DOI:
10.1039/C8RA05499A
(Paper)
RSC Adv., 2018,
8, 33432-33444
Encapsulation of erythromycin and bacitracin antibiotics into natural sporopollenin microcapsules: antibacterial, cytotoxicity, in vitro and in vivo release studies for enhanced bioavailability†
Received
27th June 2018
, Accepted 19th September 2018
First published on 27th September 2018
Abstract
Nature produces large quantities of superbly complex and highly reliable microcapsules. The micrometre-sized Lycopodium clavatum spores are one example of these robust capsules. The encapsulation of erythromycin (EM) and bacitracin (BAC) antibiotics into the Lycopodium clavatum sporopollenin (LCS) extracted from these spore species is explored for the first time. The LCS microparticles are extensively characterised before and after loading using SEM, CLSM, TGA and FTIR techniques. The loading capacity and entrapping efficiency of EM were 16.2 and 32.4%, respectively. The antibacterial activities of pure antibiotics, empty LCS and the antibiotic-loaded LCS were evaluated against Staphylococcus aureus (Gram-positive), Pseudomonas aeruginosa (Gram-negative), and Klebsiella pneumoniae (Gram-negative) human pathogenic bacterial strains. A remarkable increase in the antibacterial fold activity of both EM- and BAC-loaded LCS compared to that of the pure antibiotics is observed. Crucial for drug delivery applications, empty LCS, EM- and BAC-loaded LCS were found to be nontoxic against human epithelial colorectal adenocarcinoma cells Caco-2 as revealed by the cytotoxicity evaluation. The in vitro release mechanism of EM in pH 7.4 showed a deviation from Fick's law. In vivo release of EM from EM-loaded LCS (an oral dose of 50 mg kg−1) revealed high values of the area under the plasma concentration–time curve (AUC0–6 h and AUC0–∞ were 1620 and 2147 μg h L−1, respectively) indicative of the enhanced EM bioavailability. The successful loading of antibiotics into the nontoxic LCS and the enhanced bioavailability can open up intriguing applications in oral and topical drug delivery strategies.
1. Introduction
In nature, pollen grains protect the reproductive genetic material of male flowers from harsh environmental conditions such as dehydration, high temperatures, UV light and microbial attack.1 The male plant's contribution is encapsulated within the cytoplasmic core of the pollen grain until the germination process where certain cells transfer to the female flower via pollen tubes.2 The cytoplasmic area is surrounded by two layers; the intine and the exine shells. The latter outer layer represents the sporopollenin biopolymer, which has been described as the “diamond of the plant world”.3 Growing suggestions are in support of sporopollenin being a highly cross-linked biopolymer of carbon, hydrogen, and oxygen with complex building blocks of straight and branched aliphatic chains.4 The nitrogen-free sporopollenin extraction protocols vary and can involve either harsh chemical treatments utilising strong bases and acids at raised temperatures5 or using mild conditions.6–8 It was suggested that harsh chemical extraction can denature sporopollenin,9 therefore, possible structure and/or morphology alternations have to be taken into account when one makes a comparison between empty sporopollenin originated from different plant species and extracted through different protocols. Due to their resilience, consistency in size and unique morphology, sporopollenin have recently attracted intriguing attention for a wide range of applications acting as either microparticles (as solid support)10–16 or microcapsules (for diverse encapsulation of materials).3,17–22 The majority of research concerning sporopollenin has arisen from the thick-shelled (∼1.5 μm) S-type species of Lycopodium clavatum L. spores (common club moss or ground pine). Moreover, sporopollenin from L. clavatum have recently shown high efficiencies in encapsulation of drugs,21,23 live cells,20 oils,18,24 proteins19 and inorganic nanoparticles.17 Synergetic microencapsulation using raw L. clavatum25 and sunflower26 spores, to preserve their herbal therapeutic benefits, has been recently explored.
Although various types of inorganic and organic materials have been encapsulated into sporopollenin, encapsulation, antibacterial activity, in vitro and in vivo release of antibiotics loaded inside L. clavatum sporopollenin (LCS) has not been reported so far. We focus in this article on erythromycin (EM) and bacitracin (BAC) as macrolide and polypeptide antibiotics, respectively. Erythromycin, produced from Saccharopolyspora erythraea strains, is extensively used for the treatment of several bacterial infections and is frequently prescribed as an alternative for patients allergic to penicillin.27–30 However, EM was considered as a labile antibiotic since it degrades in the acidic medium of the stomach via an intramolecular dehydration reaction producing inactive by-products such as anhydroerythromycin, a more toxic derivative of the drug.27,30 This limits the efficacy and bioavailability of this important antibiotic as well as increasing its gastrointestinal side effects. Several methods have been developed to enhance the acidic stability and therapeutic effectiveness of erythromycin.31–35 The use of EM analogs (azithromycin and clarithromycin) showed a nuance increase in acidic stability.31,32 Enteric EM coating has been utilised for delayed-release tablets that protect the acidic degradation in the stomach.33 Another route for improving the acidic stability of EM was to use pH-responsive polymers34 or via encapsulation within biodegradable gelatine microcapsules35 or in nanoemulsions.36 Bacitracin is a peptide antibiotic, produced by Bacillus subtilis, that can strongly inhibit Gram-positive organisms.37 Bacitracin has progressively become a widespread component in diverse topical products such as cosmetics, skin ointments, and powders.38 It was reported that BAC is one of the most common topical antibiotics utilised in the irrigation solution for neurosurgical cases.39
Unlike the aforementioned studies, we report here the use of naturally occurring LCS as microcapsules for loading EM or BAC antibiotics in an effort to protect these drugs from the harsh environment and to enhance their bioavailability and pharmacokinetic properties. Since the sporopollenin microcapsules are shown to be highly resilient to strong acid, strong alkali and a wide range of various enzymes40,41 found in the gastrointestinal tract (GIT), the release of labile antibiotics throughout the GIT using these LCS microcapsules is deemed interesting. To the best of our knowledge, the in vitro and in vivo EM and/or BAC release from LCS and the cytotoxicity evaluation of empty and loaded LCS against human epithelial Caco-2 cells have not been reported before. We also demonstrate, for the first time, the in vitro antibacterial activity of EM and BAC loaded LCS microcapsules. Our technique involves loading the extracted LCS microcapsules with the desired antibiotic via vacuum-assisted passive diffusion through the nanochannels of the LCS. Physico-chemical characterisations of the LCS before and after the encapsulation process were investigated using several techniques. Our protocol showed an enhanced EM bioavailability via oral delivery as the sporopollenin exines have been reported to cross the gut wall.3 Our findings also shed the light for the potential applications of these versatile LCS biopolymers for BAC topical formulations and taste masking of antibiotics. Moreover, similar results are expected with most commonly used antibiotics using these versatile natural biomaterials to meet the ever increasing demand for drug microencapsulation.
2. Experimental
2.1. Materials
Lycopodium clavatum L. (common club moss) spores (27 μm type) were purchased from Fagron, UK. Mueller Hinton broth (MHB) or Mueller Hinton agar (MHA), Lysogeny broth (LB), ethanol and standard antibiotics (erythromycin and bacitracin), were purchased from Sigma-Aldrich (St. Louis, MO, USA). Eosin Yellow disodium salt was purchased from SRL (India). All chemicals were purchased from Sigma Aldrich (St. Louis, MO, USA) unless otherwise stated. Double sterilised Milli-Q water was used throughout the experiments.
2.2. Source of microorganisms and cultures media
Three different human pathogenic bacterial strains namely Staphylococcus aureus (Gram-positive), Pseudomonas aeruginosa (Gram-negative), and Klebsiella pneumoniae (Gram-negative) strains were obtained from the Spanish Type Culture Collection (CECT; Valencia, Spain) and the species-level confirmation was identified using the microbial identification system (BioMèrieux, mini API, Roma, Italy). All bacterial strains were stored in Phosphate Buffer Saline (PBS) with 10% Tryptone Soy Broth (TSB, Conda Laboratories, Madrid, Spain) and 10% glycerol at −80 °C until employed. The microbiological study was performed in three different culture media: Mueller-Hinton broth (MHB), or Mueller Hinton agar (MHA), Lysogeny broth and Luria–Bertani (LB) in both liquid and solid phase. The MHB was prepared according to the instructions of the supplier by mixing 21 g of ready-made MH powder with 1 L of distilled water. The MH broth prepared by this way contains beef infusion, casein hydrolysate and starch. The LB broth was prepared according to the recipe by mixing distilled water (1 L) with tryptone (10 g), yeast extract (5 g) and sodium chloride (10 g). The pH was balanced to 7.2 ± 0.2 in both cases. The agars were prepared from the broth liquids by adding 20 g of the agar powder per 1 L of the broth.
2.3. Encapsulation of antibiotics into LCS
The LCS microcapsules were extracted from raw Lycopodium clavatum L. spores using a method described elsewhere.20 Briefly, raw spores were consecutively treated with acetone, KOH and orthophosphoric acid to get rid of the core materials and eventually obtain nitrogen-free LCS exines. Loading of this type of sporopollenin species with different materials can be achieved by passive diffusion or vacuum loading, or via using compressed tablets.19,20 We used in the current study a method employing a passive diffusion followed by a vacuum loading. A 125 mg of either EM or BAC antibiotic powder was dissolved in 5 mL of a (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) v/v water/ethanol solution in a 10 mL glass tube and stirred for 10 minutes for complete dissolution. Then 125 mg of dry LCS were suspended in the antibiotic solution and vortexed for 15 minutes at 500 rpm for passive loading. This gives a (1:1) w/w (50% w/w) antibiotic loading. The glass tube was then placed in a vacuum desiccator to allow a vacuum loading technique for another 2 hours. The EM or BAC loaded LCS microparticles were collected by filtering through a Whatman filter paper 11 μm, washed with water and pure ethanol to remove surface adhered antibiotics. Finally, the EM or BAC loaded LCS were dried at 60 °C until constant weight and stored at 2–8 °C until further use. For confocal laser scanning microscopy (Section 2.6), eosin Y was used to form a fluorescent binary complex with EM before loading into the LCS microparticles (details in ESI†). The loading capacity was determined by suspending 10 mg of EM-loaded LCS in 3.5 mL PBS:methanol (1:1 v/v), stirred for 5 min and sonicated for 1 min in three cycles at 24% amplitude using Ivymen ultrasonic homogeniser CY-500 (Spain). The LCS were then removed by filtration and the absorbance of the filtrate was measured at λ = 330 nm (Unicam Heλios UV-Vis Spectrometer) for EM content.
:1) v/v water/ethanol solution in a 10 mL glass tube and stirred for 10 minutes for complete dissolution. Then 125 mg of dry LCS were suspended in the antibiotic solution and vortexed for 15 minutes at 500 rpm for passive loading. This gives a (1:1) w/w (50% w/w) antibiotic loading. The glass tube was then placed in a vacuum desiccator to allow a vacuum loading technique for another 2 hours. The EM or BAC loaded LCS microparticles were collected by filtering through a Whatman filter paper 11 μm, washed with water and pure ethanol to remove surface adhered antibiotics. Finally, the EM or BAC loaded LCS were dried at 60 °C until constant weight and stored at 2–8 °C until further use. For confocal laser scanning microscopy (Section 2.6), eosin Y was used to form a fluorescent binary complex with EM before loading into the LCS microparticles (details in ESI†). The loading capacity was determined by suspending 10 mg of EM-loaded LCS in 3.5 mL PBS:methanol (1:1 v/v), stirred for 5 min and sonicated for 1 min in three cycles at 24% amplitude using Ivymen ultrasonic homogeniser CY-500 (Spain). The LCS were then removed by filtration and the absorbance of the filtrate was measured at λ = 330 nm (Unicam Heλios UV-Vis Spectrometer) for EM content.
2.4. Antibacterial activity
The antibacterial activity of EM-loaded LCS and BAC-loaded LCS was investigated by the standard disc diffusion assay.42 Three bacterial strains were used, namely Staphylococcus aureus (Gram-positive), Pseudomonas aeruginosa (Gram-negative), and Klebsiella pneumoniae (Gram-negative). The standard bacterial inoculum suspension (106 CFU mL−1) (colony forming unit) were firstly evaluated by measuring optical density (OD) at 600 nm and then 100 μL of the bacterial inoculum were over streaked on the surface of the Gibcos LB broth/agar medium using sterile cotton swab and grown overnight at 37 °C on a rotary shaker (150 rpm). A 6 mm diameter double sterilised paper discs (Model SR-24A-ADVPB, Consolidated, Machine Corporation, Boston, MA, USA) were placed on the prepared Gibcos LB agar plates. A 10 μL from EM-loaded and BAC-loaded LCS suspension (with MIC conc. Value for each) were dropwise added into the filter discs. All discs were completely dried and placed onto the plates and incubated at 37 °C for 24 h. The antibacterial activity for each EM-loaded and BAC-loaded LCS was then evaluated by measuring the zone of inhibition around the filter discs for the tested bacteria. Discs of erythromycin, bacitracin and empty LCS were used alone as controls. All experiments were performed in triplicates. The fold increase in the antibacterial activity was also measured in triplicate using the formula:43
| Fold increase (%) = (b − a)/a × 100 |
where a is the mean of inhibition zone for antibiotic alone, b is the mean of inhibition zone of antibiotic-loaded LCS.
2.5. Determination of the minimum inhibitory concentration (MIC)
The minimum inhibitory concentration (MIC) of the EM-loaded and BAC-loaded LCS was recorded for each Staphylococcus aureus, Pseudomonas aeruginosa, and Klebsiella pneumoniae using the micro-dilution method as described elsewhere.44,45 In general, Stock suspension of each EM-loaded and BAC-loaded LCS (1 mg mL−1) were prepared in sterile distilled water and each tested bacteria were grown in liquid Luria–Bertani (LB) medium at 37 °C and 250 rpm over 9 hours, before they were diluted to 5 × 108 colony forming units (CFU mL−1) as the inoculum. Each bacterial strain was then re-grown in fresh LB medium alone and in the presence of different gradient concentrations of the EM-loaded and BAC-loaded LCS (0.5, 1, 2, 3, 4, 8, 10, 20 and 30 μg mL−1). The MIC for each one was determined by measuring the optical density (O.D.) at 600 nm of the culture broths. Controls containing only bacterial inoculum and the empty LCS sporopollenin was used. The lowest concentration which completely inhibited the growth of microbes was recorded as MIC. From the above assay, a loopful of inoculum was taken from each well showing no visual growth after incubation and spotted onto MHA plates to validate the MIC assay. All experiments were performed in triplicates.
2.6. Cytotoxicity assay
The MTT (3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to measure the cytotoxicity or biocompatibility of LCS, EM-loaded and BAC-loaded LCS against human epithelial colorectal adenocarcinoma cells Caco-2. The cells were cultured in high glucose Dulbecco's modified Eagle's medium supplemented with antibiotic agents (penicillin 120 IU mL−1 and streptomycin 100 IU mL−1) and 10% (v/v) fetal bovine serum (FBS) and non-essential amino acids. The cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C. Briefly, the cells were grown to 1 × 105 cells per well in 96-well plates and then incubated at 37 °C with 5% CO2 with various concentrations of LCS, EM-loaded and BAC-loaded LCS for 24 h. After treatments, the medium was changed and the cells were washed three times with PBS (pH 7.4) to remove the dead cells. The cells were subsequently incubated with 300 μL (1 mg mL−1) of MTT for 3 h in 5% CO2 incubator for analysis of cytotoxicity and 50% inhibitory concentration (IC50) was calculated. The experiments were performed in triplicates. Cell viability (%) related to the control wells containing cell culture medium without the samples was calculated based on the average of three replicates. The results were expressed as means ± standard error (SE) of three separate experiment cultures.
2.7. Confocal laser scanning microscopy (CLSM) analysis
CLSM of the empty LCS and EM-loaded LCS sporopollenin analysis was performed using a Carl Zeiss LSM710 (Germany) confocal microscope. Laser excitation lines 405 nm, 488 nm and 543 nm with a Plan-Abochromat 63 × /1.4 oil DIC (differential interference contrast) M27 objective lens were used. Atleast three images were captured for each sample. All images and videos were processed under the same conditions using ZEN 2009 and LSM image browser software (ZEISS, Germany).
2.8. FTIR, TGA and SEM analyses
The infrared spectra were obtained in the 650–4000 cm−1 range by a Perkin-Elmer 100 FTIR spectrometer. Samples were ground with anhydrous potassium bromide (spectrosol grade) to obtain disks to a ratio of 1/9 (w/w). FTIR spectra were a result of 3 scans against a background. Thermogravimetric analysis (TGA) curves were obtained using TA Instruments (TGA Q50 Thermogravimetric Analyzer) with a heating rate of 10 °C min−1 up to 700 °C under N2 flow. Scanning electron microscope (SEM) analysis obtained using JSM-5400 LVJEOL (Japan) after coating all samples with 20 nm gold with gold sputter (JEOL JFC-1100E). Images were taken with an acceleration voltage of 5 kV at various magnifications. Optical images were taken by a Nikon microscope fitted with PHD-5MP digital camera (MicroCam, Egypt) and the images were processed using microvision software.
2.9. In vitro release study
Release study of EM antibiotic was carried out using dialysis bag method. 10 mg of EM-loaded LCS microcapsules were dispersed in 5 mL of PBS (pH 7.4) solution and subsequently poured into a dialysis bag (MW cut off of 5 kDa). The dialysis bag was then immersed in 50 mL of PBS receptor media with mild stirring (100 rpm) at 37 ± 0.4 °C. Aliquot of 4 mL was withdrawn from the receptor media at specified time intervals (up to 48 h) and an equivalent volume of a fresh PBS media was replenished into the receptor media. The release sample was analysed with UV-Vis spectroscopy at 330 nm and the amount of EM released was calculated using the EM standard curve.
2.10. In vivo release study
Adult male albino rats weighing 110–130 g were obtained from the Laboratory Animal Center (Dokki, Giza, Egypt). All animals had access to a standard diet and water ad libitum. The rats were left in plastic cages in a well-ventilated room (temperature 25 °C ± 3) for 2 weeks prior the start of the experiment as an acclimatization period. Prior the administration of the EM-loaded LCS oral doses, the rats were fasted overnight for at least 12 h with free access to water. The rats then received an oral dose (50 mg kg−1) of EM-loaded LCS microcapsules (as a suspension in 1 mL of pure water) without constraining the animals. Oral administration was carried out with a rigid gavage tube. The rats were sacrificed via cervical decapitation and blood samples were collected into heparinised 1.5 mL Eppendorf tubes at 0.5, 1, 2, 3, 5 and 6 hours after oral administration. Plasma samples were separated through centrifugation of the collected blood for 15 min at 3000 rpm and stored at −80 °C for subsequent analysis. The EM plasma concentrations in rats were measured chromatographically using an HPLC, Agilent Technologies 1200 Series, G1315D DAD (USA) with a Zorbax NH2 Analytical 4.6 × 250 mm 5-micron column. The mobile phase composition was constant (isocratic elution) containing (A: 10% 10 mM dibasic hydrogen phosphate buffer, pH 6.5 and B: 90% methanol). Erythromycin was measured at 25 °C at a flow rat 1.5 mL min−1 employing UV detector at a wavelength 210 nm. A typical chromatogram is shown in Fig. S9 (ESI).†
3. Results and discussion
3.1. Encapsulation of antibiotics into sporopollenin
Drug stability, targeting and controlled release are desirable properties that can be achieved via microencapsulation which gains great importance. Generally, conventional microencapsulation strategies are pricey and problematic, particularly in producing uniform, monodisperse and efficient biodegradable microcapsules. Sporopollenin extracted from natural pollens showed great advantages over the synthetic microcapsules owing to, inter alia, their unique structural and morphological properties.21 Prior to antibiotics loading process, we have extracted the inner cytoplasmic materials from the raw Lycopodium clavatum L. spores to obtain the outer exines shells (LCS) free from allergens. The extraction protocol used in the current study was known to produce sporopollenin, from the same pollen species studied here, with 0% N2.17,19,20,46 Fig. 1a represents SEM images of the extracted empty LCS showing their surface morphology and microstructure at different magnifications. The extracted LCS microcapsules have retained their native morphology with consistent size (ca. 25 μm in diameter) when compared to the raw precursor spores (Fig. S1 and S2 in ESI†). This indicative of the robust resistance of these unique biopolymer shells to extensive chemical attacks as previously reported.21 This in turn might help withstanding the harsh stomach environment, permitting oral drug administration and better release into the gastrointestinal (GI) tract. As can be seen from Fig. 1a, the surface morphology of the LCS is characterised by obvious reticular microstructure and ornamentation, having hexagonal cells resembling somewhat the honey comb. The trilete scars (Y shaped) can also be seen which are characteristic for this particular pollen species. The EM or BAC loaded LCS microparticles retained their surface morphology after the passive-vacuum encapsulation process. Fig. 1b shows SEM images of an example of EM-loaded LCS where the structural integrity is evident which is of greatest importance for encapsulants (additional SEM images can be seen in Fig. S3 in ESI†). Moreover, the surface of the EM-loaded LCS showed no evidence of residual EM accumulating within their reticular cells in contrast to other reported studies.25,26
 |
| | Fig. 1 SEM images of LCS microcapsules showing the surface morphological characterisations. (A) Empty LCS before loading and (B) after passive-vacuum EM loading. | |
The possible routes for the EM or BAC antibiotics to diffuse into the cavities of the LCS are probably via the nanochannels or the trilete scars which are important for the in/out transfer of nutrition to pollens or for the formation of the pollination tube, respectively.2 A proposed mechanism of encapsulation of the antibiotics into LCS microcapsules is shown in Scheme 1a and b. As seen in Scheme 1a, the microstructure of an empty LCS microcapsule is shown and depicting the uniform ridges surrounding its shell surface and an empty core, ready for loading. The passive-vacuum loading protocol was carried out by applying a vacuum to a LCS suspension in antibiotic solution where the antibiotics diffuse into the empty LCS via the nanochannels present on the shell surface of the LCS microcapsules (Scheme 1b). It was reported that the transfer of the male genetic materials occurs through the apertures of the mature pollens with aperturate while it takes place through the whole exine sporopollenin shell for the nonaperturate pollens.47,48 It was found that, using tracer of colloidal iron, the exine shell layer were crossed in various species of mature spores by radial microchannels of around 25 nm in diameter.47–49 In this respect, the mechanism by which different materials transfer in or out the sporopollenin through the apertures and/or the nanochannels is not fully known since the full chemical structure of the sporopollenin is still largely unidentified.3 It is worth mentioning that the chemical and physical properties of these nanochannels, as a possible way for loading of active, are varied depending on the plant species. Bohne et al.50 reported that, for tripartite sporopollenin extracted from Pine pollens, there was a physical barrier to non-electrolytes with a Stokes' radius > 4 nm resulting from the inhomogeneity in nanochannels' sizes. They also concluded that there was another electrostatic barrier to electrolytes as a result of the specific functional groups within these nanochannels, hence their charge. Nevertheless, in contracts to the results of Bohne et al.,50 the LCS extracted from Lycopodium clavatum species did not show such barriers as revealed in several studies where various materials were successfully loaded inside them.19–22,46
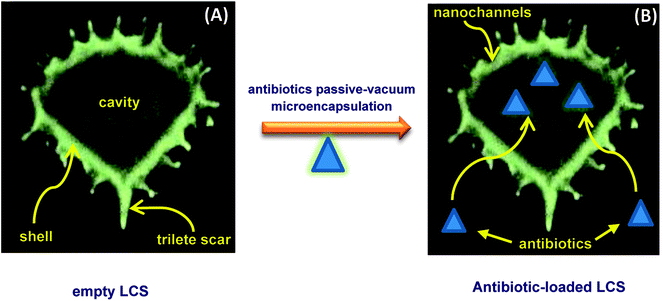 |
| | Scheme 1 Proposed mechanism of passive-vacuum antibiotic encapsulation into empty LCS microcapsules. | |
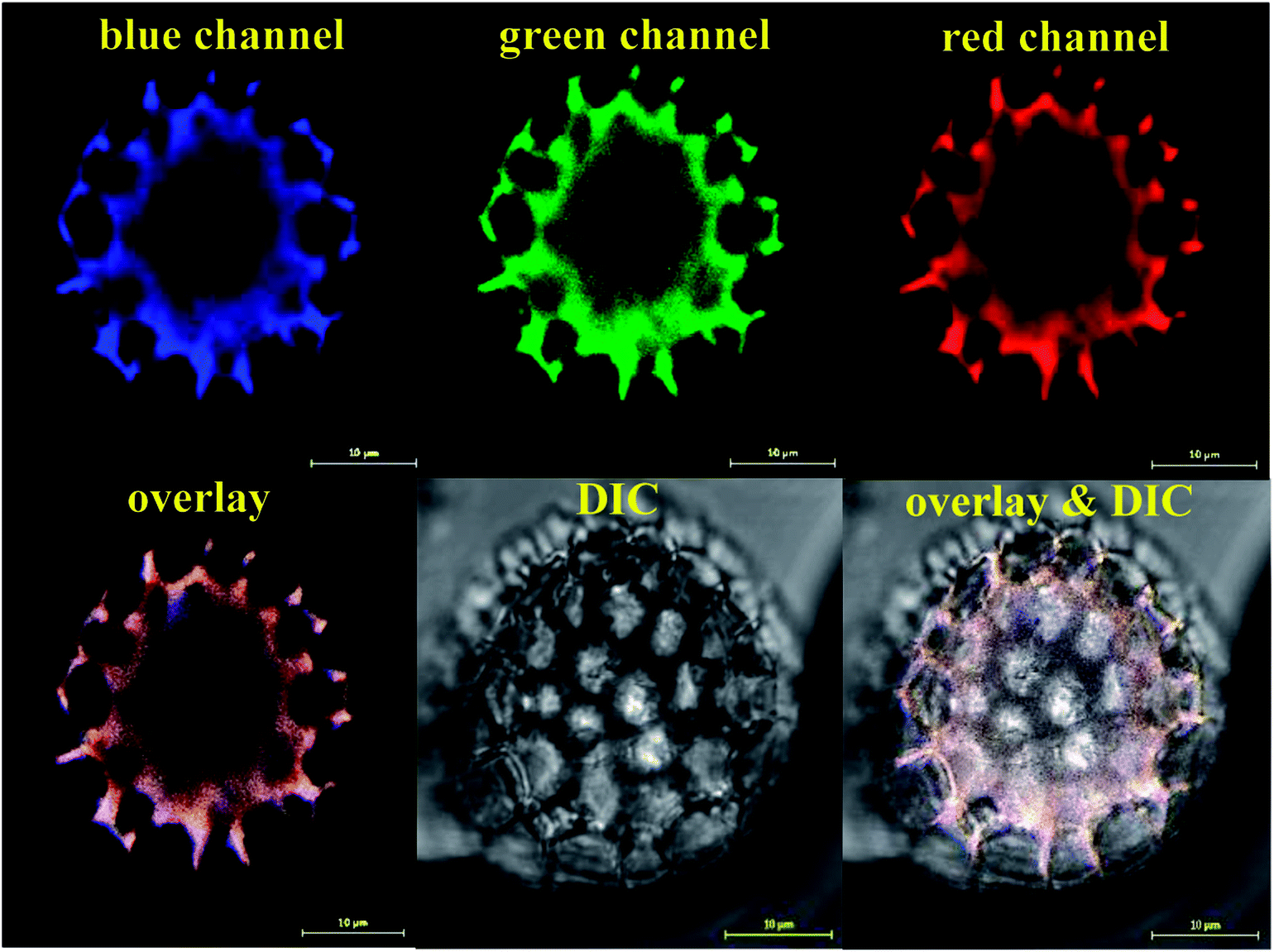
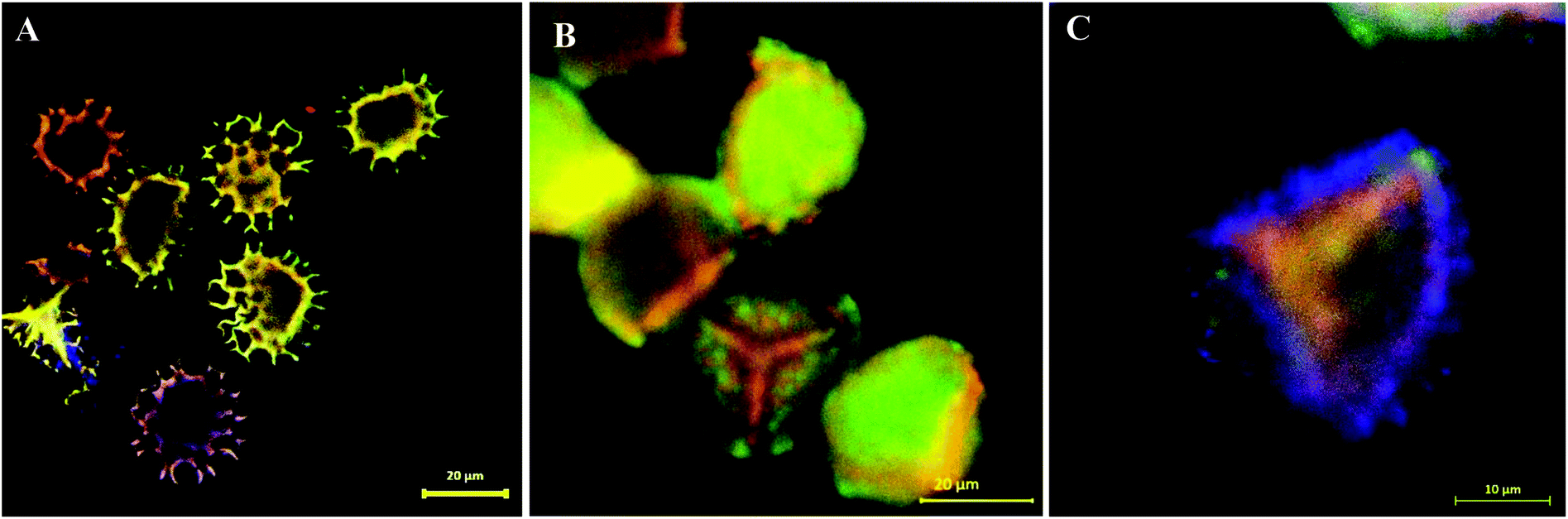
Following the SEM morphological and structural characterisations, which cannot solely give loading confirmation, we have used CLSM analysis to verify the encapsulation of EM or BAC within the LCS. Since these antibiotics do not have conjugated structure (Scheme S1†) particularly EM, labelling with a fluorescent dye is required for proper CLSM detections. We have used a modified simple method described for the formation of binary complexes between some macrolide antibiotics and the eosin yellow as a xanthene dye in aqueous buffered medium at pH 3.0 for detecting these antibiotics spectrophotometrically in pure or commercial forms.51,52 The binary complex of EM–eosin Y showed a strong maximum absorption in the range of 542–544 nm, creating a bathochromic shift of around 30 nm from the pure eosin Y.51 We have verified the formation of the EM–eosin Y complex simply by examining, under a UV lamp, two filter papers sprayed with aqueous solution of eosin Y with and without a spot of the EM (Fig. S7, ESI†) where the formed conjugated complex emitted strong orange-yellow colour. We present here an example of CLSM results obtained for LCS loaded with the EM–eosin Y binary complex. For the CLSM technique, we have captured 2d and z-stacked images and videos at fixed setting conditions for all samples. Fig. 2 shows CLSM images at blue, green and red channels as well as DIC and overlay images at a middle slice (at ∼12.5 μm depth in z-direction) of an empty single LCS microcapsule before encapsulation. An empty large cavity, ready for loading, can be clearly seen without any autofluorescence, indicative of the effectiveness of the extraction protocol used. However, the remaining exine shells fluoresced since it is known that natural spores exhibit autofluorescence over a wide excitation wavelength.18,21 Also, we have captures CLSM images at random depth and a movie for z-stack scanning direction to further verify the cavities in empty LCS microparticles which is exemplified by the image presented in Fig. 3a and other split channels and a movie shown (Fig. S4 and Video V1 in ESI†). It can be seen in Fig. 3a that 5 LCS microparticles showed empty cavities while the other two were scanned at either the upper or lower slice of the microparticles.
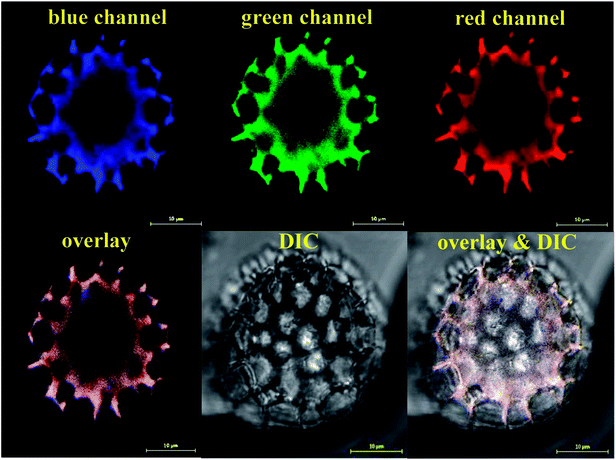 |
| | Fig. 2 CLSM images of single empty LCS microparticle before the loading process. The microparticle was scanned with three laser excitation wavelengths represented by the blue, green and red channels. An image for the overlay of the three channels and the improved contrast DIC image are shown. Large empty black cavity ready for loading is obvious. Images were captured on a slice of a depth (z-direction) of around 12.5 μm. | |
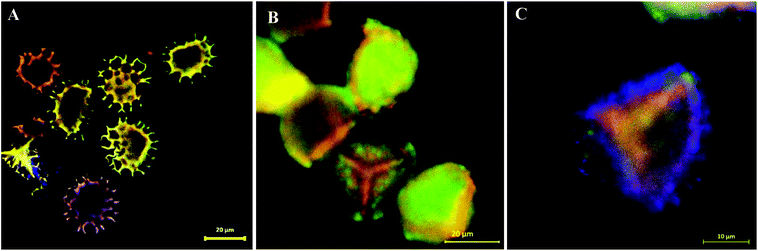 |
| | Fig. 3 Overlay CLSM images of (A) multiple empty LCS microcapsules. (B and C) LCS microparticle loaded with EM–eosin Y binary complex via passive-vacuum technique showing the successful loading of the EM antibiotic inside the cavity of the sporopollenin microparticles. | |
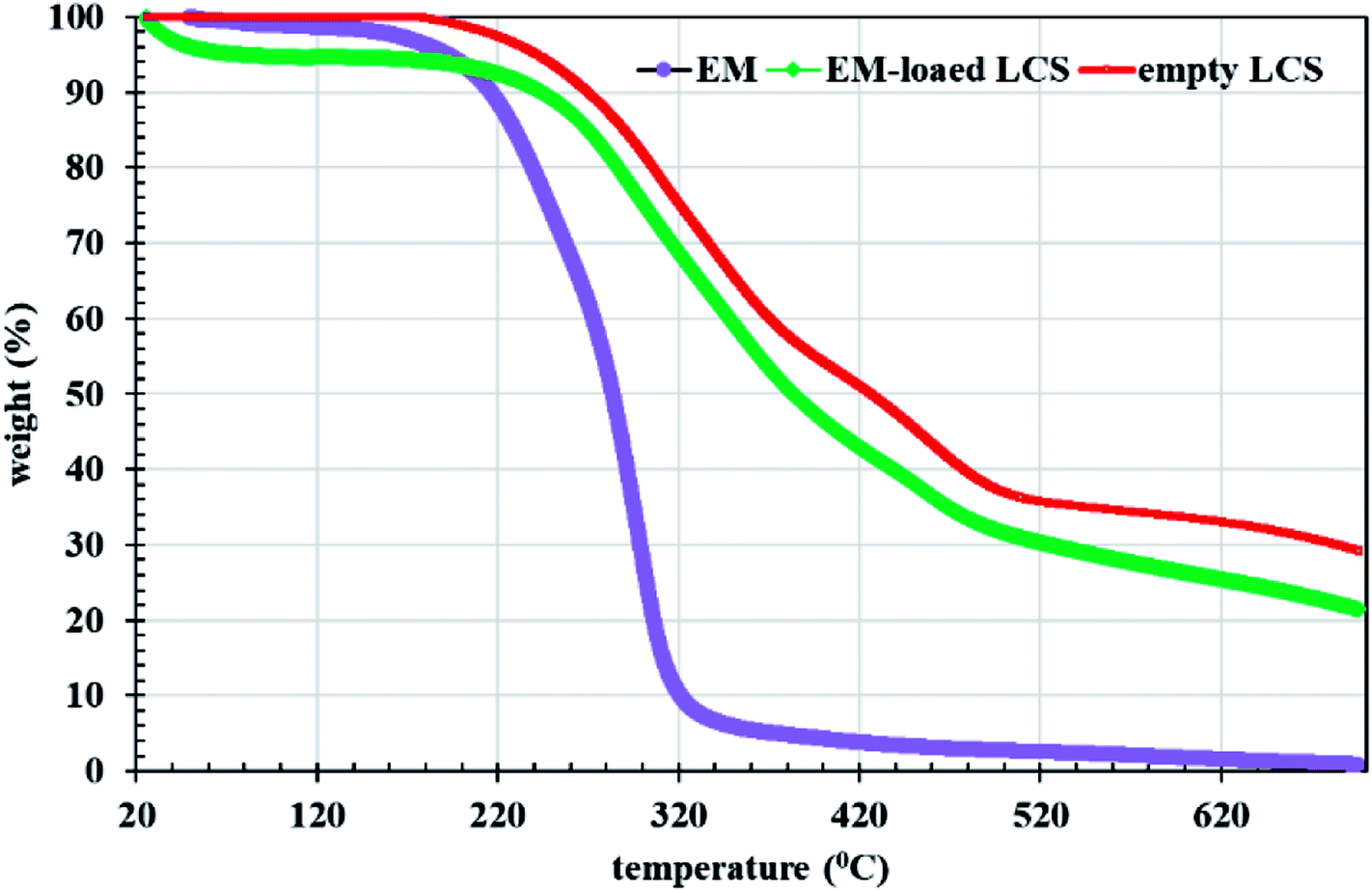
Fig. 3b and c show overlay CLSM images of LCS after loading with the EM–eosin Y binary complex where a strong orange-green fluorescence is evident, indicative of successful encapsulation of the stained EM. It is worth stressing that the encapsulated LCS microparticles were washed carefully with water/ethanol to remove any residual materials deposited on their surface (Fig. 1b and S3†). To further support EM–eosin Y encapsulation inside the LCS, more 2d and 3d CLSM images along with a z-stack video are provided in Fig. S5, S6 and Movie V2 (ESI).† An attempt has been made to visualise the BAC (without labelling) loaded inside the LCS, but the overlay image showed weak fluorescent as shown in Fig. S8 (ESI).† The efficiency of EM loading of (1:1) w/w (50% w/w) into LCS was evaluated by UV and TGA analysis based on a theoretical drug loading (TDL) of 50%. Results from UV analysis revealed that the EM loading and drug entrapping efficiency (DEE) were 16.2 and 32.4%, respectively. TGA data can be used to estimate the drug loading as reported by Kotcherlakota et al.53 The percentage of the drug content was estimated from the differences between the % mass loss of EM-loaded LCS and the unloaded LCS calculated from TGA data (Fig. 4).
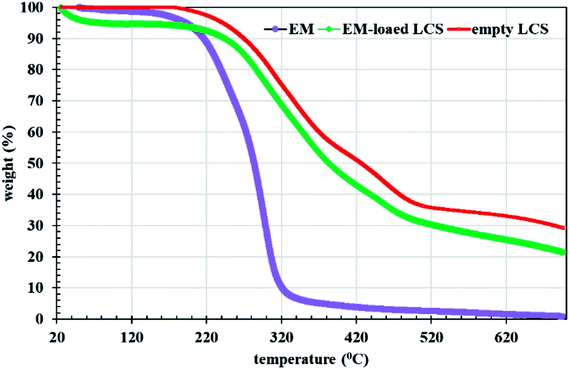 |
| | Fig. 4 TGA analysis for EM, empty and EM-loaded LCS. | |
Analysis of TGA data given in Fig. 4 in the range of 20–700 °C, revealed that the EM loading was 11% and the DEE was 22%. The difference between the methods might be due to the difference in the thermal stability between the loaded and unloaded LCS. Nevertheless, these values are competitive when compared to that obtained for other erythromycin microencapsulation strategies.35,36,54 We next performed a comparison of FTIR superimposed spectra of pure erythromycin, empty LCS and EM-loaded LCS (Fig. 5) to further confirm the encapsulation of the EM into the inside cavities of LCS microparticles and whether an interaction exists between the components. It can be seen from the Fig. 5 that there was no significant change in the spectra of EM-loaded LCS and the empty LCS, indicative of the successful encapsulation of EM within the LCS and not on their surface. Similar observations were reported for ascorbic acid55 and ibuprofen23 encapsulated into similar sporopollenin. In addition, it was reported that pollen grains exhibited a strong light scattering, owing to their size and spatial orientation, resulted in anomalous FTIR spectra which can significantly interfere with and alter the signals of chemical absorption.56 In this respect, our FTIR results revealed that the characterise surface functional groups of the LCS have not been significantly altered before and after EM encapsulation process. Concomitantly, the mass gain obtained from the TGA analysis and the clean surface of the EM-loaded LCS (Fig. 1b and 3b and c) further give an indication of the validity of the encapsulation protocol used.
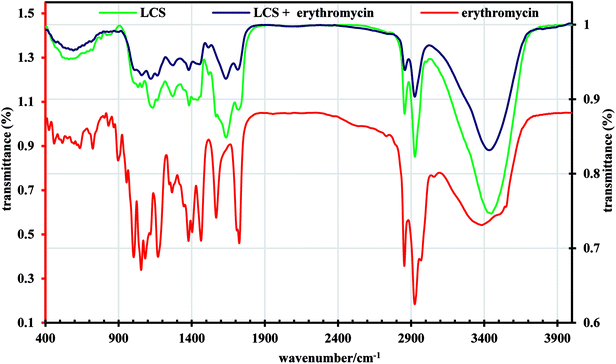 |
| | Fig. 5 FTIR spectra of pure erythromycin, empty LCS and EM-loaded LCS. | |
3.2. Antibacterial activity
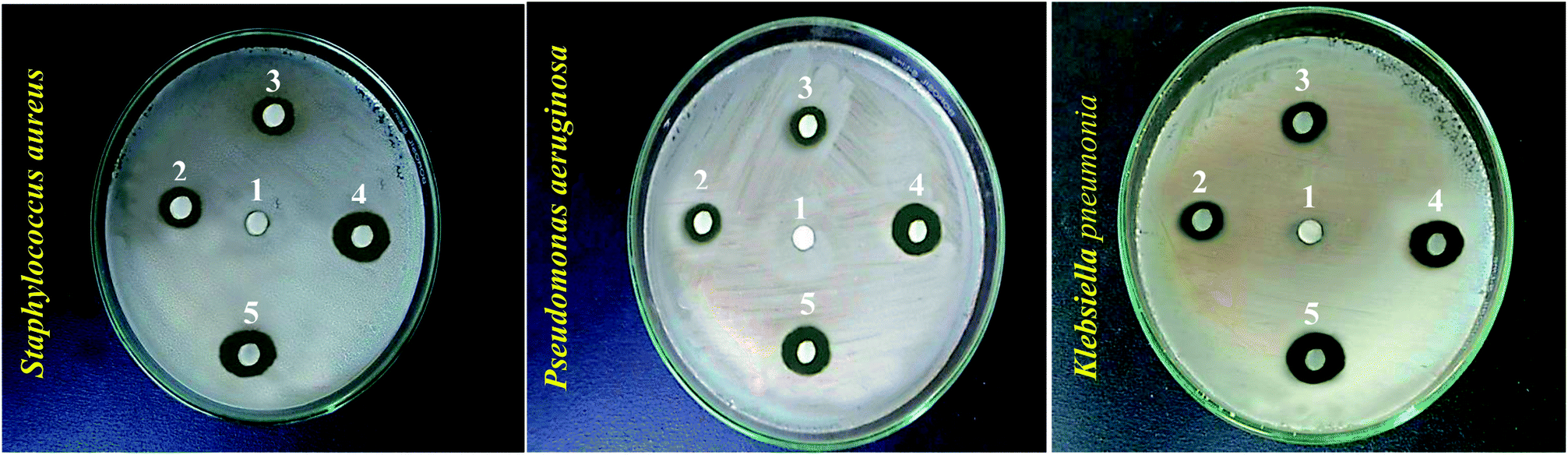
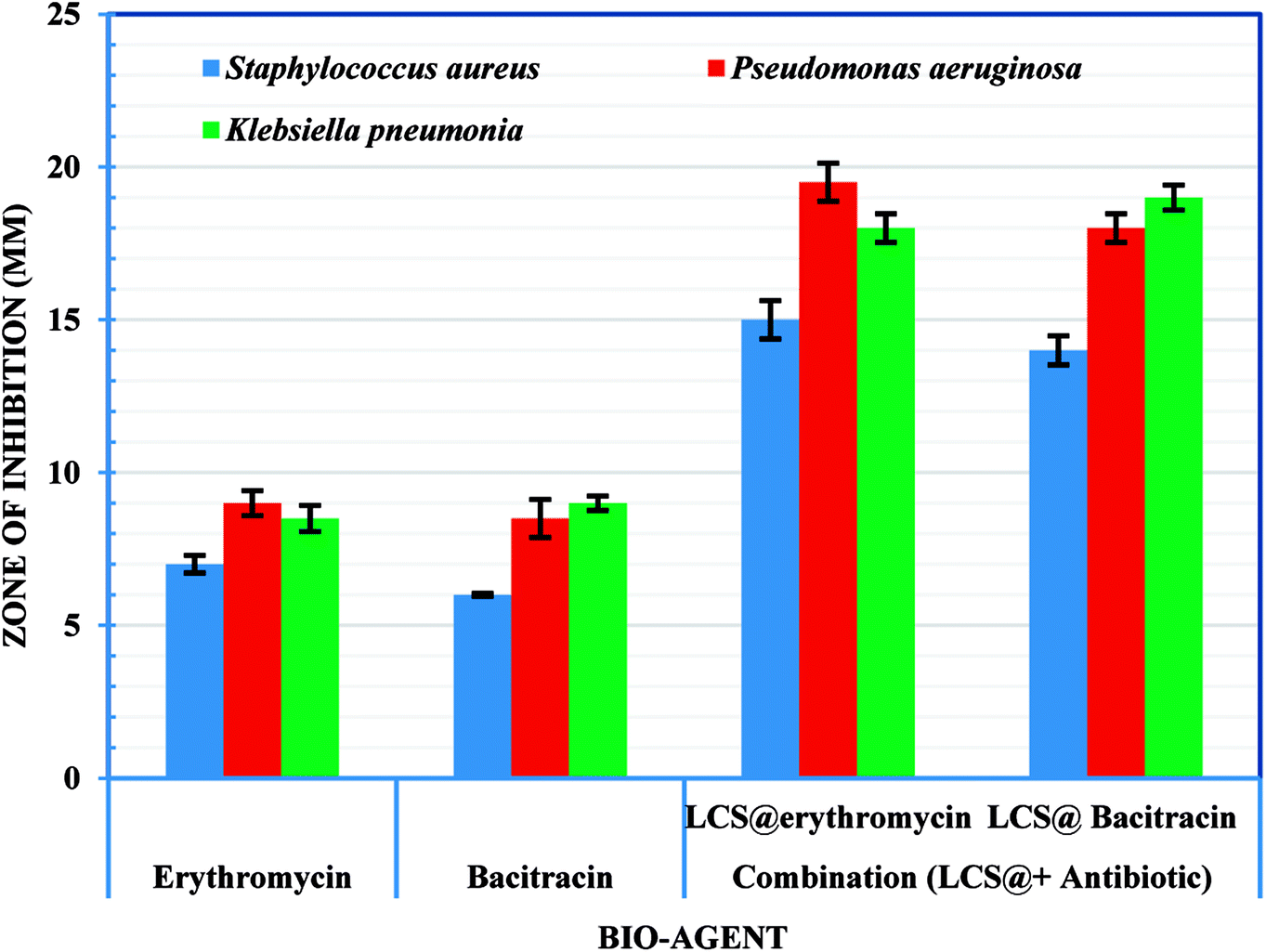
Following the confirmation of antibiotic encapsulation into the LCS microparticles by the aforementioned techniques, it was pertinent to evaluate the antibacterial activity of antibiotic-loaded LCS. To the best of our knowledge, there were no previous reports examined the antibacterial activity of EM or BAC-loaded L. clavatum sporopollenin. The antibacterial activity of both EM-loaded LCS and BAC-loaded LCS as well as the pure antibiotics was investigated by disc diffusion method (Fig. 6, 7 and Table S1†). The observed MIC values for EM-loaded LCS were 1.5, 2.0, and 3 μg mL−1 and 2.0, 2.0, and 4 μg mL−1 for BAC-loaded LCS for K. pneumonia, S. aeruginosa and S. aureus, respectively. The antibacterial results showed that no antibacterial activity was noticed for empty LCS microcapsules. Intriguingly, a better significant increase in the antibacterial fold activity of EM-loaded LCS and BAC-loaded LCS against both Gram positive and Gram negative bacteria compared to that of the pure antibiotics (Fig. 6 and 7), (Table S1 in ESI†). The mechanism by which such a remarkable enhanced antibacterial activity of the antibiotics loaded LCs is unclear. However, we envisage that the unique natural structural morphology of LCS surfaces might help the adherence with the bacterial cell wall leading to an increase in the time of contact, thus enhancing the antibacterial activity. This is somewhat similar to the de novo contacts of pollen grains to the female stigma cells which was reported to be governed possibly by specific interactions involved the lipophilic molecules on the pollen exine.57
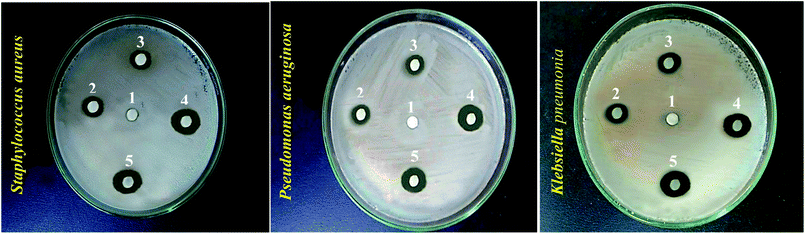 |
| | Fig. 6 Digital images showing antibacterial activity obtained using the disc diffusion method for different bioagents against different bacteria strains (given). Numbers given above the different inhibition zones represent: (1) empty LCS; (2) erythromycin; (3) bacitracin; (4) EM-loaded LCS and (5) BAC-loaded LCS. | |
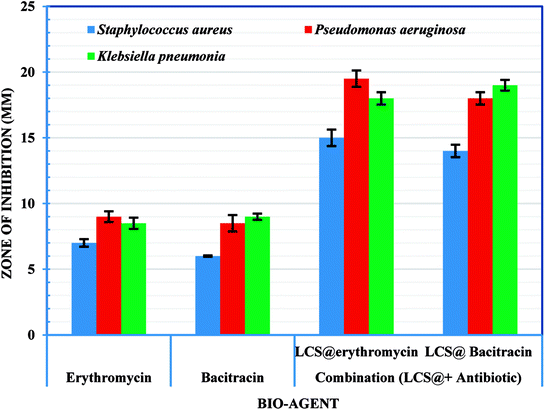 |
| | Fig. 7 Antibacterial and synergistic activity of empty LCS, EM-loaded LCS and BAC-loaded LCS. | |
On the other hand, the noticeable better bactericidal effect of EM-loaded LCS and BAC-loaded LCS against Gram negative bacteria than Gram positive bacteria is likely caused by the difference in the cell wall composition between these two bacterial groups, where the presence of peptidoglycan layer in Gram positive bacteria could be a reason for the less activity as it can prevent antibiotic molecules to enter the cell wall easily.58,59
3.3. Cytotoxicity assay
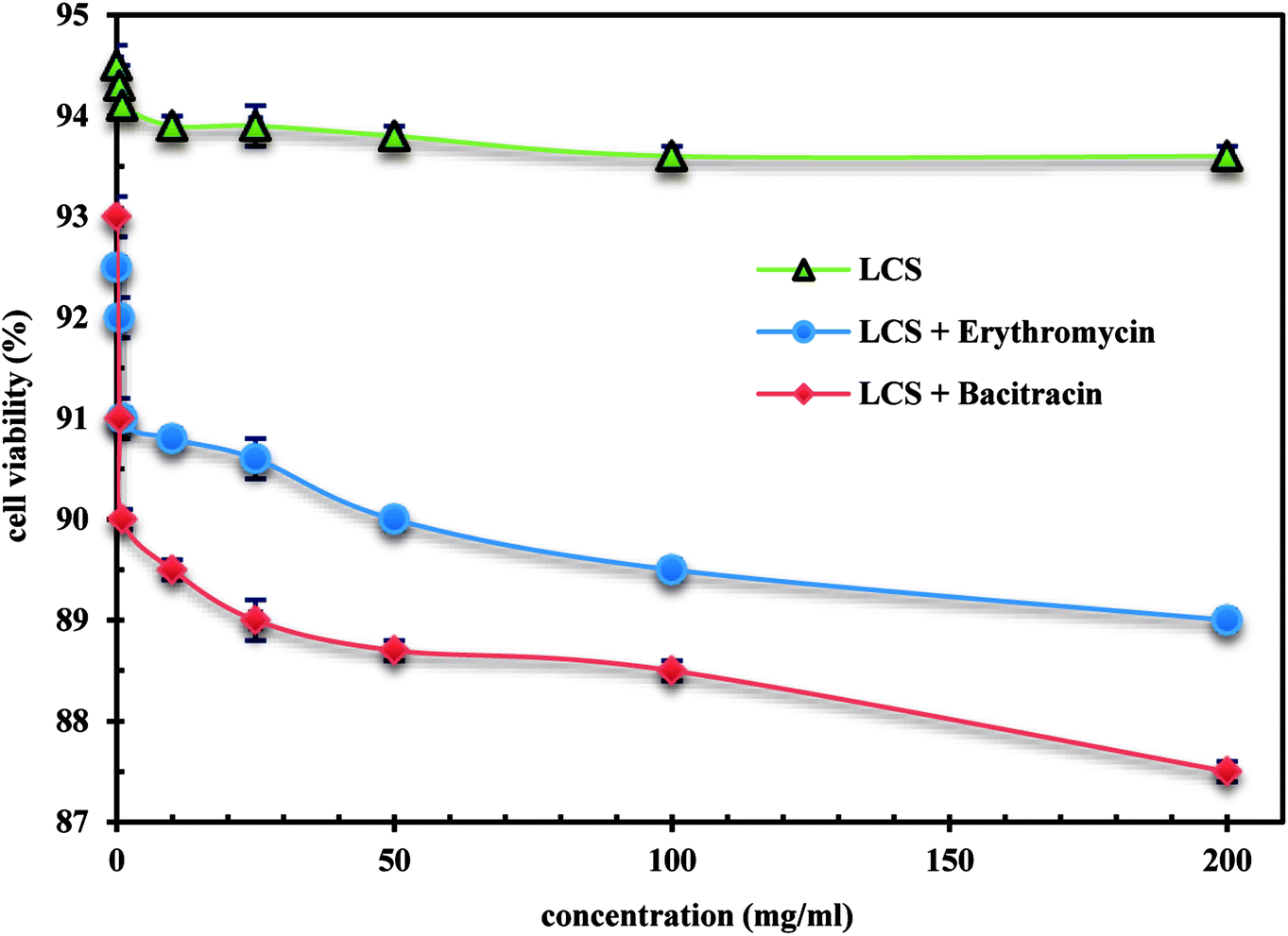
We demonstrate a first toxicological evaluation study of empty and EM- or BAC-loaded LCS microparticles against the Caco-2 cells. The cytotoxicity of the empty LCS was firstly evaluated before enhancing their therapeutic values to secure a remarkable and safe use of LCS for drug delivery applications. MTT was used to investigate the cytotoxicity of the LCS, EM-loaded LCS and BAC-loaded LCS against human epithelial colorectal adenocarcinoma cells Caco-2 cells. The MTT assay results showed metabolically that the human epithelial active cells can reduce the MTT tetrazolium salt to dark blue formazan crystals compared to the dead cells.60,61 The changing in colour might be backed to the ability of mitochondrial dehydrogenase enzyme from viable cells to cleave the tetrazolium rings of the pale yellow MTT and form dark blue formazan crystals, largely impermeable to the cell membranes.62 The cell viability with respect to the concentration of LCS, EM-loaded LCS and BAC-loaded LCS showed no significant toxicity as given in Fig. 8. The cell viability 93.0, 91.0, 90.0, 89.5, 89.0, 88.7, 88.5 and 87.5% was obtained with 0.1, 0.5, 1.0, 10, 25, 50, 100 and 200 mg mL−1, respectively for BAC-loaded LCS. While LCS showed the superior cell viability alone along a broad range of concentration. Taken all together, the cytotoxicity results clearly suggests that LCS, EM-loaded LCS and BAC-loaded LCS were biocompatible with Caco-2 cells, thus increasing their effective use for drug delivery.
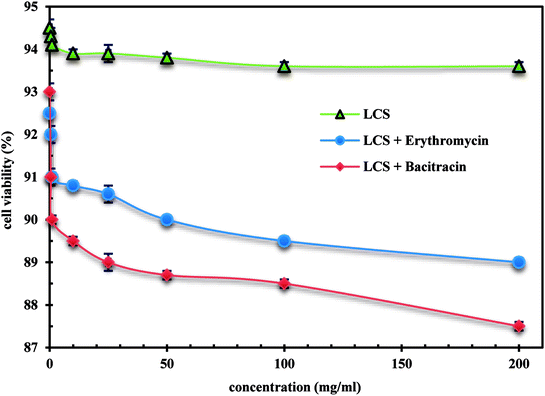 |
| | Fig. 8 Cytotoxicity evaluation of empty LCS, EM-loaded LCS and BAC-loaded LCS. | |
3.4. In vitro release study
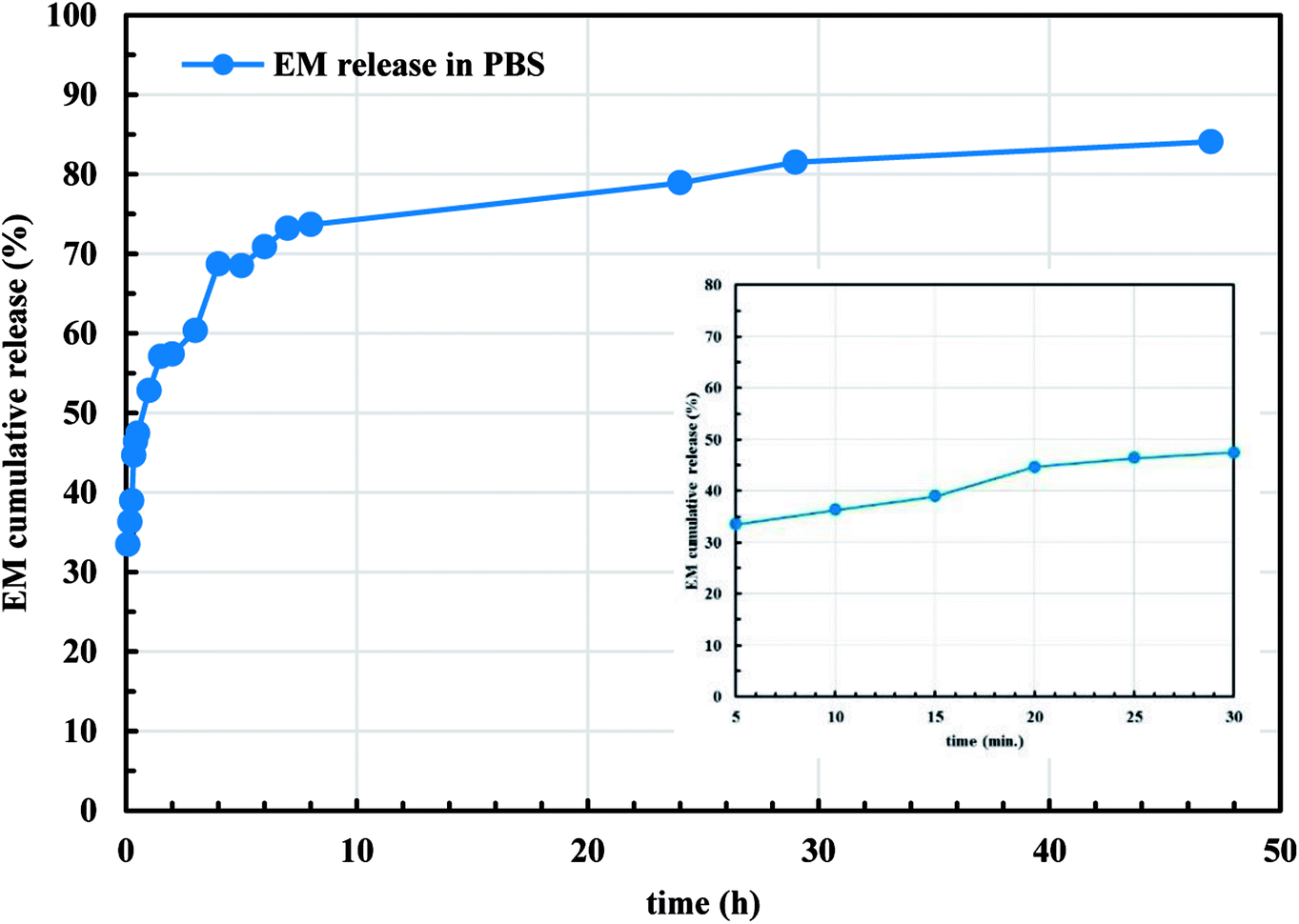
Since the microencapsulation of antibiotics into LCS microcapsules was evident, we explored the in vitro release profile of EM from EM-loaded LCS microcapsules in PBS (pH 7.4) as shown in Fig. 9. As seen in Fig. 9, the in vitro release profiles of EM demonstrated a cumulative percentage release in biphasic profile where initial burst release followed by sustained release are seen. The inset of Fig. 9 represents the initial release profile of EM within the first 30 minutes where 47.4% of the EM released after 30 minutes. Initially, 57.3% of total EM drug was released in first 2 h and 84.1% of the EM was released after around 47 h. The initial burst release might be attributed to the high solubility of EM in the receptor media for the EM drug available close to the inside surface of the LCS microcapsules which released quickly. On the other hand, the sustained profile might be due to the presence of EM molecules deep inside the cores of the LCS. In addition, the receptor media might take time to enter and release the EM drug entrapped inside the LCS microcapsules through the nanochannels on their surfaces. The in vitro release profile shown in Fig. 9 suggested a significant slower release when compared to other reported studies utilising sunflower pollens26 or the release of ibuprofen from L. clavatum sporopollenin.23 We have applied different in vitro release kinetic models such as zero order, first order, Higuchi model, Korsmeyer-Peppas to explore the possible release mechanism of EM from EM-loaded LCS microspheres.63,64 We found that the more general version of the power law equation of Korsmeyer-Peppas64 gave the best fit to the cumulative release data (R2 = 0.9617). The applicability of Korsmeyer-Peppas equation is limited to the first 60% of the release profile (Mt/M∞ < 0.60), where the Mt and M∞ are the mass of drug released at a specific time and at equilibrium, respectively. The value of the drug release exponent “n” in the Korsmeyer-Peppas equation is used to determine the possible release mechanism of the drug in one-dimensional way for non-swellable devises with different shapes.63 The value of “n” was found to be (n = 0.21) which is lower than the expected values64 indicating a deviation from Fick's law as a possible release mechanism of EM from the EM-loaded LCS. This deviation might be as a results of the presence of porosity in the LCS microcapsules where few studies reported lower “n” values for drug release from porous hydrogel.65
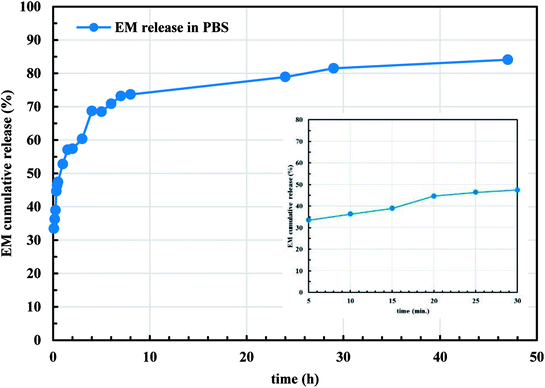 |
| | Fig. 9 In vitro release profile of EM from EM-loaded LCS into phosphate buffered saline (PBS), pH = 7.4, at 37 °C. The inset is the initial release profile within 30 minutes. | |
3.5. In vivo release study
We further studied in vivo release profiles of EM since its encapsulation into the LCS microparticles was evident in an attempt to address the pharmacokinetic and the bioavailability of the drug for oral use. As discussed earlier, the widely used erythromycin degrades in the acidic medium of the stomach, therefore enhancing its bioavailability is deemed important in the pharmaceutical industry.55,66 Moreover, the scant attention in the literature regarding the bioavailability of the materials encapsulated into LCS sporopollenin is unfortunate despite they exhibited promising unique properties. It was reported that the encapsulation of the ethyl ester derivative of EPA (eicosapentaenoic acid) from fish oil within L. clavatum sporopollenin has enhanced the EPA bioavailability in bloodstream.66 The area under the plasma concentration–time curve (AUC) is used to determine the total drug exposure over a period of time and the Cmax is the maximum concentration of the drug. These two parameters are frequently used in bioequivalence studies. Time profile for EM plasma concentration in rats is shown in Fig. 10. The EM-loaded LCS were orally administered to the rats in a dose of 50 mg kg−1. The AUC0–6 h and AUC0–∞ for EM was estimated using the linear trapezoid rule and the concentration at time = zero, Cp(0), was estimated by extrapolating the best fitted curve back to zero. The estimated AUC0–6 h and AUC0–∞ values were 1620 and 2147 μg h L−1, respectively. The time of maximum concentration (Tmax) for EM in plasma was 2 h, whereas the Cmax was 343.4 μg mL−1.
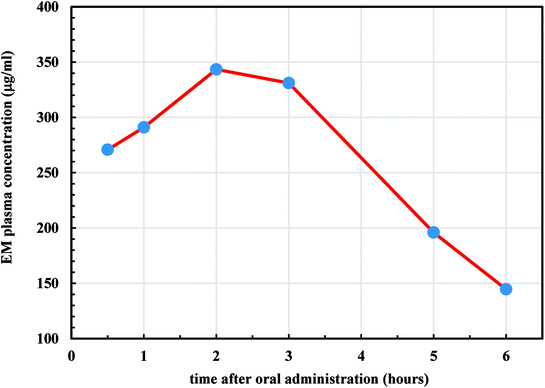 |
| | Fig. 10 Plasma concentration–time profile of EM-loaded LCS after oral administration of 50 mg kg−1 to male albino rats. | |
The obtained results for AUC0–6 h and AUC0–∞ were significantly higher when compared to the previously reported values for EM, orally administered, to rats but without encapsulation (control values).67 The apparent half-life (t1/2) is the time taken for the drug plasma concentration to be reduced by 50% and depends on how quickly the drug is eliminated from the plasma.68 Also, the t1/2 indicates whether accumulation of the drug will occur under a multiple dosage regimen that help determining the suitable dosing interval. The t1/2 of EM in the plasma was found to be (2.53 h), estimated using the linear trapezoid to calculate the elimination rate constant (kel) where (t1/2 = ln(2)/kel). From the results estimated from Fig. 10, it transpired that the microencapsulation of EM in the LCS exines significantly enhanced its bioavailability which has not been studied before. The mechanism underlying the improved release of the EM throughout the GI from the LCS in vivo is not clear, thus more investigations are required which are currently undertaken. The remarkable bioavailability of the EM might be attributed to the mucosal adhesion of the LCS microparticles allowing improved release into the gastrointestinal tract. The unique natural architecture of the LCS surface might also play a fundamental rule by increasing the contact time of adhesion between the loaded microcapsules and the GI.
4. Conclusions
A departure from conventional microencapsulation techniques, we have successfully encapsulated erythromycin (EM) and bacitracin (BAC) antibiotics into the Lycopodium clavatum sporopollenin (LCS) microcapsules for the first time. The EM or BAC antibiotic was loaded into the empty LCS via a combination of passive-vacuum technique in a (1:1) w/w loading. The surface of the antibiotic loaded LCS did not show any significant accumulation of residual antibiotics, indicative of the efficient encapsulation as revealed from SEM and FTIR analysis. The percentage of EM loading was 16.2 and the entrapping efficiency was 32.4%, calculated from the UV data. The CLSM analysis further confirms the encapsulation of EM or BAC into the LCS. In order to visualise the EM within the LCS using CLSM, a binary complex between EM and the eosin yellow dye was prepared in aqueous buffered medium at pH 3.0 which shows λmax in the range of 542–544 nm. We demonstrated that the antibacterial activity EM- and BAC-loaded LCS against Gram-positive and Gram-negative bacterial strains revealed a significant increase in the antibacterial fold activity when compared to that of the pure antibiotics. Crucial for drug delivery applications, the plain LCS, EM- and BAC-loaded LCS were found to be nontoxic against Caco-2 cells. The in vitro release profile of EM indicated a deviation from Fick's law as a possible release mechanism of EM. The in vivo release study of EM-loaded LCS revealed enhanced EM bioavailability in rats' plasma as estimated from the AUC0–6 h and AUC0–∞. The estimated t1/2 of EM in rat's plasma was found to be 2.53 h. Our results suggested that these versatile LCS microcapsules played a crucial rule for the enhanced antibacterial activity and bioavailability of the encapsulated antibiotics owing to their unique morphological and encapsulation properties, enabling them to withstand the harsh stomach conditions and provide a better GI release. In addition, our preliminary results indicated that the LCS and feasibly other spore species, can be used in several pharmaceutical applications with a variety of antibiotics, in particular where the therapeutic materials are labile.
Animal use
All work was conducted with the formal approval of the Faculty of Science, Minia University's policy on animal use and ethics. All mandatory laboratory health and safety procedures have been complied with in the course of conducting the experimental work reported in this study.
Conflicts of interest
There are no conflicts of interest to declare.
Notes and references
- T. Ariizumi and K. Toriyama, Annu. Rev. Plant Biol., 2011, 62, 437–460 CrossRef CAS PubMed.
- R. Wiermann, F. Ahlers and I. Schmitz-Thom, in Biopolymers Online, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, DOI:10.1002/3527600035.bpol1007.
- G. Mackenzie, A. N. Boa, A. Diego-Taboada, S. L. Atkin and T. Sathyapalan, Front. Mater., 2015, 2, 66 Search PubMed.
- F. Ahlers, H. Bubert, S. Steuernagel and R. Wiermann, Z. Naturforsch, 2000, C55, 129–136 Search PubMed.
- F. Zetzsche and K. Huggler, Justus Liebigs Ann. Chem., 1928, 461, 89–109 CrossRef CAS.
- P. E. Jardine, W. T. Fraser, B. H. Lomax and W. D. Gosling, J. Micropalaeontol., 2015, 34, 139–149 CrossRef.
- R. C. Mundargi, M. G. Potroz, J. H. Park, J. Seo, E. L. Tan, J. H. Lee and N. J. Cho, Sci. Rep., 2016, 6, 19960 CrossRef CAS PubMed.
- E. Domínguez, J. A. Mercado, M. A. Quesada and A. Heredia, Grana, 1998, 37, 93–96 CrossRef.
- K. Schulze Osthoff and R. Wiermann, J. Plant Physiol., 1987, 131, 5–15 CrossRef.
- G. Mackenzie and G. Shaw, Int. J. Pept. Protein Res., 1980, 15, 298–300 CrossRef CAS PubMed.
- S. Barrier, A. Löbbert, A. J. Boasman, A. N. Boa, M. Lorch, S. L. Atkin and G. Mackenzie, Green Chem., 2010, 12, 234–240 RSC.
- N. Unlu and M. Ersoz, Sep. Purif. Technol., 2007, 52, 461–469 CrossRef.
- B. P. Binks, J. H. Clint, G. Mackenzie, C. Simcock and C. P. Whitby, Langmuir, 2005, 21, 8161–8167 CrossRef CAS PubMed.
- S. P. de Souza, J. Bassut, H. V. Marquez, I. I. Junior, L. S. M. Miranda, Y. Huang, G. Mackenzie, A. N. Boa and R. O. M. A. de Souza, Catal. Sci. Technol., 2015, 5, 3130–3136 RSC.
- K. Yusuf, A. Aqel, A. K. F. Dyab, Z. A. Alothman and A. Y. Badjah-Hadj-Ahmed, J. Liq. Chromatogr. Relat. Technol., 2016, 39, 752–761 CrossRef CAS.
- A. K. F. Dyab, Micro-structured material and method for the preparation thereof, EP2769995 (B1), 2016.
- V. N. Paunov, G. Mackenzie and S. D. Stoyanov, J. Mater. Chem., 2007, 17, 609 RSC.
- S. Barrier, A. S. Rigby, A. Diego-Taboada, M. J. Thomasson, G. Mackenzie and S. L. Atkin, LWT–Food Sci. Technol., 2010, 43, 73–76 CrossRef CAS.
- S. Barrier, A. Diego-Taboada, M. J. Thomasson, L. Madden, J. C. Pointon, J. D. Wadhawan, S. T. Beckett, S. L. Atkin and G. Mackenzie, J. Mater. Chem., 2011, 21, 975–981 RSC.
- S. A. Hamad, A. K. F. Dyab, S. D. Stoyanov and V. N. Paunov, J. Mater. Chem., 2011, 21, 18018–18023 RSC.
- A. Diego-Taboada, S. T. Beckett, S. L. Atkin and G. Mackenzie, Pharmaceutics, 2014, 6, 80–96 CrossRef PubMed.
- A. K. F. Dyab and K. U. Sadek, RSC Adv., 2018, 8, 23241–23251 RSC.
- A. Diego-Taboada, L. Maillet, J. H. Banoub, M. Lorch, A. S. Rigby, A. N. Boa, S. L. Atkin and G. Mackenzie, J. Mater. Chem. B, 2013, 1, 707–713 RSC.
- A. Diego-Taboada, P. Cousson, E. Raynaud, Y. Huang, M. Lorch, B. P. Binks, Y. Queneau, A. N. Boa, S. L. Atkin, S. T. Beckett and G. Mackenzie, J. Mater. Chem., 2012, 22, 9767 RSC.
- R. C. Mundargi, M. G. Potroz, S. Park, J. H. Park, H. Shirahama, J. H. Lee, J. Seo and N.-J. Cho, Adv. Funct. Mater., 2016, 26, 487–497 CrossRef CAS.
- R. C. Mundargi, M. G. Potroz, S. Park, H. Shirahama, J. H. Lee, J. Seo and N. J. Cho, Small, 2016, 12, 1167–1173 CrossRef CAS PubMed.
- E. L. Cyphert, J. D. Wallat, J. K. Pokorski and H. A. von Recum, Antibiotics, 2017, 6(2), 11 CrossRef PubMed.
- S. Alvarez-Elcoro and M. J. Enzler, Mayo Clin. Proc., 1999, 74, 613–634 CrossRef CAS PubMed.
- R. White, Thorax, 1994, 49, 944 CrossRef CAS PubMed.
- E. F. Fiese and S. H. Steffen, J. Antimicrob. Chemother., 1990, 25, 39–47 CrossRef CAS PubMed.
- W. M. Scheld, M. S. Whitman and A. R. Tunkel, Infect. Control Hosp. Epidemiol., 1992, 13, 357–368 CrossRef.
- J. M. Pereira, R. Mejia-Ariza, G. A. Ilevbare, H. E. McGettigan, N. Sriranganathan, L. S. Taylor, R. M. Davis and K. J. Edgar, Mol. Pharm., 2013, 10, 4640–4653 CrossRef CAS PubMed.
- S. Ogwal and T. Xide, Afr. Health Sci., 2001, 1, 90–96 CAS.
- H. Zhang, H. Wu, L. Fan, F. Li, C. h. Gu and M. Jia, Polym. Compos., 2009, 30, 1243–1250 CrossRef CAS.
- F. Wang, P. Liu, D. Jiang, C. Liu, F. Zhang and X. Chen, Chem. Res. Chin. Univ., 2008, 24, 196–199 CrossRef CAS.
- L. T. C. Tran, C. Gueutin, G. Frebourg, C. Burucoa and V. Faivre, Biochem. Biophys. Res. Commun., 2017, 493, 146–151 CrossRef CAS PubMed.
- J. Yao, S. Zhang, W. Li, Z. Du and Y. Li, RSC Adv., 2016, 6, 515–521 RSC.
- L. Bommarito, S. Mietta and G. Cadario, Ann. Allergy Asthma Immunol., 2015, 115, 74–75 CrossRef PubMed.
- K. H. Reid, C. B. Shields, M. J. Raff, L. S. Cowles and J. D. Johnson, Neurosurgery, 1987, 20, 868–870 CrossRef CAS PubMed.
- R. Wiermann and S. Gubatz, in International Review of Cytology, ed. S. D. Russell and C. Dumas, Academic Press, 1992, vol. 140, pp. 35–72 Search PubMed.
- S. Herminghaus, S. Gubatz, S. Arendt and R. Wiermann, Z. Naturforsch., C: J. Biosci., 1988, 43, 491–500 CAS.
- A. Bauer, W. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol., 1966, 45, 493 CrossRef CAS PubMed.
- A. M. Fayaz, K. Balaji, M. Girilal, R. Yadav, P. T. Kalaichelvan and R. Venketesan, Nanomedicine, 2010, 6, 103–109 CrossRef CAS PubMed.
- L. Qi, Z. Xu, X. Jiang, C. Hu and X. Zou, Carbohydr. Res., 2004, 339, 2693–2700 CrossRef CAS PubMed.
- I. M. Mukisa, D. Porcellato, Y. B. Byaruhanga, C. M. Muyanja, K. Rudi, T. Langsrud and J. A. Narvhus, Int. J. Food Microbiol., 2012, 160, 1–10 CrossRef CAS PubMed.
- A. K. F. Dyab, E. M. Abdallah, S. A. Ahmed and M. M. Rabee, J. Encapsulation Adsorpt. Sci., 2016, 06, 109–131 CrossRef CAS.
- J. Pettitt, Protoplasma, 1976, 88, 117–131 CrossRef.
- B. M. Gullvåg, Grana, 1966, 6, 435–475 Search PubMed.
- J. R. Rowley, J. J. Skvarla and G. El-Ghazaly, Can. J. Bot., 2003, 81, 1070–1082 CrossRef.
- G. Bohne, E. Richter, H. Woehlecke and R. Ehwald, Ann. Bot., 2003, 92, 289–297 CrossRef CAS PubMed.
- M. I. Walash, M. S. Rizk, M. I. Eid and M. E. Fathy, J. AOAC Int., 2007, 90, 1579–1587 CAS.
- A. M. El-Brashy, M. El-Sayed Metwally and F. A. El-Sepai, Farmaco, 2004, 59, 809–817 CrossRef CAS PubMed.
- R. Kotcherlakota, A. K. Barui, S. Prashar, M. Fajardo, D. Briones, A. Rodriguez-Dieguez, C. R. Patra and S. Gomez-Ruiz, Biomater. Sci., 2016, 4, 448–459 RSC.
- Y. Fan, W. Shan-Guang, P. Yu-Fang, S. Feng-Lan and L. Tao, Drug Dev. Ind. Pharm., 2009, 35, 639–645 CrossRef PubMed.
- L. J. Blackwell, Sporopollenin exines as a novel drug delivery system, PhD thesis, University of Hull, 2007.
- B. Zimmermann, M. Bagcioglu, C. Sandt and A. Kohler, Planta, 2015, 242, 1237–1250 CrossRef CAS PubMed.
- G. M. Zinkl, B. I. Zwiebel, D. G. Grier and D. Preuss, Development, 1999, 126, 5431–5440 CAS.
- Y. Wu, F. Shi, M. Hamid and Y. Zhu, Acta Microbiol. Sin., 2014, 54, 926–935 CAS.
- S. Shrivastava, T. Bera, A. Roy, G. Singh, P. Ramachandrarao and D. Dash, Nanotechnology, 2007, 18, 225103 CrossRef.
- S. Rehman, A. S. Shawl, S. Sultana, A. Kour, S. Riyaz-ul-Hassan and G. N. Qazi, Ann. Microbiol., 2009, 59, 157 CrossRef CAS.
- W. Zhao, T. Wang, S. Liu, Q. Chen and R. Qi, Microb. Pathog., 2015, 87, 51–58 CrossRef CAS PubMed.
- R. Konwarh, N. Karak, C. E. Sawian, S. Baruah and M. Mandal, Carbohydr. Polym., 2011, 83, 1245–1252 CrossRef CAS.
- P. Costa and J. M. Sousa Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS.
- J. Sauri, D. Millan, J. M. Sune-Negre, H. Colom, J. R. Tico, M. Minarro, P. Perez-Lozano and E. Garcia-Montoya, Int. J. Pharm., 2014, 477, 431–441 CrossRef CAS PubMed.
- A. Wakil, G. Mackenzie, A. Diego-Taboada, J. G. Bell and S. L. Atkin, Lipids, 2010, 45, 645–649 CrossRef CAS PubMed.
- T. Hori, H. Hashimoto and M. Konishi, Biomed. Chromatogr., 2006, 20, 917–923 CrossRef CAS PubMed.
- D. A. Smith, K. Beaumont, T. S. Maurer and L. Di, J. Med. Chem., 2018, 61, 4273–4282 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05499a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mohamed A. Mohamed
*a,
Mohamed A. Mohamed