DOI:
10.1039/C8RA03679A
(Paper)
RSC Adv., 2018,
8, 30950-30956
Retracted Article: ASIC1a involves acidic microenvironment-induced activation and autophagy of pancreatic stellate cells†
Received
29th April 2018
, Accepted 29th August 2018
First published on 4th September 2018
Abstract
Acid-sensing ion channel 1a (ASIC1a), as a member of the proton-gated cation channel family, can be activated by low extracellular pH, and takes part in many acidity-associated physiopathological processes. However, whether ASIC1a is expressed in human pancreatic stellate cells (PSCs) and involved in acid-induced physiopathological events has not been reported yet. In this study, we investigated the expression of ASIC1a in PSCs and its possible role in the activation and autophagy of PSCs evoked by extracellular acid. Our results show that ASIC1a is present in PSCs, and an enhanced expression of ASIC1a occurs under acid stimuli. More importantly, the activation and autophagy of PSCs can be induced in acidic medium, and inhibition of ASIC1a by ASIC1a-specific blocker psalmotoxin-1 (PcTx1) or siRNA knockdown could suppress these two acid-associated processes. Collectively, our present study reports for the first time that ASIC1a is expressed in PSCs, and provides evidence for the involvement of ASIC1a in the acidic microenvironment-induced activation and autophagy of PSCs.
Introduction
PSCs are located in the periacinar spaces in a quiescent state in normal healthy pancreas, but can be activated by many extracellular stimuli including pro-inflammatory cytokines, oxidative stress, hypoxia and increased interstitial pressure.1,2 The activated PSCs can express α-smooth muscle actin (α-SMA) and synthesize a large amount of extracellular matrix (ECM) proteins leading to excessive fibrosis of the pancreas. For example, in human pancreatic cancer, the fibrotic stroma can account for up to 90% of the tumor volume, and the fibrotic microenvironment has been proven to support tumorigenesis, chemoresistance, invasion and progression of tumors.3,4 Owing to the altered cancer cell glucose metabolism, hypoxia and insufficient blood perfusion, the microenvironment of solid tumors is acidic, which means that PSCs are exposed to low pH conditions in pancreatic cancer. However, the effect of acidic environment on the biological behaviour of PSCs and the underlying molecular mechanisms are far from clear.
Acid-sensing ion channels (ASICs) are H+-gated cation channels, which belong to the degenerin/epithelial Na+ channel (DEG/ENaC) superfamily, and at least six ASICs subunit proteins, namely 1a, 1b, 2a, 2b, 3 and 4, have been identified.5 Among them, ASIC1a attracts special attention because of its important biological functions in the pathogenesis of acid-associated diseases. Previous studies show that ASIC1a is mainly expressed in mammalian central and peripheral nervous systems and plays an important role in pain perception, learning and memory, and fear conditioning.6 Since an acidic extracellular pH is a major feature of malignant tumor tissues, the expression and role of ASIC1a in many kinds of tumor cells including liver cancer,7 gastric cancer,8 lung cancer9 and gliomas,10 pancreatic cancer11 have been investigated, and the results show that ASIC1a involves the proliferation, invasion or autophagy of tumor cells, suggesting a novel therapeutic role for ASIC1a in these tumors. Interestingly, recent studies suggest that hepatic stellate cells (HSCs) and rat articular chondrocytes both can express ASIC1a, and inhibition of ASIC1a could reduce acid or PDGF-induced activation of HSCs and liver fibrosis,12,13 and ASIC1a can promote acid-induced autophagy of rat articular chondrocytes through AMPK/FoxO3a pathway.14
PSCs are very like HSCs, but whether ASIC1a is expressed in PSCs has not been reported yet. Given that ASIC1a plays an irreplaceable role in acidity-associated events, it is reasonable to relate ASIC1a with the possible changed physiological function of PSCs. In the present study, we aim to investigate the expression of ASIC1a in PSCs cultured in pH 7.4 (the normal tissue pH) and pH 6.5 medium (one of the most widely used pH values minicing an acidic tumor microenvironment in vitro) respectively, and then explore the effect of inhibition of ASIC1a on the acid-induced pathophysiological processes including activation and autophagy of PSCs.
Experimental section
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco. Psalmotoxin-1 (PcTx1) was purchased from ApexBio Technology. Cell RIPA lysis buffer was obtained from Solarbio Company. Primary antibodies of ASIC1a, α-SMA and beclin 1 were obtained from Abcom. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) and collagen 1 antibodies, FITC-conjugated goat anti-rabbit IgG (H+L), Alexa Fluor 555-conjugated donkey anti-rabbit IgG (H+L) and HRP-conjugated secondary antibodies were obtained from Beyotime Biotechnology. Anti-GAPDH was obtained from CST. Other general agents were commercially available.
Cell culture and treatment
Human PSCs were purchased from the Cell Bank of Shanghai Tongpai Biological Technology Company and cultured in DMEM containing 10% FBS at 37 °C with 5% CO2. To simulate the extracellular acid environment, the medium was adjusted to acidic pH (pH 6.5) with HCl. PSCs were cultured in two different pHs, including pH 7.4 and pH 6.5 for 4 h, then the medium was replaced with complete medium for another 3 h. To observe the influence of ASIC1a on the acid-induced physiological function, PSCs, pretreated with 50 nM of ASIC1a-specific inhibitor PcTx1 for 30 min, or PSCs with ASIC1a knockdown using small interfering RNA (siRNA), were cultured in pH 6.5.
Immunofluorescence assay
After treatment as indicated above, PSCs were fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.3% Triton X-100 in PBS for 15 min, and then blocked with 3% BSA for 1 h. After this, PSCs were divided into three groups and incubated with 1: 100 anti-ASIC1a, anti-α-SMA, anti-collagen 1 antibody overnight at 4 °C respectively, followed by incubation with 1: 100 FITC-conjugated goat anti-rabbit IgG (for ASIC1a and collagen 1) or Alexa Fluor 555-conjugated donkey anti-rabbit IgG (for α-SMA) in the dark for 1 h at room temperature. Nuclear staining was counterstained with DAPI for 15 min. Finally, the cells were washed with PBS, mounted with coverslips and imaged using an inverted fluorescence microscope.
Knockdown of ASIC1a in PSCs by siRNA
Based on siRNA design principles and the sequence of ASIC1a in Genebank, siRNA duplexes targeting human ASIC1a and negative control (NC) were designed and synthesized, and then the adenoviral vector Ad-ASIC1a-siRNA and Ad-NC-siRNA were constructed by JRDUN Biotechnology (Shanghai) Co., Ltd. PSCs were transfected with Ad-ASIC1a-siRNA or Ad-NC-siRNA according to the manufactures' instructions, and after 48 h of transfection, ASIC1a protein and mRNA expression levels were analyzed by western blotting and qRT-PCR to confirm the ASIC1a knockdown efficiency (Fig. S1†). All transfections were performed in triplicate.
Western blotting
Cells collected from each group were washed twice with ice-cold PBS, and then were lysed in RIPA lysis buffer for 30 min on ice to extract total protein. The concentration of protein was determined by BCA assay. Equal amounts of protein lysates (15 μL) from each group was separated by 10% SDS-PAGE and transferred to a PVDF membrane. The membrane was washed with TBST and then blocked with 5% non-fat milk for 1 h at room temperature. After this, the membranes were incubated in the appropriate primary antibodies and GAPDH monoclonal antibodies overnight at 4 °C, followed by HRP-conjugated secondary antibodies at room temperature for 1 h. Last, the membranes were visualized using Tanon 5200 enhanced chemiluminescence (ECL) detection system, and the density of target bands was quantified with a computerized digital imaging system. The above procedure was repeated three times using GAPDH as control.
Quantitative real-time polymerase chain reaction analysis (qRT-PCR)
Total RNA was extracted from PSCs using Trizol reagent. The first-strand cDNA was synthesized using Thermoscript RT-PCR System according to the manufacturer's instructions. The primers were designed and synthesized by JRDUN Biotechnology (Shanghai) Co., Ltd according to the serial number from Genbank, as shown in Table 1. GAPDH was used as an internal control. qRT-PCR was performed using 0.1 L of cDNA with SYBR Green dye detection of amplification products on a real-time PCR Detection System. The cycle threshold value was defined as the PCR cycle number at which the reporter fluorescence crossed the threshold. The relative expression levels of target genes were normalized by GAPDH and calculated by 2−ΔΔCt.
Table 1 Primer sequences for qRT-PCR
| Primer |
Primer sequences |
Product length/bp |
| ASIC1a |
F: 5′-TGACCCGCTATGGCAAAG-3′ |
121 |
| R: 5′-TCCAGCACCAGGATGTTC-3′ |
| α-SMA |
F: 5′-ATTGCCGACCGAATGCAG-3′ |
193 |
| R: 5′-TGCGGTGGACAATGGAAG-3′ |
| Collagen 1 |
F: 5′-CTTCGTGTAAACTCCCTCCATCC-3′ |
136 |
| R: 5′-AAGTCCATGTGAAATTGTCTCCCA-3′ |
| Beclin 1 |
F: 5′-AGGGATGGAAGGGTCTAAG-3′ |
155 |
| R: 5′-GGGCTGTGGTAAGTAATGG-3′ |
| GAPDH |
F: 5′-AATCCCATCACCATCTTC-3′ |
218 |
| R: 5′-AGGCTGTTGTCATACTTC-3′ |
Statistical analysis
All experiments were repeated a minimum of three times, and the data are expressed as mean ± SD. The comparison among the different groups was performed using analysis of variance (ANOVA) and unpaired Student's t-test, and P < 0.05 was considered statistically significant.
Results and discussion
ASIC1a is expressed in PSCs and acid upregulate its expression
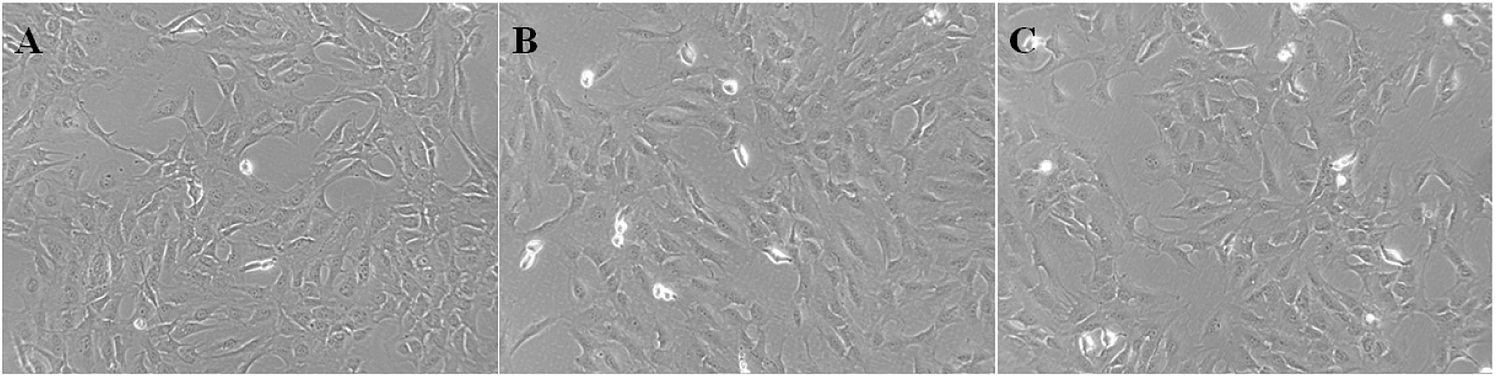
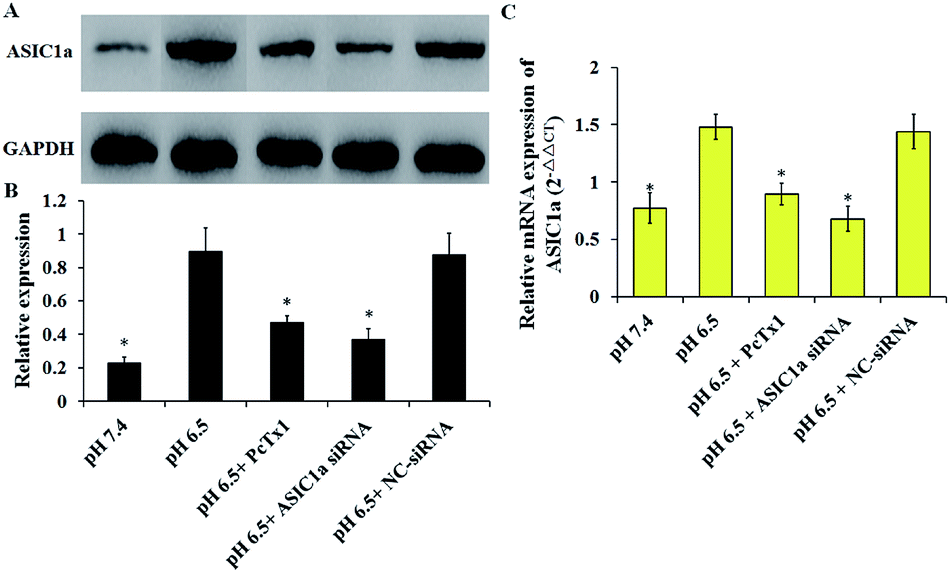
ASIC1a has been found to be expressed in a variety of non-neuronal tissues and plays an important role in pH changes related to tissue acidosis. Besides tumor cells, it is a remarkable fact that ASIC1a is also expressed in HSCs and involves the activation of HSCs through MAPK pathway, which benefit the formation of hepatic fibrosis.12,13 PSCs and HSCs share many common features, but whether ASIC1a is present in PSCs has not been reported. Considering that the typical pH value of tumor ECM is about 6.5–6.8, in this study, PSCs were cultured in physiological conditions (pH 7.4) and pH 6.5 medium, and then the expression of ASIC1a was detected. First, an observable morphology change of PSCs occured in acidic medium. In pH 7.4 medium, PSCs had a stellate, thin and angular appearance (Fig. 1A), and the acid exposure made PSCs shorter and thicker (Fig. 1B), but ASIC1a-specific inhibitor PcTx1 can lessen this change (Fig. 1C). Then, we investigated the cellular distribution of ASIC1a in PSCs using immunofluorescence assay, which is a very useful method for imaging subcellular localization, exploring the interaction of PSCs and tumor cells.15,16 As shown in Fig. 2A, ASIC1a was mainly present in plasma membrane, and the fluorescence intensity of ASIC1a in pH 6.5 group was stronger than that in pH 7.4 group, indicating this protein expression increased. The phenomenon was further confirmed by the results from western blotting and qRT-PCR, which showed that, compared with pH 7.4 group, there was around 4-fold and 2-fold significant increase of ASIC1a protein and ASIC1a mRNA under acidic stimuli, respectively (Fig. 3).
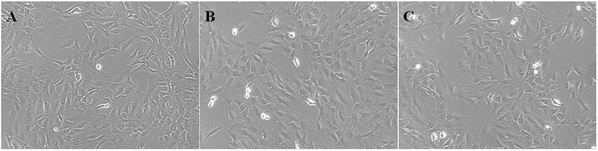 |
| | Fig. 1 Morphology of PSCs at pH 7.4 medium (A), pH 6.5 medium (B) or pretreatment with PcTx1 and then culture in acid medium (C) was observed (400 X). | |
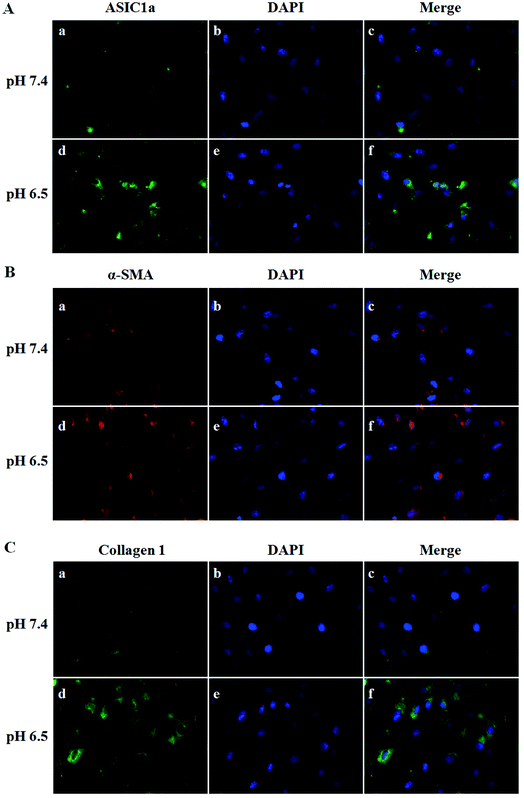 |
| | Fig. 2 Immunofluorescence staining of ASIC1a (A), α-SMA (B) and collagen 1 (C) in PSCs cultured in different pH medium. ASIC1a, collagen 1 and α-SMA were labeled with FITC (green) and Alexa Fluor 555 (red), respectively, and the nuclei were counterstained with DAPI (400 X). | |
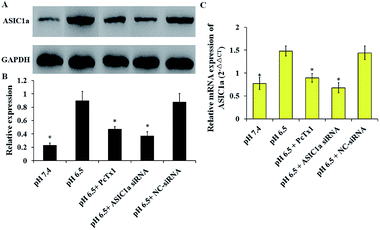 |
| | Fig. 3 Expression of ASIC1a in PSCs in different culture condition was assessed by western blotting and qRT-PCR. Western blotting results show that ASIC1a protein is expressed in PSC, and extracellular acid could increase its expression, but PcTx1 or siRNA knockdown of ASIC1a could inhibit the acid-induced expression of ASIC1a (A and B), which are consistent with the results of ASIC1a mRNA detected by qRT-PCR (C). Data are expressed as mean ± SD of three independent experiments. *P < 0.05 versus pH 6.5 group. | |
The pH sensitivity of ASIC1a, namely pH50, is 5.8–6.8, and as expected, the acidic medium (pH 6.5) promoted ASIC1a expression.6 PcTx1 is a 40-residue peptide isolated from the venom of the South American tarantula Psalmopoeus cambridgei, and has a very high selectivity for ASIC1a but does not affect other ion channels including ASIC1b.17 Therefore, PcTx1 has become a potent pool for the functional study of ASIC1a. Gene silencing using siRNA, as a powerful tool for the study on molecular function, was used to knock down ASIC1a expression in our study. As shown in Fig. 3, the expression of ASIC1a protein and ASIC1a mRNA in pH 6.5 medium was inhibited to 52.3% and 60.5% by PcTx1, respectively, while the corresponding data in ASIC1a knockdown group was 41.3% and 45.9%, respectively, but the effect can not be observed in NC-siRNA group. In a word, the present data identified that ASIC1a is expressed in PSCs, and extracellular acid can upregulate ASIC1a expression. It has been reported that tissue acidosis can occur in several pathological conditions such as inflammation, lesions and cancer.12 PSCs play an integral role in the pathogenesis of pancreatitis and pancreatic cancer, and according to the present in vitro study, an acidic environment will contribute to enhance the expression of ASIC1a in PSCs, but this effect on the development and progression of these two major diseases of the pancreas is still unknown and worth further study. At least, in analogy with HSCs, the role of ASIC1a in the mechanism of pancreatic fibrosis need to be addressed in the further.
Inhibition of ASIC1a suppresses the activation of PSCs induced by extracellular acid
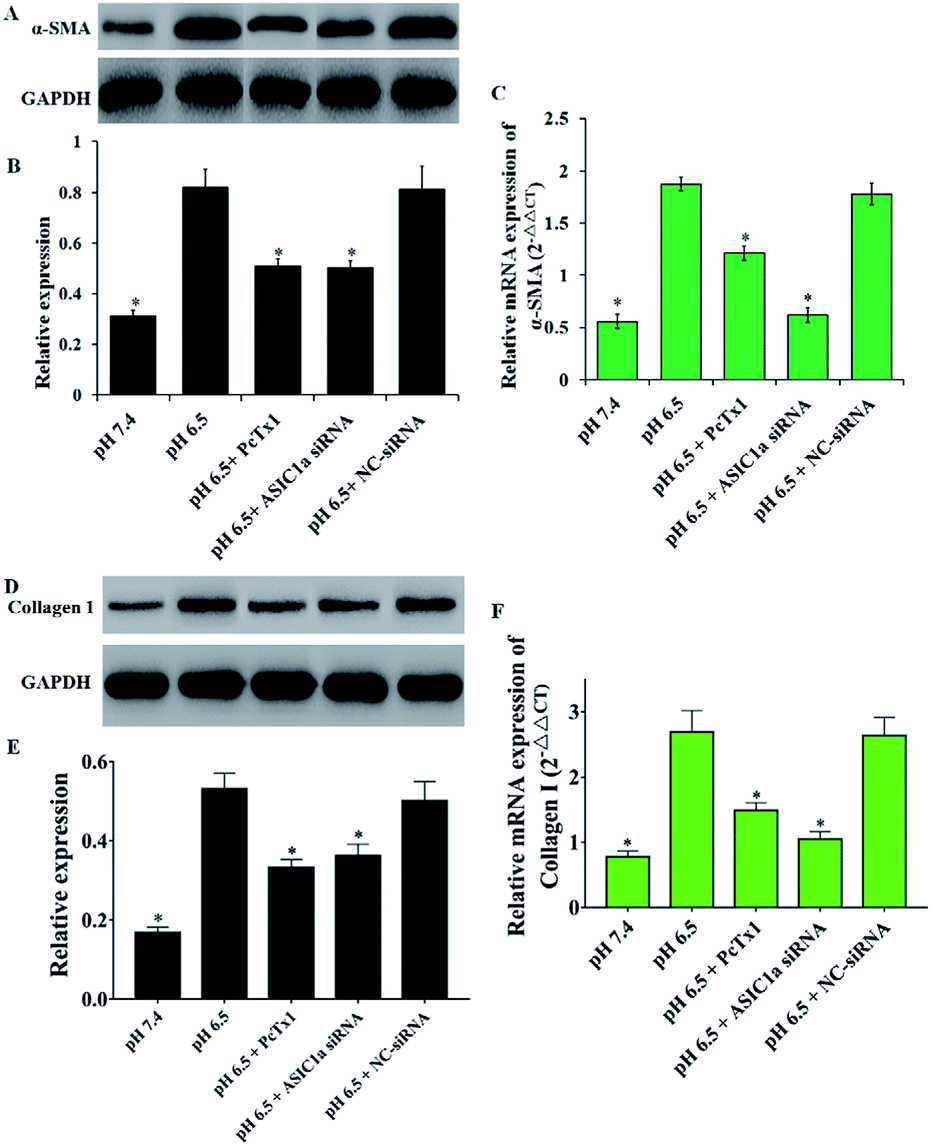
Activated PSCs play a pivotal role in the pathologic processes of pancreatic diseases, and inhibition of PSCs activation, or even the transformation of activated PSCs into quiescence PSCs, has been found to be beneficial to the treatment of pancreatic cancer and pancreatitis.18–20 The activated PSCs are positive for α-SMA, collagen 1, so this two molecules are often used as the marker of PSCs activation.21 As shown in Fig. 2B and C, compared with pH 7.4, PSCs cultured in pH 6.5 have stronger red fluorescence (α-SMA) and green fluorescence (collagen 1), suggesting that extracellular acid can promote the activation of PSCs. Then, the protein and mRNA expression of α-SMA, collagen 1 was detected to further assess the activation of PSCs in different pH medium. Consistent with the results from the immunofluorescence assay, the western blotting and qRT-PCR results showed that acid solution exposure significantly increased the expression of α-SMA and collagen 1 both at protein level and mRNA level (Fig. 4). It is conceivable that an acidic environment existed in pancreatic cancer or pancreatitis can induce PSCs activation, and the activated PSCs in turn will produce extensive desmoplasia. Not only that, activated PSCs also have been found in all stages of pancreatic cancer, including premalignant lesions such as IPMNs and PanINs. However, the molecular mechanisms of PSC activation have been not fully understood. Considering that ASIC1a takes part in many acid-induced pathophysiological events, the role of ASIC1a in PSCs activation was explored. As shown in Fig. 4B and C, compared with pH 7.4 medium, the acid treatment made the expression of α-SMA protein and mRNA increase to 2.6 and 3.3 folds, respectively. The similar phenomena was observed in the results of collagen 1 expression (Fig. 4D–F). It is notable that the expression of α-SMA and collagen 1 in PcTx1 pretreatment or ASIC1a knockdown group only has a tiny increase even in acidic medium, and is markedly lower than that in pH 6.5 group (P < 0.05). Based on these results, we can conclude that ASIC1a is involved in the acid-induced activation of PSCs, which can be weakened, at least partly, through the inhibition of ASIC1a.
 |
| | Fig. 4 ASIC1a involves the acid-induced activation of PSCs. The expression of α-SMA (A–C) and collagen 1 (D–F) at protein and mRNA level in PSCs in pH 6.5 medium both increase compared with that in pH 7.4 medium, but PcTx1 or siRNA knockdown of ASIC1a could reduce the enhanced expression of α-SMA and collagen 1. Data are expressed as mean ± SD of three independent experiments. *P < 0.05 versus pH 6.5 group. | |
Inhibition of ASIC1a attenuates the autophagy of PSCs induced by extracellular acid
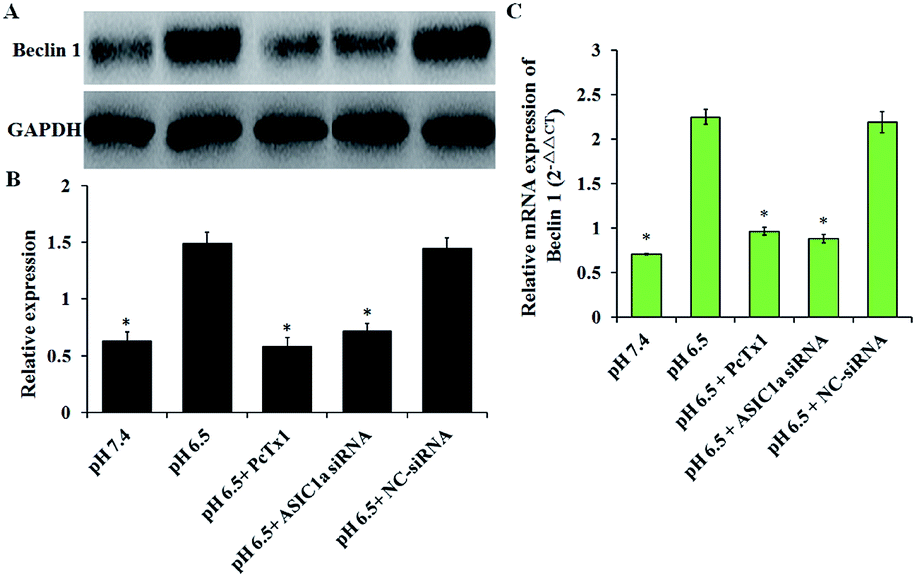
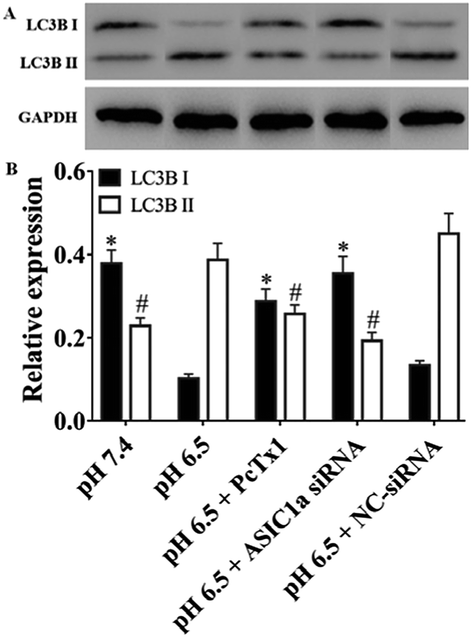
Under stress condition, e.g. acidic environment, starvation, hypoxia, autophagy might be initiated. Autophagy is a major catabolic pathway in which the cell degrades macromolecules and damaged organelles, and beclin 1 has been well-characterized to play a crucial role in the initial stage of autophagy.22 As shown in Fig. 5, the expression of beclin 1 protein and mRNA, was upregulated in pH 6.5 medium compared with pH 7.4 medium, while PcTx1 or ASIC1a knockdown by siRNA could attenuate the acid-induced up-regulation of beclin 1 expression, suggesting that ASIC1a work in the autophagy initiation in acid medium. But, autophagy is a dynamic and complicated process. During the formation of autophagosome, the soluble form of LC3B I is converted to the autophagic vesicle-associated form of LC3B II, and the amount of LC3B II is usually considered to correlate well with the number of autophagosomes.23 So, to further evaluate the effect of ASIC1a on the autophagy flux, the conversion from endogenous LC3B I to LC3B II in different conditions was investigated by western blotting. As shown in Fig. 6, compared with pH 7.4 group, LC3B I expression appeared an obvious downtrend in pH 6.5 group, while LC3B II expression showed an opposite character, and the ratio of LC3B II/LC3B I increased from 0.6 to 3.8. Meanwhile, PcTx1 pretreatment or siRNA knockdown of ASIC1a had a noticeable negative effect on the acid-induced enhanced expression of LC3B II, indicating the formation of autophagosome is almost completely blocked (Fig. 6). All told, the above results exhibit that PSCs autophagy occur in acidic medium, and inhibition of ASIC1a can impair the autophagy largely at the initiation and maturation stage.
 |
| | Fig. 5 ASIC1a involves the acid-induced expression of beclin 1 in PSC. The semiquantitative analyses of the western blotting gels (A and B) and the results of qRT-PCR (C) both show that, compared with pH 7.4 group, an up-regulated expression of beclin 1 occurred at protein and mRNA level in PSCs in pH 6.5 group, which can be suppressed by PcTx1 or siRNA knockdown of ASIC1a. Data are expressed as mean ± SD of three independent experiments. *P < 0.05 versus pH 6.5 group. | |
 |
| | Fig. 6 The protein expression of LC3B II and LC3B I in PSCs with different treatment. Results show the expression of LC3B II increases and the expression of LC3B I decreases after PSCs treated with acid, but remained constant when PSCs were pretreated with PcTx1 or siRNA knockdown of ASIC1a. *,#P < 0.05 versus the expression of LC3B I and LC3B II in pH 6.5 group, respectively. | |
In fact, the autophagy of PSCs has a particular significance for pancreatic disease, especially pancreatic cancer. It has been reported that PSCs can secrete alanine through autophagy and provide an alternative carbon source for pancreatic cancer cells, which is critical for pancreatic cancer metabolism;24 in addition, autophagy is required for activation of PSCs, and autophagic PSCs are associated with shorter survival times and disease recurrence in patients with pancreatic cancer.25 These studies suggest that autophagy of PSCs may become a unique target for the treatment of pancreatic cancer. Since ASIC1a involves the acid-induced autophagy of PSCs, blockade of ASIC1a in PSCs may be useful for the prognosis of pancreatic cancer. However, an in-depth research on the mechanism of ASIC1a-mediated autophagy is certainly need.
It is interesting and noteworthy that inhibition of ASIC1a in PSCs by PcTx1 or siRNA knockdown both can suppress the acid-induced upregulation of ASIC1a, activation- and autophagy-related molecules at protein level and mRNA level. ASIC1a is the only family member that is reported to be permeable to Ca2+, and intracellular Ca2+ might increase under acid environment where ASIC1a is activated by the extracellular H+ and then Ca2+ influx happen. In fact, ASIC1a can work via adjusting intracellular Ca2+ concentration, which has been reported by a lot of literatures.9,11,14,26,27 Ca2+ serves as one of the most important second messengers regulating many important cellular functions, and it may have a positive effect on the expression of ASIC1a and activation and autophagy of PSCs. So, once ASIC1a is blocked, ASIC1a-mediated Ca2+ influx response to acid stimulation is limited, and the downstream processes are affected accordingly. This is one possible explanation for the phenomenon that we have observed in present study, but the exact mechanism need to explore further.
Conclusions
Our present study reports for the first time that ASIC1a is expressed in PSCs, and acid medium can upregulate its expression. Meanwhile, acid exposure also offers a condition for PSCs activation and autophagy, which can be attenuated partly by inhibiting ASIC1a, indicating acid-activated ASIC1a may involve these two important physiopathological events. Considering that the activation and autophagy of PSCs is of great significance for the development and progression of pancreatic diseases, targeting ASIC1a maybe provide a new strategy for the treatment of pancreatic cancer and pancreatitis. However, further efforts should be made to reveal the underlying molecular mechanism on the role of ASIC1a in the interaction between PSCs and pancreatic cancer cells or pancreatic cells.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (Grant No. 81471714, 81170435) and Staring Foundation for Young Researcher of Changhai Hospital (CH201714).
References
- A. Allam, A. R. Thomsen, M. Gothwal, D. Saha, J. Maurer and T. B. Brunner, Pancreatology, 2017, 17, 514–522 CrossRef PubMed.
- S. P. Pothula, Z. Xu, D. Goldstein, R. C. Pirola, J. S. Wilson and M. V. Apte, Cancer Lett., 2016, 381, 194–200 CrossRef PubMed.
- J. A. McCarroll, S. Naim, G. Sharbeen, N. Russia, J. Lee, M. Kavallaris, D. Goldstein and P. A. Phillips, Front. Physiol., 2014, 5, 141 Search PubMed.
- M. Erkan, S. Hausmann, C. W. Michalski, A. A. Fingerle, M. Dobritz, J. Kleeff and H. Friess, Nat. Rev. Gastroenterol. Hepatol., 2012, 9, 454–467 CrossRef PubMed.
- H. J. Kweon and B. C. Suh, BMB Rep., 2013, 46, 295–304 CrossRef PubMed.
- J. A. Wemmie, R. J. Taugher and C. J. Kreple, Nat. Rev. Neurosci., 2013, 14, 461–471 CrossRef PubMed.
- C. Jin, F. L. Yuan, Y. L. Gu, X. Li, M. F. Liu, X. M. Shen, B. Liu and M. Q. Zhu, Oncotarget, 2017, 8, 25977–25988 Search PubMed.
- Q. Zhang, S. Wu, J. Zhu, D. Chai and H. Gan, Gene, 2017, 608, 79–85 CrossRef PubMed.
- Y. Wu, B. Gao, Q. J. Xiong, Y. C. Wang, D. K. Huang and W. N. Wu, Tumor Biol., 2017, 39, 1–8 Search PubMed.
- N. Kapoor, R. Bartoszewski, Y. J. Qadri, Z. Bebok, J. K. Bubien, C. M. Fuller and D. J. Benos, J. Biol. Chem., 2009, 284, 24526–24541 CrossRef PubMed.
- S. Zhu, H. Y. Zhou, S. C. Deng, S. J. Deng, C. He, X. Li, J. Y. Chen, Y. Jin, Z. L. Hu, F. Wang, C. Y. Wang and G. Zhao, Cell Death Dis., 2017, 8, e2806 CrossRef PubMed.
- F. R. Wu, C. X. Pan, C. Rong, Q. Xia, F. L. Yuan, J. Tang, X. Y. Wang, N. Wang, W. L. Ni and F. H. Chen, Mol. Cell. Biochem., 2014, 395, 199–209 CrossRef PubMed.
- C. X. Pan, F. R. Wu, X. Y. Wang, J. Tang, W. F. Gao, J. F. Ge and F. H. Chen, Cell Biol. Int., 2014, 38, 1003–1012 Search PubMed.
- B. Dai, F. Zhu, Y. Chen, R. Zhou, Z. Wang, Y. Xie, X. Wu, S. Zu, G. Li, J. Ge and F. Chen, Int. J. Mol. Sci., 2017, 18, 2125 CrossRef PubMed.
- A. Suetsugu, C. S. Snyder, H. Moriwaki, S. Saji, M. Bouvet and R. M. Hoffman, Anticancer Res., 2015, 35, 2545–2551 Search PubMed.
- C. S. Snyder, A. R. Harrington, S. Kaushal, E. Mose, A. M. Lowy, R. M. Hoffman and M. Bouvet, Pancreas, 2013, 42, 952–958 CrossRef PubMed.
- N. J. Saez, E. Deplazes, B. Cristofori-Armstrong, I. R. Chassagnon, X. Lin, M. Mobli, A. E. Mark, L. D. Rash and G. F. King, Br. J. Pharmacol., 2015, 172, 4985–4995 CrossRef PubMed.
- A. T. Lee, Z. Xu, S. P. Pothula, M. B. Patel, R. C. Pirola, J. S. Wilson and M. V. Apte, Alcohol.: Clin. Exp. Res., 2015, 39, 2123–2133 CrossRef PubMed.
- M. V. Apte, R. C. Pirola and J. S. Wilson, Front. Physiol., 2012, 3, 344 Search PubMed.
- D. Yeo, P. Phillips, G. S. Baldwin, H. He and M. Nikfarjam, Int. J. Cancer, 2017, 140, 2101–2111 CrossRef PubMed.
- H. Song and Y. Zhang, BMC Cancer, 2018, 18, 36 CrossRef PubMed.
- L. L. Fu, Y. Cheng and B. Liu, Int. J. Biochem. Cell Biol., 2013, 45, 921–924 CrossRef PubMed.
- D. J. Klionsky, K. Abdelmohsen, A. Abe, M. J. Abedin, H. Abeliovich and A. A. Arozena, Autophagy, 2016, 12, 1–222 CrossRef PubMed.
- C. M. Sousa, D. E. Biancur, X. Wang, C. J. Halbrook, M. H. Sherman, L. Zhang, D. Kremer, R. F. Hwang, A. K. Witkiewicz, H. Ying, J. M. Asara, R. M. Evans, L. C. Cantley, C. A. Lyssiotis and A. C. Kimmelman, Nature, 2016, 536, 479–483 CrossRef PubMed.
- S. Endo, K. Nakata, K. Ohuchida, S. Takesue, H. Nakayama, T. Abe, K. Koikawa, T. Okumura, M. Sada and K. Horioka, Gastroenterology, 2017, 152, 1492–1506 CrossRef PubMed.
- Y. Zhang, T. Zhang, C. Wu, Q. Xia and D. Xu, Lab. Invest., 2017, 97, 53–69 CrossRef PubMed.
- Y. Mari, C. Katnik and J. Cuevas, Cell Calcium, 2010, 48, 70–82 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03679a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Qian-qian Wang‡b,
Gui-xia Pan‡a,
Guo-rong Jiaa,
Xiao Lia,
Chao Wangb,
Li-ming Zhang*b and
Chang-jing Zuo
a,
Qian-qian Wang‡b,
Gui-xia Pan‡a,
Guo-rong Jiaa,
Xiao Lia,
Chao Wangb,
Li-ming Zhang*b and
Chang-jing Zuo