
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a lignin-based phosphorus-containing flame retardant and its application in polyurethane
Y. M. Zhanga,
Q. Zhaoa,
L. Lib,
R. Yana,
J. Zhanga,
J. C. Duana,
B. J. Liuc,
Z. Y. Sund,
M. Y. Zhanga,
W. Hu *a and
N. N. Zhang*a
*a and
N. N. Zhang*a
aCollege of Chemical Engineering, Changchun University of Technology, 2055 Yan'an Street, Changchun 130012, P. R. China. E-mail: huwei@ccut.edu.cn; Fax: +86-431-85717216; Tel: +86-431-85717216
bSchool of Life Science and Technology, Harbin Institute of Technology, Harbin 150080, P. R. China
cCollege of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China
dState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
First published on 18th September 2018
Abstract
In this work, new lignin-based flame retardant LHDs were successfully synthesized through the reaction between lignin, 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO) and hexamethylene diisocyanate (HDI). The chemical structure of LHD was characterized by FTIR, 1H NMR, 31P NMR. The thermal stability of LHD was studied by TGA. The results showed that the residual carbon content of L15HD (15% of lignin in LHD) at 600 °C reached 16.55%, indicating that this prepared flame retardant can be a type of good char forming agent. LHDs were then applied to prepare flame-retardant lignin-based polyurethane (FLPU). Lignin-based polyurethane (LPU) was synthesized by the reaction between lignin, polyethylene glycol 200 (PEG 200) and hexamethylene diisocyanate (HDI). The limiting oxygen index (LOI) value of the FLPU reached 30.2% when the addition content of L15HD (15% lignin in LHD) in L20PU (20% lignin in LPU) was 25%, exhibiting excellent flame-retardant properties. Scanning electron microscopy (SEM) analysis of the FLPU char residual showed that there was a continuous dense outer carbon layer on the residue surface, and the inner carbon layer had many expansion bubbles, indicating the LHDs have an excellent flame retardant effect for PU. In addition, FLPU presented better hardness and adhesion than PU. The hardness of FL15-25L20PU (lignin content in LPU was 20%, and added content of L15HD in LPU was 25%) reached 4H, and its adhesion was 0. These excellent properties illustrated that the LHDs are ideal flame retardants and reinforcing agents for LPU because of the co-curing and strong interface between LHD and LPU.
Introduction
Lignin, a complex natural aromatic polymer, is the second most abundant biomass next to cellulose. At present, lignin is widely available as a major by-product of the pulp and paper industries as well as bio-refineries.1–3 It is composed of three different types of phenylpropane units, p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol.2,4 They form a rigid, three-dimensional network structure.5 With the impressive properties of renewability, biodegradability, environmental friendliness and low price, lignin has become an excellent sustainable green chemical raw material.2–4,6 Lignin has various functional groups, such as hydroxyl units (both phenolic and aliphatic), which make it easy to be modified.4,5,7 On the other hand, thanks to its aromatic structure, lignin could be advantageously applied in polymeric matrices as a char promotor agent that can allow some reduction of the combustion rate. As a consequence, the enormous potential of lignin in value-added products is its application as the precursor of different classes of polymeric materials, including epoxies,8,9 polyesters,10,11 phenolics12 and polyurethanes.13,14 One strategy to broaden the practical application of lignin in industry for environmental protection and the utilization of natural resources has outstanding economic and social benefits.Polyurethane (PU) contained urethane linkage (–NH–CO–O–), which was synthesized by a reaction of diisocyanate with diol (with functionality higher than 2). Polyurethane has been widely capitalized on aerospace, automotive, coatings, and other fields because of their unique merits, such as low density, high tensile strength, and high dimensional stability.15 PU has rapidly grown to be one of the most diverse and widely used plastics with a continuously increasing global market. Nonetheless, diol, one of the most important compounds dependent on petrochemical resources, is crucial for the expenses of PU. There are phenol and hydroxyl groups on lignin, which could also react with the isocyanate. It is very promising to replace the fossil sources by the renewable lignin as additives or part of the reactant monomer to obtain eco-friendly bio-polyurethane.
On the other hand, the inherently high flammability of PU has restricted its application. Hence, to extend the durable applications of PU, its flame retardant property need to be improved. To improve the fire-resistant property of PU is an interesting research subject. To improve the fire-resistant property of PU is an interesting research subject. Adding the flame-retardant (FR) additives into the polymers is the most popular approach to improve the flame retardancy.16–18 Many literatures had reported that lignin and its derivatives were used as flame retardants of polyurethanes and other materials.
For example, Lu et al. used lignosulfonate to substitute part of diethylene glycol (DEG) to copolymerize with isocyanate to produce lignosulfonate based rigid polyurethane (LRPU) foams. Lignosulfonate was employed as carbonization agent and combined with APP to improve the flame retardancy of LRPU.19 Gao et al. obtained liquefied lignin-based polyol by liquefying calcium lignosulphonate with a polyhydroxy alcohol mixture. And it was simultaneously incorporated with commercial flame retardant polyol, polyurethane microencapsulated ammonium polyphosphate and organically modified layered double hydroxide into rigid polyurethane foam (RPUF). The results indicated that the mechanical property, thermal performance, flame retardancy and fire behaviour of RPUF composites were improved.20 Zhu et al. prepared a flame retardant modified lignin (LPMC) containing chemically grafted phosphorus-nitrogen by liquefaction, esterification and salt formation. And then the LPMC was applied to prepare polyurethane foam. The LOI value of polyurethane foam was 26.7%, passing the UL-94 measurement V-1 grade with no dropping.21 However, the lignin in many studies was necessary to be purified before use, and the modification process were so complicated in the experiments. Others also applied the modified lignin to polypropylene,22 polylactic acid,23 and acrylonitrile butadiene styrene, etc.,24 but the properties were always not so preferable. The specific comparison of some related reports are shown in Table 1.
| References | Modified method | Flame retardancy |
|---|---|---|
| 19 | Lignosulfonate/APP | The heat release rate (HRR) and total heat release (THR) was reduced; the LOI value increased to be over 30% |
| 20 | Liquefied lignin-based polyol (LBP)/MAPP/organically modified layered double hydroxide (OLDH) | LOI was up to 28.1%; the initial degradation temperature of modified foam increased from 228 °C to 265 °C, and the mass residue of foam at 900 °C increased from 0.6 to 15.4% |
| 21 | Lignin was chemically grafted with phosphorus–nitrogen-containing groups via a liquefaction–esterification–salification process to prepare lignin-based phosphate melamine compound (LPMC) | LOI values reached to be 28.3%; the UL-94 tests were achieved V-1 rate |
| 23 |  |
The samples reached V-0 in UL-94 test |
| 28 |  |
BKZ tests were reached the highest to HF-2; thermal stability was improved |
Nowadays, 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO) and its derivatives are considered as suitable alternative to halogenated flame retardants in the light of its effective extinguishing behaviour in gas phase and condensed phase, and its environmentally friendliness.25 And so far, there was no report on the use of HDI as a bridge to link commercial lignin molecular with DOPO to produce a flame retardant with low expenses.
In this work, a lignin-based flame-retardant noted as LHD was synthesized by the reaction of DOPO, hexamethylene diisocyanate (HDI) and lignin. Diisocyanate was used as a bridge to connect lignin and DOPO. This could also be an effective way for solving the compatibility between the flame retardant and the matrix PU or lignin-based PU (LPU). The mechanical properties of the flame-retardant lignin-based polyurethane (FLPU) composites can be reinforced by LHD because it contained the same urethane structure with polyurethane and the phenyl groups in lignin. Thus, the FLPU films could contain not only the flame retardant element phosphorus but also the bioproduct lignin, which could act as the reinforcing agent and carbonizing agent. The performance of the flame retardant lignin based PU films in terms of thermal stability, flame retardancy and properties as coatings were studied in detail.
Experimental
Material
DOPO (99%) was purchased from Zhengzhou Alpha Chemical Co., Ltd. Organosolv lignin was obtained from Yanghai Chemical Co., Ltd. Hexamethylene diisocyanate (HDI) (99%) was provided by Macklin. PEG200 (Mr = 190–210 g mol−1) was brought from Tianjin Institute of Fine Chemical rehabilitation. Dibutyltin dilaurate (95%) as a catalyst was purchased from Macklin. Triethylamine (AR) was obtained from West Long Chemical Co., Ltd. N,N-dimethylformamide (95%, AR) was provided by Tianjin Tiantai Fine Chemicals Co. Ltd. Ethanol (AR, 98 v%) was purchased from Sinopharm Chemical Reagent Co., Ltd.Synthesis of lignin-based flame retardant (LHDs)
DOPO was dried at 80 °C overnight before use to remove water. To a three-necked round-bottom, DOPO, HDI, lignin (total hydroxyl content is 3.225 mmol g−1) at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of NCO: (–OH and –P–H). The mass of catalyst triethylamine was 0.4% and a certain amount of DMF was added. The amount of DOPO used in all formulations was 0.1 mol. The mixture was reacted at 90 °C for 12 h. Then, the reaction mixture was cooled to room temperature, washed with ethanol thoroughly, and dried at 80 °C for 12 h in a vacuum oven and then ground into powder. The synthesis route was shown in Scheme 1.
:1 molar ratio of NCO: (–OH and –P–H). The mass of catalyst triethylamine was 0.4% and a certain amount of DMF was added. The amount of DOPO used in all formulations was 0.1 mol. The mixture was reacted at 90 °C for 12 h. Then, the reaction mixture was cooled to room temperature, washed with ethanol thoroughly, and dried at 80 °C for 12 h in a vacuum oven and then ground into powder. The synthesis route was shown in Scheme 1.
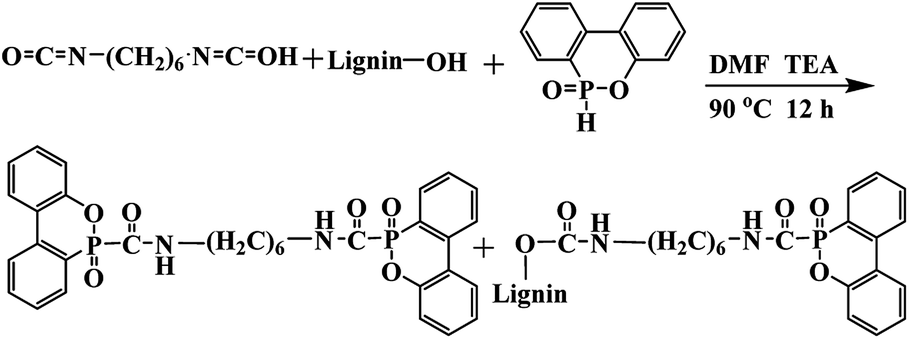 | ||
| Scheme 1 Synthesis of LHDs. | ||
Preparation of the flame-retarding PU composites (FLPU films)
Polyurethanes were prepared through condensation reactions between –NCO in hexamethylene diisocyanate (HDI) and the hydroxyl groups in polyethylene glycol (PEG 200) and lignin. The lignin content was 20%. PEG 200 was used to be the soft segment in polyurethane. In our work, the molar ratio of NCO/OH was 1.3, which was calculated according to the isocyanate group from HDI and total hydroxyl content from lignin (phenolic and aliphatic group) and polyethylene glycol.Typically, FL15-25L20PU (content of lignin in the flame-retardant lignin-based polyurethane was 20%, content of lignin in the LHD was 15%, and the mass fraction of the LHD was 25%) was synthesized as following: 4 g lignin was dissolved in 30 ml of DMF under magnetic stirring. Subsequently, 6.97 g PEG200 was added, then 9.03 g HDI and 0.4 g dibutyltin dilaurate was added. After stirring at 80 °C about 90 min, 6.67 g L15HD powder was added into the prepared polyurethane prepolymer and mixed well. Then the mixture was poured into molds carefully and cured at room temperature overnight, followed by heating at 50 °C, 100 °C for 12 h, respectively. The thickness of the films were about 1 mm.
Characterization
Results and discussion
Chemical characterization of lignin-based flame-retardant (LHDs)
A lignin based flame-retardant noted as LHD was synthesized by the reaction of DOPO, hexamethylene diisocyanate (HDI) and lignin as shown in Scheme 1. Diisocyanate was used as a bridge to connect lignin and DOPO. The flame retardant prepared by the method of the present study was expected to form a conjugation O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–PO, which was helpful to increase the thermal stability and flame retardancy of the product. More OC–PO structure was produced with the increased temperature. The chemical structure of the synthesized LHDs was characterized by FTIR, 1H NMR and 31P NMR.
C–PO, which was helpful to increase the thermal stability and flame retardancy of the product. More OC–PO structure was produced with the increased temperature. The chemical structure of the synthesized LHDs was characterized by FTIR, 1H NMR and 31P NMR.
FTIR characterization of LHDs
Fig. 1 showed the FTIR spectra of DOPO and LHDs. From the FTIR spectrum of DOPO, the peak at 2434 cm−1 was belonged to the stretching vibration of P–H group.21 In case of LHDs, there were several absorption peaks. The peak at 3280 cm−1 was corresponded to N–H group; the peaks at 1593 cm−1 and 1475 cm−1 was attributed to P–C, and the peak at 1200 cm−1 was related to the PO group,25 while the characteristic absorption peak of P–H group in DOPO disappeared. The absorption peaks around 2932 cm−1 and 2857 cm−1 corresponded to the vibration of –CH2 in HDI. Furthermore, in the FTIR spectrum of LHDs, the peak at 1650 cm−1 was assigned as the stretching vibration of CO bond of HDI.25 It was indicated that LHDs have been successfully synthesized.
 | ||
| Fig. 1 FTIR spectra of DOPO and LHDs. | ||
1H NMR characterization of LHDs
The 1H NMR spectra of the synthesized LHDs were presented in Fig. 2. As to DOPO, the peak at 8.83 ppm was attributed to the proton in P–H.26 In the case of LHDs, the chemical shift at 8.83 ppm disappeared and the peaks at 9.3 ppm belonged to N–H appeared.26 This further suggested that the LHDs was synthesized successfully. The peaks between 7.0 and 8.3 ppm were corresponded to the protons in the benzene ring. Additionally, the peaks at 2.5–3.4 ppm were assigned to the CH2 in HDI. The two peaks at 1.1 ppm and 1.4 ppm should be corresponded to the –CH3 of lignin.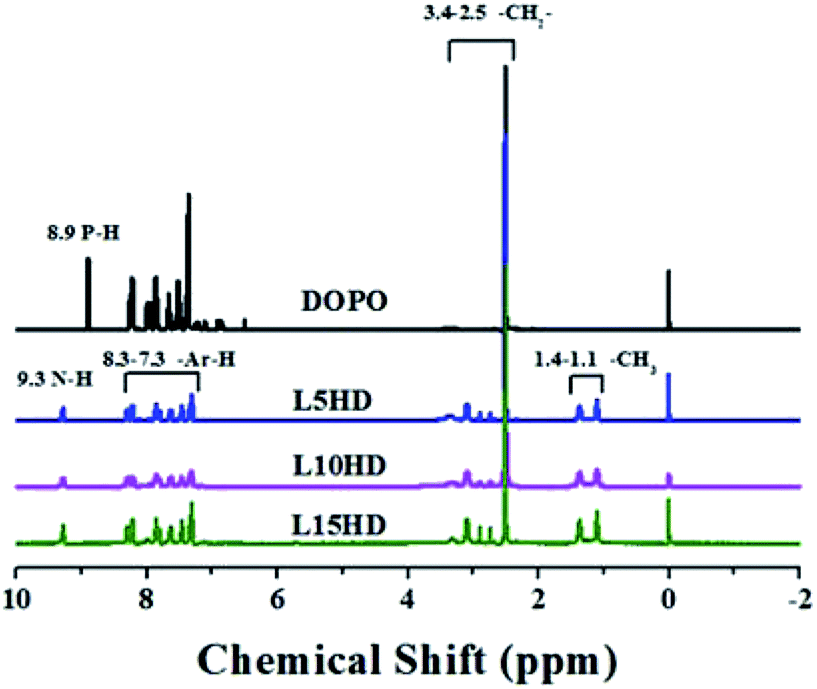 | ||
| Fig. 2 1H NMR of DOPO and LHDs. | ||
31P NMR characterization of LHDs
The 31P NMR spectra of DOPO and LHDs were described in Fig. 3. The peaks at 12.6 ppm and 16.4 ppm were attributed to the phosphorus atoms in DOPO group. The peak at 11.3 ppm was assigned to the phosphorus atoms in the LHDs. All the above characterization results confirmed the chemical structure of the prepared LHDs. It was reasonable to conclude that the LHDs were successfully synthesized.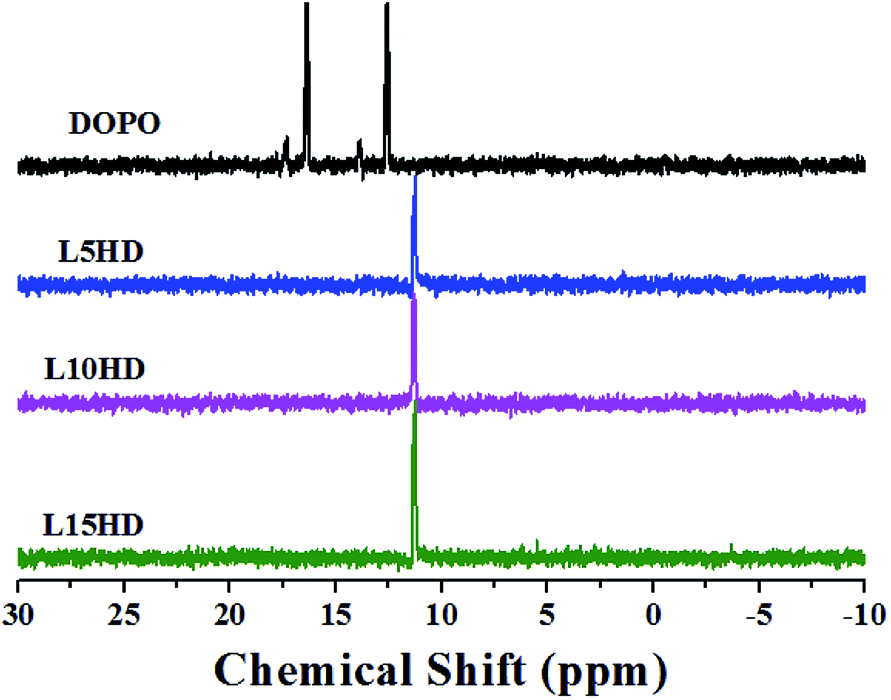 | ||
| Fig. 3 31P NMR of DOPO and LHDs. | ||
TGA analysis of DOPO and LHDs
The detailed information about thermal stability and thermal degradation behaviour of LHDs was investigated by TG analysis test. Fig. 4 shows the TGA curves of samples. The data results are summarized in Table 2.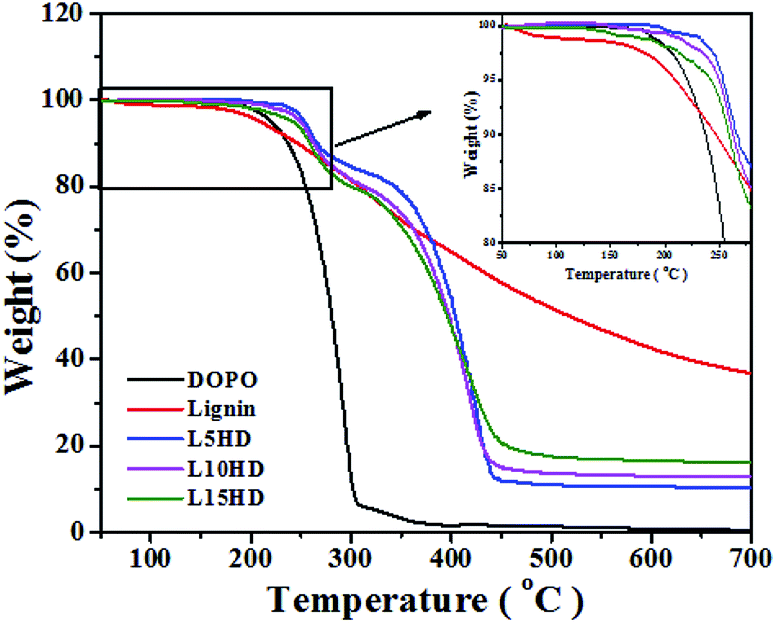 | ||
| Fig. 4 TGA curves of DOPO and LHDs. | ||
| Samples | T5 (°C) | T10 (°C) | Char residue (%) | ||
|---|---|---|---|---|---|
| 500 °C | 600 °C | 700 °C | |||
| a T5: initial degradation temperature (temperature at 5% weight loss). T10: temperature at 10% weight loss. | |||||
| DOPO | 220 | 237 | 1.34 | 0.78 | 0.42 |
| Lignin | 210 | 247 | 51.89 | 42.59 | 36.72 |
| L5HD | 253 | 266 | 10.94 | 10.49 | 10.42 |
| L10HD | 250 | 262 | 13.66 | 13.00 | 12.82 |
| L15HD | 242 | 258 | 17.54 | 16.55 | 16.13 |
It can be seen from Fig. 4 that DOPO started to decompose (5% weight loss temperature (T5)) at 220 °C, and there was no major residue left after 300 °C. This was consistent with another report.25 The thermal degradation temperature of lignin had a broad temperature range.29,30 In the range of 100–170 °C, the loss of water happened. The decomposition of β-aryl-alkyl-ether bond occurred between 180–230 °C.31 T5 of lignin was 210 °C, which was lower than that of DOPO and showed the lower thermal stability of lignin. The degradation of the propanoid side chain of lignin occured in the temperature range of 230–260 °C, with the formation of low molecular weight products. The C–C and β–β linkages in the lignin cleaved at 275–350 °C. Above 500 °C, the formation of char was resulted by the further rearrangements and condensation of the aromatic structure. These results were consistent with other reports.23,30,32,33
The flame retardant prepared by the method of the present study was expected to form a conjugation between OC–PO, which could help to increase the thermal stability of the product. This should be attributed to the beneficial π–π interaction of the aromatic groups between DOPO itself and the additional benzyl of lignin.34 The T5 and T10 of LHDs were much higher than that of DOPO and lignin. T5 of L5HD reached 253 °C. It was shown in Table 2 that the thermal degradation temperature of LHDs gradually decreased with the increase of lignin content, which was mainly because that the lignin portion degraded at lower temperature.
Furthermore, the OC–PO structure could act as acid source and blowing agent during combustion. And it would be thermo-oxidized, dehydrated by the acid, and to act as carbonizing agent. On the other hand, the char yield of lignin was up to 42.59% at 600 °C, as shown in Table 2, indicating that it was an excellent char-forming agent owing to its high content of aromatic ring structure.21 The char was the decomposed product after the lignin was fired at high temperature. Therefore, it should be very stable. It could insulate heat and material transfer, reduce the rate of combustion of the polymer and block oxygen, which play an efficient role in flame retardancy. The ability to form char during the thermal degradation was a basic aspect of flame retardant intumescent systems.22,27
It can be observed that LHDs have higher char yield than DOPO at 600 °C as shown in Fig. 4 and Table 2. The residual carbon content increased with the lignin content. And the residual carbon content of L15HD was as high as 17.54% at 500 °C, which showed high thermal stability.35 The high residual char content was attributed to the ether bonds and aromatic ring structures in lignin.
X-ray photoelectron spectroscopy (XPS) test
The L15HD sample was treated in a muffle furnace under a nitrogen atmosphere at various temperatures, and thereafter XPS measurement were measured to check the elemental composition and nature at various temperature ranges as an example. The XPS measurement of the L15HD at different degradation step temperature: RT, 230, 350 and 500 °C were performed and analysed in detail. The XPS spectra of L15HD treated at different temperature are shown in Fig. 5, and the element content data are summarized in Table 3.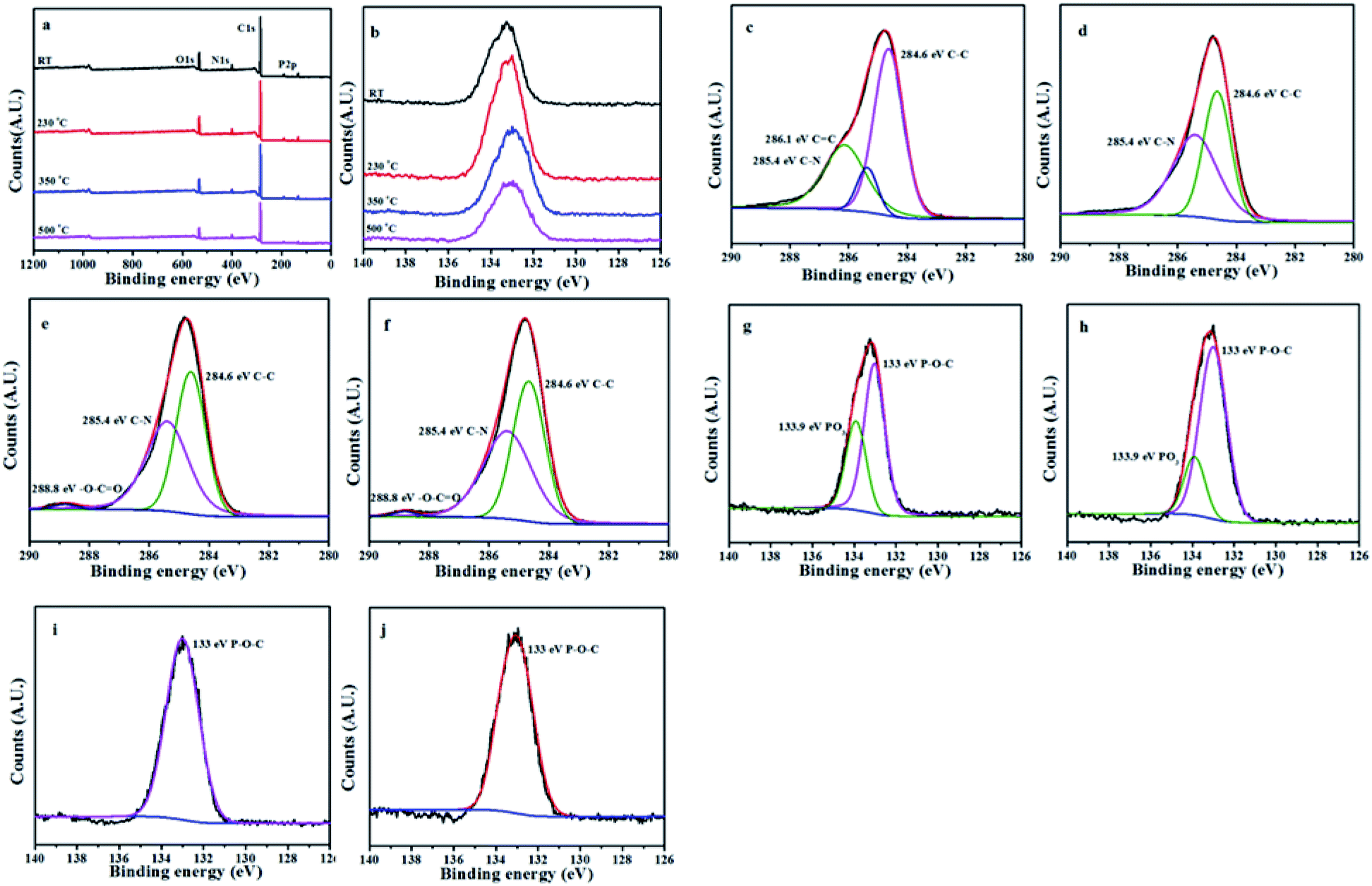 | ||
| Fig. 5 XPS spectra of L15HD at different temperature (a); P2p XPS spectra of L15HD at different temperature (b); C1s XPS spectra at room temperature (c); C1s XPS spectra at 230 °C (d); C1s XPS spectra of at 350 °C (e); C1s XPS spectra at 500 °C (f); P2p XPS spectra at room temperature (g); P2p XPS spectra at 230 °C (h); P2p XPS spectra at 350 °C (i); P2p XPS spectra at 500 °C (j). | ||
| Peak | Temperature (°C) | |||
|---|---|---|---|---|
| RT | 280 | 380 | 500 | |
| C1s | 74.45 | 76.17 | 78.96 | 78.94 |
| O1s | 18.9 | 15.25 | 13.58 | 13.4 |
| N1s | 4.25 | 4.79 | 4.01 | 4.88 |
| P2p | 2.4 | 3.80 | 3.45 | 2.79 |
It can be observed from Fig. 5a and b and Table 3 that the phosphorus content increased first and then decreased with the increasing temperature. The initial decomposition of phosphate was in the solid phase. With the increase of temperature, part of the PO˙ ions would volatilize into gas phase. The C1s spectra was shown in Fig. 5c–f. The peak at around 284.6 eV was attributed to C–C in aliphatic and aromatic species,36–38 the peak at around 285.4 eV was assigned as C–N bonds in the HDI acting as bridges,36 the peak at around 286.1 eV was assigned to CC in aromatic ring structures.39 In addition, as shown in Fig. 5e and f, a new peak appeared at around 288.8 eV which was attributed to –C(O)–O–R groups.37 This may be due to the esterification reaction of the hydroxyl group produced in the decomposition with the phosphoric acid produced from L15HD to form a P–O–C crosslinked structure.37,40 As shown in the P2p XPS spectra of Fig. 5g–j, the peaks at around 133.0 eV and 133.9 eV were assigned as P–O–C and PO3 groups, respectively.36 It further illustrated that the decomposition of flame retardant produced phosphate and P–O–C structure. And the peaks at 133.9 eV in Fig. 5i and j disappeared, which indicated that there was no phosphate left in the carbon residue.
Properties of the flame-retarding LPU (FLPU)
| Samples | LOI (%) | T5 (°C) | T10 (°C) | Residue at 600 °C (%) |
|---|---|---|---|---|
| a T5: initial degradation temperature (temperature at 5% weight loss); T10: temperature 10% weight loss occurs. FL15-25L20PU: content of lignin in the flame-retardant lignin-based polyurethane is 20%, content of lignin in the LHD is 15%, and the mass fraction of the LHD is 25%. | ||||
| PU | 17.1 | 299 | 314 | 2.48 |
| L20PU | 21.3 | 266 | 295 | 19.92 |
| FL5-15L20PU | 25.3 | 263 | 297 | 19.55 |
| FL10-15L20PU | 28.3 | 268 | 290 | 11.87 |
| FL15-15L20PU | 28.9 | 265 | 290 | 17.98 |
| FL15-20L20PU | 29.1 | 257 | 287 | 18.21 |
| FL15-25L20PU | 30.2 | 254 | 286 | 21.01 |
When 15% prepared flame retardant L5HD (5% lignin in LHD) was inserted in L20PU, the LOI value of FLPU was further increased to be 25.3%. This meant LHD was excellent flame retardant for LPU. It was shown the LOI value of FLPU increased with the lignin content in LHDs. When the addition content of LHD was 15%, the LOI value increased from 25.3% to 28.9% with the lignin content in LHD increased from 5% to 15%. This indicated that lignin in LHDs were effective to improve the flame retardancy of LPU.
With the further increase of loading content of LHD, the LOI values of PU continued to increase. The highest LOI value of 30.2% appeared in the FL15-25L20PU with 25% of L15HD inserted in L20PU. On the one hand, as an abundant polyphenol biopolymeric material with substantial aromatic structures, lignin can promote the formation of carbon layer and had a certain degree of flame retardancy.22,41 In addition, phosphorus-containing flame retardant LHD produced phosphate compounds during the decomposition process, which promoted the char forming of the soft segments.42 And then, a continuous and dense expanded carbon layer was formed on the surface of the FLPU. This could effectively prevent the internal and external combustible gases, oxygen and heat to be transferred. It is worth noting that the decomposition of FLPU produced combustible products, which could be entrapped in the interior swelling carbon layer. This helped to improve the flame retardancy of FLPU. The proposed flame retardancy mechanism of FLPU is shown in Fig. 6. As a result, the flame retardancy of LHD was improved. And thus, the flame retardant property of FLPU was improved with the increasing content of LHDs. Furthermore, all of the FLPUs in this study did not show dripping during burning. The results evidenced that lignin has a significant char formation and synergistic flame retardancy effect for LHDs and FLPU.
 | ||
| Fig. 6 Flame-retardancy mechanism for FLPU. | ||
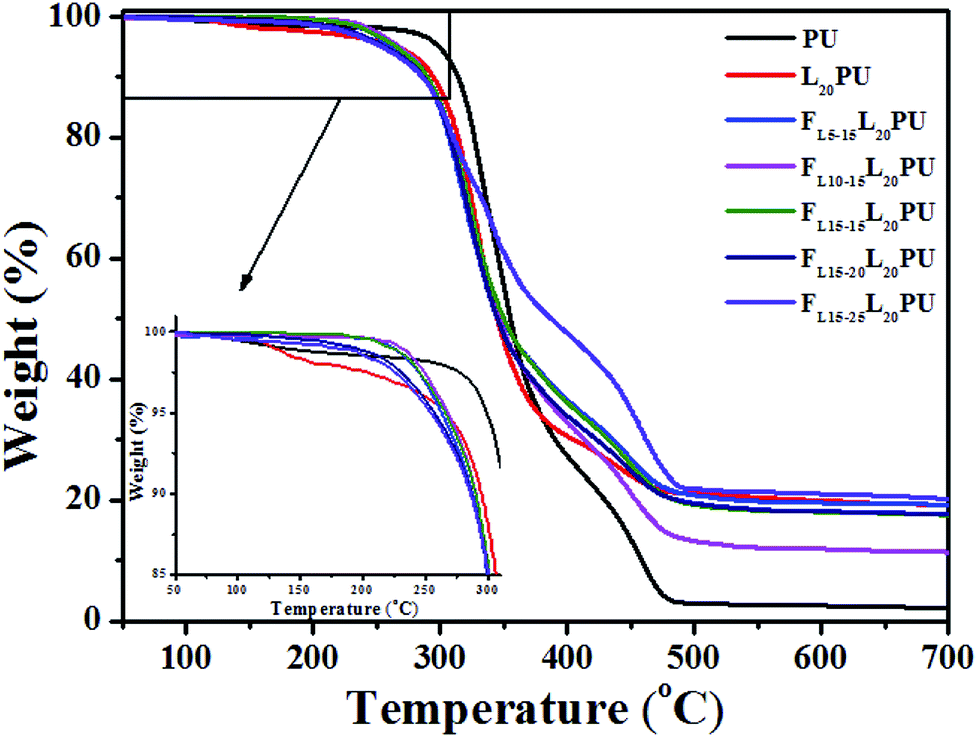 | ||
| Fig. 7 TGA curves of FLPU films. | ||
As shown in Fig. 7, the first degradation step (260 to 390 °C) of FLPU was a degradation of hard segment duo to the pyrolysis of carbamate structure.34,42–44 The second degradation step (420 to 490 °C) was a degradation of soft segment. As presented in Table 4, the 5% weight loss degradation temperature (T5) of PU and L20PU was found to be at 299 °C and 266 °C, respectively. The lower T5 of FLPU was because lignin decomposed at lower temperature. On the other hand, with the addition of LHD in L20PU matrix, the T5 of FLPU films decreased gradually, which may be attributed to the fact that the thermal stability of LHDs were lower than that of L20PU. It was reasonable that, as flame retardant, the decomposition of LHDs occurred prior to the L20PU when the samples were heated, because the flame retardants were easier to degrade to accelerate carbonization of the matrix.21,45
Furthermore, LHDs produced phosphate compounds during the degradation which promoted the char forming. Therefore, the char residues of FLPU films increased with the increase of flame retardant LHDs content. The residue char content reached 21.01%, while that of PU was only 2.48%. The TGA results proved that the inserted L15HD could enhance the flame retardancy by increasing the char yield.
XPS test of FL15-25L20PU
The FL15-25L20PU sample was treated in a muffle furnace under a nitrogen atmosphere at various temperatures. The XPS measurement of the FL15-25L20PU at different temperature ranges: RT, 280, 380 and 500 °C were performed and analysed in detail. The XPS spectra of FL15-25L20PU at different temperature are shown in Fig. 8, and the element content data are illustrated in Table 5.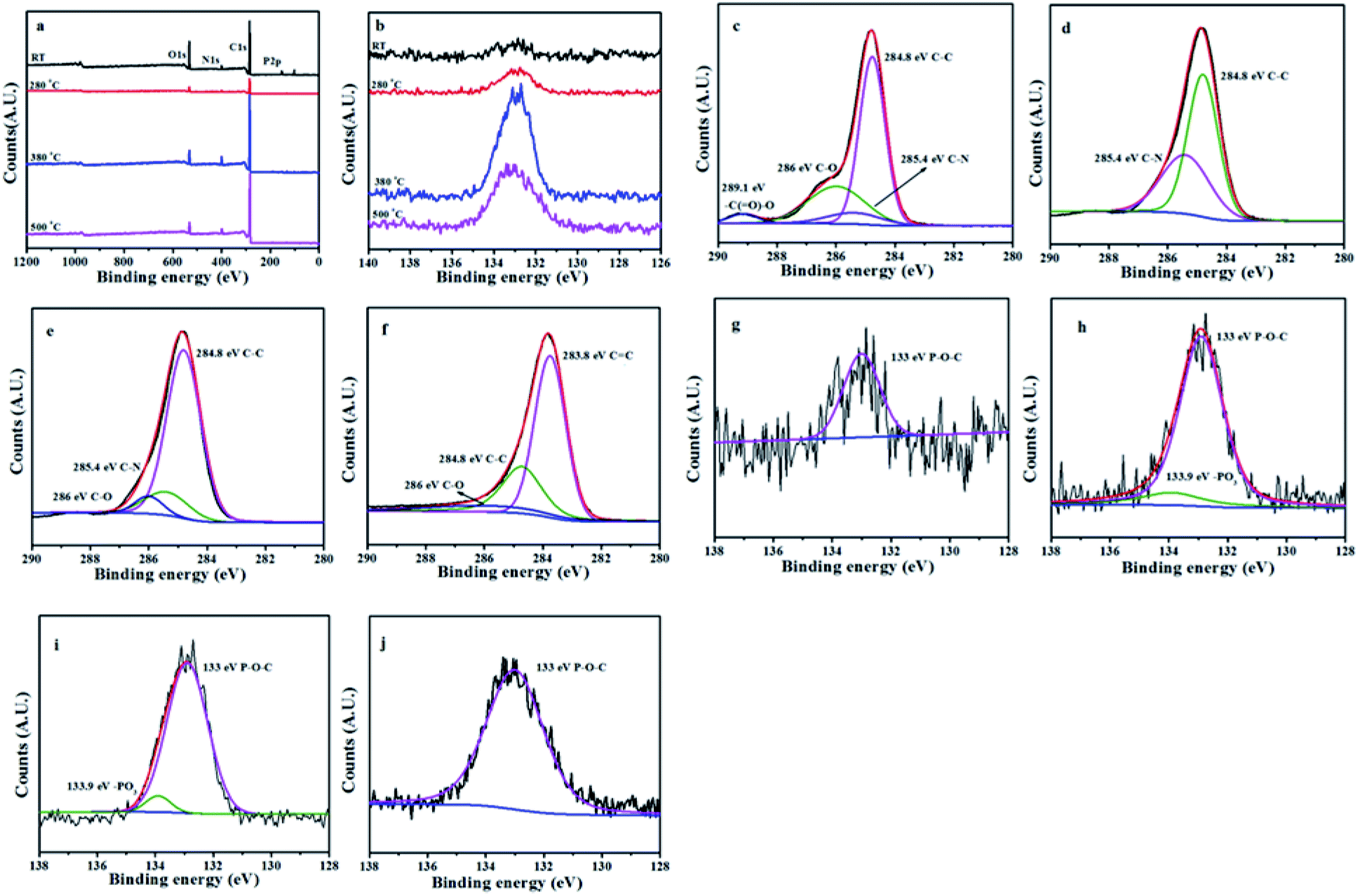 | ||
| Fig. 8 XPS spectra scan of FL15-25L20PU at different temperature (a); P2p XPS spectra of FL15-25L20PU at different temperature (b); C1s XPS spectra at room temperature (c); C1s XPS spectra at 280 °C (d); C1s XPS spectra at 380 °C (e); C1s XPS spectra at 500 °C (f); P2p XPS spectra at room temperature (g); P2p XPS spectra at 280 °C (h); P2p XPS spectra at 380 °C (i); P2p XPS spectra at 500 °C (j). | ||
| Peak | Temperature (°C) | |||
|---|---|---|---|---|
| RT | 280 | 380 | 500 | |
| C1s | 77.63 | 78.89 | 83.5 | 85.44 |
| O1s | 18.79 | 13.46 | 9.13 | 8.26 |
| N1s | 3.23 | 6.52 | 6.22 | 5.42 |
| P2p | 0.35 | 1.13 | 1.14 | 0.89 |
As shown in Table 5, the content of phosphorus also increased first and then decreased as the temperature increased, and was 0.89% at 500 °C. It can be seen from Fig. 8c–f, the C1s spectra of FL15-25L20PU, the peak at around 284.8 eV was attributed to C–C in aliphatic and aromatic species,39 the peak at around 285.4 eV was assigned to C–N bonds in FLPU. The peaks at around 286.0 eV and 289.1 eV were corresponded to C–O,49,50 and –C(O)–O–R bonding,46 respectively. Another new peak at 283.8 eV was assigned to CC in aromatic structures.47 As shown in Fig. 8g–j, the P2p XPS spectra, the peak around 133.0 eV and 133.9 eV were assigned to P–O–C and PO3 groups, respectively.36 In Fig. 8g and j, the peak at 133.0 eV was attributed as P–O–C bonds in L15HD and char residue. In Fig. 8h and i, the double peak at 133.0 eV and 133.9 eV were assigned to the phosphate produced by the decomposition of L15HD and the cross-linked structure of P–O–C. The formation of these carbon layers can compact the surface and improve the flame retardancy of FLPU.
Morphology and structure of residual char
The char residue surface and inner morphology of thermally treated LPU and FLPU samples were characterized by SEM. Fig. 9 shows the microstructures of interior and exterior char of PU and LPU formed in the limiting oxygen index test. From Fig. 9, there were lots of cracks on the outer char of pure PU, which made the char couldn't stop further combustion. On the other hand, its inner char was incompact and empty, which suggested its intrinsic poor flame retardancy. However, the carbon layer became continuous and dense after being incorporated with flame retardant LHDs. The expansion carbon layer was so continuously densified that it could protect the underlying polymer. The charring layer played a very important role in preventing the heat transfer, flame spreading, and droplet which were generated during the combustion process.40 As shown in Fig. 9d–f, the burnt surface had plenty of rugged creases, and the inner surface presented a large number of bubbles. Fig. 9g–i gave the enlarged view of FLPU for comparison. It can be more clearly observed the denseful structure of the inner and outer carbon layers. This intumescent group could slow down the heat and mass transfer between gas and the solidified phase.21 It is obvious that the compact layer was a excellent flame shield to protect the underlying composite. This could obviously improve the flame retardancy of FLPU films. This SEM characterization result was consistent with that of LOI test. | ||
| Fig. 9 SEM of exterior and interior surfaces of the char residue after LOI test: (a): PU; (b): L20PU; (c): FL5-15L20PU; (d): FL10-15L20PU; (e): FL15-15L20PU; (f): FL15-25L20PU; (g)/(h)/(i) were the enlarged view of (d)/(e)/(f). | ||
Coating properties of the FLPU films
Table 6 shows the coating properties of the FLPU films. The hardness of the coatings is critical because the wear resistance and scratch resistance of the coatings are affected by their hardness.48,49 The flexibility of the molecular chain and the crosslink density of the polymer network can affect the hardness.50 As can be seen from Table 6, with the addition of lignin, the hardness of the PU film was increased from grade B to 3H. This was because the addition of lignin can form a crosslinked structure in polyurethane and increase the crosslink density. The addition of LHD further increased the hardness of PU up to 4H. This was mainly due to the addition of LHD into the films. The interaction between the flame retardants LHD and LPU matrix was strong because of the co-existence of lignin and co-curing of LHD and LPU.| Samples | Hardness | Flexibility (mm) | Adhesion | Impact resistance(100 cm/1000g) |
|---|---|---|---|---|
| PU | B | 2 | 0 | Pass |
| L20PU | 3H | 2 | 0 | Pass |
| FL5-15L20PU | 3H | 2 | 0 | Pass |
| FL10-15L20PU | 3H | 2 | 0 | Pass |
| FL15-15L20PU | 4H | 2 | 0 | Pass |
| FL15-20L20PU | 4H | 2 | 0 | Pass |
| FL15-25L20PU | 4H | 2 | 0 | Pass |
The hydroxyl groups in the polymer chain can interact with the substrate. Therefore, the content of hydroxyl groups in the polymer can affect the adhesion of the polymer to the substrate.51 As can be seen from Table 6, all samples exhibited excellent flexibility, adhesion, and impact resistance. The film's flexibility was up to 2 mm, adhesion was 0, impact resistance passed the CJQ-II film impactor 100 cm/1000 g test. The flexibility of the flame-retarded lignin-based PU film was mainly maintained due to the presence of some aliphatic side chains in HDI and the lignin structure.52
Therefore, the mechanical properties of the FLPU film can be reinforced by LHD because both LHD and PU contained the same urethane structure and the phenyl groups of lignin. This was shown to be an effective way for solving the compatibility between the flame retardant and the matrix PU or lignin based PU. Thus, the FLPU could contain not only the flame retardant element phosphorus but also the lignin, which could act as the reinforcing agent and carbonizing agent. Its performances in terms of thermal stability, flame retardancy and properties as coatings were much improved with LHD insertion.
Conclusions
Lignin-based flame retardant was successfully synthesized in this study, and its chemical structure was confirmed by FTIR, 1H NMR and 31P NMR. The temperature of 5% mass loss of LHDs with different lignin content was higher than 242 °C, which showed excellent thermal stability. And with the increase of lignin content, the residual char of LHD were increased to be as high as 17.54% at 500 °C. LHDs were compounded with lignin-based polyurethanes to obtain FLPU, and the LOI test, TGA analysis, and SEM were performed to investigate the combustion properties and thermal stability of FLPU. The results showed that the LOI value of the flame-retardant polyurethane reached 30.2% when the L15HD content reached 25% in L20PU, illustrating excellent flame-retardant properties. The lignin-based flame retardant was beneficial to the formation of the expanded carbon layer, and could play a key role in the flame retardation of the solidified phase. FLPU also showed excellent properties for coating applications. The hardness of FLPU films reached 4H. And the flexibility was up to 2 mm. The adhesion was 0, and the impact resistance passed the CJQ-II film impactor 100 cm/1000 g test. Thus, LHD could be promising to be applied as flame retardant and reinforcing material for LPU. More importantly, the prepared lignin-based flame retardant could also be applied in other materials, such as fully biodegradable flame-retardant polylactic acid composites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China [No. 21404013], the Science and Technology Development Plan of Jilin Province, China [No. 20160101323JC, 20170101110JC, 20180201076GX, 20180201075GX], the Jilin Provincial Development and Reform Commission, China [2018C041-1], the Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences.References
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337(6095), 695–699 CrossRef PubMed.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17(11), 4862–4887 RSC.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116(4), 2275–2306 CrossRef PubMed.
- F. G. Calvo-Flores and J. A. Dobado, Chemsuschem, 2010, 3(11), 1227–1235 CrossRef PubMed.
- H. Chung and N. R. Washburn, ACS Appl. Mater. Interfaces, 2012, 4(6), 2840–2846 CrossRef PubMed.
- W. Liu, R. Zhou, H. L. S. Goh, S. Huang and X. Lu, ACS Appl. Mater. Interfaces, 2014, 6(8), 5810–5817 CrossRef PubMed.
- T. Saito, R. H. Brown, M. A. Hunt, D. L. Pickel, J. M. Pickel, J. M. Messman, F. S. Baker, M. Keller and A. K. Naskar, Green Chem., 2012, 14(12), 3295–3303 RSC.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J. P. Pascault, Chem. Rev., 2014, 114(2), 1082–1115 CrossRef PubMed.
- G. Sun, H. Sun, Y. Liu, B. Zhao, N. Zhu and K. Hu, Polymer, 2007, 48(1), 330–337 CrossRef.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5(9), 3119–3141 RSC.
- N. D. Luong, N. T. T. Binh, D. D. Le, O. K. Dong, D. S. Kim, D. S. Kim, S. H. Lee, J. K. Baek, Y. S. Lee and J. D. Nam, Polym. Bull., 2012, 68(3), 879–890 CrossRef.
- Y. Kang, Z. Chen, B. Wang and Y. Yang, Ind. Crops Prod., 2014, 56(56), 105–112 CrossRef.
- B. L. Xue, J. L. Wen, M. Q. Zhu and R. C. Sun, RSC Adv., 2014, 4(68), 36089–36096 RSC.
- B. L. Xue, J. L. Wen and R. C. Sun, ACS Sustainable Chem. Eng., 2014, 2(6), 1474–1480 CrossRef.
- H. Lim, eXPRESS Polym. Lett., 2008, 2(3), 194–200 CrossRef.
- L. Song, Y. Hu, Y. Tang, R. Zhang, Z. Chen and W. Fan, Polym. Degrad. Stab., 2005, 87(1), 111–116 CrossRef.
- C. K. Ranaweera, M. Ionescu, N. Bilic, X. Wan, P. K. Kahol and R. K. Gupta, J. Renewable Mater., 2017, 5(suppl. 1), 1–12 Search PubMed.
- H. B. Chen, P. Shen, M. J. Chen, H. B. Zhao and D. A. Schiraldi, ACS Appl. Mater. Interfaces, 2016, 8(47), 32557–32564 CrossRef PubMed.
- W. M. Lu, Q. Li, Y. Zhang, H. W. Yu, S. Hirose, H. Hatakeyama, Y. Matsumoto and Z. F. Jin, J. Wood Sci., 2018, 64(3), 287–293 CrossRef.
- L. P. Gao, G. Y. Zheng, Y. H. Zhou, L. H. Hu and G. D. Feng, J. Therm. Anal. Calorim., 2015, 120(2), 1311–1325 CrossRef.
- H. B. Zhu, Z. M. Peng, Y. M. Chen, G. Y. Li, L. Wang, R. Pang, Z. U. H. Khan and P. Wan, RSC Adv., 2014, 4(98), 55271–55279 RSC.
- A. D. Chirico, M. Armanini, P. Chini, G. Cioccolo, F. Provasoli and G. Audisio, Polym. Degrad. Stab., 2003, 79(1), 139–145 CrossRef.
- L. Costes, F. Laoutid, M. Aguedo, A. Richel, S. Brohez, C. Delvosalle and Ph. Dubois, Eur. Polym. J., 2016, 84, 652–667 CrossRef.
- B. Prieur, M. Meub, M. Wittemann, R. Klein, S. Bellayer, G. Fontaine and S. Bourbigot, Polym. Degrad. Stab., 2016, 127, 32–43 CrossRef.
- C. H. Lin, C. C. Feng and T. Y. Hwang, Eur. Polym. J., 2007, 43(3), 725–742 CrossRef.
- P. Wang, F. Yang, L. Li and Z. Cai, Polym. Degrad. Stab., 2016, 129, 156–167 CrossRef.
- N. Mandlekar, A. Cayla, F. Rault, S. Giraud, F. SalaÜn, G. Malucelli and J. P. Guan, An Overview on the Use of Lignin and Its Derivatives in Fire Retardant Polymer Systems, In Tech Open, 2018 Search PubMed.
- S. Gaan, S. Liang, H. Mispreuve, H. Perler, R. Naescher and M. Neisius, Polym. Degrad. Stab., 2015, 113, 180–188 CrossRef.
- Z. Jia, C. Lu, P. Zhou and L. Wang, RSC Adv., 2015, 5(66), 53949–53955 RSC.
- M. Brebu and C. Vasile, Cellul. Chem. Technol., 2010, 44(9), 353–363 Search PubMed.
- Z. M. Wang, X. H. Yang, Y. H. Zhou and C. G. Liu, BioResources, 2013, 8(3), 3833–3843 Search PubMed.
- W. Fiddler, W. E. Parker, A. E. Wasserman and R. C. Doerr, J. Agric. Food Chem., 1967, 15(5), 757–761 CrossRef.
- C. Branca, A. Paola Giudicianni and C. D. Blasi, Ind. Eng. Chem. Res., 2003, 42(14), 3190–3202 CrossRef.
- N. M. Neisius, M. Lutz, D. Rentsch, P. Hemberger and S. Gaan, Ind. Eng. Chem. Res., 2014, 53(8), 2889–2896 CrossRef.
- K. Srikulkit, C. Iamsamai and S. T. Dubas, J. Met., Mater. Miner., 2006, 16(2), 41–45 Search PubMed.
- X. L. Chen, Y. Hu, C. M. Jiao and L. Song, Polym. Degrad. Stab., 2007, 92(6), 1141–1150 CrossRef.
- W. C. Zhang, X. M. Li and R. J. Yang, Polym. Degrad. Stab., 2014, 99(1), 118–126 CrossRef.
- W. C. Zhang, X. M. Li and R. J. Yang, Polym. Degrad. Stab., 2011, 96(10), 1821–1832 CrossRef.
- Z. H. Zheng, Y. Liu, L. Zhong and H. Y. Wang, J. Mater. Sci., 2016, 51(12), 5857–5871 CrossRef.
- Z. H. Zheng, Y. Liu, L. Zhang, B. Y. Dai, X. D. Yang and H. Y. Yan, J. Therm. Anal. Calorim., 2017, 127(3), 2013–2023 CrossRef.
- B. Prieur, M. Meub, M. Wittemann, R. Klein, S. Bellayer, G. Fontaine and S. Bourbigot, RSC Adv., 2017, 7, 16866–16877 RSC.
- R. Yang, W. Hu, L. Xu, Y. Song and J. Li, Polym. Degrad. Stab., 2015, 122, 102–109 CrossRef.
- W. Zhou, F. Chen, H. Zhang and J. Wang, J. Wood Chem. Technol., 2017, 37(5), 323–333 CrossRef.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34(10), 1068–1133 CrossRef.
- H. Ma, L. Tong, Z. Xu, Z. Fang, Y. Jin and F. Lu, Polym. Degrad. Stab., 2007, 92(4), 720–726 CrossRef.
- O. Ivashenko, J. T. van Herpt, B. L. Feringa, W. R. Browne and P. Rudolf, Chem. Phys. Lett., 2013, 559(2), 76–81 CrossRef.
- W. Zhu, X. L. Cai, Z. W. Xu, L. Zhou and Z. Y. Wang, Integr. Ferroelectr., 2017, 180(1), 1–11 CrossRef.
- J. Sun, X. Wang and D. Wu, ACS Appl. Mater. Interfaces, 2012, 4(8), 4047–4061 CrossRef PubMed.
- R. Schwalm, L. Häußling, W. Reich, E. Beck, P. Enenkel and K. Menzel, Prog. Org. Coat., 1997, 32(1–4), 191–196 CrossRef.
- G. Bayramoğlu, M. V. Kahraman, N. Kayaman-Apohan and A. Güngör, Prog. Org. Coat., 2006, 57(1), 50–55 CrossRef.
- T. H. Chiang and T. E. Hsieh, Int. J. Adhes. Adhes., 2006, 26(7), 520–531 CrossRef.
- J. Dai, S. Ma, X. Liu, L. Han, Y. Wu, X. Dai and J. Zhu, Prog. Org. Coat., 2015, 78, 49–54 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |