
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoptisine suppresses tumor growth and progression by down-regulating MFG-E8 in colorectal cancer
Qianyu Cao†
a,
Shengwei Hong† a,
Yuanyuan Li†a,
Heng Chena,
Yining Shena,
Kang Shaoa,
Mengjie Lua,
Hui Daia,
Shitang Ma*b and
Guoliang Dai*c
a,
Yuanyuan Li†a,
Heng Chena,
Yining Shena,
Kang Shaoa,
Mengjie Lua,
Hui Daia,
Shitang Ma*b and
Guoliang Dai*c
aThe First Clinical College, Nanjing University of Chinese Medicine, Nanjing 210023, P. R. China
bCollege of Life and Health Sciences, Anhui Science and Technology University, Fengyang 233100, P. R. China. E-mail: mast@ahstu.edu.cn
cDepartment of Clinical Pharmacology, Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing 210029, P. R. China. E-mail: daiguoliang2@qq.com
First published on 3rd September 2018
Abstract
Treating colorectal cancer (CRC) continues to be a clinical challenge. Coptisine, an alkaloid derived from Coptis chinensis Franch. shows toxic effects on CRC cells, but its underlying mechanism remains elusive. MFG-E8 is involved in tumor growth and progression. Herein, we evaluated the effects of coptisine on MFG-E8 in CRC, and explored the mechanism. The expression of MFG-E8 in CRC and adjacent normal colon tissue samples from patients was detected. The effects of coptisine on CRC cells HCT116 in vitro were evaluated by CCK-8, adhesion and transwell assays. A xenograft tumor model was used to assess the effects of coptisine in vivo. The morphology of CRC tissue was observed by HE staining. Cell signaling was tested using western blotting and immunohistochemical assay. The expression of MFG-E8 in human CRC tissue samples significantly increased compared with that of adjacent normal ones. Coptisine significantly reduced the expressions of MFG-E8 in HCT116 cells and tumor-bearing mice. Moreover, coptisine suppressed the growth, adhesion and metastasis of CRC cells. Coptisine also suppressed the expression of MMP-2 and MMP-9 via the PI3K/AKT signaling pathway. Furthermore, it inhibited epithelial–mesenchymal transition in vivo and in vitro. Coptisine inhibited CRC growth and progression by down-regulating MFG-E8, and is a potential candidate for treatment.
1. Introduction
Colorectal cancer (CRC), as one of the most common tumors in the world, seriously threatens human health. Surgery is preferred for early cases. However, patients are primarily diagnosed at the advanced stage and have already suffered from metastasis.1,2 Therefore, it is urgent to treat CRC with a safer and more effective compound.The phosphatidylinositol (PI) 3-kinase-AKT (PI3K-AKT) pathway is critical for the growth and survival of cancer cells,3 being related to matrix metalloproteinases (MMPs) and implicated in cancer progression.4 MMPs cleave many different targets, and modulate important signaling pathways participating in cell growth, migration, angiogenesis and inflammation. Especially, MMP-2 and MMP-9 play key roles in the metastasis of CRC, such as promotion of tumor-induced angiogenesis and tumor invasion.5 Furthermore, the PI3K-AKT signaling pathway dominantly regulates the epithelial–mesenchymal transition (EMT) process,6 a crucial step in the migration and invasion of many cancers including CRC.7 During EMT, the expression of E-cadherin decreases, resulting in the loss of connection between epithelial cells that then acquire mesenchymal characteristics and increase migratory and invasive capacities.8 Therefore, studying the underlying mechanism of CRC to find the upstream signals of PI3K/AKT may be helpful to develop new therapies.
Milk fat globular-epidermal growth factor 8 protein (MFG-E8) is a secreted pleiotropic glycoprotein that promotes breast morphogenesis, angiogenesis and tumor development. Besides regulating tumor growth, invasion, angiogenesis and EMT, MFG-E8 is also involved in the progression of a variety of tumors including breast cancer, ovarian cancer and CRC.9–11 MFG-E8 overexpression has been positively correlated with the clinical stage of CRC tissues.9 In addition, knocking down MFG-E8 gene can inhibit tumor growth in a murine CRC model.12 Recently, Zhao et al. found that MFG-E8 overexpression promoted CRC progression via the AKT/MMPs signaling pathway.13 In short, MFG-E8 plays a key role in the onset and progression of CRC, inferring that CRC may be treated by inhibiting MFG-E8.
Coptis chinensis Franch., which has antibacterial, hypolipidemic, antitumor and many other pharmacological effects, has been widely used in China. As one of the main bioactive components in C. chinensis, coptisine exhibits similar pharmacological activities.14 Recently, Huang et al. reported that coptisine suppressed HCT116 cell-related tumor growth by inhibiting the RAS-ERK pathway.15 However, the underlying mechanism by which coptisine treats CRC remains largely unknown.
Herein, we detected the expressions of MFG-E8 in human CRC samples, and assessed the effects of coptisine on human CRC cells. Coptisine effectively inhibited MMP-2, MMP-9, EMT and PI3K-AKT by suppressing MFG-E8, as well as CRC migration, invasion and metastasis, as a feasible candidate for treatment.
2. Experimental
2.1. Patient selection and tissue preparation
30 patients with CRC were treated at the National Center of Colorectal Surgery, the Third Affiliated Hospital of Nanjing University of Chinese Medicine. All patients underwent radical resection; no patients received either chemotherapy or radiotherapy before surgery. This study followed the tenets of the Declaration of Helsinki, and informed written consent was obtained from all patients and controls after clinicians explained the purpose, nature and possible consequences of the study. The study protocol was approved by the Medical Ethics Committee of the Third Affiliated Hospital of Nanjing University of Chinese Medicine (KY2014004).2.2. Reagents
Coptisine was purchased from Sigma-Aldrich (St. Louis, MO, USA). MFG-E8 antibody was purchased from Santa Cruz (USA). Antibodies for PI3K, AKT, p-AKT, MMP-9, MMP-2, E-cadherin, snail, N-cadherin and vimentin were purchased from Abcam (CA, USA). Antibody for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was from Sigma-Aldrich (St. Louis, MO, USA). CCK8 kit was purchased from Biosharp (Hefei, China). All other chemicals were from Sigma-Aldrich.2.3. Hematoxylin–eosin (HE) staining
The HE staining method was as reported previously.16 Briefly, paraffin-embedded colon tissue was sectioned (4–5 μM). Slides were dewaxed and rehydrated. Hematoxylin staining for 4 min, eosin staining for 90 s, dehydration from low to high concentrations of alcohol gradient, and then transparent with xylene. Finally, use neutral resin to seal.2.4. Western blot
The tumor tissues from human or tumor-bearing mice added with RIPA lysis buffer, homogenized and lysed for 30 min on ice, and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to prepare a protein sample. Protein concentration was detected by BCA method.17 According to the molecular weight of the target protein, an appropriate concentration of SDS-PAGE gel was prepared. After the sample was applied, it was electrified and subjected to vertical electrophoresis. The protein was properly separated and stopped. Wet transfer film. Blocked for more than 1 hour. Add diluted primary antibody and incubate overnight at 4 °C on a shaking shaker. Diluted secondary antibody was added and incubated at room temperature for two hours. Wash 3 times. Run the gel imaging system software, chemiluminescence imaging. Take a picture and save it. The primary antibodies used included glyceraldehyde 3-phosphate dehydrogenase, 1:5000; MFG-E8, 1:100; PI3K, 1:500; AKT, 1:500; p-AKT, 1:500; MMP-9, 1:500; MMP-2, 1:500; E-cadherin 1:1000; snail 1:500, N-cadherin 1:1000 and vimentin 1:1000.
000 rpm to prepare a protein sample. Protein concentration was detected by BCA method.17 According to the molecular weight of the target protein, an appropriate concentration of SDS-PAGE gel was prepared. After the sample was applied, it was electrified and subjected to vertical electrophoresis. The protein was properly separated and stopped. Wet transfer film. Blocked for more than 1 hour. Add diluted primary antibody and incubate overnight at 4 °C on a shaking shaker. Diluted secondary antibody was added and incubated at room temperature for two hours. Wash 3 times. Run the gel imaging system software, chemiluminescence imaging. Take a picture and save it. The primary antibodies used included glyceraldehyde 3-phosphate dehydrogenase, 1:5000; MFG-E8, 1:100; PI3K, 1:500; AKT, 1:500; p-AKT, 1:500; MMP-9, 1:500; MMP-2, 1:500; E-cadherin 1:1000; snail 1:500, N-cadherin 1:1000 and vimentin 1:1000.
2.5. Cell culture
Human colon cancer cell line HCT116 was purchased from the American Type Culture Collection. Cells were cultured with DMEM containing 10% fetal bovine serum (FBS, Life Technologies), 100 U ml−1 penicillin and 100 mg ml−1 streptomycin and incubated in a cell incubator containing 5% CO2, at 37 °C.2.6. CCK assay for cell proliferation
Cell Count Kit-8 (CCK-8) was used to test cell proliferation. HCT116 cells were seeded at 2 × 104 cells per well in 96-well plates. Then incubated with medium containing different concentrations of coptisine for 24 hours. After that, 10 μl of CCK-8 reagent was added to each well and incubated for 4 hours. The microplate reader measures absorbance (OD) at a wavelength of 450 nm. The cell viability of the control group was defined as 100%, and the cell viability was calculated according to the formula: cell viability = (OD of test group/OD of control group) × l00%.2.7. Cell adhesion assays
Cell adhesion assay was done as described previously.18 In brief, HCT116 cells were treated with different concentrations of coptisine for 24 h. Before adhesion, 96-well plates were treatment with FN (20 μg ml−1) -coated for 30 min. Then Cells (1 × 104 cells per well) were added to FN-coated 96-well plates and labeled with FDA (5 μg ml−1) for 20 min; unbound cells were removed by 2 washes with PBS. Immunofluorescence images were taken with a DMI3000B inverted microscope (Leica, Germany).2.8. In vitro migration and invasion assays
In brief, serum-free medium containing different concentrations of coptisine (20, 40, 80 μg ml−1) was added to the upper chamber of the transwell (Corning, USA), and the complete medium was added to the lower chamber and cultured for 24 hours. The cells in the upper chamber were removed and the cells on the underside of the membrane were fixed, stained with crystal violet and observed under the microscope.2.9. Xenograft tumor models
HCT116 cells (2 × 106) were injected subcutaneously into the right flank of nude mice. Four days after HCT116 vaccination, each mouse was injected with coptisine (30, 60, 90 mg kg−1, i.p.) daily for 14 d. Control mice were administered saline. The tumor volume was measured with a caliper every 3 d and the tumor volume was calculated according to the formula [(length × width2)/2]. 21 d after inoculation of HCT116 cells, all mice were sacrificed and the tumors were removed and weighed.2.10. Immunohistochemical staining
Take tissue, quickly place in paraformaldehyde solution and fix and prepare wax block. After sectioning, the wax block was dewaxed and rehydrated. Endogenous peroxidases in tissue sections were inactivated with 3% H2O2 and washed after antigen retrieval. The goat serum was used for blocking, followed by incubation of the primary antibody at 4 °C overnight and the secondary antibody incubated at 37 °C for 1 hour. DAB color solution was added for staining. Hematoxylin counterstained. Dehydrated after sealing, using light microscopy.2.11. Statistical analysis
Statistical analysis was carried out using t test or ANOVA. Results are expressed as mean ± SD. p < 0.05 was considered as a significant difference. All analyses were performed with GraphPad Prism Version 5.01 (GraphPad Software Inc., San Diego, CA, USA).3. Results
3.1. MFG-E8 was significantly up-regulated in CRC tissues
First, we detected the expressions of MFG-E8 in human CRC and adjacent normal colon tissue samples. The hematoxylin and eosin (HE) staining of 30 pairs of samples showed moderately differentiated adenocarcinomas (Fig. 1A). Immunohistochemical assay presented that the expression of MFG-E8 in human CRC tissue was significantly increased (Fig. 1B and C). Furthermore, western blot revealed that the expression of MFG-E8 in human CRC tissue was significantly up-regulated than that of adjacent normal colon tissue (Fig. 1D and E). Collectively, MFG-E8 was significantly up-regulated in CRC tissues.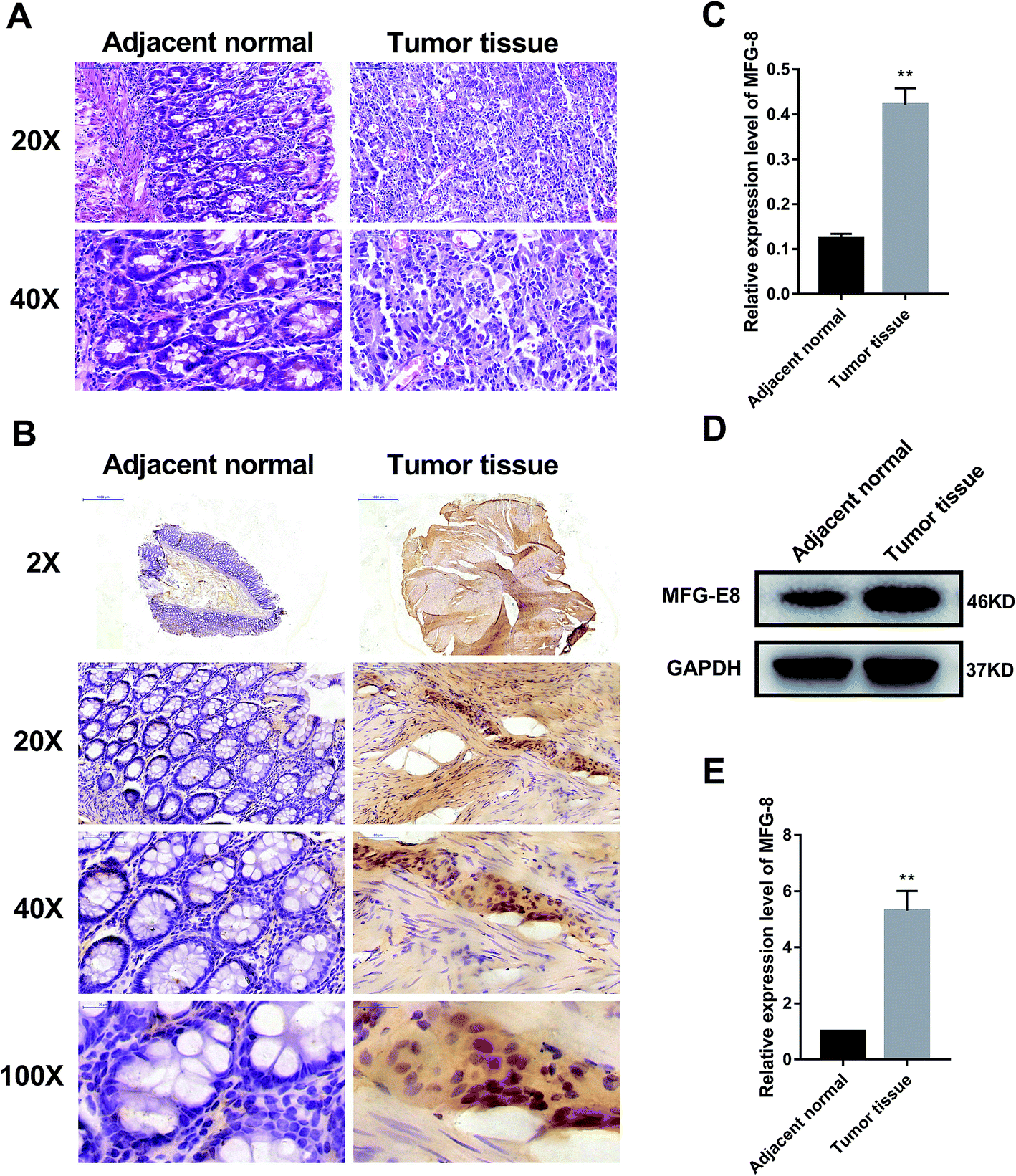 | ||
| Fig. 1 MFG-E8 was significantly activated in CRC tissues. (A) HE staining of human adjacent normal and CRC tissue samples from patients with CRC. (B) and (C) MFG-E8 expressions in human tissue samples. The protein levels of MFG-E8 were detected by immunohistochemical assay. (D) and (E) Protein expressions of MFG-E8 in human tissue samples detected by western blot. *p < 0.05, **p < 0.01, versus adjacent normal tissue sample. | ||
3.2. Coptisine reduced MFG-E8 expression and showed potent cytotoxicity towards HCT116 cells
Afterwards, we studied whether coptisine reduced the expression of MFG-E8. Western blotting revealed that coptisine markedly reduced MFG-E8 expression (Fig. 2A).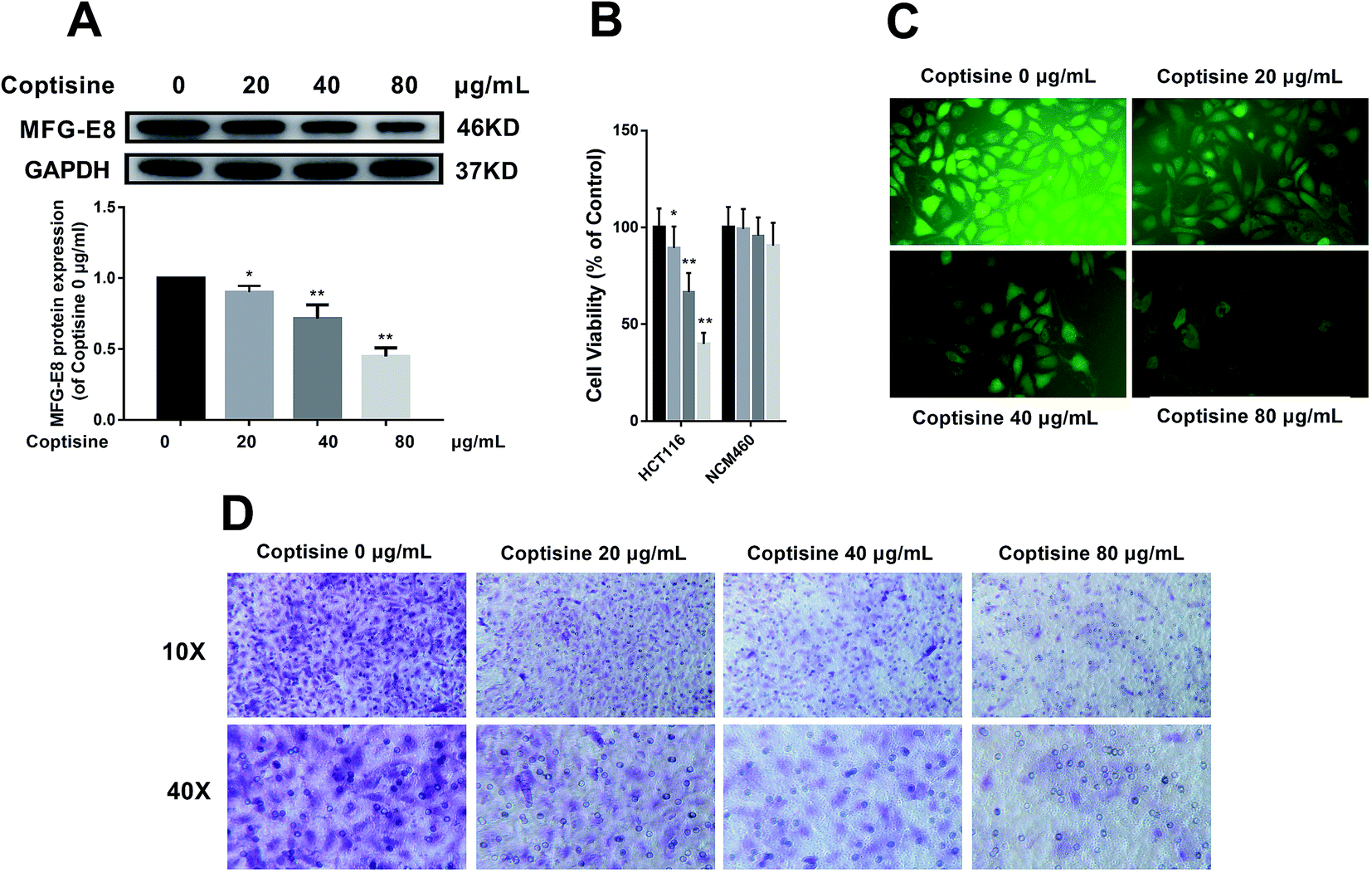 | ||
| Fig. 2 Coptisine inhibited MFG-E8 expression, viability, adhesion and metastasis of CRC cells. (A) Effects of coptisine on the protein expression of MFG-E8 in CRC cells. The protein level of MFG-E8 was detected by western blot. *p < 0.05, **p < 0.01 versus control group without any treatment. (B) Cytotoxicity of coptisine against HCT116 cells and NCM460 cells. (C). HCT116 cells treated with or without coptisine (20, 40, 80 μg ml−1) were employed for the adhesion assay. (D) HCT116 cells treated with or without coptisine (20, 40, 80 μg ml−1) were used for the transwell assay. | ||
It has previously been reported that knockdown of MFG-E8 had significant inhibitory effects on tumors,19 so we next studied whether coptisine functioned as a novel chemotherapeutic agent for human CRC. Coptisine significantly suppressed CRC HCT116 cell viability dose-dependently compared with that of the control group, while coptisine had no significant effect on NCM460 cell viability, as evidenced by CCK-8 assay (Fig. 2B). Also, coptisine significantly inhibited the adhesion of HCT116 cells (Fig. 2C). Moreover, it managed to suppress the migration of these cells obviously (Fig. 2D). Overall, coptisine was able to inhibit the growth, adhesion and metastasis of human CRC cells in vitro.
3.3. Coptisine down-regulated expressions of MMP-2 and MMP-9 in HCT116 cells
For CRC, increased MMP-2 and MMP-9 expressions have been correlated with poor treatment outcomes.20 Since knocking down MFG-E8 can significantly inhibit MMP-2 and MMP-9,13 we investigated whether coptisine functioned identically. As shown in Fig. 3, coptisine down-regulates MMP-2 and MMP-9 in dose-dependent manners (20, 40, 80 μg ml−1).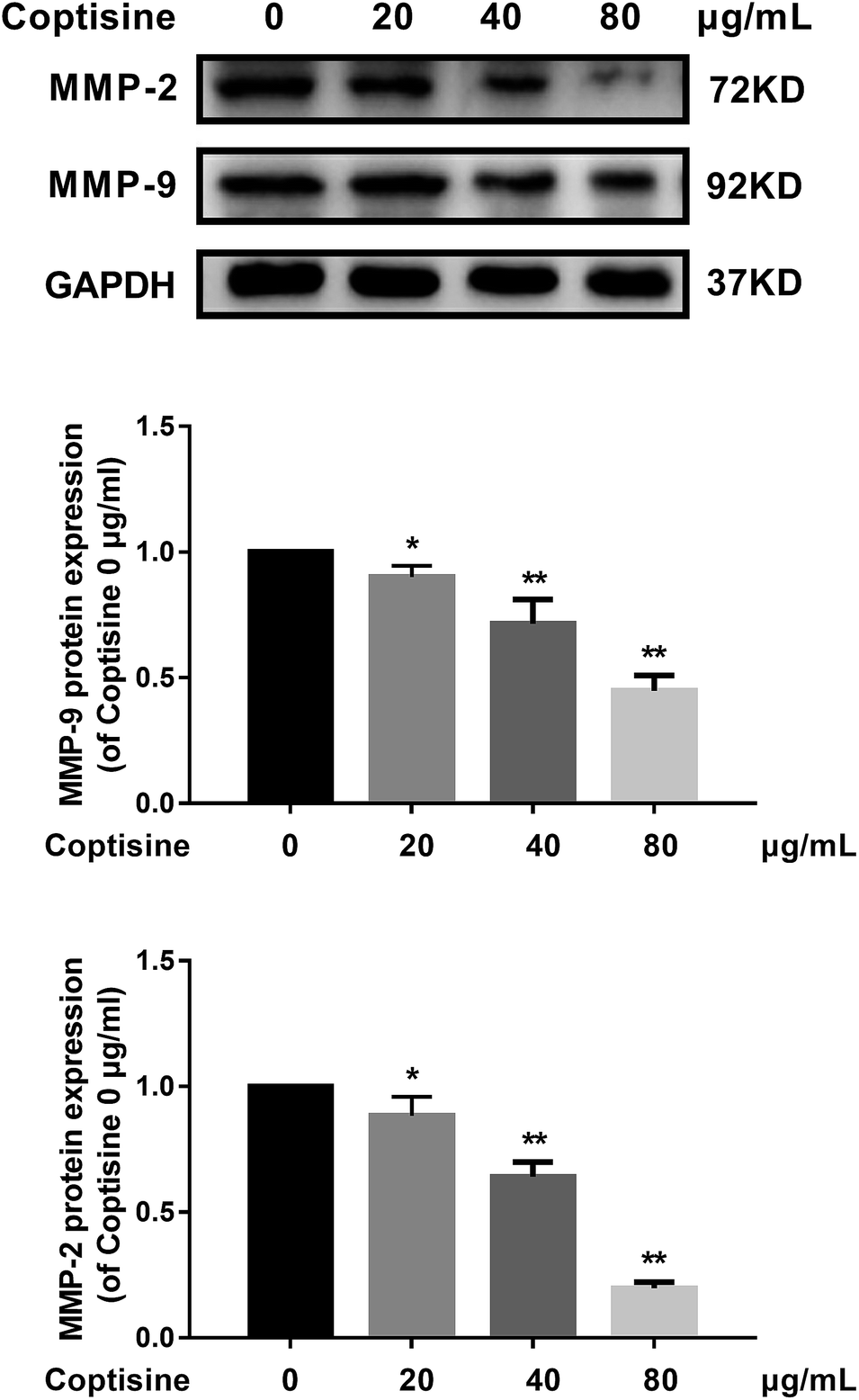 | ||
| Fig. 3 Coptisine inhibited MMP-2 and MMP-9 in CRC cells. MMP-2 and MMP-9 expressions in HCT116 cells after treatment with coptisine (0, 20, 40, 80 μg ml−1) for 24 h were detected by western blot. GAPDH was used as internal control. *p < 0.05, **p < 0.01 versus control group without any treatment. | ||
3.4. Coptisine inhibited EMT of HCT116 cells
MFG-E8 has been reported to induce EMT in multiple tumor cells.13,19 We next examined the ability of coptisine to inhibit EMT in HCT116 cells. HCT116 cells were treated with different concentration of coptisine (0, 20, 40, 80 μg ml−1) for 24 h. The effects of coptisine on EMT markers, including E-cadherin, N-cadherin, vimentin and snail, were confirmed by western blot. The results showed that coptisine significantly increased the expression of E-cadherin and inhibited the expression of N-cadherin, vimentin and snail (Fig. 4). Clearly, coptisine effectively prevented EMT of HCT116 cells in a dose-dependent manner.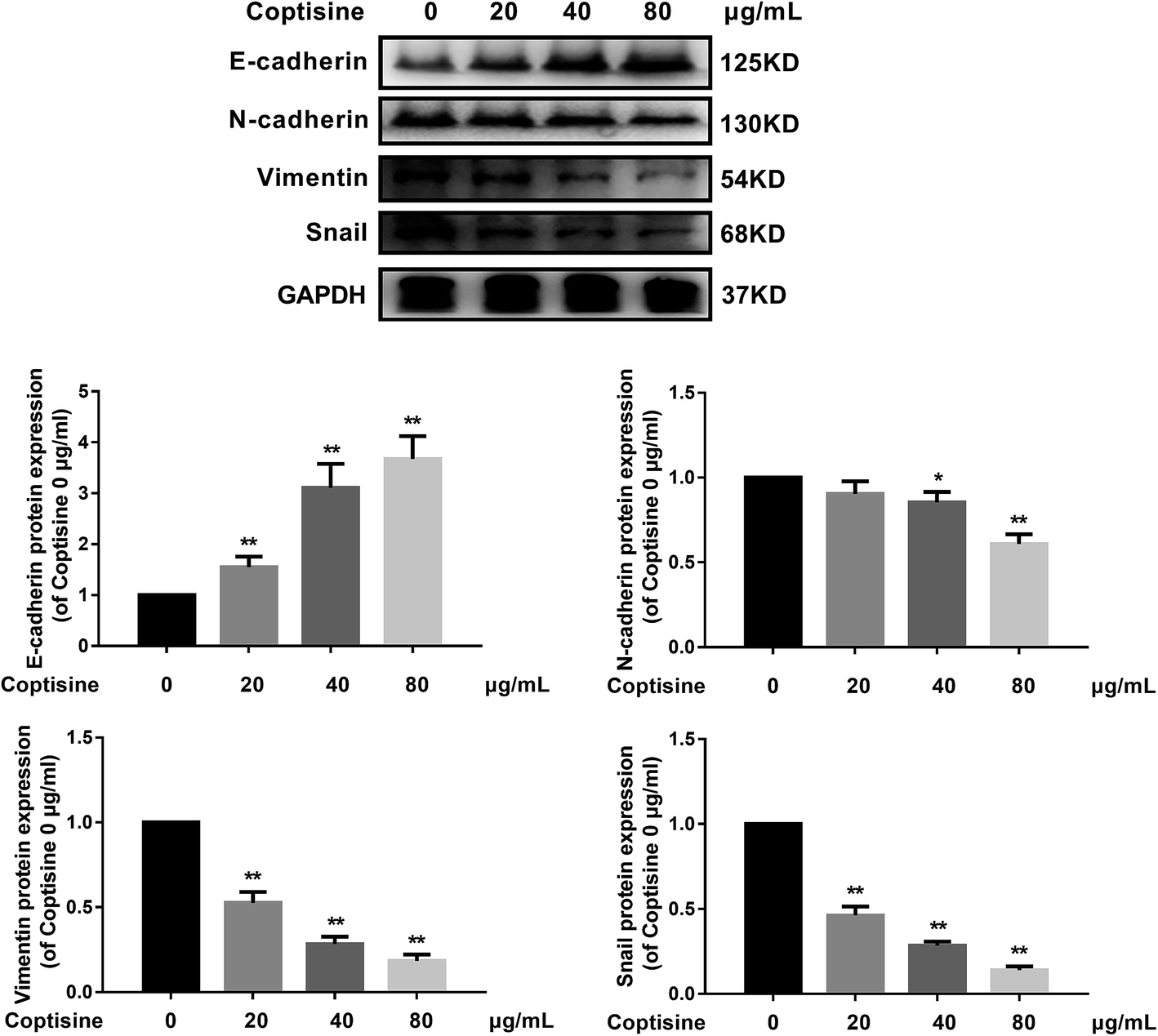 | ||
| Fig. 4 Coptisine inhibited EMT of CRC cells. E-cadherin, N-cadherin, vimentin and snail expressions in HCT116 cells after treatment with coptisine (0, 20, 40, 80 μg ml−1) for 24 h were detected by western blot. GAPDH was used as internal control. *p < 0.05, **p < 0.01 versus control group without any treatment. | ||
3.5. PI3K/AKT signaling pathway was involved in the antitumor effects of coptisine
MFG-E8 can enhance tumorigenicity and metastatic capacity via AKT-dependent pathways.19 We herein detected total AKT and phosphorylated AKT (p-AKT) by western blotting, and found that HCT116 cells treated with coptisine had lower p-AKT level than that of the control group (Fig. 5A). To confirm the role of AKT activation in the antitumor effects of coptisine, we employed a PI3K/AKT inhibitor LY294002. LY294002 inhibited the protein expressions of N-cadherin, vimentin and snail, whereas up-regulated that of E-cadherin in HCT116 cells (Fig. 5B). Moreover, it suppressed the protein expressions of PI3K and p-AKT (Fig. 5C). Accordingly, inhibition of the PI3K/AKT pathway was involved in the antitumor effects of coptisine.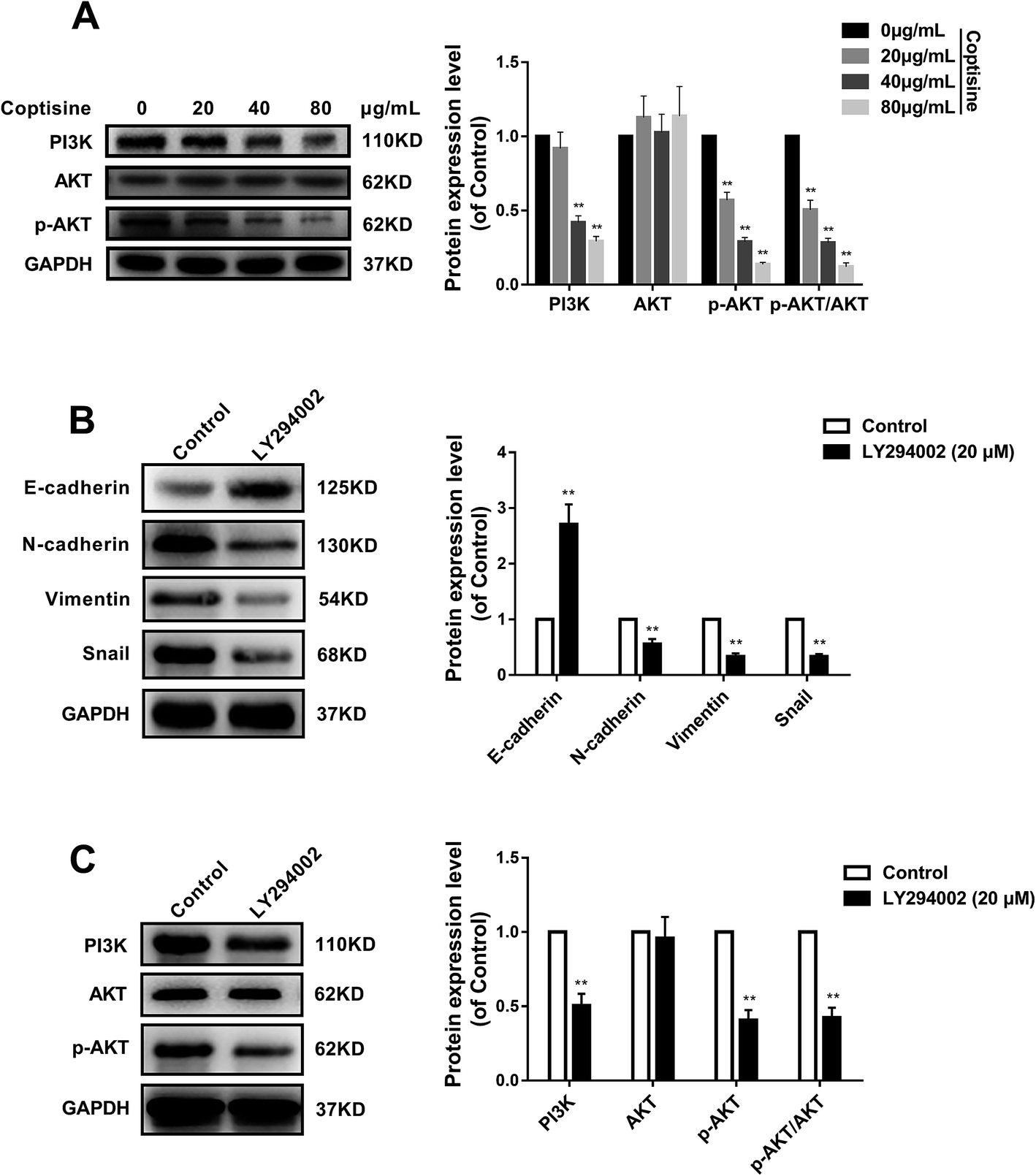 | ||
| Fig. 5 Coptisine inhibited the PI3K/AKT pathway in MFG-E8-induced downstream molecule expressions in CRC cells. (A) Western blotting revealed p-AKT was significantly down-regulated in coptisine-treated HCT116 cells compared with that of control group. (B) HCT116 cells were pretreated with LY294002 (20 μM) or DMSO for 24 h of incubation. Western blotting was then performed to detect E-cadherin, N-cadherin, vimentin and snail protein levels. (C) HCT116 cells were pretreated with LY294002 (20 μM) or DMSO for 24 h of incubation. Western blotting was then performed to detect AKT and p-AKT protein expression levels. Band densities were quantified by Quantity One software and normalized to that of GAPDH. *p < 0.05, **p < 0.01 versus control group. *p < 0.05, **p < 0.01 versus control group without any treatment. | ||
3.6. Coptisine inhibited tumor growth by down-regulating MFG-E8 in xenograft model
To further investigate the inhibition of coptisine in MFG-E8 in vivo, HCT116 cells were inoculated into nude mice. The tumor volumes of coptisine treatment groups were markedly smaller than those of control group. The tumor volume and weight were analyzed in the control and coptisine treatment group (Fig. 6A–C). Coptisine treatment significantly reduced tumor volume and weight in xenografts. The HE analysis of coptisine treatment groups and model group of tumor samples demonstrated tumor growth was inhibited (Fig. 6D).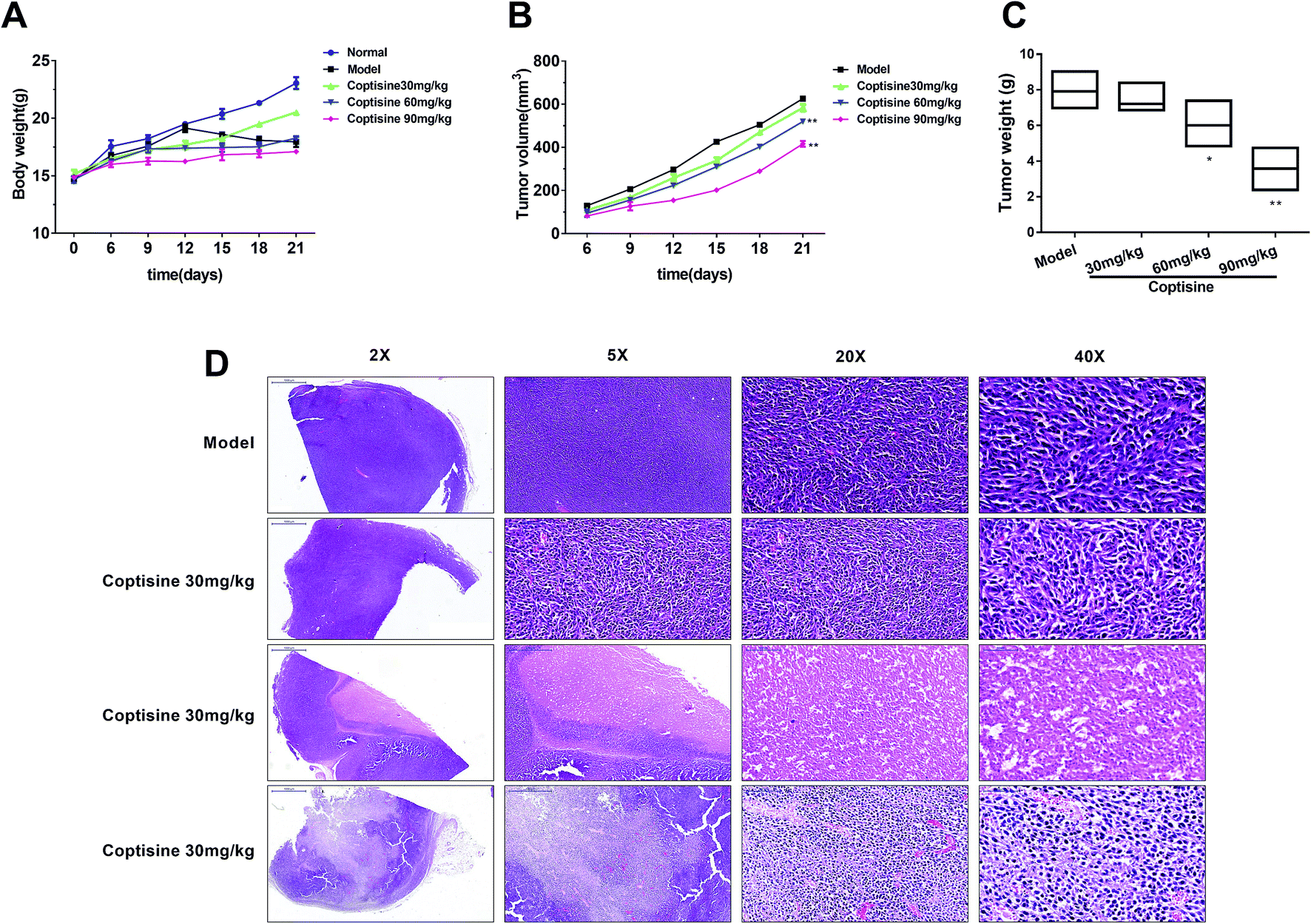 | ||
| Fig. 6 Coptisine inhibited xenograft tumor growth in vivo. HCT116 cells (2 × 106) were injected subcutaneously into the right flanks of nude mice. HCT116 cell-xenografted nude mice (n = 6) were injected with PBS (control) and coptisine (30, 60, 90 mg kg−1 body weight) for 21 days. (A) Tumor size was measured every 3 days after tumor cell inoculation for 6 days. (B) Body weight was measured every 3 days after tumor cell inoculation for 6 days. (C) Tumor weight was measured after treatment for 21 days. (D) Images of HE-stained mouse tumor segments in control and coptisine treatment groups. HE staining of tumor segments. *p < 0.05, **p < 0.01 versus control group. | ||
Then we measured the expression of MFG-E8. Western blotting revealed that coptisine significantly inhibited the expression of MFG-E8, being consistent with the in vitro results (Fig. 7A). Subsequently, we studied whether coptisine was able to inhibit MMP-2 and MMP-9 in vivo. As shown in Fig. 7B, coptisine down-regulates MMP-2 and MMP-9 obviously in the xenograft model. As to EMT in vivo, coptisine significantly up-regulated the expression of E-cadherin but down-regulated those of N-cadherin, vimentin and snail (Fig. 7C), suggesting that the EMT process was effectively prevented. Given that coptisine decreased p-AKT level compared with that of the control group (Fig. 7D), it significantly inhibited tumor growth by down-regulating MFG-E8.
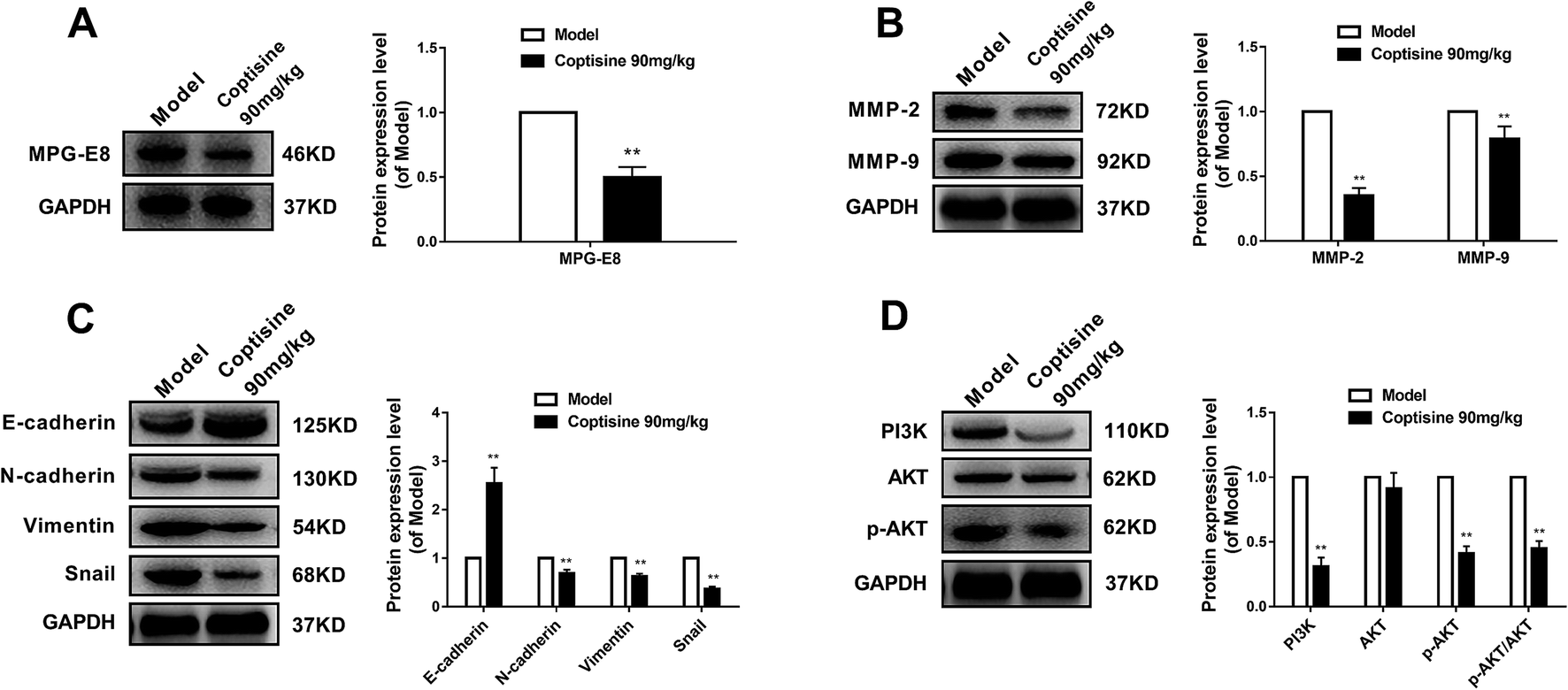 | ||
| Fig. 7 Coptisine down-regulated MFG-E8 expressions of xenograft tumors in vivo. HCT116 cells (2 × 106) were injected subcutaneously into the right flanks of nude mice. HCT116 cell-xenografted nude mice (n = 6) were injected with PBS (control) and coptisine (90 mg kg−1 body weight) for 21 days. (A) Effects of coptisine on MFG-E8 expression. GAPDH was used as internal control. (B) Effects of coptisine on the expressions of MMP-2 and MMP-9. GAPDH was used as internal control. (C) Effects of coptisine on the expressions of E-cadherin, N-cadherin, vimentin and snail. GAPDH was used as internal control. (D) Effects of coptisine on the expressions of AKT and p-AKT. GAPDH was used as internal control. *p < 0.05, **p < 0.01 versus control group. | ||
4. Discussion
In this study, the expression of MFG-E8 in human CRC tissue samples significantly increased compared with that of adjacent normal ones. Coptisine significantly reduced the expressions of MFG-E8 in HCT116 cells and tumor-bearing mice. Besides, coptisine suppressed the growth, adhesion and metastasis of CRC cells. It also suppressed the expressions of MMP-2 and MMP-9 via the PI3K/AKT signaling pathway. Additionally, coptisine inhibited EMT in vivo and in vitro. Taken together, these results may help clarify the role of coptisine in CRC through MFG-E8 and provide an alternative option for future treatment.Upon initial diagnosis, 20–25% of patients with colon cancer and 18% of cases with rectal cancer have already undergone metastasis.2 Advanced cancers are routinely treated by chemotherapy which, nevertheless, induces nausea, vomiting and pain, probably requiring drug dose reduction or discontinuation. Therefore, it is of great significance to develop new therapies based on the unraveling of pathogenic mechanisms.
In this study, western blotting and immunohistochemical assay demonstrated that MFG-E8 protein was overexpressed in CRC tissues (Fig. 1B–E), being in accordance with previous literatures.9,13 It is well-documented that MFG-E8 was highly expressed in CRC, exerting pro-proliferative effects.11,12 Jinushi et al. reported that MFG-E8 significantly increased in melanoma, and promoted tumor invasion, EMT and angiogenesis via an AKT-dependent pathway.19 Meanwhile, Yang et al. found that MFG-E8 increased in triple-negative (ER (−), PgR (−), erbB2 (−)) breast cancer, but decreased in ER (+) and erbB2 (+) breast cancer as a tumor suppressor.21 Hence, MFG-E8 may play different roles in various cells. In addition, overexpressing MFG-E8 can facilitate CRC progression, whereas knocking it down can inhibit CRC cell growth.13 Thus, MFG-E8 may be a marker for CRC progression, and also a promising target for anti-CRC drug development.
It has recently been verified that MFG-E8 overexpression promoted melanoma and CRC progression through AKT signaling.13,19 The PI3K/AKT signaling pathway can suppress CRC cell apoptosis, stimulate growth and proliferation, and facilitate malignant growth.22 This pathway has also been involved in the direct induction of EMT, which both inhibits the degradation of snail and directly up-regulates its expression in cells.6 Oral cancer has persistent AKT activation and low E-cadherin expression. The expression of snail, an important molecule in EMT, reduces at both transcriptional and translational levels after treatment with AKT inhibitors. In head and neck squamous cell carcinoma, activation of PI3K/AKT increases MMP9 expression, degrades E-cadherin as well as promotes cell invasion and migration. We herein found that treatment with PI3K/AKT inhibitor LY294002 significantly inhibited MMP-2, MMP-9 and EMT in HCT116 cells. Notably, the PI3K/AKT signaling pathway also activates the αVβ3 integrin receptor pathway.23 MFG-E8 is a ligand for αVβ3/5 integrin and a key activator of the PI3K/AKT pathway in some cells.13,19 Therefore, inhibition of MFG-E8 may suppress CRC progression by inhibiting the PI3K/AKT signaling pathway.
Coptisine evidently down-regulated the protein expression of MFG-E8 in a dose-dependent manner, exerting antitumor effects like those of MFG-E8 knockdown. It significantly suppressed EMT via the PI3K/AKT pathway that has been considered to predominantly drive the EMT process in CRC. EMT is a complex manifestation of epithelial plasticity and contributes to tumor metastasis. The loss of epithelial marker E-cadherin in CRC has been associated with high histological grade and metastatic capacity. Herein, coptisine was able to inhibit the growth, adhesion and metastasis together with the protein expressions of N-cadherin, vimentin and snail of HCT116 cells (Fig. 3 and 6). In contrast, it up-regulated the expression of E-cadherin protein (Fig. 4). Moreover, coptisine treatment significantly reduced the tumor volume and weight of xenografts (Fig. 6A–C). Furthermore, it significantly inhibited the protein expressions of N-cadherin, vimentin and snail, whereas up-regulated that of E-cadherin in vivo (Fig. 7C). Meanwhile, coptisine notably down-regulated the expressions of MMP-2 and MMP-9 via the PI3K/AKT pathway in vivo and in vitro. Since treatment of HCT116 cells with LY294002 showed identical effects on PI3K and p-AKT expressions to those of coptisine, the PI3K/AKT signaling pathway participated in the antitumor effects of coptisine. The findings suggested that coptisine inhibited EMT of CRC by suppressing MFG-E8 as well as cell invasion and migration.
Coptisine, as a safe and effective compound, is the main anticancer component of C. chinensis, with an LD50 value of over 800 mg kg−1. In this study, the optimal dose for suppressing CRC in mice was 90 mg kg−1 which was exceedingly safe. Therefore, CRC may be safely and effectively treated by coptisine.
In summary, MFG-E8 was expressed in CRC tissue samples. Coptisine inhibited the growth, adhesion, EMT and metastasis of human CRC cells by suppressing MFG-E8/PI3K/AKT signaling. Thus, MFG-E8 inhibition may be studied by using coptisine which may be an eligible candidate for treating CRC.
Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 81573685), Medical Technology Development Foundation of Nanjing China (grant no. ZKX16056), Postdoctoral Scientific Research Foundation (grant no. BSH201501) and Medical Technology Development Foundation of Nanjing China (grant no. ZKX14046).References
- D. Cunningham, W. Atkin, H. J. Lenz, H. T. Lynch, B. Minsky, B. Nordlinger and N. Starling, Lancet, 2010, 375, 1030–1047 CrossRef.
- M. De Rosa, U. Pace, D. Rega, V. Costabile, F. Duraturo, P. Izzo and P. DelrioGenetics, Oncol. Rep., 2015, 34, 1087–1096 CrossRef PubMed.
- J. A. Fresno Vara, E. Casado, J. de Castro, P. Cejas, C. Belda-Iniesta and M. Gonzalez-Baron, Cancer Treat. Rev., 2004, 30, 193–204 CrossRef PubMed.
- B. Z. Guan, R. L. Yan, J. W. Huang, F. L. Li, Y. X. Zhong, Y. Chen, F. N. Liu, B. Hu, S. B. Huang and L. H. Yin, Cell Adhes. Migr., 2018, 12, 109–117 CrossRef PubMed.
- R. F. Araujo Jr, G. A. Lira, J. A. Vilaca, H. G. Guedes, M. C. Leitao, H. F. Lucena and C. C. Ramos, Pathol., Res. Pract., 2015, 211, 71–77 CrossRef PubMed.
- W. Xu, Z. Yang and N. Lu, Cell Adhes. Migr., 2015, 9, 317–324 CrossRef PubMed.
- A. Loboda, M. V. Nebozhyn, J. W. Watters, C. A. Buser, P. M. Shaw, P. S. Huang, L. Van't Veer, R. A. Tollenaar, D. B. Jackson, D. Agrawal, H. Dai and T. J. Yeatman, BMC Med. Genomics, 2011, 4, 9 CrossRef PubMed.
- B. P. Wijnhoven, W. N. Dinjens and M. Pignatelli, Br. J. Surg., 2000, 87, 992–1005 CrossRef PubMed.
- M. Jia, H. Yao, C. Chen, Y. Wang, H. Wang, T. Cui and J. Zhu, Arch. Med. Res., 2017, 48, 270–275 CrossRef PubMed.
- C. Yang, D. Sgroi and E. Schmidt, Cancer Res., 2007, 14–18 Search PubMed.
- Y. S. Yi, Mediators Inflammation, 2016, 2016, 5628486 Search PubMed.
- R. Kusunoki, S. Ishihara, Y. Tada, A. Oka, H. Sonoyama, N. Fukuba, N. Oshima, I. Moriyama, T. Yuki, K. Kawashima, M. M. Ansary, Y. Tajima, R. Maruyama, T. Nabika and Y. Kinoshita, J. Gastroenterol., 2015, 50, 862–875 CrossRef PubMed.
- Q. Zhao, L. Xu, X. Sun, K. Zhang, H. Shen, Y. Tian, F. Sun and Y. Li, Tumour Biol., 2017, 39 DOI:10.1177/1010428317707881.
- F. Tang, W. Mei, D. Tian and D. Huang, Evidence-based Anticancer Materia Medica, 2011, vol. 26, pp. 111–130 Search PubMed.
- T. Huang, Y. Xiao, L. Yi, L. Li, M. Wang, C. Tian, H. Ma, K. He, Y. Wang, B. Han, X. Ye and X. Li, Sci. Rep., 2017, 7, 38524 CrossRef PubMed.
- G. Wang, G. Dai, J. Song, M. Zhu, Y. Liu, X. Hou, Z. Ke, Y. Zhou, H. Qiu, F. Wang, N. Jiang, X. Jia and L. Feng, Front. Pharmacol., 2018, 9, 301 CrossRef PubMed.
- R. C. Krieg, Y. Dong, K. Schwamborn and R. Knuechel, J. Biochem. Biophys. Methods, 2005, 65, 13 CrossRef PubMed.
- Y. T. Tai, K. Podar, L. Catley, Y. H. Tseng, M. Akiyama, R. Shringarpure, R. Burger, T. Hideshima, D. Chauhan, N. Mitsiades, P. Richardson, N. C. Munshi, C. R. Kahn, C. Mitsiades and K. C. Anderson, Cancer Res., 2003, 63, 5850–5858 Search PubMed.
- M. Jinushi, Y. Nakazaki, D. R. Carrasco, D. Draganov, N. Souders, M. Johnson, M. C. Mihm and G. Dranoff, Cancer Res., 2008, 68, 8889–8898 CrossRef PubMed.
- A. H. Said, J. P. Raufman and G. Xie, Cancers, 2014, 6, 366–375 CrossRef PubMed.
- C. Yang, T. Hayashida, N. Forster, C. Li, D. Shen, S. Maheswaran, L. Chen, K. S. Anderson, L. W. Ellisen, D. Sgroi and E. V. Schmidt, Cancer Res., 2011, 71, 937–945 CrossRef PubMed.
- S. A. Danielsen, P. W. Eide, A. Nesbakken, T. Guren, E. Leithe and R. A. Lothe, Biochim. Biophys. Acta, 2015, 1855, 104–121 Search PubMed.
- A. S. Antonov, G. N. Antonova, D. H. Munn, N. Mivechi, R. Lucas, J. D. Catravas and A. D. Verin, J. Cell. Physiol., 2011, 226, 469–476 CrossRef PubMed.
Footnote |
| † These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |