
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of Ru(II)@oligonucleotide nanosized polymers as potential tumor-imaging luminescent probes†
Geng-Nan Yu‡
a,
Jun-Chao Huang‡b,
Li Lia,
Ruo-Tong Liua,
Jie-Qiong Caoc,
Qiong Wu *ac,
Shuang-Yan Zhanga,
Cheng-Xi Wang*a,
Wen-Jie Mei*ad and
Wen-Jie Zhengc
*ac,
Shuang-Yan Zhanga,
Cheng-Xi Wang*a,
Wen-Jie Mei*ad and
Wen-Jie Zhengc
aSchool of Pharmacy, Guangdong Pharmaceutical University, Guangzhou, 510006, China. E-mail: wenjiemei@126.com; wuqiongniu.1113@163.com; hiwcx@hotmail.com
bTraditional Chinese Medicine College, Guangdong Pharmaceutical University, Guangzhou, 510006, China
cSchool of Chemistry, Jinan University, Guangzhou, 510632, China
dGuangdong Province Engineering Technology Centre for Molecular Probe and Biomedicine Imaging, Guangzhou, 510006, China
First published on 30th August 2018
Abstract
The development of Ru(II) complexes as luminescent probes has attracted increasing attention in recent decades. In this study, the nanosized polymers of two Ru(II) complexes [Ru(phen)2(dppz)](ClO4)2 (1, phen = 1,10-phenanthrolin; dppz = dipyrido[3,2-a:2′,3′-c]phenazine) and [Ru(phen)2(Br-dppz)](ClO4)2 (2, Br-dppz = 11-bromodipyrido[3,2-a:2′,3′-c]phenazine) with oligonucleotides were prepared and investigated as potential tumor-imaging probes. The formation of the nanosized polymers, which had an average width of 125–438 nm and an average height of 3–6 nm, for 1 and 2@oligonucleotides were observed through atomic force microscopy. The emission spectra indicated that the luminescence of 1 and 2 markedly increased after binding to oligonucleotides and double-strand DNA (calf thymus DNA), respectively. Moreover, further studies indicated that 1@oligonucleotides and 2@oligonucleotides can easily enter into tumor cells and selectively highlight the tumor area in the zebrafish bear xenograft tumor (MDA-MB-231). In summary, this study demonstrated that 1@oligonucleotides and 2@oligonucleotides could be developed as potential tumor-imaging luminescent probes for clinical diagnosis and therapy.
Introduction
Fluorescence imaging has become an increasingly significant approach among modern medical treatments for tumor diagnosis and surgery.1,2 The development of confocal fluorescence microscopy has enabled the early detection of tumor tissues by medical professionals through fluorescence imaging,3 and fluorescence-guided surgery can aid surgeons in identifying and removing malignant lesions.4 Although current clinical imaging agents are excellent for cellular uptake and have low toxicity,5 they suffer from short stokes shifts, short luminescence lifetimes, and photobleaching.6 An ideal luminophore for cellular imaging should be photostable, have a high quantum yield, possess good cell permeability, and exhibit low cytotoxicity.7 Novel luminescence probes should satisfy these requirements in the future.In the past two decades, Ru(II) complexes have been frequently investigated as luminescence probes because of their excellent photostability, large stokes shifts, excellent photophysical properties, and long lifetimes.8 Ru(dppz) systems reported as DNA probes,9,10 such as complex 1, have attracted considerable interest because of the outstanding photophysical and photochemical properties of Ru(II) complexes and the light switch-on effect. Although this complex has no luminescence in water solutions, it can exhibit strong luminescence when intercalating with DNA.11 Moreover, several studies have reported that Ru(II) complexes can be utilized in imaging sub-cellular structures, such as membranes,12 endoplasmic reticulum,13 lysosomes,14 mitochondria,15 and nuclei.16 In previous works, Ru(II) complexes have been modified with cell-penetrating peptides,17 biotins,18 estradiols,19 and oligonucleotides20 to improve their cellular uptake and decrease their cytotoxicity. An increasing number of studies on the use of Ru(II) complexes as luminescence probes have confirmed the great potential of such complexes in clinical imaging. Nonetheless, the clinical application of Ru(II) complexes is still hampered by several limitations, particularly the lack of tumor targeting and location.
Drug targeting is an attractive concept because it can enhance the therapeutic effects and reduce the system toxicity.21 Drug targeting can be divided into two methods: active targeting and passive targeting. Active targeting can be achieved through several strategies, such as antibodies,22 aptamers,23 and peptides,24 which are the ligands in angiogenic blood vessels or the cell membrane. Drugs can be modified with these ligands to target tumor cells. However, these active targeting methods are unsuitable for solid tumor tissues, thereby limiting their clinical applications. In recent years, the use of nanoparticles for passive targeting25 has attracted considerable attention in cancer therapy and diagnosis.26 Nanoparticles are defined as submicron (<1 mm) colloidal systems, which can be classified as nanospheres or nanocapsules depending on the preparation process.27 Nanocapsules are vesicular systems that encapsulate drugs in an aqueous or oily cavity formed from a single polymeric membrane. Nanospheres are matrix systems from which drugs are dispersed throughout the particles. Nanoparticles deliver drugs to tumor tissues in vivo via physiological barriers, and the size of the nanoparticles affects the biodistribution in vivo.28 Owing to solid tumors with abundant abnormal blood vessel and great membrane permeability, the nano-sized macromolecular anticancer drugs can easily penetrated into tumor cells and largely enriched in tumor tissues through EPR-based selective anticancer nanotherapy. Then, the selective high local concentration of nano-sized anticancer drugs have been consider as a potential tumor targeting inhibitor with minimal side effects in both preclinical and clinical settings.29 Recent reports have demonstrated that drugs incorporated into polymeric particles display high selectivity and local concentration.30 For example, the anticancer drug DOX, when carried by nanoparticles, shows distribution in whole body for heart, liver, spleen, lung, kidney, and so on, but also enhanced accumulation31 and selective delivery in solid tumors. As such, DOX-containing nanoparticles have been in clinical trials.
Therefore, to reduce the side effects of Ru(II) complexes as luminescent imaging agents and increase their concentration in tumor tissues, two Ru(II) complexes (Scheme 1) [Ru(phen)2(dppz)](ClO4)2 (1, phen = 1,10-phenanthrolin; dppz = dipyrido[3,2-a:2′,3′-c]phenazine) and [Ru(phen)2(Br-dppz)](ClO4)2 (2, Br-dppz = 11-bromodipyrido[3,2-a:2′,3′-c]phenazine) were designed as Ru(II)@oligonucleotide nanosized polymers and constructed. The atomic force microscopy (AFM) results of the Ru(II)@oligonucleotide indicated that Ru(II) complexes can selectively induce oligonucleotides to transform into nanosized polymers. The luminescence of the Ru(II) complexes was enhanced after the addition of oligonucleotides. The stability of the Ru(II)@oligonucleotide nanosized polymers was assessed through electronic spectroscopy and circular dichroism (CD) spectroscopy. The results revealed that both 1 and 2 could selectively stabilize and bind to oligonucleotides, and that the Ru(II)@oligonucleotide nanosized polymers should be stable. Further study shown that Ru(II)@oligonucleotide nanosized polymers was selectively localized in tumor cells with strong phosphorescence in cellular and zebrafish xenografts model. These findings may suggest that 1 and 2@oligonucleotide nanosized polymers can be utilized as potential luminescence probes in tumor diagnosis and therapy.
 | ||
| Scheme 1 Molecular structures of 1 and 2. | ||
Results and discussion
Synthesis and characterization
The target compounds 1 and 2 have been prepared by heating at reflux for 6 h under N2 atmosphere. The yield for 1 and 2 is about 67 and 61%, respectively. The mass spectra in acetonitrile exhibited a peak at m/z 372.5 (100%) for 1 and at 412.3 for 2 (100%) which was ascribed to [M-2ClO4−]2+ and agreed with the theoretical value. The chemical shift (δ) of 1 in 1H NMR at 9.61 (d, J = 1.2 Hz, 1H), 9.59–9.59 (m, 1H), 8.81 (d, J = 1.2 Hz, 1H), 8.80 (t, J = 1.3 Hz, 2H), 8.78 (d, J = 1.3 Hz, 1H), 8.41 (s, 4H), 8.28 (dt, J = 4.5, 2.3 Hz, 2H) and 8.21–8.18 (m, 4H) ppm can be attributed to eight H atoms in phen ligands, respectively. The chemical shift attributed to the phenanthroline ring appeared at (7.78–8.52) of 7.78 (dd, J = 8.3, 5.3 Hz, 2H), 7.90 (dd, J = 6.8, 4.0 Hz, 2H) and 8.52 (dt, J = 6.4, 3.2 Hz, 2H) ppm. The chemical shift at 8.07 (dd, J = 5.2, 1.2 Hz, 2H) and 7.82 (dd, J = 8.3, 5.3 Hz, 2H) ppm can be attributes to H5 and H6 in co-ligand phenanthroline. For complex 2, the chemical shift (δ) at 9.52 (d, J = 1.2 Hz, 1H), 9.50 (dd, J = 2.4, 1.3 Hz, 1H), 9.48 (d, J = 1.3 Hz, 1H), 8.83–8.80 (m, 2H), 8.80 (d, J = 1.0 Hz, 1H), 8.41 (d, J = 1.2 Hz, 3H), 8.28 (dd, J = 5.3, 1.2 Hz, 1H), 8.25 (t, J = 3.0 Hz, 1H), 8.23 (dd, J = 5.4, 1.7 Hz, 1H), 8.21 (dd, J = 4.5, 2.5 Hz, 1H), 8.20 (d, J = 1.3 Hz, 1H), and 8.08 (ddd, J = 5.2, 2.5, 1.2 Hz, 2H) ppm can be attributed to eight H atoms in phen ligands, respectively. The chemical shift attributed to the phenanthroline ring appeared at (7.90–8.52) of 7.78 (dd, J = 8.3, 5.3 Hz, 2H), 7.92–7.87 (m, 2H) and 8.32 (dt, J = 5.4, 2.7 Hz, 1H) ppm. The chemical shift at 8.78 (s, 1H), 7.83 (td, J = 8.1, 5.3 Hz, 2H) and 8.63 (d, J = 2.2 Hz, 1H) ppm can be attributed to H4, H5 and H6 in co-ligand phenanthroline (Fig. S2†).Preparation and characterization of the nanosized polymers
Ru(II) complexes were dissolved by dimethyl sulfoxide and then diluted to 100 μM solution by Tris–HCl KCl or NaCl buffer (pH = 7.2) ([KCl] = 0.1 M, [NaCl] = 0.1 M). The oligonucleotides or CT-DNA were dissolved and diluted to 100 μM solution by Tris–HCl KCl or NaCl buffer (pH = 7.2) ([KCl] = 0.1 M, [NaCl] = 0.1 M). The oligonucleotides or CT-DNA solution (100 μM) and Ru(II) complexes (100 μM) were well mixed in an Eppendorf tube.AFM was conducted to investigate the nanosized polymers formed from 1 and 2 with oligonucleotides. The sequence of the oligonucleotides was 5′-TGGGGAGGGTGGGGAGGGTGGGGAAGG-3′. The morphologies and sizes of the oligonucleotide nanosized polymers that were treated by the complexes are shown in Fig. 1A and B. Abundant lacunose DNA particles and Ru(II) molecules associated with each other into a higher ordered conglomerate and self-assembled into nanosized polymers.32 Both 1 and 2@oligonucleotides displayed an irregular rotundity shaped. The 1@oligonucleotide nanosized polymers had an average width of 116–170 nm and an average height of 3–5 nm. The 2@oligonucleotide nanosized polymers had an average width of 375–422 nm and an average height of 5–6 nm (Fig. S4†). As 1 and 2 were added, the CT-DNA condensed into vast amorphous and irregular particles. These particles, which were loose and net-like, had an average width of 50–400 nm and an average height of 1–3 nm (Fig. S5†).33–35 The sphere-like morphology of the Ru(II)@oligonucleotides considerably differed from the net-like morphology of the Ru(II)@CT-DNA particles, suggesting that the conformation of oligonucleotides or CT-DNA was changed after being treated by 1 and 2. Furthermore, the size of the oligonucleotide nanosized polymers treated by complexes was larger than that of the CT-DNA presumably because of the higher affinity of 1 and 2 binding to oligonucleotides.36 Moreover, the size of the 2@oligonucleotides was larger than that of the 1@oligonucleotides possibly because of the effects of the substituents on the ligand. These results unambiguously differentiated the morphological characteristics of oligonucleotides and CT-DNA in the presence of 1 and 2, respectively, confirming that the complexes can selectively induce oligonucleotides to form nanosized polymers.
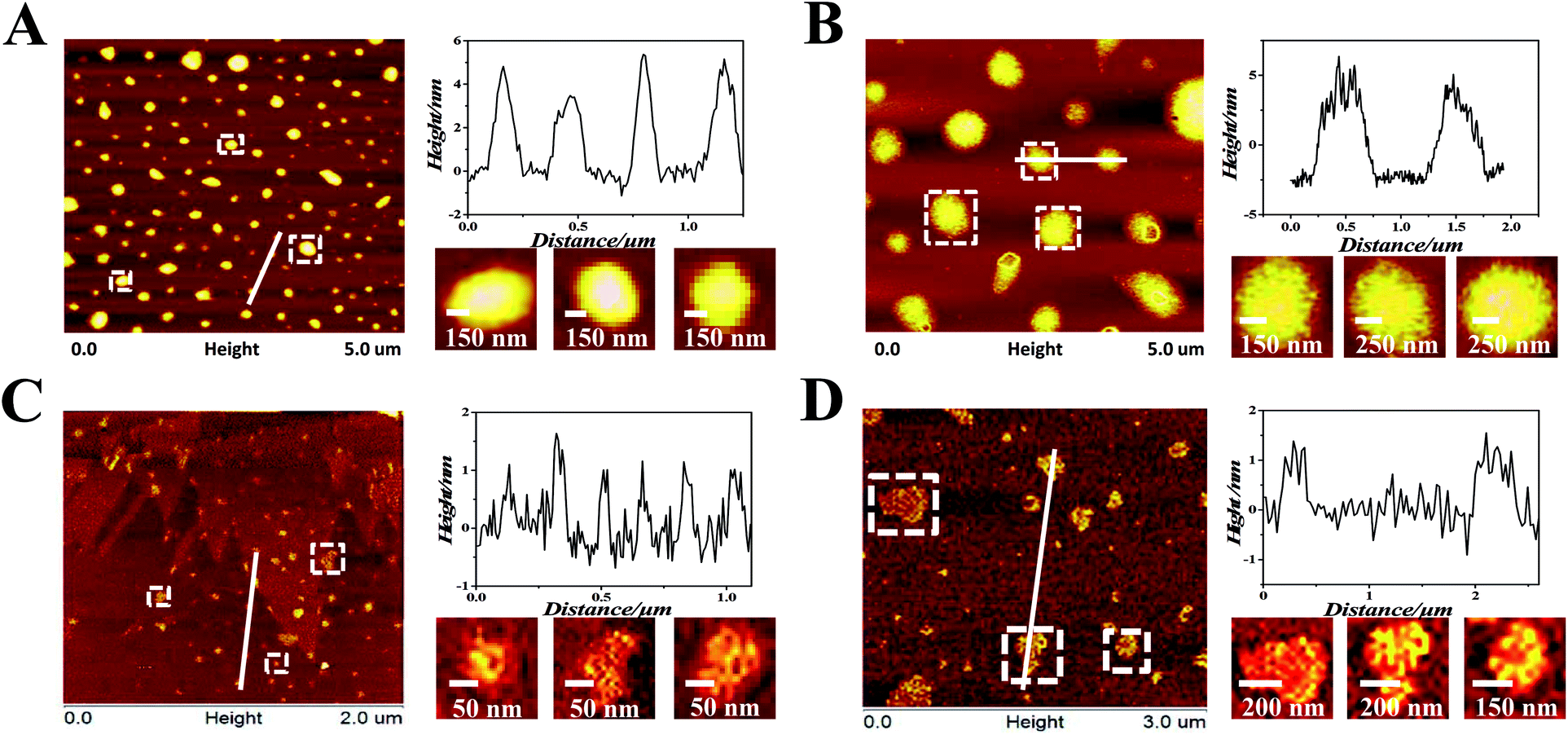 | ||
| Fig. 1 (A) AFM images of oligonucleotides after being treated by 1 (in pH 7.2 Tris–HCl KCl buffers). (B) AFM images of oligonucleotides after being treated by 2 (in pH 7.2 Tris–HCl KCl buffers). (C) AFM images of CT-DNA after being treated by 1 (in pH 7.2 Tris–HCl NaCl buffers). (D) AFM images of CT-DNA after being treated by 2 (in pH 7.2 Tris–HCl NaCl buffers). [Ru] = 100 μM, [oligonucleotides] = 100 μM, [CT-DNA] = 100 μM. | ||
To investigate the surface properties and stability of the polymers, the TEM (Fig. S7†) and particles size (Fig. S8†) of Ru(II)@oligonucleotides and Ru(II)@CT-DNA were tested. It can be observed that the size of Ru(II)@oligonucleotides, especially the 2@oligonucleotides, was larger than that of Ru(II)@CT-DNA. For oligonucleotides, upon the addition of 1 and 2, DNA condensates exerted more tight nanosized polymers, especially for 1@oligonucleotides with excellent dispersivity which complex 1 exhibited greater binding affinity to oligonucleotides than 2. However for CT-DNA, Ru@CT-DNA presented smaller size and loose polymers with the addition of 1 and 2. It is suggested that 1 and 2 showed weak binding affinity to CT-DNA. Besides, it is found that the size of 1@CT-DNA and 2@CT-DNA exerted big fluctuation in 7 days, but the size of 1@oligonucleotides and 2@oligonucleotides exhibited some certain degree changes and kept a excellent stability in 7 days. These results were agreement with above result, and it is indicated that 1 and 2 may display great affinity to oligonucleotide.
DNA-binding behaviour of Ru(II) complexes with oligonucleotides
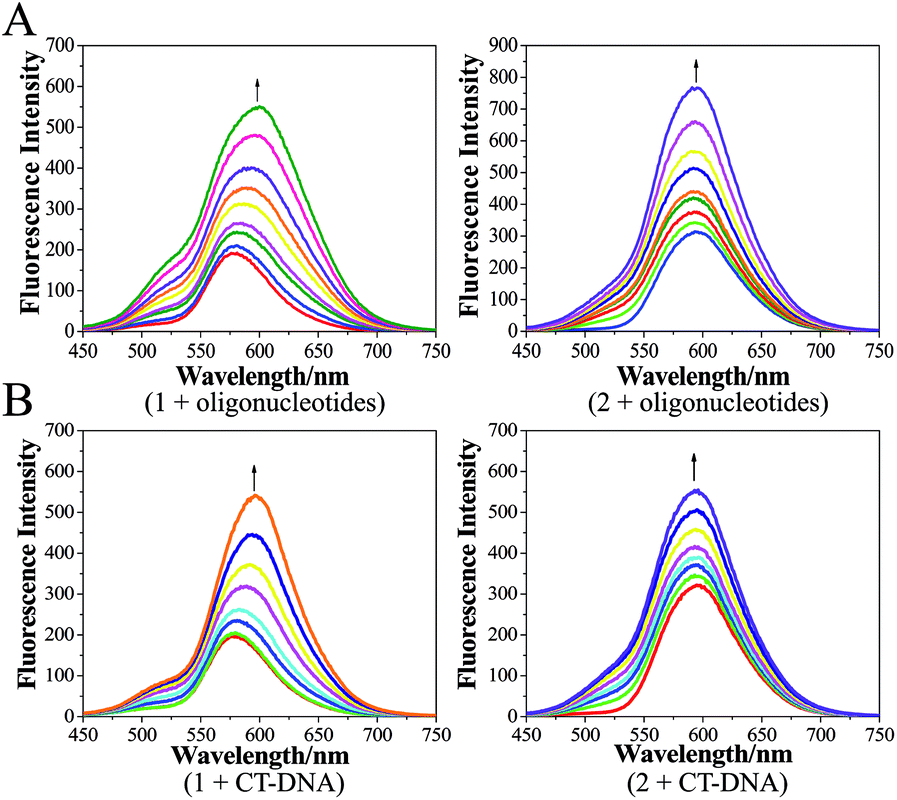 | ||
| Fig. 2 (A) Changes in the luminescence intensities of 1 and 2 in the absence and presence of oligonucleotides. [Ru] = 100 μM, [oligonucleotides] = 0.067n μM, n = 0, 1, 2…, (in pH 7.2 Tris–HCl KCl buffers). (B) Changes in the luminescence intensities of 1 and 2 in the absence and presence of CT-DNA. [Ru] = 100 μM, [CT-DNA] = 0.067n μM, n = 0, 1, 2…, (in pH 7.2 Tris–HCl NaCl buffers). | ||
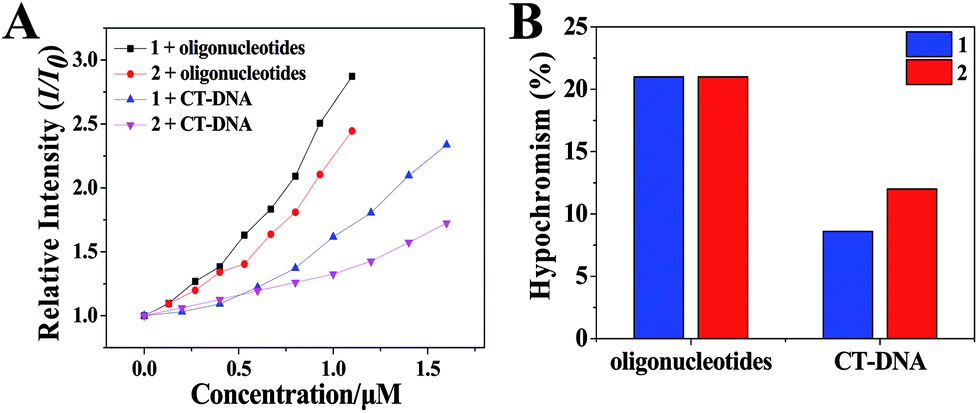 | ||
| Fig. 3 (A) The relative intensity (I/I0) of 1 and 2 with increasing concentration of oligonucleotides or CT-DNA. [Ru] = 20 μM, [oligonucleotides] = 0.067n μM, n = 0, 1, 2…, [CT-DNA] = 0.067n μM, n = 0, 1, 2…, (in pH 7.2 Tris–HCl KCl or NaCl buffers). (B) Hypochromic effect of the changes of 1 and 2 between oligonucleotides and CT-DNA, [Ru] = 20 μM, [oligonucleotides] = 100 μM, [CT-DNA] = 100 μM, (in pH 7.2 Tris–HCl KCl or NaCl buffers). | ||
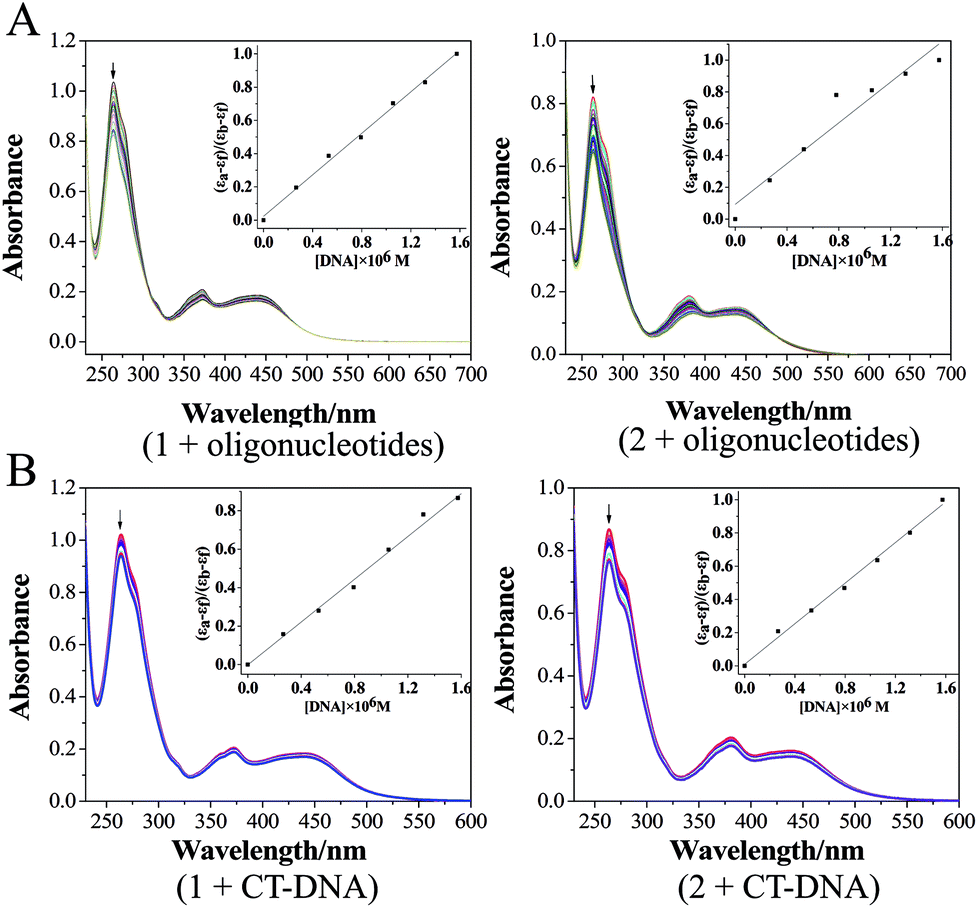 | ||
| Fig. 4 (A) Electronic spectra of 1 and 2 in the absence and presence of oligonucleotides in the Tris–HCl KCl buffer (pH 7.2). ([Ru] = 20 μM, [oligonucleotides] = 0.067n μM, n = 0, 1, 2…); (B) electronic spectra of 1 and 2 in the absence and presence of CT-DNA in the Tris–HCl NaCl buffer (pH 7.2). ([Ru] = 20 μM, [CT-DNA] = 0.067n μM, n = 0, 1, 2…). | ||
 | ||
| Fig. 5 (A) CD titration spectra of oligonucleotides with increasing amounts of 1 and 2 in the incubation buffer, [Ru] = 0, 2.660, 5.305, 7.937, 10.554, 13.158, 15.748, 18.325 μM, [oligonucleotides] = 100 μM. (B) CD titration spectra of CT-DNA with increasing amounts of 1 and 2 in the incubation buffer, [Ru] = 0, 2.660, 5.305, 7.937, 10.554, 13.158, 15.748, 18.325 μM, [CT-DNA] = 100 μM. | ||
In general, intercalation of a compound into DNA is known to cause an observable increase in the viscosity of a DNA solution due to an increase in the distance of base pairs at the intercalation site. Viscosity changes were measured using CT-DNA with increasing concentrations of the complex 1 and 2, as shown in Fig. S9.† The effects of complex 1 and 2 versus [Ru(bpy)3]2+ on the relative viscosity of rod-like DNA are shown in Fig. S9.† It is well known that [Ru(bpy)3]2+ interacts with DNA in a classical electronic effect. On increasing the concentration of complex 1, the relative viscosity of the DNA increased steadily compared to [Ru(bpy)3]2+. Moreover, a number of studies have reported that this kind of Ru(II) complex interact with oligonucleotides through groove binding mode by using molecular docking calculation.8 This suggests that complex 1 and 2 insert into the base-pairs of CT-DNA via an intercalative mode and interact with oligonucleotides through groove binding mode, which is in agreement with the spectroscopic studies.
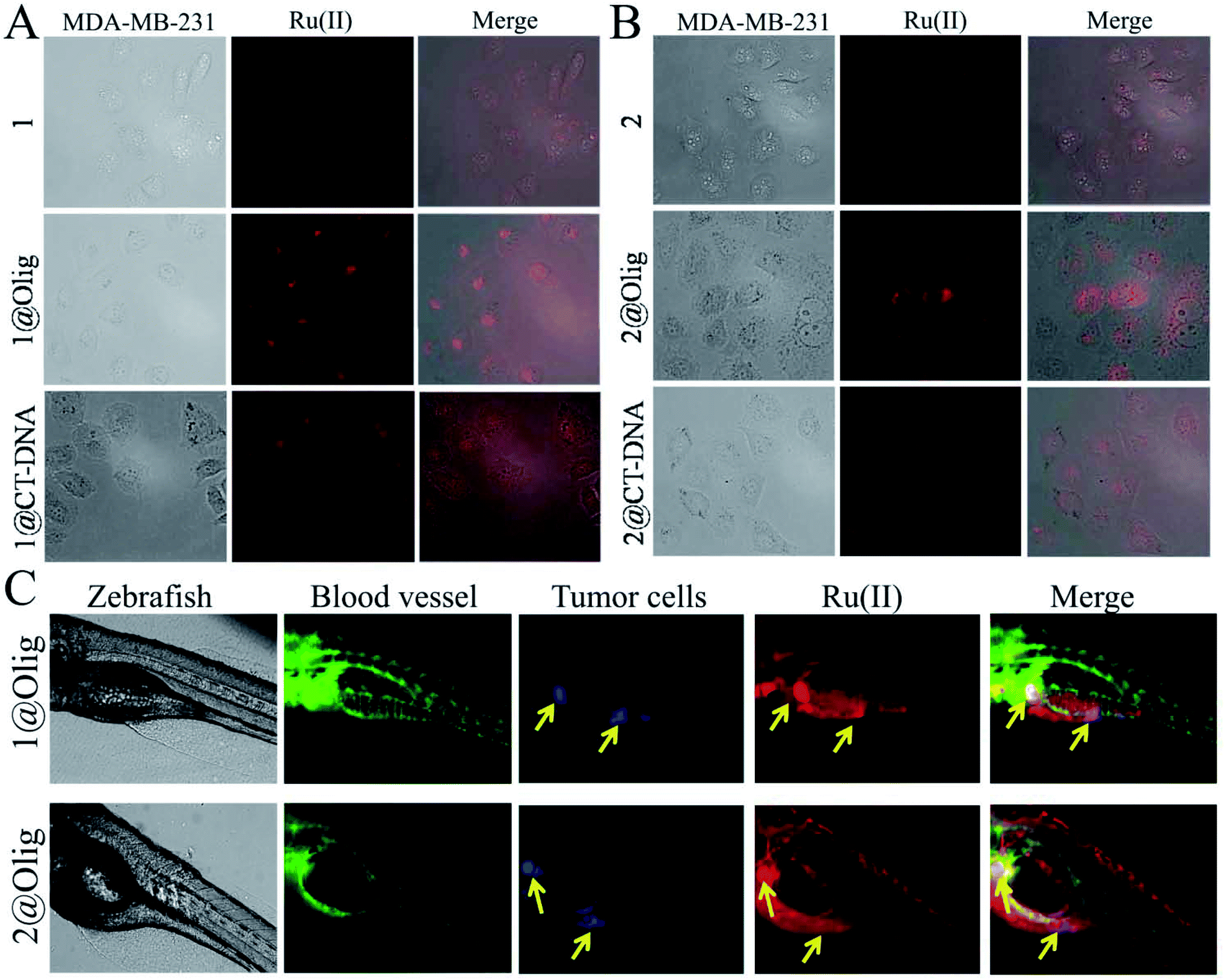 | ||
| Fig. 6 Selectively imaging tumor cells by Ru(II)@oligonucleotide in vitro and in vivo. (A) MDA-MB-231 cells imaging of 1, 1@oligonucleotide and 1@CT-DNA at 37 °C for 24 h. (B) MDA-MB-231 cells imaging of 2, 2@oligonucleotide and 2@CT-DNA at 37 °C for 24 h. (C) Tumor area highlighted by 1@oligonucleotide and 2@oligonucleotide in zebrafish xenografts model. ([Ru] = 2.5 μM, [oligonucleotides] = 2.5 μM). | ||
The zebrafish is an excellent model to study the bioimaging properties of fluorescent probe. Here, building on work with a transgenic zebrafish (fil1:EGFP), MDA-MB-231 breast cancer bearing model, which the blood vessels were labeled green fluorescence, blue fluorescently labelled MDA-MB-231 cells were serially transplanted in limiting dilutions to identify the tumor cells near sub intestinal vessel (SIV) of zebrafish.50 Then, the ability of 1@oligonucleotide and 2@oligonucleotide to monitor the tumor bioimaging in zebrafish xenografts model was investigated. The 72 hour post fertilization (hpf) zebrafish larvae were incubated with 1@oligonucleotide and 2@oligonucleotide (2.5 μM) for 96 h at 28.5 °C. As shown in Fig. 6C, when treatment with 1@oligonucleotides and 2@oligonucleotides for 96 h, it is discovered that 1@oligonucleotides and 2@oligonucleotides primarily distributed in intestine, but more distinctly highlight tumor area. That means the nanosized 1@oligonucleotides and 2@oligonucleotides could effectively enriched in tumor cells in vivo. These findings inspired us to develop oligonucleotides modified Ru(II) complex as an efficient phosphorescent probe to track living tumor cells for future clinical applications.
Conclusions
In conclusion, the present study established a novel method to construct a Ru(II)@oligonucleotide nanosized polymers to highlight tumor cells through EPR effect in vitro and in vivo. Then, a nanosized functionalized Ru(II) complexes with great phosphorescence imaging ability can be obtained by oligonucleotide modification. Moreover, it is found that the binding behavior of two Ru(II) complexes plays a key role in the condensation of DNA to form nanosized polymers. Further studies shown that Ru(II)@oligonucleotide nanosized polymers can distinctly highlight tumor cells in vitro and obviously enrich in tumor area in zebrafish xenografts model. In a word, the present nanosized polymers developed as a potential tumor selective tracker, which is expected to have potential applications of tumor targeting imaging and tracking in vitro and in vivo.Experimental
Reagents and materials
All reagents were purchased from commercial suppliers and used without further purification. Solvents were dried and purified through conventional methods prior to use. Ru(III) chloride hydrate, o-phenylenediamine, and 4-bromo-1,2-phenylenediamine were purchased from Aladdin. 1,10-Phenanthroline-5,6-dione was prepared by using a previously reported method.51 The ligands dppz and Br-dppz were prepared as described by Norden et al.52 All chemicals, including solvents, were obtained from commercial vendors and used as received. Oligonucleotides (5′-TGGGGAGGGTGGGGAGGGTGGGGAAGG-3′) and calf thymus DNA were purchased from Sangon Biotech (Shanghai) Co. All aqueous solutions were prepared with doubly distilled water. The Tris–HCl buffer (which was utilized in the CD spectroscopy, emission titration, UV titration, and AFM) consisted of 10 mM Tris and 100 mM NaCl or 100 mM KCl, and the pH was adjusted to 7.2 by using HCl solution.Instruments
The ESI-MS spectra were obtained in methanol on Agilent 1100 ESI-MS system operated at room temperature. The 1H NMR and 13C NMR spectra were recorded in a dimethyl-d6 sulfoxide (DMSO-d6) solution on a Bruker Avance III 500 spectrometer operated at room temperature. The UV-vis absorption spectra were obtained on a Shimadzu UV-2550 spectrophotometer. The steady-state emission spectra were recorded on a RF-5301 fluorescence spectrophotometer. The CD spectra were obtained on a Jasco J810 CD spectrophotometer. AFM imaging was performed with an atomic force microscope (AFM; Bruker, Dimension FastScanTM). Tumor cell imaging was observed on fluorescence microscopy (Leica, DMI8).Synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 glycol and water mixture. The reaction mixture was diluted with 80 mL of water after the solution was cooled to room temperature. Then, the mixture was filtered, and excess sodium perchlorate was added to obtain an orange solid. The crude products were filtered and washed with cooled water and ether, and purified by flash silica gel column chromatography to afford a red powder. The reaction yield was 67%. ESI-MS (in CH3CN, m/z): 843.1 [M-ClO4−]+ and 372.5 [M-2ClO4−]2+(Fig. S1A†). 1H NMR (500 MHz, DMSO-d6) δ 9.61 (d, J = 1.2 Hz, 1H), 9.59–9.59 (m, 1H), 8.81 (d, J = 1.2 Hz, 1H), 8.80 (t, J = 1.3 Hz, 2H), 8.78 (d, J = 1.3 Hz, 1H), 8.52 (dt, J = 6.4, 3.2 Hz, 2H), 8.41 (s, 4H), 8.28 (dt, J = 4.5, 2.3 Hz, 2H), 8.21–8.18 (m, 4H), 8.07 (dd, J = 5.2, 1.2 Hz, 2H), 7.90 (dd, J = 6.8, 4.0 Hz, 2H), 7.82 (dd, J = 8.3, 5.3 Hz, 2H), 7.78 (dd, J = 8.3, 5.3 Hz, 2H) (Fig. S2A†). 13C NMR (126 MHz, DMSO-d6) δ 153.96 (s), 153.31 (s), 152.69 (s), 150.76 (s), 147.15 (s), 141.93 (s), 140.21 (s), 136.97 (s), 133.21 (s), 132.60 (s), 130.49 (d, J = 2.8 Hz), 130.10 (s), 129.45 (s), 128.09 (s), 127.55 (s), 126.32 (d, J = 13.0 Hz) (Fig. S3A†).
:1 glycol and water mixture. The reaction mixture was diluted with 80 mL of water after the solution was cooled to room temperature. Then, the mixture was filtered, and excess sodium perchlorate was added to obtain an orange solid. The crude products were filtered and washed with cooled water and ether, and purified by flash silica gel column chromatography to afford a red powder. The reaction yield was 67%. ESI-MS (in CH3CN, m/z): 843.1 [M-ClO4−]+ and 372.5 [M-2ClO4−]2+(Fig. S1A†). 1H NMR (500 MHz, DMSO-d6) δ 9.61 (d, J = 1.2 Hz, 1H), 9.59–9.59 (m, 1H), 8.81 (d, J = 1.2 Hz, 1H), 8.80 (t, J = 1.3 Hz, 2H), 8.78 (d, J = 1.3 Hz, 1H), 8.52 (dt, J = 6.4, 3.2 Hz, 2H), 8.41 (s, 4H), 8.28 (dt, J = 4.5, 2.3 Hz, 2H), 8.21–8.18 (m, 4H), 8.07 (dd, J = 5.2, 1.2 Hz, 2H), 7.90 (dd, J = 6.8, 4.0 Hz, 2H), 7.82 (dd, J = 8.3, 5.3 Hz, 2H), 7.78 (dd, J = 8.3, 5.3 Hz, 2H) (Fig. S2A†). 13C NMR (126 MHz, DMSO-d6) δ 153.96 (s), 153.31 (s), 152.69 (s), 150.76 (s), 147.15 (s), 141.93 (s), 140.21 (s), 136.97 (s), 133.21 (s), 132.60 (s), 130.49 (d, J = 2.8 Hz), 130.10 (s), 129.45 (s), 128.09 (s), 127.55 (s), 126.32 (d, J = 13.0 Hz) (Fig. S3A†).Preparation of the nanosized polymers
Ru(II) complexes were dissolved by dimethyl sulfoxide and then diluted to 100 μM solution by Tris–HCl KCl or NaCl buffer (pH = 7.2) ([KCl] = 0.1 M, [NaCl] = 0.1 M). The oligonucleotides or CT-DNA were dissolved and diluted to 100 μM solution by Tris–HCl KCl or NaCl buffer (pH = 7.2) ([KCl] = 0.1 M, [NaCl] = 0.1 M). The oligonucleotides or CT-DNA solution (100 μM) and were well mixed with the same volume of Ru(II) complexes (100 μM) in an Eppendorf tube at room temperature for 72 h.AFM observation
A 100 μL mixed solution was added to a copper wire mesh and naturally volatilized for 2 h. The mixture of oligonucleotide solution (100 μM) or CT-DNA solution (100 μM) was incubated with Ru(II) complexes (100 μM) for 3 days. Next, a 10 μL mixed solution was removed to a mica plate and naturally volatilized for 2 h. An image of the sample was captured by AFM (Bruker, Dimension FastScanTM).Viscosity measurements
The viscosity measurement is an effective method to judge the interaction mode of Ru(II) complexes with DNA. Fixed solutions of complexes and DNA in different concentrations were prepared in Tris–HCl buffer media, with [Ru]/[DNA] = 0, 0.02, 0.04, 0.06, 0.08, 0.1. Before testing, the solutions were stored in a thermostatic water bath at (30 ± 0.1 °C) 1 h. The formula calculated viscosity is: η = (t − t0)/t0. Viscosity curves were obtained using (η/η0)1/3 as the Y-axis and with r (r = [Ru]/[DNA]) as the X-axis.Transmission electron microscope (TEM) observation
The mixture of oligonucleotide solution (100 μM) or CT-DNA solution (100 μM) was incubated with Ru(II) complexes (100 μM) for 24 h. A 20 μL mixed solution was added to a copper wire mesh and naturally volatilized for three times. The images of the samples were obtained on TEM (Hitachi Ltd, Tokyo, Japan).Particle size observation
The mixture of oligonucleotide solution (10 μM) or CT-DNA solution (10 μM) was incubated with Ru(II) complexes (10 μM). The particle size change of the samples varied with time, were obtained on Zetasizer Nano ZS (Malvern, Britain).Fluorescence emission titrations
Fluorescence spectroscopy was performed on a RF-5301 fluorescence spectrophotometer using a 1 cm path length quartz cell. Luminescence titrations of Ru(II) complexes were measured by using a fixed Ru(II) concentration (100 μM) with increasing CT-DNA or oligonucleotides. The first spectrum was obtained after 5 min to allow the sample (3000 μL) to equilibrate. Then, 2 μL of the 100 μM DNA solution was added to the sample cell with thorough mixing. The titration processes were repeated until no apparent change occurred in the spectra for at least three titrations, which indicated that binding saturation was achieved.54Electronic absorption spectra
The electronic absorption spectra were obtained on a Shimadzu UV-2550 spectrophotometer using 1 cm path length quartz cuvettes (3 mL). The absorption titration of the Ru(II) complex in Tris–HCl buffer was performed by using a fixed complex concentration, to which increments of the DNA stock solution were added. The concentration of the complex solution was 20 μM, and CT-DNA or oligonucleotides were added by degrees. Complex-DNA solutions were incubated for 3 min before the absorption spectra were recorded.51CD spectroscopy
The CD spectra were obtained on a Jasco J810 circular dichroism (CD) spectrophotometer with a thermoelectrically controlled cell holder. The cell path length was 1 cm. The CD spectra were obtained in the range of 230–600 nm at 0.5 nm increments with an averaging time of 0.5 s.55Cellular imaging
MDA-MB-231 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cell lines of MDA-MB-231 were maintained in DMEM with fetal bovine serum (10%), penicillin (100 units per mL), and streptomycin (50 units per mL) at 37 °C in a CO2 incubator (95% relative humidity, 5% CO2). MDA-MB-231 cells were resuspended in 10% FBS DMEM at a density of 2.5 × 104 cells per mL, which were then treated with drugs and incubated for 24 h at 37 °C, unless otherwise stated. Cells were washed thrice in PBS. Cell morphology was observed by laser confocal microscope.Selectively imaging tumor cells by Ru(II)@oligonucleotide in zebra fish xenografts model
The establishment of zebrafish embryo breast cancer model was performed by the description of the related literature.50 Juvenile zebrafish of breast cancer model (72 h old) were incubated in 24-well plates (10 fishes in every well) with 1 mL solutions with 1@oligonucleotides and 2@oligonucleotides (2.5 μM) in aquaculture water. The localization of 1@oligonucleotides and 2@oligonucleotides in breast cancer zebrafish were observed every 24 h with a fluorescence microscopy. The relevant ethical protocols used for the in vivo study for zebrafish were performed in compliance with relevant laws or guidelines and were followed institutional guidelines. The animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals issued by the National Institutes of Health, and approved by the Laboratory Animal Ethics Committee of Guangdong Pharmaceutical University.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by National Nature Science Foundation of China (81572926, 81703349), the Provincial Major Scientific Research Projects in Universities of Guangdong Province (2014KZDXM053), the Science and Technology Project of Guangdong Province (2014A020212312), the Innovation Projects in Universities of Guangdong Province (2015cxqx151), Tradition Chinese Medicine Bureau of Guangdong Province (20151265), the Innovation Team Projects in Universities of Guangdong Province (2016KCXTD018), Provincial College Students Innovation and Entrepreneurship Training Program of Guangdong Province (51307173), National College Students Innovation and Entrepreneurship Training Program (51307164).Notes and references
- T. Vo-Dinh, J. Wang, J. R. Lakowicz, P. T. Vernier, Y. Sun, Z. K. Gryczynski, M. A. Gundersen and L. Marcu, Proc. SPIE, 2005, 5703, 127 CrossRef.
- R. J. Antcliff, M. R. Stanford, D. S. Chauhan, E. M. Graham, D. J. Spalton, J. S. Shilling, T. J. Ffytche and J. Marshall, Ophthalmology, 2000, 107, 593–599 CrossRef PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef PubMed.
- Q. R. Tummers, C. E. Hoogstins, K. N. Gaarenstroom, C. D. de Kroon, M. I. van Poelgeest, J. Vuyk, T. Bosse, V. T. Smit, C. J. van de Velde, A. F. Cohen, P. S. Low, J. Burggraaf and A. L. Vahrmeijer, Oncotarget, 2016, 7, 32144–32155 CrossRef PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef PubMed.
- W. R. Freeman, D. U. Bartsch, A. J. Mueller, A. S. Banker and R. N. Weinreb, Clin. Sci., 1998, 116, 455–463 Search PubMed.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2795 CrossRef PubMed.
- M. R. Gill, H. Derrat, C. G. Smythe, G. Battaglia and J. A. Thomas, ChemBioChem, 2011, 12, 877–880 CrossRef PubMed.
- A. E. Friedman, J.-C. Chambron, J.-P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef.
- X. Q. Guo, F. N. Castellano, L. Li and J. R. Lakowicz, Biophys. Chem., 1998, 71, 51–62 CrossRef PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef PubMed.
- C. Qian, J. Q. Wang, C. L. Song, L. L. Wang, L. N. Ji and H. Chao, Metallomics, 2013, 5, 844–854 RSC.
- Z. P. Zeng, Q. Wu, F. Y. Sun, K. D. Zheng and W. J. Mei, Inorg. Chem., 2016, 55, 5710–5718 CrossRef PubMed.
- U. Neugebauer, Y. Pellegrin, M. Devocelle, R. J. Forster, W. Signac, N. Moran and T. E. Keyes, Chem. Commun., 2008, 42, 5307–5309 RSC.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2009, 131, 8738–8739 CrossRef PubMed.
- K. Kam-Wing Lo, T. K.-M. Lee, J. S.-Y. Lau, W.-L. Poon and S.-H. Cheng, Inorg. Chem., 2008, 47, 200–208 CrossRef PubMed.
- L. Blackmore, R. Moriarty, C. Dolan, K. Adamson, R. J. Forster, M. Devocelle and T. E. Keyes, Chem. Commun., 2013, 49, 2658–2660 RSC.
- R. Langer, Nature, 1998, 392, 5–10 Search PubMed.
- D. Schrama, R. A. Reisfeld and J. C. Becker, Nat. Rev. Drug Discovery, 2006, 5, 147–159 CrossRef PubMed.
- S. M. Nimjee, C. P. Rusconi and B. A. Sullenger, Annu. Rev. Med., 2005, 56, 555–583 CrossRef PubMed.
- A. Corti and M. Ponzoni, Ann. N. Y. Acad. Sci., 2004, 1028, 104–112 CrossRef PubMed.
- M. Goldberg, R. Langer and X. Jia, J. Biomater. Sci., Polym. Ed., 2007, 18, 241–268 CrossRef PubMed.
- E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef PubMed.
- C. D. Irene Brigger and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631–651 CrossRef.
- J. C. E. A. Sykes, G. Zheng and W. C. W. Chan, ACS Nano, 2014, 8, 5696–5706 CrossRef PubMed.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Nano Lett., 2009, 9, 1909–1915 CrossRef PubMed.
- B. Subia and S. C. Kundu, Nanotechnology, 2013, 24, 035103 CrossRef PubMed.
- L. Wu, L. Zhang, G. Shi and C. Ni, Mater. Sci. Eng., C, 2016, 61, 278–285 CrossRef PubMed.
- Q. Wu, W. Mei, K. Zheng and Y. Ding, Sci. Rep., 2016, 6, 28582 CrossRef PubMed.
- K. Qiu, B. Yu, H. Huang, P. Zhang, J. Huang, S. Zou, Y. Chen, L. Ji and H. Chao, Sci. Rep., 2015, 5, 10707 CrossRef PubMed.
- B. Yu, C. Ouyang, K. Qiu, J. Zhao, L. Ji and H. Chao, Chem.–Eur. J., 2015, 21, 3691–3700 CrossRef PubMed.
- B. Yu, Y. Chen, C. Ouyang, H. Huang, L. Ji and H. Chao, Chem. Commun., 2013, 49, 810–812 RSC.
- L. Liu, H. Zhang, X. Meng, J. Yin, D. Li and C. Liu, Biomaterials, 2010, 31, 1380–1391 CrossRef PubMed.
- G. L. Liao, X. Chen, L. N. Ji and H. Chao, Chem. Commun., 2012, 48, 10781–10783 RSC.
- Y. Jenkins, A. E. Friedman, N. J. Turro and J. K. Barton, Biochemistry, 1992, 31, 10809–10816 CrossRef PubMed.
- S. A. Poteet, M. B. Majewski, Z. S. Breitbach, C. A. Griffith, S. Singh, D. W. Armstrong, M. O. Wolf and F. M. MacDonnell, J. Am. Chem. Soc., 2013, 135, 2419–2422 CrossRef PubMed.
- H. J. Yu, L. Yu, Z. F. Hao and Y. Zhao, Spectrochim. Acta, Part A, 2014, 124, 187–193 CrossRef PubMed.
- M. Balaz, B. C. Li, J. D. Steinkruger, G. A. Ellestad, K. Nakanishi and N. Berova, Org. Biomol. Chem., 2006, 4, 1865–1867 RSC.
- S. Mahadevan and M. Palaniandavar, Inorg. Chem., 1998, 37, 3927–3934 CrossRef PubMed.
- X. Qu, J. O. Trent, I. Fokt, W. Priebe and J. B. Chaires, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12032–12037 CrossRef PubMed.
- D. Mazumdar, N. Nagraj, H.-K. Kim, X. Meng, A. K. Brown, Q. Sun, W. Li and Y. Lu, J. Am. Chem. Soc., 2009, 131, 5506–5515 CrossRef PubMed.
- N. Shimada, A. Kano and A. Maruyama, Adv. Funct. Mater., 2009, 19, 3590–3595 CrossRef.
- M. Balaz, A. E. Holmes, M. Benedetti, G. Proni and N. Berova, Bioorg. Med. Chem., 2005, 13, 2413–2421 CrossRef PubMed.
- M. Balaz, A. E. Holmes, M. Benedetti, P. C. Rodriguez, N. Berova, K. Nakanishi and G. Proni, J. Am. Chem. Soc., 2005, 127, 4172–4173 CrossRef PubMed.
- A. H.-J. Wang, G. J. Quigley, F. J. Kolpak, J. L. Crawford, J. H. V. Boom, G. van der Marel and A. Rich, Nature, 1980, 282, 680–686 CrossRef.
- S. S. Bhat, A. S. Kumbhar, A. A. Kumbhar, A. Khan, P. Lonnecke and E. Hey-Hawkins, Chem. Commun., 2011, 47, 11068–11070 RSC.
- J. Wang, Z. Cao, X. M. Zhang, M. Nakamura, M. Sun, J. Hartman, R. A. Harris, Y. Sun and Y. Cao, Cancer Res., 2015, 75, 306–315 CrossRef PubMed.
- V. G. Vaidyanathan and B. U. Nair, J. Inorg. Biochem., 2002, 91, 405–412 CrossRef PubMed.
- C. Hiort, P. Lincoln and B. Norden, J. Am. Chem. Soc., 1993, 115, 3448–3454 CrossRef.
- E. Amouyal, A. Homsi, J.-C. Chambron and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1990, 6, 1841–1845 RSC.
- X. H. Lu, S. Shi, J. L. Yao, X. Gao, H. L. Huang and T. M. Yao, J. Inorg. Biochem., 2014, 140, 64–71 CrossRef PubMed.
- P. U. Maheswari, V. Rajendiran, M. Palaniandavar, R. Parthasarathi and V. Subramanian, J. Inorg. Biochem., 2006, 100, 3–17 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05454a |
| ‡ Equally contribution. |
| This journal is © The Royal Society of Chemistry 2018 |