
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceB(C6F5)3 catalyzed direct nucleophilic substitution of benzylic alcohols: an effective method of constructing C–O, C–S and C–C bonds from benzylic alcohols†
Shan-Shui Meng *,
Qian Wang,
Gong-Bin Huang,
Li-Rong Lin,
Jun-Ling Zhao* and
Albert S. C. Chan*
*,
Qian Wang,
Gong-Bin Huang,
Li-Rong Lin,
Jun-Ling Zhao* and
Albert S. C. Chan*
School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: mengshsh@mail.sysu.edu.cn; zhaojling3@mail.sysu.edu.cn; chenxz3@mail.sysu.edu.cn
First published on 3rd September 2018
Abstract
An efficient and general method of nucleophilic substitution of benzylic alcohols catalyzed by non-metallic Lewis acid B(C6F5)3 was developed. The reaction could be carried out under mild conditions and more than 35 examples of ethers, thioethers and triarylmethanes were constructed in high yields. Some bioactive organic molecules were synthesized directly using the methods.
Alcohols are ideal building blocks for high-value chemicals because of their high stability, low toxicity and wide availability.1 However, due to the poor leaving ability of the hydroxyl group, in most cases, alcohol substitution requires hydroxyl preactivation or the use of excess Brønsted acids or stoichiometric amounts of Lewis acids. In 2005, the OH-activation for nucleophilic substitution was considered a central issue by the ACS Green Chemistry Institute2 because the direct catalytic nucleophilic substitutions of alcohols are highly desirable in constructing high-value organic compounds such as ethers, thioethers and a variety of bioactive molecules. Furthermore, the reaction produces only H2O as a byproduct, which meets the requirements of green chemistry and atom-economy.3
In recent years, the direct catalytic nucleophilic substitution of alcohol has been explored, however, many urgent, difficult problems are still waiting to be solved. The catalytic nucleophilic substitutions of π-activated alcohols such as allylic/propargyl alcohols have been developed as well as the double bond or triple bond of the substrates being able to coordinate to the metal catalysts (Au, Bi, In, Al, Fe etc.), stabilizing the carbocations.4 In contrast, for benzylic alcohols, the catalytic substitution reactions are much less explored.5 Furthermore, harsh conditions and excess nucleophiles are two major defects in the existing reports, and for different nucleophiles, varied catalysts are always required. Therefore, a mild, green, efficient and general method of benzylic alcohol nucleophilic substitution is highly desirable (Scheme 1).
 | ||
| Scheme 1 The difference between traditional method and catalytic method. | ||
In recent years, B(C6F5)3 as a non-metallic Lewis acid has drawn notable attention because of its strong Lewis acidity, high-stability and environmental friendliness.6 Furthermore, the borane hydrate is also a strong Brønsted acid.7 And these inspired us that the borane may offer another chance to realize the direct substitution reaction of benzylic alcohols such as etherification, thioetherification and arylation (Scheme 2).
 | ||
| Scheme 2 Catalytic substitution of alcohols. | ||
For the etherification of alcohols, the difficulty is that how to realize the high selectivity between cross-etherification and homo-etherification. Initially we chose diphenylmethanol 1a and BnOH as the model alcohol compounds. The reaction was carried out with 10 mol% B(C6F5)3 in toluene at 60 °C (Table 1). To our delight, the cross-etherification product was obtained in high yield and the homo-etherification product could not be detected (85%, entry 1). In the examination of several common solvents for the cross-etherification of 1a, 1,2-dichloroethane was found to be superior (95%, entry 2). The reaction could not give any products in the THF (entry 4) because a “Frustrated Lewis Pair”8 was formed between B(C6F5)3 and THF. When the catalyst loading was decreased to 5 mol%, the yield had no erosion (entry 6), and a further decrease led to a longer reaction time and a slight lower yield (entry 7). Longer time was needed when the reaction was carried out at room temperature.
|
|||
|---|---|---|---|
| Entry | X% | Solvent | Yieldb% |
| a Unless otherwise noted, all reactions were conducted using 1.2 equiv. BnOH with a substrate concentration 0.2 M in the indicated solvent at 60 °C on a 0.2 mmol scale, 12 h.b Isolated yield.c Room temperature, 48 h. | |||
| 1 | 10 | Toluene | 85 |
| 2 | 10 | 1,2-Dichloroethane | 95 |
| 3 | 10 | Ethyl acetate | 30 |
| 4 | 10 | THF | <5 |
| 5 | 10 | DCM | 88 |
| 6 | 5 | 1,2-Dichloroethane | 95 |
| 7 | 1 | 1,2-Dichloroethane | 93 |
| 8c | 10 | 1,2-Dichloroethane | 89 |

After we optimized the reaction conditions, we decided to investigate the etherification substrates scope (Table 2). Diphenylmethanol with different substitutes coupled with BnOH always provided the desired ethers in excellent yields (2b–e), the electron-rich groups gave slightly higher yields compared to the electron-withdrawing groups such as F, Cl. But for the substrate with 2-CF3, no products could be obtained. Even for a bulky substrate, the etherification also occurred smoothly (2f, 88%). A little longer reaction-time was required when less reactive substrates were tested, the yields were still satisfactory (2g–j, 93–96%). All the reaction did not give any product of homo-etherification products. When a secondary alcohol iPrOH was used as coupling reagent, the reaction still gave good yields and excellent selectivities (2k–m, 91–96%). The homo-etherification of alcohol was also tested in the reaction conditions, e.g. diphenylmethanol, the product could be isolated in a high yield (2n, 95%).
|
|---|
 |
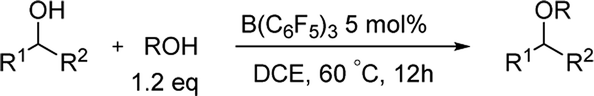
As we had realized the cross-etherification of alcohols, we decided to look into the possibility of applying the system in the C–S bonds construction as the thioether-containing compounds have a unique significance in drugs.9 To our delight, the product of thioetherification between 1a and 4-methylbenzenethiols was isolated in 97% yield (Table 3) when the reaction was carried in the optimal conditions, and the different types of substitutes on the benzene ring had slight effect on the yields (3b–e). For the substrate containing heteroaromatic-ring, the thioetherification product could be isolated in a quantitative yield (3f). The reaction finished in high yields for phenethyl alcohols (3g–i) and the cyclic substrates also gave the similar results (3j, 3k).
|
|---|
 |

Triarylmethanes are well-known structures in several areas, including photochromic agents, dye precursors and applications in materials,10 they also play an important role in medicinal chemistry.11 Naturally we applied our catalytic system in the synthesis of triarylmethanes (Table 4). The substitution reaction between diphenylmethanols and 2,6-di-tert-butylphenol worked quite well and gave the desired triarylmethanes in 91–99% yields (5a–f). For the benzylic alcohols, the target products were isolated in high yields (5g–j) as well. A similarity of antitubercular compound 5k was also synthesized effectively. Different aromatic nucleophiles were tested in the reaction, the indole substitution product 5l was obtained in 91% yield. Isoerianin as an excellent anti-cancer small molecule and its isomerism (5m and 5n) were also synthesized in 92% yield totally.12
|
|---|
 |

To give more insight of the reaction, a SN1 mechanism was proposed (Scheme 3). When the B(C6F5)3 was under moisture conditions, it could generate a strong Brønsted acid B(C6F5)3·nH2O, and the Brønsted acid could activated the alcohol substrates, the hydroxyl group leaved as one molecular H2O giving a carbocation, then the nucleophile attacked the carbocation forming the product.
 | ||
| Scheme 3 Plausible mechanism. | ||
Conclusions
In summary, via a rational design, a general, practical, green and efficient method of direct nucleophilic substitution of benzylic alcohols was developed by our group. The reaction only needs a mild temperature (60 °C), a low catalyst loading (as low as 1 mol%) and a slight excess of nucleophile (1.2 equivalent). A series of unsymmetrical ethers were synthesised by the method in high yields without any homo-etherification products. Furthermore, the catalytic system was also suitable for the thioetherification and arylation of benzylic alcohols, and a wide range of thioethers and triarylmethanes were obtained in high efficiency. Further application and extension of B(C6F5)3-catalyzed substitution reactions is still being explored by our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Research Foundation for Advanced Talents of Sun Yat-sen University (36000-18821101) and the National Natural Science Foundation of China (Grant No. 21772199 and 21372221) for financial support of this program.Notes and references
- (a) E. Emer, R. Sinisi, M. G. Capdevila, D. Petruzziello, F. De Vincentiis and P. G. Cozzi, Eur. J. Org. Chem., 2011, 647–666 CrossRef; (b) J. Moran, M. Dryzhakov and E. Richmond, Synthesis, 2016, 48, 935–959 CrossRef; (c) C. Nájera and A. Baeza, Synthesis, 2013, 46, 25–34 CrossRef; (d) R. Ortiz and R. P. Herrera, Molecules, 2017, 22, 574–585 CrossRef PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- (a) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC; (b) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef PubMed.
- (a) T. Ohshima, Y. Nakahara, J. Ipposhi, Y. Miyamoto and K. Mashima, Chem. Commun., 2011, 47, 8322–8324 RSC; (b) M. Yasuda, T. Somyo and A. Baba, Angew. Chem., Int. Ed., 2006, 45, 793–796 CrossRef PubMed; (c) Z. P. Zhan, W. Z. Yang, R. F. Yang, J. L. Yu, J. P. Li and H. J. Liu, Chem. Commun., 2006, 3352–3354 RSC; (d) M. Georgy, V. Boucard and J. M. Campagne, J. Am. Chem. Soc., 2005, 127, 14180–14181 CrossRef PubMed; (e) M. Rueping, C. Vila and U. Uria, Org. Lett., 2012, 14, 768–771 CrossRef PubMed; (f) S. Haubenreisser and M. Niggemann, Adv. Synth. Catal., 2011, 353, 469–474 CrossRef; (g) H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409–413 CrossRef PubMed.
- (a) S. Podder, J. Choudhury and S. Roy, J. Org. Chem., 2007, 72, 3129–3132 CrossRef PubMed; (b) L. Zhang, A. Gonzalez-de-Castro, C. Chen, F. Li, S. Xi, L. Xu and J. Xiao, Mol. Catal., 2017, 433, 62–67 CrossRef; (c) S. Shirakawa and S. Kobayashi, Org. Lett., 2007, 9, 311–314 CrossRef PubMed; (d) Y.-L. Liu, L. Liu, Y.-L. Wang, Y.-C. Han, D. Wang and Y.-J. Chen, Green Chem., 2008, 10, 635 RSC; (e) A. Zhu, L. Li, J. Wang and K. Zhuo, Green Chem., 2011, 13, 1244 RSC; (f) K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 2605–2609 CrossRef PubMed; (g) J. Li, X. Zhang, H. Shen, Q. Liu, J. Pan, W. Hu, Y. Xiong and C. Chen, Adv. Synth. Catal., 2015, 357, 3115–3120 CrossRef; (h) Q. Xu, H. Xie, P. Chen, L. Yu, J. Chen and X. Hu, Green Chem., 2015, 17, 2774–2779 RSC.
- (a) T. Schwier, M. Rubin and V. Gevorgyan, Org. Lett., 2004, 6, 1999–2001 CrossRef PubMed; (b) M. Rubin and V. Gevorgyan, Org. Lett., 2001, 3, 2705–2707 CrossRef PubMed; (c) C. R. Reddy, G. Rajesh, S. V. Balaji and N. Chethan, Tetrahedron Lett., 2008, 49, 970–973 CrossRef; (d) M. Dryzhakov, M. Hellal, E. Wolf, F. C. Falk and J. Moran, J. Am. Chem. Soc., 2015, 137, 9555–9558 CrossRef PubMed; (e) M. Dryzhakov and J. Moran, ACS Catal., 2016, 6, 3670–3673 CrossRef; (f) M. Shibuya, M. Okamoto, S. Fujita, M. Abe and Y. Yamamoto, ACS Catal., 2018, 8, 4189–4193 CrossRef.
- C. Bergquist, B. M. Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 2000, 122, 10581–10590 CrossRef.
- (a) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef PubMed; (b) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef PubMed; (c) M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- (a) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef PubMed; (b) X. Han and J. Wu, Org. Lett., 2010, 12, 5780–5782 CrossRef PubMed; (c) R. A. Croft, J. J. Mousseau, C. Choi and J. A. Bull, Chem. - Eur. J., 2018, 24, 818–821 CrossRef PubMed; (d) Y. Yang, Z. Ye, X. Zhang, Y. Zhou, X. Ma, H. Cao, H. Li, L. Yu and Q. Xu, Org. Biomol. Chem., 2017, 15, 9638–9642 RSC.
- Y.-Q. Xu, J.-M. Lu, N.-J. Li, F. Yan, X.-W. Xia and Q.-F. Xu, Eur. Polym. J., 2008, 44, 2404–2411 CrossRef.
- (a) Shagufta, A. K. Srivastava, R. Sharma, R. Mishra, A. K. Balapure, P. S. Murthy and G. Panda, Bioorg. Med. Chem., 2006, 14, 1497–1505 CrossRef PubMed; (b) M. K. Parai, G. Panda, V. Chaturvedi, Y. K. Manju and S. Sinha, Bioorg. Med. Chem. Lett., 2008, 18, 289–292 CrossRef PubMed; (c) R. Palchaudhuri, V. Nesterenko and P. J. Hergenrother, J. Am. Chem. Soc., 2008, 130, 10274–10281 CrossRef PubMed; (d) K. Risberg, I. J. Guldvik, R. Palchaudhuri, Y. Xi, J. Ju, O. Fodstad, P. J. Hergenrother and Y. Andersson, J. Immunother., 2011, 34, 438–447 CrossRef PubMed.
- (a) M. A. Soussi, O. Provot, G. Bernadat, J. Bignon, J. Wdzieczak-Bakala, D. Desravines, J. Dubois, J. D. Brion, S. Messaoudi and M. Alami, Eur. J. Med. Chem., 2014, 78, 178–189 CrossRef PubMed; (b) S. Messaoudi, A. Hamze, O. Provot, B. Treguier, J. Rodrigo De Losada, J. Bignon, J. M. Liu, J. Wdzieczak-Bakala, S. Thoret, J. Dubois, J. D. Brion and M. Alami, ChemMedChem, 2011, 6, 488–497 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05811c |
| This journal is © The Royal Society of Chemistry 2018 |