 761, 112–117 CrossRef.
- S. Y. Han, T. Y. Du, L. M. Lai, X. R. Jiang, C. S. Cheng, H. Jiang and X. M. Wang, RSC Adv., 2016, 6, 82033–82039 RSC.
- Y. Zhang, W. Gao, L. Zuo, L. Zhang, Y. Huang, H. Lu, W. Fan and T. Liu, Adv. Mater. Interfaces, 2016, 3, 1600137 CrossRef.
- A. S. Adekunle, B. O. Agboola, J. Pillay and K. I. Ozoemena, Sens. Actuators, B, 2010, 148, 93–102 CrossRef.
- J. Salamon, Y. Sathishkumar, K. Ramachandran, Y. S. Lee, D. J. Yoo, A. R. Kim and G. GnanaKumar, Biosens. Bioelectron., 2014, 64, 269–276 CrossRef PubMed.
- O. E. Fayemi, A. S. Adekunle, B. E. Kumara Swamy and E. E. Ebenso, J. Electroanal. Chem., 2018, 818, 236–249 CrossRef.
- G. J. Rani, K. J. Babu, G. G. kumar and M. A. J. Rajan, J. Alloys Compd., 2016, 688, 500–512 CrossRef.
- T. Peik-See, A. Pandikumar, H. Nay-Ming, L. Hong-Ngee and Y. Sulaiman, Sensors, 2014, 14, 15227–15243 CrossRef PubMed.
- H. Bagheri, A. Afkhami, P. Hashemi and M. Ghanei, RSC Adv., 2015, 5, 21659–21669 RSC.
- H. Teymourian, A. Salimi and S. Khezrian, Biosens. Bioelectron., 2013, 49, 1–8 CrossRef PubMed.
- D. Wu, Y. Li, Y. Zhang, P. Wang, Q. Wei and B. Du, Electrochim. Acta, 2014, 116, 244–249 CrossRef.
- H. Song, G. Xue, J. Zhang, G. Wang, B. C. Ye, S. Sun, L. Tian and Y. Li, Microchim. Acta, 2017, 184, 843–853 CrossRef.
- H. Bagheri, N. Pajooheshpour, B. Jamali, S. Amidi, A. Hajian and H. Khoshsafar, Microchem. J., 2017, 131, 120–129 CrossRef.
- D. M. Fernandes, M. Costa, C. Pereira, B. Bachiller-Baeza, I. Rodríguez-Ramos, A. Guerrero-Ruiz and C. Freire, J. Colloid Interface Sci., 2014, 432, 207–213 CrossRef PubMed.
- Y. Liu, W. Zhu, D. Wu and Q. Wei, MeasurementMater. Sci. Eng., C, 2015, 60, 1–5 Search PubMed.
- R. Kuchi, V. Dongquoc, S. Surabhi, D. Kim, S.-G. Yoon, S.-Y. Park, J. Choi and J.-R. Jeong, Phys. Status Solidi A, 2018, 1701032 CrossRef.
- A. K. Das, S. Sahoo, P. Arunachalam, S. Zhang and J.-J. Shim, RSC Adv., 2016, 6, 107057–107064 RSC.
- J. Liu, J. Cheng, R. Che, J. Xu, M. Liu and Z. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 2503–2509 CrossRef PubMed.
- M. B. Rooney, D. C. Coomber and A. M. Bond, Anal. Chem., 2000, 72, 3486–3491 CrossRef PubMed.
- M. Arvand and M. Hassannezhad, Mater. Sci. Eng., C, 2014, 36, 160–167 CrossRef PubMed.
- M. Hsu, Y. Chen, C. Lee and H. Chiu, ACS Appl. Mater. Interfaces, 2012, 4, 5570–5575 CrossRef PubMed.
- S. Sundar, R. Mariappan, K. Min and S. Piraman, RSC Adv., 2016, 6, 77133–77142 RSC.
- N. A. Al Abass, G. Denuault and D. Pletcher, Phys. Chem. Chem. Phys., 2014, 16, 4892 RSC.
- S. Ku, S. Palanisamy and S. Chen, J. Colloid Interface Sci., 2013, 411, 182–186 CrossRef PubMed.
- Y. S. Hsieh, B. D. Hong and C. L. Lee, Microchim. Acta, 2016, 183, 905–910 CrossRef.
- Y. Wang, Y. Li, L. Tang, J. Lu and J. Li, Electrochem. Commun., 2009, 11, 889–892 CrossRef.
- S. F. Rahman, K. Min, S.-H. Park, J.-H. Park, J. C. Yoo and D.-H. Park, Biotechnol. Bioprocess Eng., 2016, 21, 627–633 CrossRef.
- A. K. Das and C. R. Raj, J. Electroanal. Chem., 2010, 638, 189–194 CrossRef.
- P. Ramesh and S. Sampath, Electroanalysis, 2004, 16, 866–869 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05885g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Rambabu Kuchi‡
a,
Rambabu Kuchi‡