
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlumbagin induces ROS-mediated apoptosis and cell cycle arrest and inhibits EMT in human cervical carcinoma cells
Ankita Jaiswala,
Akash Sabarwal ab,
Jai Prakash Narayan Mishraa and
Rana P. Singh*ab
ab,
Jai Prakash Narayan Mishraa and
Rana P. Singh*ab
aSchool of Life Sciences, Central University of Gujarat, Gandhinagar, Gujarat, India
bCancer Biology Laboratory, School of Life Sciences, Jawaharlal Nehru University, New Delhi 110067, India. E-mail: rana_singh@mail.jnu.ac.in
First published on 17th September 2018
Abstract
Plumbagin, an important phytochemical from the roots of the medicinal plant Plumbago zeylanica L. has shown many biological activities. The roots of this plant have been in use in the Indian system of medicine for more than twenty five centuries for treatments of various ailments. It has shown anticancer activities, however, the anticancer and anti-metastatic effects of plumbagin are largely unknown against cervical cancer cells. Herein, we investigated the molecular alterations associated with plumbagin-mediated inhibition of growth, survival and epithelial to mesenchymal transition of human cervical cancer SiHa and HeLa cells. Plumbagin (1–4 μM) caused a significant decrease in the cell viability and increased the cell death in SiHa and Hela cells after 24 and 48 h. Plumbagin also caused strong G2/M and S-G2/M phase cell cycle arrest in SiHa and HeLa cells, respectively which was accompanied by a decrease in the expression of cyclin and CDK levels. The expression levels of both mRNAs and proteins of cyclin B1, A and E2 and CDK 1 and 2 decreased after 24 and 48 h. Plumbagin strongly induced apoptosis along with increased ratio of Bax![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bcl2 and cleavage of caspase 3, 9, and PARP. Plumbagin caused a significant increase in reactive oxygen species generation which mediated cell death as it was attenuated by pre-treatment with N-acetyl cysteine. Additionally, we also report for the first time that plumbagin possesses an anti-metastatic effect at non-cytotoxic doses that was accompanied by the modulation of MMP-2, 9, E-cadherin, N-cadherin, β-catenin and vimentin. Taken together, our findings suggest that plumbagin has strong anticancer and anti-metastatic effects against human cervical cancer cells.
:Bcl2 and cleavage of caspase 3, 9, and PARP. Plumbagin caused a significant increase in reactive oxygen species generation which mediated cell death as it was attenuated by pre-treatment with N-acetyl cysteine. Additionally, we also report for the first time that plumbagin possesses an anti-metastatic effect at non-cytotoxic doses that was accompanied by the modulation of MMP-2, 9, E-cadherin, N-cadherin, β-catenin and vimentin. Taken together, our findings suggest that plumbagin has strong anticancer and anti-metastatic effects against human cervical cancer cells.
Introduction
Cervical cancer is one of the leading cancers among women worldwide, and accounted for 7.5% of all female cancer deaths in 2012.1 Unlike other cancers, nearly 90% of cervical cancer deaths occur in developing parts of the world,2 cervical cancer strikes early in the reproductive period of females, and incidence rises at 30–34 years of age and peaks at 50–65 years. In the context of India, approximately 132000 new cervical cancer cases and 74000 deaths occur annually, accounting for one-third of total cervical cancer-related deaths worldwide.3
Incidence and mortality from cervical cancer are attributed to geographical variations in population and prevalence of human papillomavirus (HPV), which is the major risk factor responsible for cervical cancer.4 Many co-factors enhance the risk in HPV-infected females including usage of hormonal contraceptives, high parity, high sexual activity, co-infection with HIV and smoking.4 Globally, HPV 16 and 18 genotypes contribute more than 70% of all cervical cancer cases, among these two HPV 16 is responsible alone for nearly 50–60% cancers.
Although HPV vaccines are commercially available for the control of cervical cancer, their use remains limited due to unawareness and high cost involved, furthermore, the long-run effectiveness remains yet to be proven. Thus, there's an imperative need to explore potent, cost-effective and less cytotoxic agent with fewer side effects.
Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), from the roots of Plumbago zeylanica Linn. (Plumbaginaceae) possess numerous pharmacologic activities in a traditional system of medicine and employed clinically for their prophylactic, germicidal, anti-leprotic, anti-cancer and anti-inflammatory activities.5 The plant is additionally reported to possess stimulatory effects on central nervous system, hepatoprotective, antioxidant, hypo-lipidaemic and anti-atherosclerotic properties.6,7 Plumbagin has been shown to exert anticancer activities against a wide type of tumor cells including breast cancer, lung cancer, ovarian cancer, acute promyelocytic leukemia, melanoma, and prostate cancer.8–13 Plumbagin studies related to cervical cancer are limited showing generation of reactive oxygen species in ME-180 cervical cancer cells, mitochondrial membrane depolarization and apoptotic cell death. Plumbagin mediated cell death was found through both caspase-dependent and independent pathways. Pre-treatment with N-acetyl cysteine (NAC) has blocked plumbagin mediated activation of caspases and apoptosis.14 Similarly, plumbagin induced cytotoxicity in HeLa, SiHa and CC3A cells in time and dose-dependent fashion, CC3A cells were found to be more sensitive to plumbagin treatment and radiation. In case of prostate cancer DU 145 and PC-3 cells, plumbagin decreased proliferation and apoptosis was ROS mediated, additionally it has also caused lethal endoplasmic reticulum stress. Blockage of ROS generation significantly inhibited ER stress activation and apoptosis.15 Similarly, in human MCF-7 breast cancer cells, plumbagin caused ROS generation via 1-4-phosphatidylinositol 5-kinase (P15K)-1B, and knock-down of this gene caused increase in ROS generation and decrease in cell viability indicating ROS mediated cell death.16 In human melanoma A375.S2 cells plumbagin caused cell cycle arrest and apoptosis that was mediated by ROS generation. Vitamin C and other antioxidants blocked the activation of JNK activation and apoptosis further confirming plumbagin has pro-oxidant activity in most type of cancers.12
Plumbagin has caused cell cycle arrest in many cancer cells, however, no studies were done to investigate the effect of plumbagin treatment on cell cycle regulation, mechanism of apoptosis and its effects on epithelial to mesenchymal transition in SiHa and HeLa cervical cancer cells.
Herein, for the first time, we report that plumbagin caused cell cycle arrest by downregulating cyclin–CDKs, ROS-mediated cell death which was attenuated by pre-treatment of NAC, and anti-metastatic effects in SiHa and HeLa cervical cancer cells and its mechanism of action. These findings suggest that plumbagin could be developed as potential anticancer agent against cervical cancer.
Materials and methods
Cell lines and reagents
SiHa and HeLa cervical cancer cell lines were obtained from NCCS, Pune, India. Minimum essential media (MEM)-with glutamine cell culture medium, antibiotic–antimycotic cocktail and other culture reagents were obtained from HiMedia Laboratories, India. Fetal bovine serum (FBS) was from GIBCO, Invitrogen, USA. Plumbagin, NAC, ethylenediaminetetraacetic acid (EDTA), propidium iodide, saponin, bromophenol blue, molecular biology grade DMSO were from Sigma Aldrich, USA, and ethanol was from Merck Biosciences, USA. RNaseA was procured from QIAGEN Hilden, Germany. Antibodies for CDK1, cleaved PARP, cleaved caspase-3, cleaved caspase-9, MMP-2, MMP-9, β-catenin, vimentin, N-cadherin, E-cadherin, peroxidase-conjugated secondary anti-rabbit antibody were from Cell Signaling Technology, USA, secondary anti-mouse antibody, β-actin and cyclin-A were from Sigma Aldrich, and cyclin E, CDK2, Bax, Bcl-2 were from Santa Cruz Biotechnology, USA. PVDF membrane and ECL HRP-linked substrate solution were from Merck Millipore (Billerica, MA). TRIZOL kit for RNA extraction was from Life Technologies, Carlsbad, CA, USA. cDNA synthesis kit was from Takara-Bio, Japan. Other common chemicals and laboratory reagents were from SD Fine-Chem. Ltd and SRL Pvt. Ltd. India.Cell culture and treatments
SiHa and HeLa cells were grown in MEM supplemented with 10% FBS and 1% penicillin–streptomycin–amphotericin B cocktail in standard culture conditions (37 °C, 95% humidified air and 5% CO2). The cells were grown as adherent monolayer and fed every alternate day and passaged when reached ∼80% confluency. Cell cultures were treated with desired concentrations of plumbagin dissolved in DMSO at 1, 2, 4 μM for cell cycle and apoptosis, and at 0.25, 0.5 and 0.75 μM for EMT experiments, for different time-points (24–48 h). DMSO concentration did not exceeded 0.1% (v/v) in any treatment.Cell growth and death assay
Cells were seeded at a density of 1 × 105 per 60 mm culture plate and treated with either DMSO or indicated concentrations of PB for 24 and 48 h. After incubation periods were over, cells were collected by brief trypsinization and centrifugation. The cell pellets were gently washed with ice-cold PBS, and the cell suspension was mixed with trypan blue dye and counted using hemocytometer. After each treatment time, cells were collected by brief trypsinization followed by addition of trypan blue dye and counted using hemocytometer under a phase contrast microscope. Live cells were appeared bright whereas dead cells were blue under the microscope. Each sample was in triplicate and counted twice sample and experiment were repeated at least 3 times. Similarly, cell counting was done for plumbagin-mediated ROS cell death study, in which cells were pre-treated with NAC for two hours before plumbagin treatment.17,18Flow cytometric analysis for cell cycle phase distribution
Cells were seeded in a similar manner, as in cell growth assay mentioned above and treated with plumbagin (1, 2, 4 μM) for specific time periods. At the end of each treatment, total cells were collected by brief trypsinization and centrifugation at 1500 rpm followed by processing for cell cycle analysis as reported recently.19,20 Briefly, the cell pellet was washed in PBS twice and saponin–PI (0.3% saponin, 25 mg mL−1 PI, 0.1 mM EDTA, and 10 mg mL−1 RNase A) solution was added. Cells were incubated in dark at 4 °C, followed by flow cytometric analysis using FACS Aria III flow cytometer (BD Biosciences).Semi-quantitative RT-PCR
Cells were seeded at 5 × 105 per 100 mm plate and treated with desired concentrations of plumbagin. After 24 and 48 h, cells were collected by TRIZOL reagent using manufacturer's protocol. Five μg of RNA was quantified for each sample to prepare cDNA using Takara cDNA synthesis kit. For PCR reaction mixture, template (3 μL), dNTP mix (10 mM) forward and reverse primers (10 pmol μL−1), 10× PCR buffer containing Mg++ (25 mM), and Taq DNA polymerase (3U μL−1) was added and final volume was made up to 25 μL. Thermocycler (Hamburg, Germany) was set to 25 cycles and PCR tubes were kept for reactions. After completing of PCR cycles, products were resolved on 1% agarose gel electrophoresis containing EtBr and visualised in GelDoc system (Applied Biosystems, CA, USA).21 The following primer sequences were used for RT-PCR.(1) GAPDH
Forward primer – 5′-GCC TTC CGT GTC CCC ACT GC 3′
Reverse primer – 5′-CAA TGC CAG CAG CGT CA 3′
(2) CDK1
Forward primer – 5′-ACT GGC TGA TTT CCT TGC C 3′
Reverse primer – 5′ TGA GTA ACG TGA CCC CAG CA 3′
(3) CDK2
Forward primer – 5′ AGG CCC GTG ATC CCC ACA GT 3′
Reverse primer – 5′TGG TGG GGG TGC CTT GTC CA 3′
(4) Cyclin E2
Forward primer – 5′ ACT GCT GCT GCC TTG TGC CA 3′
Reverse primer – 5′ GCC TCC ATT GCA CAC TGG TGA CA 3′
(5) Cyclin A2
Forward primer – 5′ GCA GAC GCT CCA AGA GG 3′
Reverse primer – 5′ AGG GGT GCA ACC CGT CTC GT 3′
(6) Cyclin B1
Forward primer – 5′GCG CAA AGC GCC TTC CTA CG 3′
Reverse primer – 5′CAC TGG CAC TGG CAG CAT 3′
(7) Bax
Forward primer – 5′AGG ATG CGT CCA CCA AGA AG3′
Reverse primer – 5′ CCA GTT GAA GTT GCC GTC 3′
(8) Bcl2
Forward primer – 5′AAG CGG TCC CGT GGA TAG A3′
Reverse primer – 5′TCC GGT ATT CGC AGA AGT CC 3′
Apoptosis assay
To quantify apoptosis, 50 thousand cells were seeded in similar manner, as in cell growth assay. After the completion of desired treatment time points (24 and 48 h) cells were collected by trypsinization and centrifuged at 1200 rpm for 5 min and pelleted, and mixed with Acridine Orange and Ethidium Bromide (AO/EB) (100 μg mL−1 – 1:1) working solution. Cells were visualized under 400× magnification under a fluorescent microscope. A minimum of 100 cells were scored into one of the four categories:
(i) Live cells (normal green nucleus), (ii) early apoptotic cells (bright green nucleus with condensed or fragmented chromatin), (iii) late apoptotic cell (orange stained nuclei with chromatin condensation or fragmentation), (iv) necrotic cells (uniformly stained nuclei with nuclear morphology resembling that of the viable with no condensed chromatin).
Immunoblot analysis
After treatment conditions for specific time periods, whole cell lysates were prepared.22 Bradford assay was done to estimate protein content. Samples were boiled for 5 minutes and loaded on to 10–12% denaturing SDS-PAGE gels and proteins were resolved at a constant voltage. Proteins from the gel were transferred on to PVDF membrane using transfer assembly. Membranes were blocked in blocking buffer for 1 h and incubated with specific primary antibody followed by appropriate HRP-linked secondary antibody and processed for ECL detection. Bands on films were scanned using a high-resolution scanner.17,23 Densitometric analysis of bands was done using ImageJ program from the National Institutes of Health (Bethesda, MD).DCF-DA assay
Plumbagin-mediated ROS generation was studied by DCFDA assay. Twelve thousand cells per well were seeded in 96 well tissue culture treated plates. After 24 h, cells were washed with pre-warm Krebs-ringer bicarbonate buffer solution. 10 μM DCFDA dye, dissolved in Krebs-ringer buffer was added to each well and plates were incubated for 30 minutes at 37 °C. After incubation period, buffer containing DCFDA was removed and wells were treated with buffer alone, 10 mM NAC, 1 mM H2O2 alone and in combination with NAC, 1–4 μM plumbagin, alone and in combination with NAC. Two hour NAC pre-treatment was done for wells which were to be treated with combination treatments of H2O2 or plumbagin. Finally, the fluorescence was measured at different time intervals (0, 3, 6, 12 and 24 h) using multiple reader (Thermo Scientific) with excitation filter set at 485 nm and emission filter set at 530 nm.19Wound-healing assay
SiHa and HeLa cells were inoculated in 6-well plates at 1 × 105 cells per well, and at around 70% confluency, 2 h pretreatment of mitomycin-C (0.5 μM) was given to inhibit the cell proliferation. Wounds were created with a 200 μL sterile tip, by scratching on the confluent monolayer and floating cells were removed immediately by washing two times with serum-free media. Fresh media (3 mL) containing desired plumbagin concentrations and mitomycin-C were added to each well and plates were incubated for specific time periods. Cell migration into wound areas was observed and photographed using an inverted microscope with the camera. The number of cells migrated into the wound areas was counted. The experiments were independently performed for three times.Cell invasion assay
The ability of cells to invade was evaluated using Trans-well invasion assay. After 24 h, cells were detached by trypsin and resuspended in a serum-free medium and 500 μL of cell suspension (3 × 104 cell per mL) was seeded into the upper chambers and 500 μL of MEM medium containing 10% FBS was placed in the lower chambers. Plumbagin (0.75 μM), was added in the suspension of cells in the upper chamber. After 20 h, non-migrated cells on the upper side of the membrane were carefully removed with a cotton swab. Invaded cells on the lower side were fixed in ethanol: acetic acid solution (95:5) and stained with 0.5% crystal violet solution for 15 minutes. Cells were counted in 5 microscopic fields from each membrane; and treatments were done in duplicate.
Statistical analysis
The data were statistically analyzed using the GraphPad Prism version 5. Student's t-test was employed to assess the statistical significance of the difference between control and treatment groups. A statistically significant difference was considered to be present at P < 0.05.Results
Plumbagin inhibits cell growth and decreases the viability of SiHa and HeLa cells
We first explored the effect of plumbagin on the cell growth and viability of human cervical carcinoma cells, SiHa and HeLa. Treatments with plumbagin (1–4 μM) caused a decrease in total cell number in SiHa (Fig. 1A) by 4–54% (P < 0.05 & P < 0.001) after 24 h and 7–69% (P < 0.05 & P < 0.001) after 48 h. Similar treatment decreased the cell proliferation in HeLa cells (Fig. 1C) by 8–56% (P < 0.05 & P < 0.001) and 14–65% (P < 0.05, P < 0.001) after 24 and 48 h, respectively. In SiHa cells, cell number was not significantly affected by the lowest (1 μM) plumbagin treatment.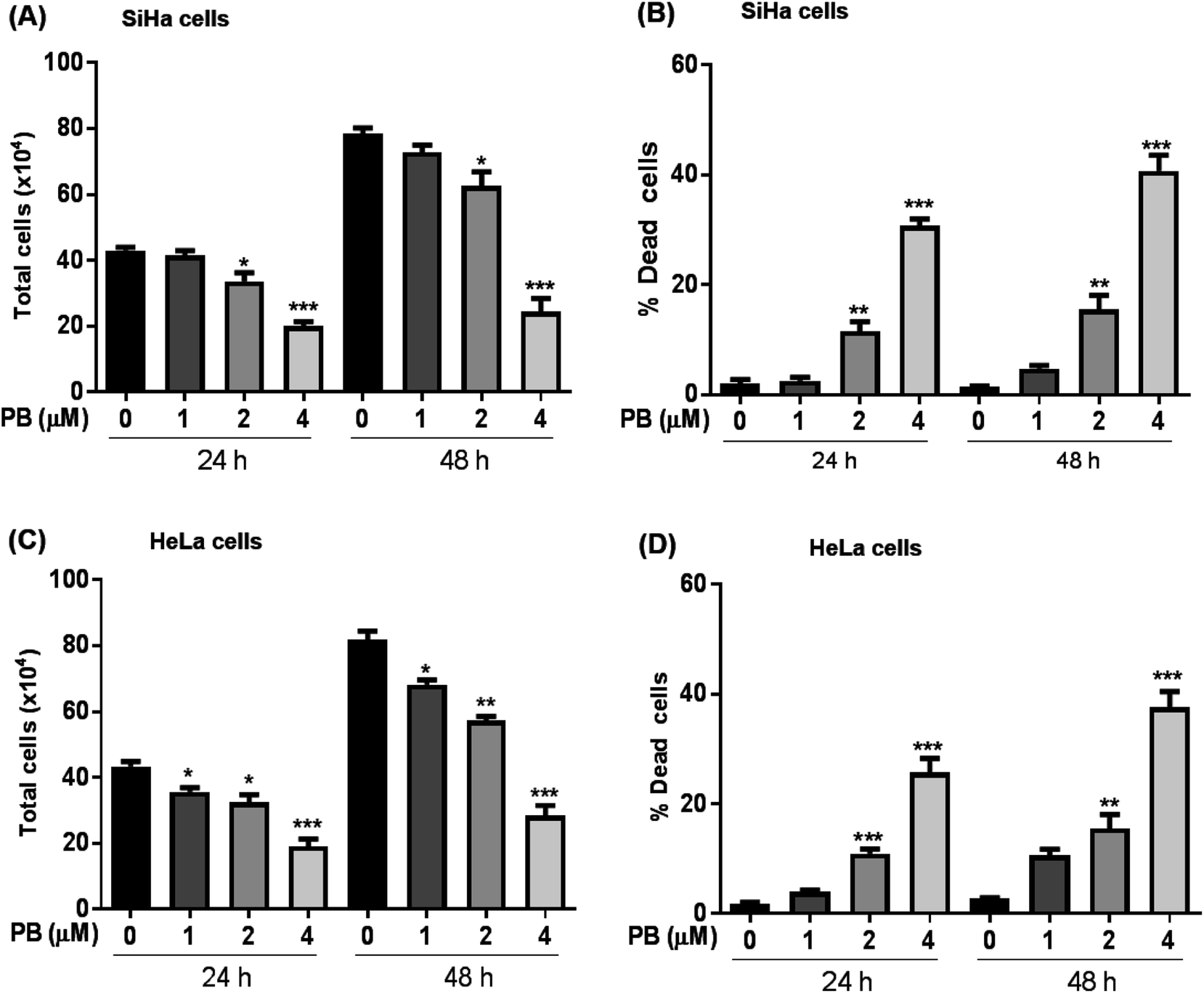 | ||
| Fig. 1 Effect of plumbagin (PB) on cell growth and death in human cervical carcinoma SiHa & HeLa cells. Cells were treated with DMSO (0) or 1, 2 and 4 μM PB for 24 and 48 h. Cell viability was assessed by trypan blue dye exclusion assay. (A and C) represent total cell number and (B and D) represent dead cells in SiHa & HeLa cells at indicated time points. Results are representative of three independent experiments. The data are presented as means of triplicate samples for each treatment. Bars, s.e.m, *P < 0.01, *P < 0.001, ***P < 0.0001. The P-value is determined by comparing each treatment with the control group. | ||
We also assessed the cell death and found that it increased manifold with 2 and 4 μM concentration of plumbagin after 24 and 48 h of treatments (P < 0.0001) in SiHa (Fig. 1B) and HeLa cells (Fig. 1D). In SiHa cells plumbagin treatment (2 & 4 μM) increased dead cells by 11–30% (P < 0.01 & P < 0.001) and 15–40% (P < 0.01 & P < 0.001) after 24 and 48 h respectively compared to control. Similarly, in HeLa cells, plumbagin treatment increased the dead cells by 10–25% (P < 0.001) and 10–35% (P < 0.001) after 24 and 48 h of treatment at 2 and 4 μM plumbagin. These results show that plumbagin strongly inhibits the cell growth and proliferation, and induces death in SiHa and HeLa cells.
Plumbagin induces G2/M and S-G2/M cell cycle arrest in cervical cancer cells
The cell cycle is a tightly regulated process that ensures proper duplication, distribution of genetic material and division of daughter cells, the cell cycle is found to be dysregulated in all cancers. Many phytochemicals and conventional drugs cause cell cycle arrest as one of the events to inhibit proliferation of cancer cells. Hence, cell cycle analysis was done to examine the effect of plumbagin on the cell cycle of SiHa and Hela cells. Cell cycle progression was determined by flow cytometry after saponin–PI staining. In SiHa cells 24 h treatment showed increased number of cells in G2/M phase i.e. 10–12% (2 & 4 μM) compared (P < 0.01 & P < 0.001) to cells in control and 10–14% (P < 0.01) after 48 h. The G2/M arrest induced by plumbagin was mainly at the expense of G1 phase cell population; at both 24 h and 48 h, a marginal increase in S population in SiHa cells was also found (Fig. 2A and C).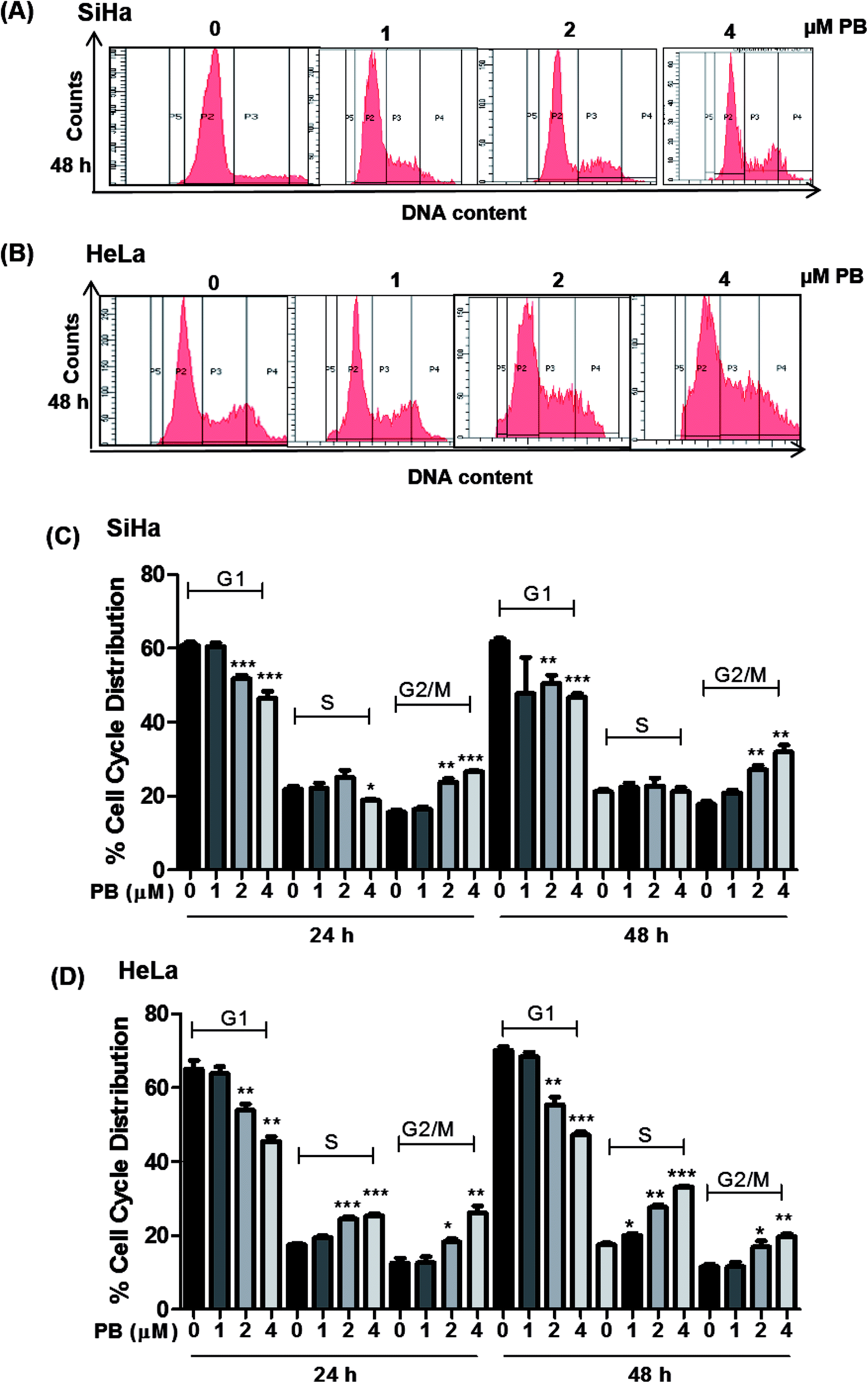 | ||
| Fig. 2 Effect of plumbagin (PB) on cell cycle progression of human cervical carcinoma SiHa & HeLa cells. The cells were treated with DMSO (0), 1, 2 and 4 μM PB for 24 h & 48 h. At the end of these treatments, cells were collected and incubated overnight with saponin/PI solution at 4 °C as detailed in Materials and methods. The percentage of cells in the different phases of the cell cycle was determined by flow cytometry. Representative histograms of SiHa (A) and HeLa (B) after 48 h. Bar graph representation of cell cycle distribution of SiHa (C) and HeLa (D) are depicted. Results are representative of three independent experiments. The data are presented as means of triplicate samples for each treatment. Bars, s.e.m, *P < 0.05, **P < 0.001, ***P < 0.001. The P-value is determined by comparing each treatment with the control group. | ||
In HeLa cells, both S and G2/M phase population was found to be increased. In HeLa cells, 2 and 4 μM concentrations of plumbagin caused 8–10% in cell population in S phase (P < 0.001) and 6–14% (P < 0.05 & P < 0.01) in G2/M phase compared to control. After 48 h plumbagin treatments caused 3 (P < 0.05), 10 (P < 0.01), and 16% (P < 0.001) S-phase arrest at 1, 2 & 4 μM, respectively and 6–10% (P < 0.05 & P < 0.01) in G2M phase at 2 & 4 μM of plumbagin. Overall plumbagin treatment showed G2-M arrest at 24 and 48 h in SiHa cells and S/G2-M arrest at 24 and 48 h in HeLa cells (Fig. 2B and D).
Plumbagin alters the expression of cell cycle regulators
Next, we analyzed the cell cycle regulatory molecules which could be involved in the S and G2/M phase arrest caused by plumbagin. The cell cycle is controlled by the activities of the cyclin/CDK complexes. Different CDKs bind to their cyclin partners to form cyclin–CDK complexes. We assessed both mRNA and protein level of the cell cycle regulators via RT-PCR and western blotting, respectively. In SiHa cells, RT-PCR result showed that the mRNA level of cyclin B1 slightly increased (1.1 fold) at 2 μM concentrations at 24 h, however, all other concentrations of plumbagin significantly decreased its level at both 24 & 48 h. mRNA levels of CDK1 showed a concentration-dependent decrease both at 24 h and 48 h (Fig. 3A). Plumbagin did not show a decrease in the protein expression of cyclin B1 at 1 μM both at 24 and 48 h, but higher concentrations showed a concentration and time-dependent decrease in the protein expression. Similarly, CDK1 protein expression was found to increase marginally at 1 μM plumbagin after 24 h, however, all other concentrations showed a decrease in the expression levels (Fig. 3B). mRNA expression of CDK1 was slightly decreased only at 4 μM, lower concentration did not decrease its expression to a very significant level. CDK2, cyclin E and A were slightly increased (1.2 fold) on lowest concentration, however higher concentrations after 48 h were more effective in decreasing their expression, compared to 24 h treatment time point (Fig. 3C). In case of protein expression lower concentrations were not effective in causing a decrease in the levels of cyclin B1, CDK1, CDK2, and cyclin A after 24 h treatment, also the levels of cyclin B1 was significantly increased (1.9 fold) even after 48 h at the lowest concentration. Also, the CDK2 level was significantly increased at 2 μM plumbagin. The highest concentration of plumbagin caused an invariable decrease in the levels of all the cyclins and CDKs after 24 and 48 h except for cyclin E2, which was constant at 48 h (Fig. 3D). These results suggest that plumbagin induced G2/M and S-G2/M arrest in cervical cells is mediated via modulation of CDK-cyclin levels and highest concentration is more effective in the downregulating their expression levels.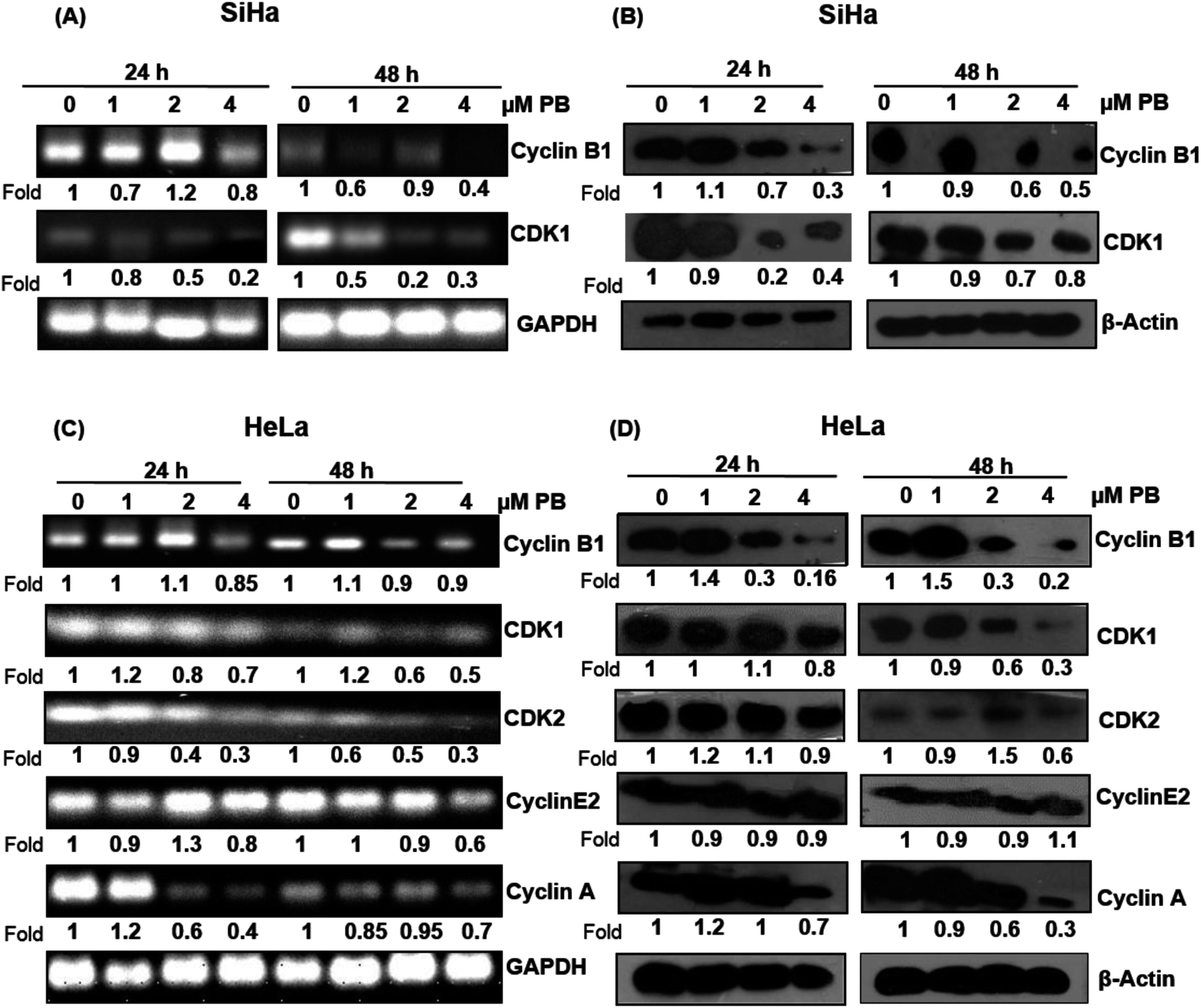 | ||
| Fig. 3 Effect of plumbagin (PB) on cell cycle regulatory mRNAs and proteins on cervical cancer SiHa and HeLa cells were studied by RT-PCR and western blot analysis. Total RNA and protein was isolated and subjected to RT-PCR and western blot analysis as described in Materials and methods. mRNA expression (A) and protein expression (B) was done for G2/M phase regulatory molecules cyclin B1 and CDK1 in SiHa cells after 24 and 48 h. Similarly, mRNA (C) and protein expression (D) of S-G2/M phase regulatory molecules including cyclin E2, cyclin A, cyclin B1 and CDK1, CDK2 in HeLa cells were studied at 24 and 48 h. Membranes were probed with different antibodies followed by peroxidase-conjugated appropriate secondary antibodies, and visualized by ECL detection system. Membranes were striped and re-probed with anti-β-actin for loading control, GAPDH was used as loading control for RT-PCR. | ||
Plumbagin induces apoptotic cell death in SiHa and HeLa cells
Next, we studied the nature of cell death caused by plumbagin in cervical cancer cells. The apoptotic cell is characterized by morphological changes in the plasma membrane, condensation of cytoplasm and nucleus and cleavage of nuclear DNA. Since there was an increase in the number of population of dead cells after plumbagin treatment in cell growth assay, hence, we checked whether plumbagin induced death was via induction of apoptosis. Morphological detection of apoptotic and necrotic cells was done by AO–EtBr double staining method. Plumbagin treatment at both 24 and 48 h showed an increase in apoptotic cells. In SiHa cells (Fig. 4A and E) plumbagin treatments increased apoptotic cells by 6–19% (P < 0.01 & P < 0.001) at 2 and 4 μm after 24 h and 5–18% (P < 0.05, P < 0.001) at 1–4 μm concentration, compared to control. While in HeLa (Fig. 4B and F) cells plumbagin treatments (1–4 μm) resulted in 6–20% (P < 0.001) and 7–20% (P < 0.05, P < 0.001) after 24 and 48 h respectively. Total cells were also counted and a dose-dependent decrease in healthy live cells was found by plumbagin treatment in both SiHa (Fig. 4C) and HeLa (Fig. 4D) cells. These results suggest that plumbagin possesses strong apoptosis effect on human cervical carcinoma cells.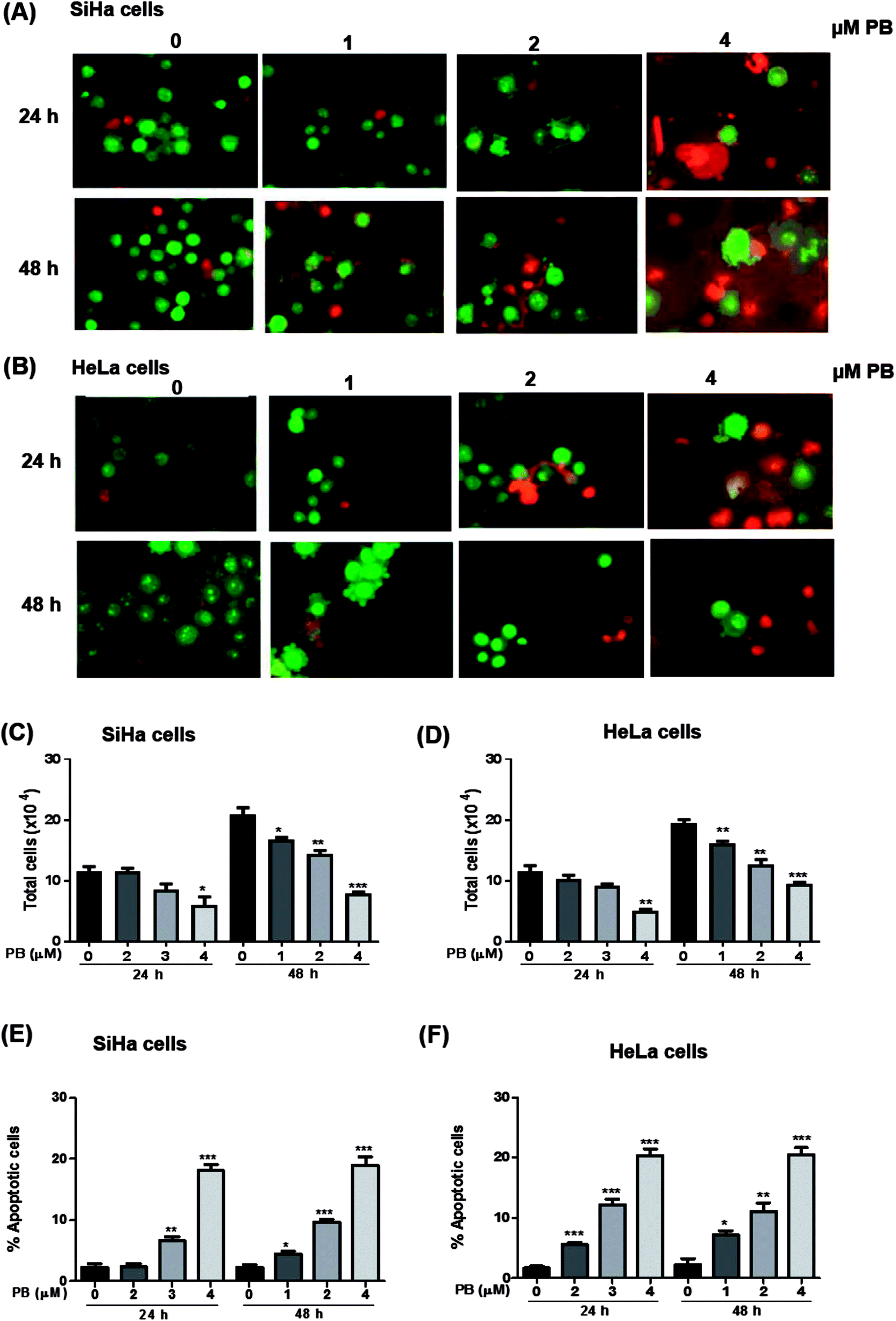 | ||
| Fig. 4 Effect of plumbagin (PB) on apoptotic cell death in human cervical carcinoma cells. SiHa (A) and HeLa (B) cells were treated with either DMSO (0), 1, 2 or 4 μM PB for 24 and 48 h. At the end of treatment time, cells were harvested and stained with acridine orange, ethidium bromide and viewed under a fluorescence microscope. Normal green nucleus represents the live cells, a bright green nucleus with condensed or fragmented chromatin represents early apoptotic cells, orange stained nuclei with chromatin condensation or fragmentation represents a late apoptotic cell. The data presented are the percentage of total no. of cells in SiHa & HeLa cells (C and D) after 24 & 48 h, and percentage of apoptotic cells in SiHa & HeLa cells (E and F) stained with acridine orange and ethidium bromide as a means of triplicate samples. Results are representative of three independent experiments. The data are presented as means of triplicate samples for each treatment. Bars, s.e.m, *P < 0.05, **P < 0.001, ***P < 0.001. The P-value is determined by comparing each treatment with the control group. | ||
Plumbagin modulates the expression of apoptosis markers
Several studies have reported that the Bcl-2 family proteins play important roles in the regulation of apoptosis by acting as either promoter (e.g. Bax) or inhibitors (Bcl-2) of cell death. We, therefore, determined their expression levels. Our results showed that plumbagin treatments in SiHa cells caused increase in the mRNA expression of Bax at 1–4 μM after 24 h in dose-dependent fashion, however, a decrease in the expression was found at 1 and 4 μM, and a slight increase was observed at 2 μM concentration (Fig. 5A). Plumbagin caused dose- and time-dependent increase in the protein expression of Bax, except for 1 μM concentration after 48 h where the expression was slightly (0.9 fold) decreased. Bcl2 expression was decreased in a time- and concentration-dependent fashion (Fig. 5C). Cleaved caspase-3, 9 and PARP were also found to be increased in presence of plumbagin (1–4 μM) in a concentration- and time-dependent manner in SiHa cells (Fig. 5C). In case of HeLa cells, mRNA expression of Bax was slightly increased and did not increase with dose and time (Fig. 5B). Similarly Bcl2 was found to be decreased with plumbagin treatment except at 2 μM after 24 h. The effect was prominent on the highest concentration after 48 h. Western blotting data revealed that plumbagin significantly increased the expression of Bax in time- and concentration fashion, except at 1 μM plumbagin after 24 h. Bcl2 expression was decreased with increase in plumbagin concentration and cleaved caspase-3, 9, PARP increased with increasing concentration and time and provides additional evidence for its apoptotic effect on cervical cancer cells (Fig. 5D). Ratio of Bax:Bcl2 is an important factor in the induction of apoptosis, a densitometry analysis showed that plumbagin effectively increased this ratio at 4 μM plumbagin in both the cell lines after 24 and 48 h. The effect was more prominent even at 2 μM plumbagin in Hela cells at both the time points (Fig. 5E and F).
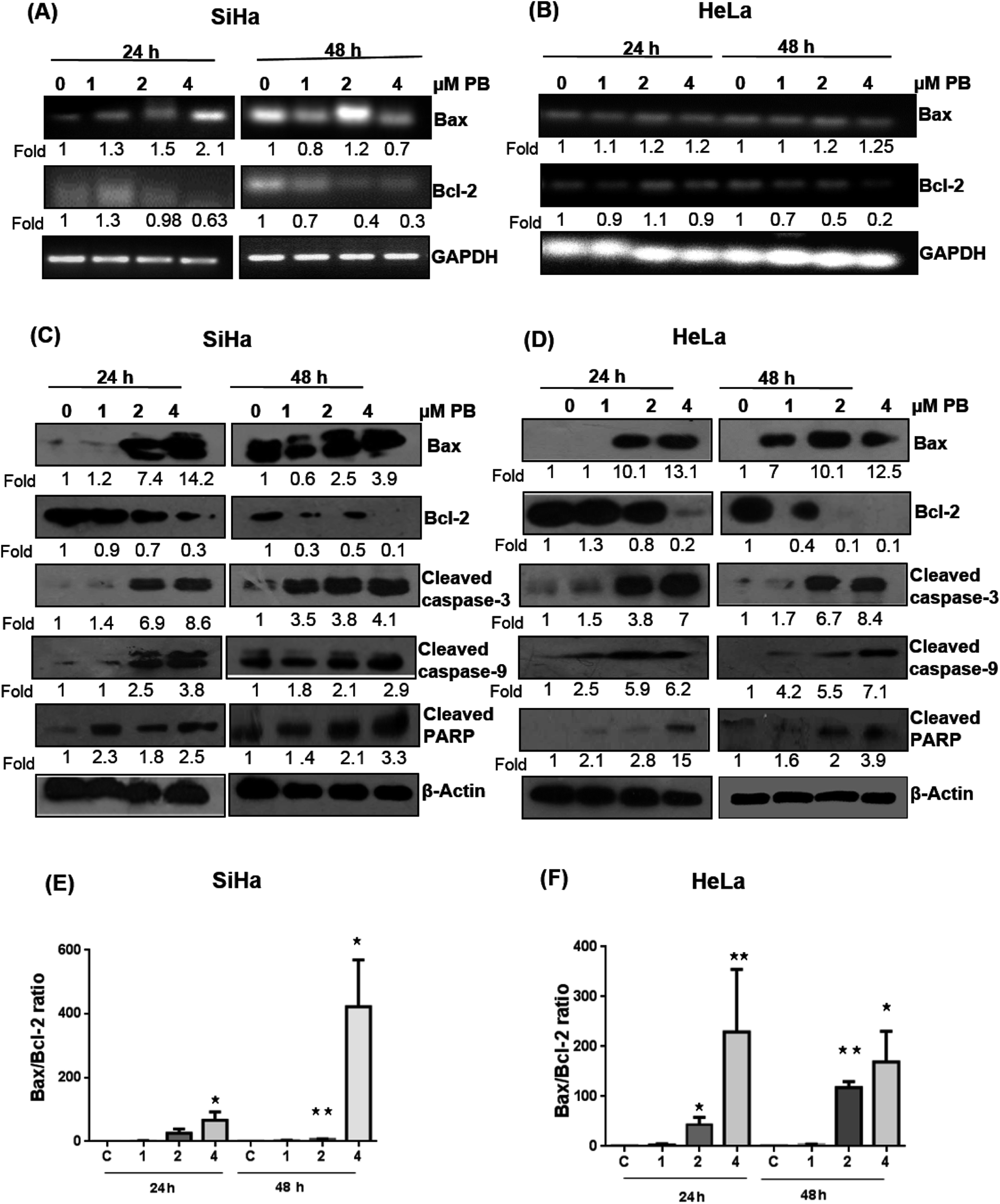 | ||
| Fig. 5 Effect of plumbagin (PB) on apoptosis regulatory mRNAs and proteins markers in SiHa and HeLa cells. Total RNA and protein was isolated and subjected to RT-PCR and western blot analysis as described in Materials and methods. mRNA (A and B) expression was done for Bax and Bcl2 in Hela (A) and SiHa (B) cells. Protein expression (C and D) was done for apoptosis regulatory proteins Bax, Bcl2, cleaved caspase 3, 9 and PARP in SiHa (C) and HeLa cells (D). Bax/Bcl-2 ratio at indicated time points and concentrations of plumbagin treatment for SiHa (E) and HeLa cells (F). Membranes were probed with different antibodies followed by peroxidase-conjugated appropriate secondary antibodies, and visualized by ECL detection system. Membranes were striped and re-probed with anti-β-actin for loading control, GAPDH was used as loading control for RT-PCR. | ||
Plumbagin induced ROS generation and NAC pre-treatment attenuated the cell proliferation and death
Many studies have shown that oxidative stress plays a crucial role in mediating various effects including DNA damage and cell death in cancer cells.19,24,25 To study the effects of plumbagin on ROS generation, cells were exposed to different concentrations and changes in DCF fluorescence was measured. Plumbagin treatment caused an increase in ROS generation in a dose-dependent manner, however, the effect was moderate on 2 μM and was manifold increased on the highest concentration after 6 h. The ROS generation increased progressively to 12 h and then started decreasing gradually to 24 h in both SiHa (Fig. 6A) and Hela (Fig. 6B) cells. NAC pretreatment significantly attenuated the ROS generation in both plumbagin and H2O2 treatment used as positive control. Next, we assessed the effect of plumbagin-mediated ROS generation on cell proliferation and death. SiHa (Fig. 6C) and HeLa (Fig. 6E) were treated either with 4 μM plumbagin or with/without pre-treatment with NAC. Interestingly, NAC pre-treatment significantly attenuated inhibition of cell proliferation caused by plumbagin in both SiHa (P < 0.001) and HeLa (P < 0.01) cells. Similarly, NAC pre-treatment also inhibited plumbagin mediated cell death by significantly in cervical cancer SiHa (P < 0.01) (Fig. 6D) and HeLa cells (P < 0.01) (Fig. 6F), indicating that most of the plumbagin mediated cell death was mediated by ROS generation. This finding provides a mechanistic view of plumbagin mediated-inhibition of cell proliferation and death is mostly by ROS generation.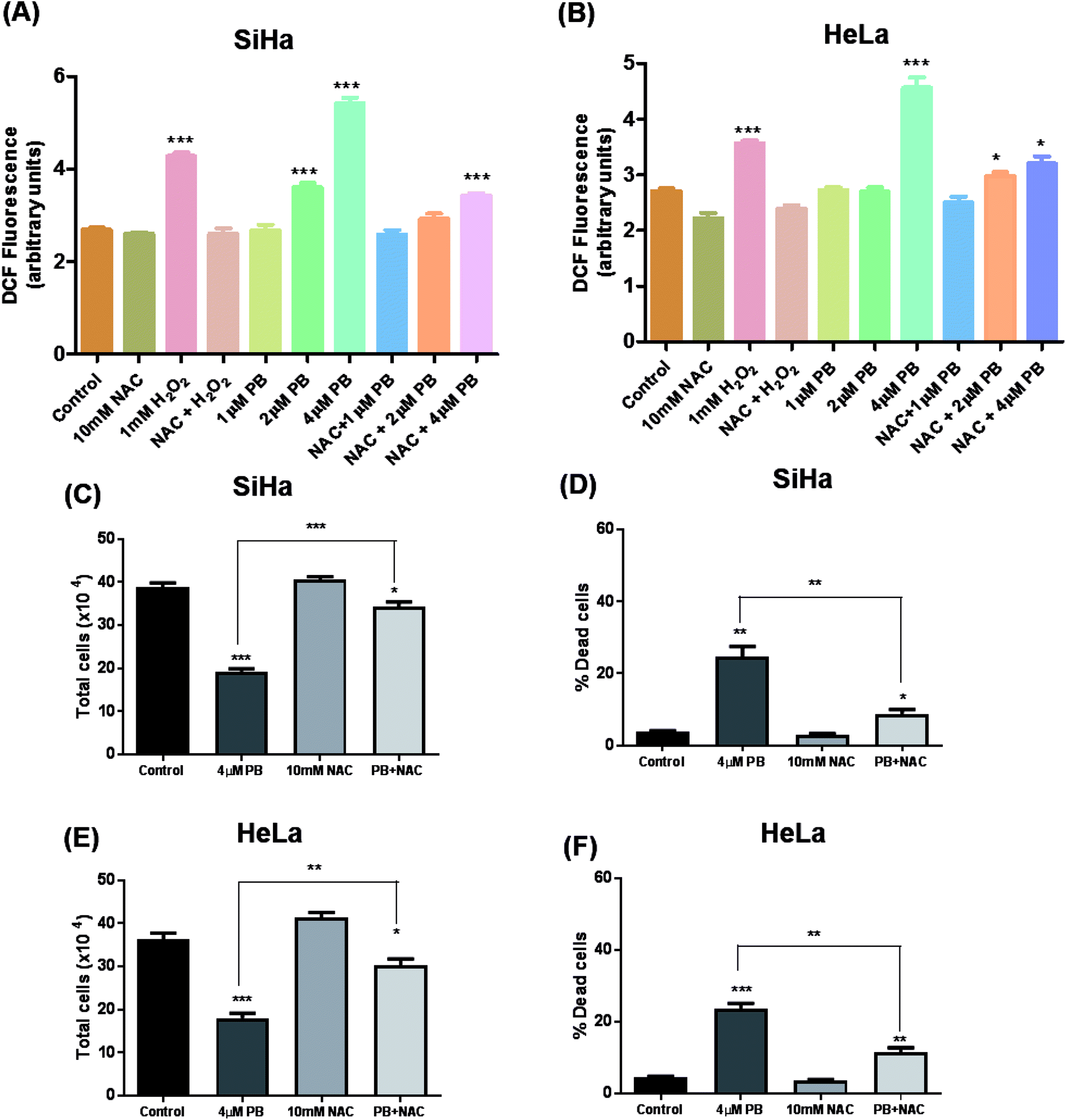 | ||
| Fig. 6 Detection of ROS using DCFDA staining. Cells were treated with different concentrations of PB alone and in combination with NAC and fluorescence was measured in SiHa (A) and HeLa (B) cells after 6 h. Cells were collected and processed for the determination of total cell number SiHa (C) and HeLa (E) and dead cells SiHa (D) and HeLa (F) by trypan blue assay. Results are representative of three independent experiments. The data are presented as means of triplicate samples for each treatment. Bars, s.e.m, *P < 0.05, **P < 0.001, ***P < 0.001. The P-value is determined by comparing each treatment with the control group. | ||
Plumbagin inhibits migration and invasion in SiHa and HeLa cells
To study the effect of plumbagin on migration efficiency, non-cytotoxic concentrations of plumbagin were used (0.25, 0.50, 0.75 μM) and the scratch assay was performed in 6 well plates. SiHa (Fig. 7A and B) and HeLa (Fig. 8A and B) cells were first treated with mitomycin-C to inhibit cell proliferation and then treated with either DMSO or indicated concentrations of plumbagin for 6, 12, 24 and 36 h. The results from wound healing assay revealed that the cells treated with plumbagin had greatly reduced the percentage of healed wound compared to control. The area of wound healed in control was taken as 100% and relative wound healing after plumbagin treatment was calculated by measuring the wound width. Treatment with plumbagin (0.25, 0.50, 0.75 μM) at 24 h resulted in inhibition of cells migration significantly (P < 0.01–0.001) by 22, 42 and 64.4% in SiHa cells (Fig. 7A and B) and by 34.7, 48.7 and 60% in HeLa cells (P < 0.01–0.001) (Fig. 8A and B). These results suggest that plumbagin is effective in preventing cervical cancer cell migration and resulted in a dose- and time-dependent decrease in the cell migration.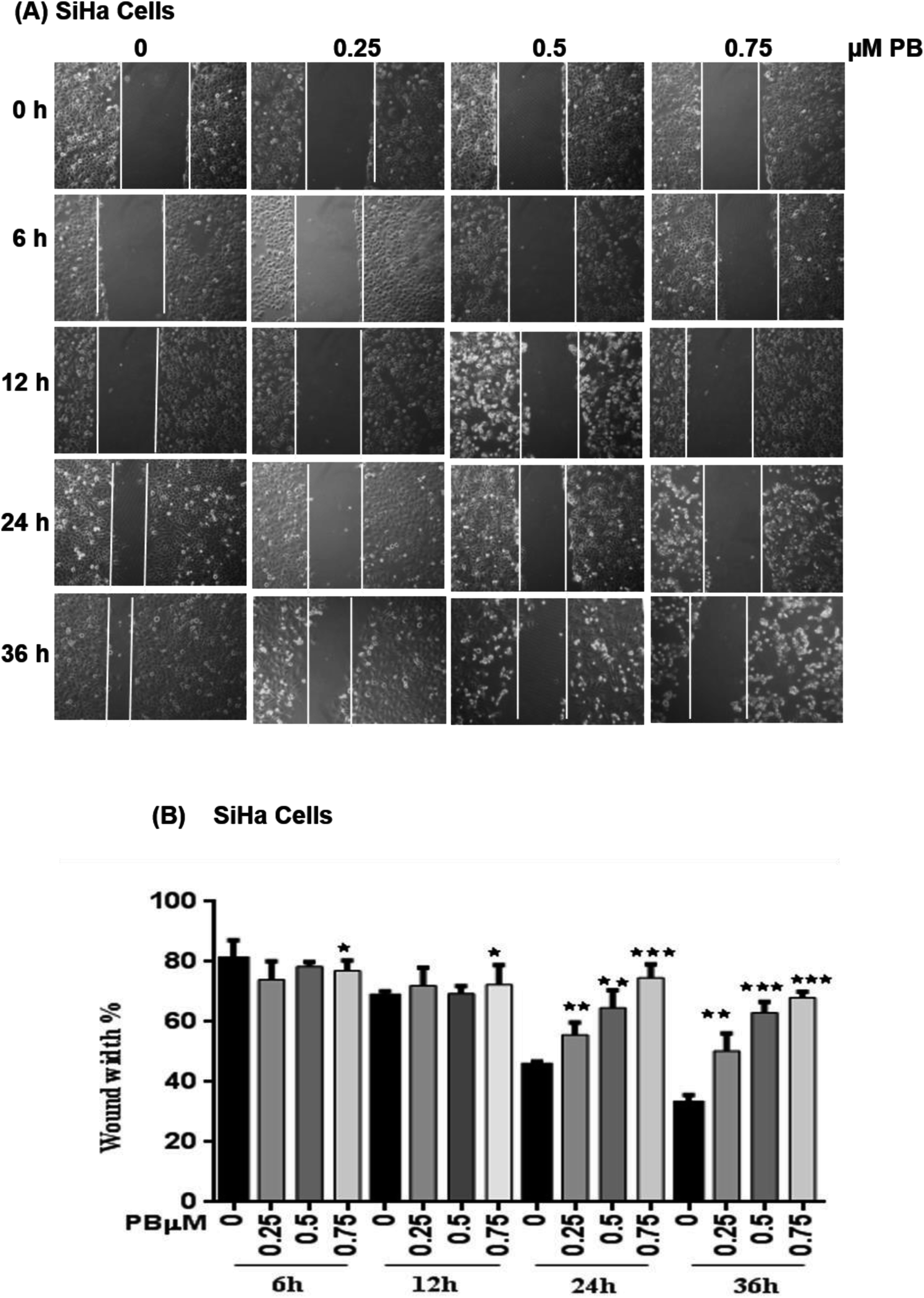 | ||
| Fig. 7 Effect of plumbagin (PB) on cell migration of SiHa cells. Wound healing assay of SiHa cells was performed, cells were treated with 0.25, 0.50 and 0.75 μM plumbagin (PB). (A) Images were captured at 0, 6, 12, 24, 36 h at 100× magnification under a phase contrast inverted microscope. (B) Following exposure to a different concentration of PB, the migration distance of SiHa cells was recorded at different time points and calculated. Results are representative of three independent experiments. Bars, s.e.m, *P < 0.05, **P < 0.001, ***P < 0.001. The P-value is determined by comparing each treatment with the control group. | ||
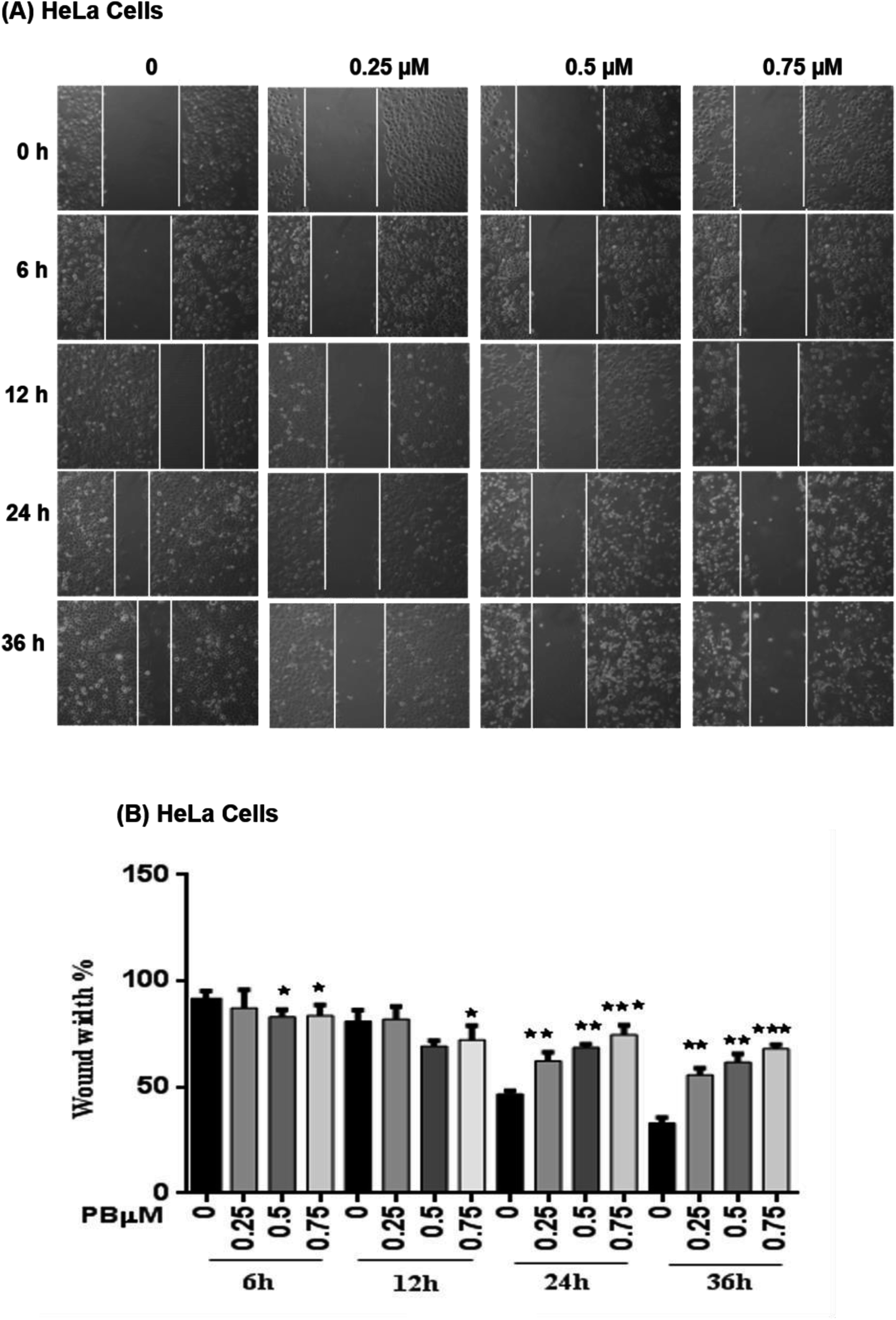 | ||
| Fig. 8 Effect of plumbagin (PB) on cell migration of HeLa cells. Wound healing assay of HeLa cells was performed, cells were treated with 0.25, 0.50 and 0.75 μM plumbagin (PB). (A) Images were captured at 0, 6, 12, 24, 36 h at 100× magnification under a phase contrast inverted microscope. (B) Following exposure to a different concentration of PB, the migration distance of HeLa cells was recorded at different time points and calculated. Results are representative of three independent experiments. Bars, s.e.m, *P < 0.05, **P < 0.001, ***P < 0.001. The P-value is determined by comparing each treatment with the control group. | ||
Next, we studied the effect of plumbagin on cell invasion potential using Boyden chamber assay. SiHa and HeLa cells were stained with crystal violet and counted under the phase contrast microscope (Fig. 9A). Plumbagin treatment (0.75 μM) at 24 h resulted in a decrease in the number of cells invaded per field by 59% (P < 0.001) compared to control in SiHa (Fig. 9B) and 66% (P < 0.001) in HeLa cells (Fig. 9C).
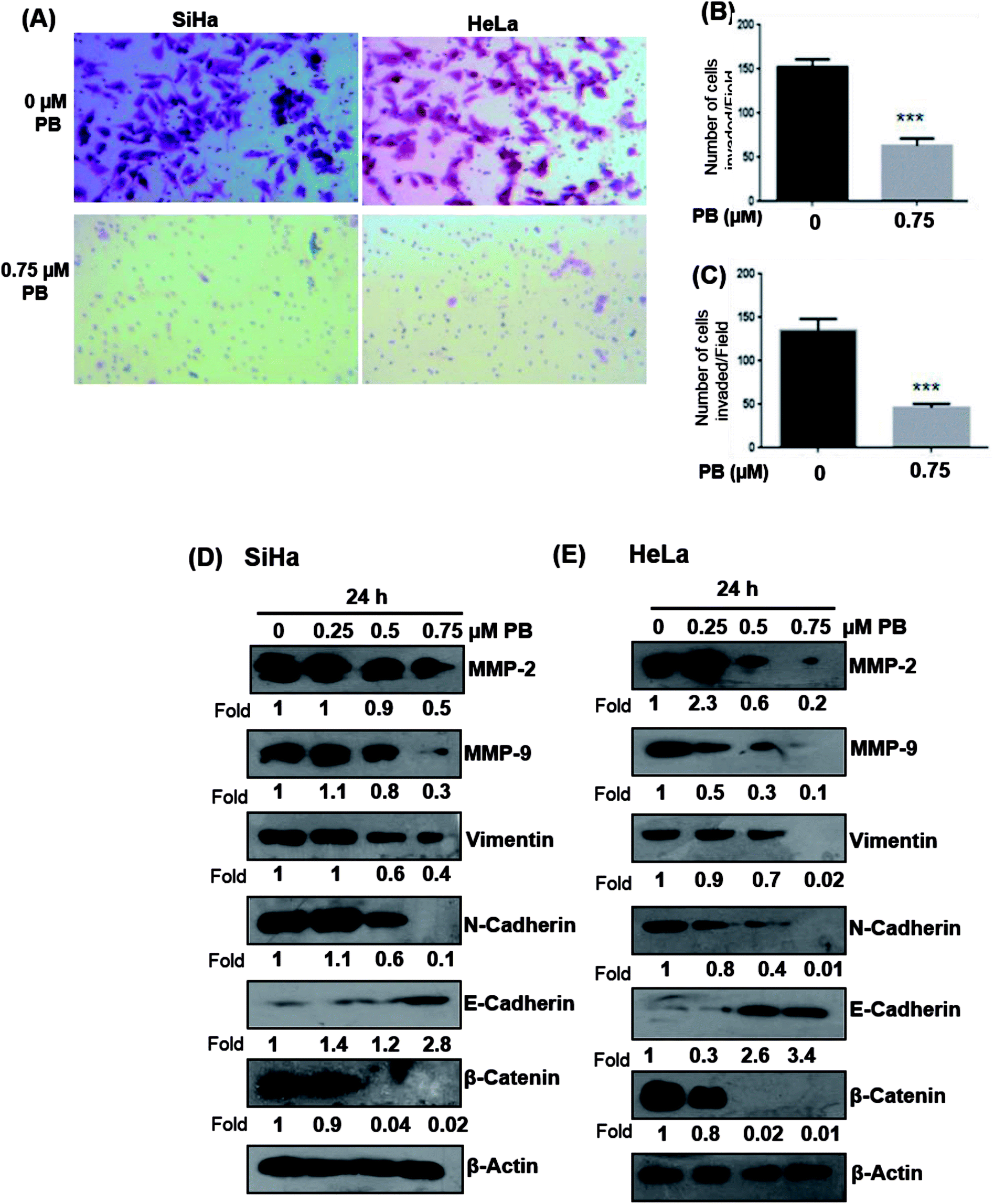 | ||
| Fig. 9 Effect of plumbagin (PB) on cell invasion and EMT regulatory proteins. SiHa and HeLa cells were treated with various concentration of plumbagin for given time points and cell invasion assay was performed and the invaded cells were photographed. The invaded cells were counted in six random fields in each treatment and data were calculated and presented as mean ± s.e.m., ***P < 0.001, by comparing each treatment with the control group. | ||
Plumbagin treatment modulates the expression of EMT markers
Upregulation of matrix metalloproteinase and Epithelial-mesenchymal transition (EMT) play a significant role in tumor invasion and metastases in different cancers.26,27 Epithelial-mesenchymal transition involves a change in the morphology of epithelial cells to a mesenchymal phenotype, that is characterized by marked decrease in the expression of epithelial cell markers like E-cadherin and an increase in the expression of mesenchymal markers, such as vimentin and N-cadherin.28 Hence, we examined whether plumbagin regulates the expression of matrix metalloproteinase (MMP) and epithelial-mesenchymal transition (EMT) markers, both SiHa and HeLa cells were treated with plumbagin at different concentrations (0.25–0.75 μM) for 24 h time period, lysates were made and western blot analysis was done to assess the effect of plumbagin on the expression of MMP-2/9, vimentin, β-catenin, N-cadherin, E-cadherin proteins in SiHa and HeLa cells. In SiHa cells, it was found that plumbagin treatment significantly reduced the expressions of MMP-2 at 0.50 & 0.75 μM and no change was observed at 0.25 μM, MMP-9 and N-Cadherin were increased slightly (1.1 Fold) increased at 0.25 μM, however, a significance decrease in the expression was observed at 0.50 & 0.75 μM plumbagin. At 0.25 μM plumbagin expression of vimentin was unchanged, however, at higher concentration a decrease was observed, in β-catenin a concentration-dependent decrease was observed. E-cadherin, an important hallmark of epithelial morphology was significantly increased (Fig. 9D). In case of HeLa cells, a significant increase was observed in the expression of MMP-2 at 0.25 μM plumbagin treatment, on 0.50 & 0.75 μM plumbagin a strong decrease in the expression was observed. Expression of MMP-9, vimentin, N-cadherin and β-catenin were decreased in concentration-dependent fashion. Similarly, a strong increase in the expression of E-cadherin was observed (Fig. 9E).Discussion
Plumbagin has been found to be a potent phytochemical that possess great therapeutic values and has been utilized to treat different ailments. Its antitumor and cytotoxic properties have been demonstrated both in vitro and in vivo studies,29 however, detailed mechanisms of plumbagin in cervical cancer are yet to be explored. Herein, we evaluated the anticancer efficacy of plumbagin in human cervical carcinoma SiHa and HeLa cells.Previous studies examining the effect of plumbagin showed a significant decrease in cell viability and cell growth in various cancer cells.30–32 In the present study, we have found that the plumbagin treatment resulted in growth inhibition of cervical cancer cells which was accompanied by an increase in the dead cell population in both concentration- and time-dependent manner. SiHa cells were found to be more sensitive as compared to HeLa cells, this could be due to the presence of a high viral load in HeLa cells making it less sensitive towards drug treatment. HeLa cells (HPV 18 positive) have 10–50 copies per cell for HPV-18 compared to 1–2 copies per cell for SiHa for HPV-16, which may result in an increased disorganization caused by HPV infection and thus, has more advanced malignancy as compared to SiHa cells.33,34
Cell growth and proliferation are synchronous events that are regulated by modulating the pace of cell cycle progression. There are numbers of signaling systems present in complex cell cycle network that controls that a cell will enter in cell cycle or not. Central features of the signaling system are the waves of cyclin expression and also the formation of cyclin/CDK complexes that occur throughout the course of the cell cycle.35 Plumbagin treatment to SiHa and HeLa cells showed a strong G2/M and S-G2/M arrest principally at the expense of cell population of G1 phase cells. These results are supported by previous studies which suggest that plumbagin treatment results in G2/M phase arrest in many cancer cells together with skin carcinoma, leukemia, breast cancer and lung cancer and S-G2/M phase arrest in myeloma cells.36 This finding suggests that the activation of checkpoint arrest can be a potential mechanism of plumbagin to inhibit the growth and proliferation of cervical cancer cells. Plumbagin was reported to modulate expression of numerous factors regulating cell cycle, like cip1/p21, cyclin B1, CDK1, Cdc25C in non-small cell lung cancer H460 cells.9 Our results also support the previous studies showing a decrease in CDK1, CDK2 and cyclin B1, cyclin A levels on plumbagin treatment. RT-PCR and western blotting results have confirmed the modification in the expression levels of each G2/M and S-G2/M phases regulating cell cycle molecules at transcriptional and protein levels. Plumbagin strongly decreased the levels of mRNA and proteins of CDK1, CDK2, cyclin B1, cyclin A. This suggests that there might be the decreased formation of active CDK2/cyclin a complex that could lead to S-phase arrest. Similarly, there will be also a decreased formation of CDK1/cyclin B1 complex which may be related to the G2/M arrest by plumbagin. These findings suggest that plumbagin could be targeting CDK–cyclin modulation for cell cycle arrest in cervical cancer cells.
Induction of apoptosis in cancer cells is a crucial feature of an anticancer agent. Chemopreventive agent halts the repair mechanism in cancer cells and prepares them for undergoing programmed cell death. Previous studies showed that plumbagin induces programmed cell death in several cancer cells including human multiple myeloma, chronic myeloid leukemia, embryonic kidney carcinoma multiple myeloma, and breast cancer cells.37
In the present study, fluorescent dyes were used to look at whether plumbagin mediated cell death is via apoptosis. Acridine orange and ethidium bromide are fluorescent DNA binding dyes we employed differentiated the cells that are in stages of apoptosis or necrosis. Treatment of SiHa and HeLa cells with plumbagin followed by AO/EB staining allowed us to morphologically distinguish the cells undergoing apoptosis. The populations of apoptotic cells were found to increase in time and dose-dependent fashion with decrease in the number of live cells. Next, to investigate the molecular event involved in plumbagin-mediated apoptosis, we analyzed the expression of both Bax, Bcl-2, cleaved-PARP, cleaved-caspase 3 and 9. PARP is a downstream target of activated caspase that gets cleaved during apoptosis induction.38 Caspase-3 plays a very important role for many key events occurring during apoptosis and act as an amplifier of the apoptotic signals.39–41 Caspase-9 is found to be located within the mitochondria, cytosol, and nucleus in humans.42 Active caspase-9 cleaves and activates downstream executioner.43 Once activated, caspase-9 initiates the caspase cascade in which goes on to cleave other caspases and many other cellular targets.44 RT-PCR and western blot result showed that plumbagin decreased the level of Bcl-2 and concomitantly increased the level of Bax. Western blot analysis revealed that plumbagin mediated apoptosis induction also involved an increase in cleavage of PARP, Caspase-3 and Caspase-9.
Many chemopreventive agents causes oxidative stress in cancer cells, which can lead to severe damage to vital macromolecules including DNA, lipids and proteins.45 Plumbagin have shown to cause ROS and can cause cell death in many cancer types.14,15,30,46 However, in cervical cancer SiHa and HeLa, ROS generation by plumbagin was not known. Our results showed that plumbagin caused strong ROS generation in SiHa and HeLa cells in dose and time and dependent fashion till 12 h after that it started decreasing, this may be due to less number of viable cells available for the analysis. Additionally, plumbagin-mediated cell death was also significantly attenuated with NAC-pretreatment. Thus, we conclude that most of the plumbagin-mediated cell death and inhibition of cell proliferation were through ROS generation.
Cancer can be prevented if inhibition of cancer-cell metastasis is achieved at an early stage. This process includes disrupting the interaction between cells and the ECM. Therefore, identifying new compounds that have the capability to modulate the expression of proteins involved in EMT may have great therapeutic value. Inhibiting MMPs synthesis is one of the crucial steps in inhibiting cancer-cell invasion and migration. So we examined the effect of plumbagin on invasion and migration of cervical cancer SiHa cells and plumbagin effectively decreased there expression levels. Epithelial-mesenchymal transition (EMT) is a fundamental event in tumor invasion and metastasis and is facilitated by reprogramming of epithelial cells.47 The decrease in epithelial E-cadherin and an increase of mesenchymal markers namely N-cadherin and vimentin are important hallmarks of EMT. In epithelial tissues, β-catenin is found to be essential for maintaining the cell layers and also links the intracellular junctions and cytoskeletal proteins.48 It is a component of the cadherin protein complex and regulates the growth of epithelial cells and intracellular adhesion.
Previous investigative studies have also revealed the anti-migratory effect of plumbagin in breast and gastric cancer cells mediated by downregulating the expression of chemokine receptor CXCR4.49 It also inhibits 12-O-tetradecanoyl phorbol-13-acetate (TPA)-induced invasion and migration inA549 cancer cells via downregulating the expression of MMPs and urokinase-type plasminogen activator (uPA).50 Recent studies revealed that plumbagin suppresses the migration and invasion of glioma cells via inactivation of PI3K/Akt signaling pathway by downregulating expression of MMP-2/9. Herein, for the first time, we have shown that plumbagin at non-cytotoxic concentrations has potential to inhibit EMT by downregulating MMP 2, 9, vimentin, β-catenin and N-cadherin and upregulating one of the most important markers of epithelial phenotype, E-cadherin in cervical cancer SiHa and HeLa cells.
Conclusions
Overall, the findings in the present study, for the first time identified the anticancer activity of plumbagin against human cervical carcinoma cells with its underlying molecular mechanisms. Plumbagin mediated growth inhibition of these cancer cells involved cell cycle arrest at S and G2/M phases, whereas death involved the induction of apoptosis through ROS, which was attenuated with NAC-pretreatment. Plumbagin significantly inhibited EMT, migration and invasion of cervical cancer cells at non-cytotoxic concentrations. Further experiments are needed to explore the target(s) of plumbagin which are involved in ROS generation. Mitochondrial respiratory chain complexes and endoplasmic reticulum are some of the important sites which are involved in ROS production, these can be explored as targets of plumbagin. In vivo studies using xenograft models are also needed to further strengthen the anti-cancer effects of plumbagin against cervical cancer.Conflicts of interest
All authors declared no conflicts of interest.Abbreviations
| CDKs | Cyclin-dependent kinases; |
| DMSO | Dimethyl sulfoxide |
| FACS | Fluorescence-activated cell sorting |
| FBS | Fetal bovine serum |
| HPV | Human papilloma virus |
| MEM | Minimum essential medium |
| PARP | Poly (ADP-ribose) polymerase |
| PB | Plumbagin |
| RT-PCR | Reverse transcriptase polymerase chain reaction |
| ROS | Reactive oxygen species |
| NAC | N-Acetyl-cysteine |
Acknowledgements
The work was supported by the funds from the Central University of Gujarat, India, and UPE-2, UGC-RN and DST-PURSE, JNU, New Delhi. AJ & AS are supported by fellowships from University Grant Commission and Department of Science & Technology New Delhi, India.References
- R. Siegel, D. Naishadham and A. Jemal, Ca-Cancer J. Clin., 2013, 63, 11–30 CrossRef PubMed
.
- F. Bray, A. Jemal, N. Grey, J. Ferlay and D. Forman, Lancet Oncol., 2012, 13, 790–801 CrossRef PubMed
- K. Kaarthigeyan, Indian J. Med. Paediatr. Oncol., 2012, 33, 7–12 CrossRef PubMed
- N. Munoz, F. X. Bosch, S. de Sanjose, R. Herrero, X. Castellsague, K. V. Shah, P. J. Snijders and C. J. Meijer, N. Engl. J. Med., 2003, 348, 518–527 CrossRef PubMed
- B. B. Hafeez, W. Zhong, J. W. Fischer, A. Mustafa, X. Shi, L. Meske, H. Hong, W. Cai, T. Havighurst, K. Kim and A. K. Verma, Mol. Oncol., 2013, 7, 428–439 CrossRef PubMed
- C. P. Bopaiah and N. Pradhan, Phytother. Res., 2001, 15, 153–156 CrossRef PubMed
- J. Itoh, T. Nabeshima and T. Kameyama, Psychopharmacology, 1990, 101, 27–33 CrossRef PubMed
- Y. L. Hsu, C. Y. Cho, P. L. Kuo, Y. T. Huang and C. C. Lin, J. Pharmacol. Exp. Ther., 2006, 318, 484–494 CrossRef PubMed
- R. Gomathinayagam, S. Sowmyalakshmi, F. Mardhatillah, R. Kumar, M. A. Akbarsha and C. Damodaran, Anticancer Res., 2008, 28, 785–792 Search PubMed
- K. A. Thasni, S. Rakesh, G. Rojini, T. Ratheeshkumar, G. Srinivas and S. Priya, Ann. Oncol., 2008, 19, 696–705 CrossRef PubMed
- Y. L. Zhao and D. P. Lu, Zhongguo Shiyan Xueyexue Zazhi, 2006, 14, 208–211 Search PubMed
- C. C. Wang, Y. M. Chiang, S. C. Sung, Y. L. Hsu, J. K. Chang and P. L. Kuo, Cancer Lett., 2008, 259, 82–98 CrossRef PubMed
- M. H. Aziz, N. E. Dreckschmidt and A. K. Verma, Cancer Res., 2008, 68, 9024–9032 CrossRef PubMed
- P. Srinivas, G. Gopinath, A. Banerji, A. Dinakar and G. Srinivas, Mol. Carcinog., 2004, 40, 201–211 CrossRef PubMed
- H. Huang, H. Xie, Y. Pan, K. Zheng, Y. Xia and W. Chen, Cell. Physiol. Biochem., 2018, 45, 267–280 CrossRef PubMed
- J.-H. Lee, J.-H. Yeon, H. Kim, W. Roh, J. Chae, H.-O. Park and D.-M. Kim, PLoS One, 2012, 7, e45023 CrossRef PubMed
- R. K. Shyanti, A. Sehrawat, S. V. Singh, J. P. N. Mishra and R. P. Singh, Toxicol. In Vitro, 2017, 44, 74–84 CrossRef PubMed
- A. Mittal, S. Tabasum and R. P. Singh, Phytomedicine, 2014, 21, 340–347 CrossRef PubMed
- A. Sabarwal, R. Agarwal and R. P. Singh, Mol. Carcinog., 2017, 56, 499–514 CrossRef PubMed
- N. Prasad, A. Sabarwal, U. C. S. Yadav and R. P. Singh, J. Biosci., 2018, 43, 249–261 CrossRef PubMed
- M. Rajput, P. K. Kujur, A. Mishra and R. P. Singh, Photodermatol., Photoimmunol. Photomed., 2018, 34, 91–101 CrossRef PubMed
- C. J. Lin, R. Sukarieh and J. Pelletier, Mol. Cancer Ther., 2009, 8, 1606–1612 CrossRef PubMed
- L. Varghese, C. Agarwal, A. Tyagi, R. P. Singh and R. Agarwal, Clin. Cancer Res., 2005, 11, 8441–8448 CrossRef PubMed
- F. Sesti, O. E. Tsitsilonis, A. Kotsinas and I. P. Trougakos, In Vivo, 2012, 26, 395–402 Search PubMed
- K. Kumar, A. Sabarwal and R. P. Singh, Mitochondrion, 2018 DOI:10.1016/j.mito.2018.06.003
- M. Rauvala, K. Aglund, U. Puistola, T. Turpeenniemi-Hujanen, G. Horvath, R. Willen and U. Stendahl, Int. J. Gynecol. Cancer, 2006, 16, 1297–1302 CrossRef PubMed
- K. Wu, J. Zeng, L. Li, J. Fan, D. Zhang, Y. Xue, G. Zhu, L. Yang, X. Wang and D. He, Oncol. Rep., 2010, 23, 1545 CrossRef PubMed
- M. Zeisberg and E. G. Neilson, J. Clin. Invest., 2009, 119, 1429–1437 CrossRef PubMed
- S. Sinha, K. Pal, A. Elkhanany, S. Dutta, Y. Cao, G. Mondal, S. Iyer, V. Somasundaram, F. J. Couch, V. Shridhar, R. Bhattacharya, D. Mukhopadhyay and P. Srinivas, Int. J. Cancer, 2013, 132, 1201–1212 CrossRef PubMed
- J. H. Lee, J. H. Yeon, H. Kim, W. Roh, J. Chae, H. O. Park and D. M. Kim, PLoS One, 2012, 7, e45023 CrossRef PubMed
- Y. Y. Cao, J. Yu, T. T. Liu, K. X. Yang, L. Y. Yang, Q. Chen, F. Shi, J. J. Hao, Y. Cai, M. R. Wang, W. H. Lu and Y. Zhang, Cell Death Dis., 2018, 9, 17 CrossRef PubMed
- H. Wu, X. Dai and E. Wang, Oncol. Lett., 2016, 12, 3614–3618 CrossRef PubMed
- G. Hu, W. Liu, J. Mendelsohn, L. M. Ellis, R. Radinsky, M. Andreeff and A. B. Deisseroth, J. Natl. Cancer Inst., 1997, 89, 1271–1276 CrossRef PubMed
- D. Zhang, Q. Zhang, L. Zhou, L. Huo, Y. Zhang, Z. Shen and Y. Zhu, BMC Cancer, 2010, 10, 650 CrossRef PubMed
- S. Lim and P. Kaldis, Development, 2013, 140, 3079–3093 CrossRef PubMed
- S. Padhye, P. Dandawate, M. Yusufi, A. Ahmad and F. H. Sarkar, Med. Res. Rev., 2012, 32, 1131–1158 CrossRef PubMed
- S. K. Sandur, H. Ichikawa, G. Sethi, K. S. Ahn and B. B. Aggarwal, J. Biol. Chem., 2006, 281, 17023–17033 CrossRef PubMed
- P. Mullen, Methods Mol. Med., 2004, 88, 171–181 Search PubMed
- A. G. Porter and R. U. Janicke, Cell Death Differ., 1999, 6, 99–104 CrossRef PubMed
- E. H. Cheng, D. G. Kirsch, R. J. Clem, R. Ravi, M. B. Kastan, A. Bedi, K. Ueno and J. M. Hardwick, Science, 1997, 278, 1966–1968 CrossRef PubMed
- D. G. Kirsch, A. Doseff, B. N. Chau, D. S. Lim, N. C. de Souza-Pinto, R. Hansford, M. B. Kastan, Y. A. Lazebnik and J. M. Hardwick, J. Biol. Chem., 1999, 274, 21155–21161 CrossRef PubMed
- B. Zhivotovsky, A. Samali, A. Gahm and S. Orrenius, Cell Death Differ., 1999, 6, 644–651 CrossRef PubMed
- D. R. McIlwain, T. Berger and T. W. Mak, Cold Spring Harbor Perspect. Biol., 2013, 5, a008656 Search PubMed
- Y. Qin, T. L. Vanden Hoek, K. Wojcik, T. Anderson, C. Q. Li, Z. H. Shao, L. B. Becker and K. J. Hamann, Am. J. Physiol.: Heart Circ. Physiol., 2004, 286, H2280–H2286 CrossRef PubMed
- T. Ozben, J. Pharm. Sci., 2007, 96, 2181–2196 CrossRef PubMed
- L. Tian, D. Yin, Y. Ren, C. Gong, A. Chen and F. J. Guo, Mol. Med. Rep., 2012, 5, 126–132 Search PubMed
- Z. Wang, Y. Li, D. Kong and F. H Sarkar, Curr. Drug Targets, 2010, 11, 745–751 CrossRef PubMed
- C. Herencia, J. M. Martínez-Moreno, C. Herrera, F. Corrales, R. Santiago-Mora, I. Espejo, M. Barco, Y. Almaden, M. de la Mata and A. Rodríguez-Ariza, PLoS One, 2012, 7, e34656 CrossRef PubMed
- K. A. Manu, M. K. Shanmugam, P. Rajendran, F. Li, L. Ramachandran, H. S. Hay, R. Kannaiyan, S. N. Swamy, S. Vali, S. Kapoor, B. Ramesh, P. Bist, E. S. Koay, L. H. Lim, K. S. Ahn, A. P. Kumar and G. Sethi, Mol. Cancer, 2011, 10, 107 CrossRef PubMed
- J. M. Shieh, T. A. Chiang, W. T. Chang, C. H. Chao, Y. C. Lee, G. Y. Huang, Y. X. Shih and Y. W. Shih, Mol. Cell. Biochem., 2010, 335, 181–193 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2018 |