
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScreening of ferrocenyl–phosphines identifies a gold-coordinated derivative as a novel anticancer agent for hematological malignancies†
Navin Kumar Verma *a,
Abdul Sadeer‡
b,
Atish Kizhakeyil‡a,
Jia Hao Pangb,
Qi Yun Angela Chiub,
Shan Wen Tayb,
Pankaj Kumara and
Sumod A. Pullarkat*b
*a,
Abdul Sadeer‡
b,
Atish Kizhakeyil‡a,
Jia Hao Pangb,
Qi Yun Angela Chiub,
Shan Wen Tayb,
Pankaj Kumara and
Sumod A. Pullarkat*b
aLee Kong Chian School of Medicine, Nanyang Technological University Singapore, Experimental Medicine Building, 59 Nanyang Drive, Singapore. E-mail: nkverma@ntu.edu.sg
bDivision of Chemistry & Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University Singapore, 21 Nanyang Link, Singapore. E-mail: Sumod@ntu.edu.sg
First published on 14th August 2018
Abstract
The development of new organometallic compounds as anticancer agents is currently an active area of research. Here, we report the design, synthesis and characterization of a panel of 10 new ferrocenyl–phosphine derivatives (FD1–FD10) and the analysis of their anti-proliferative activities in hematolymphoid cells representing non-Hodgkin cutaneous T-cell lymphoma (CTCL). The gold-coordinated ferrocenyl–phosphine complex FD10 exhibited a significant and dose-dependent cytotoxicity in 4 different CTCL cell lines – HuT78, HH, MJ and MyLa. FD10 concentrations causing 50% cell growth inhibition (IC50) of HuT78, HH, MJ and MyLa cells at 24 h were recorded to be 5.55 ± 0.20, 7.80 ± 0.09, 3.16 ± 0.10 and 6.46 ± 0.24 μM respectively. Further mechanistic studies showed that FD10 induced apoptosis in CTCL cells by an intrinsic pathway mediated via the activation of caspase-3 and poly(ADP-ribose)polymerase. It suppressed the expression and activity of STAT3 oncoprotein in CTCL cells. FD10 caused robust G0/G1 phase cell cycle arrest and reduced the expression levels of Akt S473 phosphorylation and c-Myc, both are key cell cycle regulator proteins. Taken together, this study highlights anticancer properties of the ferrocenyl–phosphine gold organometallic complex FD10 and suggests that further development of this novel class of molecule may contribute to new drug discovery for certain hematolymphoid malignancies.
Introduction
The development of anticancer organometallic complexes has been a rapidly developing and increasingly active area of research, and has shown unprecedented progress. The structural elucidation of ferrocene [Fe(C5H5)2] in the early 1950s spawned the development of a myriad of ferrocenyl compounds.1–3 Due to their interesting electrochemical properties, unique redox characteristics, innate stability in aqueous media and easy functionalization, these organometallic compounds have recently aroused increasing research interest for their potential applications in cancer therapeutics.4,5The anticancer properties of ferrocene derivatives were first studied in the late 1970s, when Brynes and co-workers reported antitumor activities of ferrocenyl compounds bearing amine or amide groups.6 While these molecules exhibited significant but low antitumor efficacy in mice, results suggested that the incorporation of the ferrocenyl group into an appropriate carrier could provide an organometallic compound with enhanced antitumor activity.6–8 Noteworthy from this perception, a novel molecule “ferrocifen” was developed by integrating ferrocene into the tamoxifen scaffold that exhibited significant cytotoxicity in ER+ MCF-7, a breast cancer cell line resistant to common anti-estrogenic drugs.9–11 Furthermore, phosphines coordinated to transition metal salts have received extensive interest in anticancer studies following the tremendous success of the blockbuster drug “cisplatin”. The deliberate incorporation of metallocenes into phosphine complexes (or vice versa, depending on synthetic ease) has been envisaged as a possible route towards new drug discovery.11,12 Indeed, the anti-proliferative activity of ferrocenyl–ethynyl phosphine metal complexes of gold in human cervix epithelioid cancer cell line, HeLa, has been reported.13 Studies suggest that certain metallocenes have high biological stability and minimal toxic impact on the normal functioning of the liver, kidney and lungs, a major area of concern with anticancer drugs.12 However, apart from the aforementioned attributes that can be traced mainly to the ferrocene backbone, the antitumor potential of ferrocene derivatives remains largely unexplored.
Hematolymphoid malignancies, such as non-Hodgkin cutaneous T-cell lymphomas (CTCL), are clinically and biologically diverse forms of cancers of the blood and immune systems, and impose a significant burden of disease globally. These diseases are (i) prevalent worldwide, (ii) often diagnosed in the elderly population in their 50s to 60s, (iii) account for ∼9% of all cancers (iv) contributes 35% of paediatric cancer, (v) being the 4th most frequently diagnosed cancer in both men (after prostate, lung, and colorectal) and women (after breast, lung, and colorectal). While the incidence and mortality for most cancers are decreasing, the number of hematolymphoid cancer cases has risen sharply worldwide over the past two decades.14,15
Over the last few years, the emergence of resistant varieties of tumors has brought forth the need for the development of new classes of anticancer agents that could also be suitable as combination therapeutics in order to circumvent drug resistance. Our sustained interest in the catalytic generation of novel ferrocenyl–phosphines via asymmetric hydrophosphination has led us to synthesize a unique class of mono-substituted ferrocenyl–phosphine derivatives bearing the aryl or alkyl ketone moiety and probe their potential effect on cancer cells.16,17 Considering the possible plethora of derivatives that could be obtained via the hydrophosphination protocol, here we narrowed the current work to less studied oxide & sulphide derivatives as well as gold(I) chloride complexes and analysed their anti-proliferative activities in hematolymphoid cells. We identified FD10, which incorporated the four chemically relevant subunits of ferrocene, ketone, phosphine and gold, as a promising drug candidate for haematological malignancies.
Experimental
Materials and physicochemical measurements
All the reagents and solvents were obtained from commercial sources and used without further purification. The compounds were characterized by standard spectroscopic techniques. All air-sensitive manipulations were carried out under dry N2 by Schlenk techniques unless otherwise stated. Solvents were degassed and/or distilled and stored over 3 Å molecular sieves prior to use. Glasswares were oven-dried and placed under vacuum for 30 min prior to use. The progress of reactions was monitored by thin-layer chromatography (TLC) on silica gel 60 F254 aluminium-backed plates (Merck). The chromatograms were visualized under ultraviolet radiation (UV) or I2. Flash column chromatography was conducted using silica gel 60 (Merck). Nuclear magnetic resonance (NMR) spectra were obtained using a Bruker AV 300 spectrometer (1H at 300 MHz, 13C at 75 MHz, 19F{1H} at 282 MHz, 31P{1H} at 121 MHz), Bruker AV 400 spectrometer (1H at 400 MHz, 13C at 100 MHz, 31P{1H} at 161 MHz) or Bruker AV 500 spectrometer (1H at 500 MHz, 13C at 125 MHz, 31P{1H} at 202 MHz). 1H spectra were referenced to an internal SiMe4 standard at δ 0 or CDCl3 at δ 7.26. All 13C spectra were referenced to CDCl3 at δ 77.16.Hydrophosphination (HP) procedure for the synthesis and NMR characterization of ferrocenyl derivatives
The racemic ferrocenyl–phosphines were synthesized from the respective ferrocenyl enones using HP procedure (Fig. 1A) as described.16,17 Briefly, HPPh2 (50.0 mg, 0.27 mmol) and degassed MeOH (2 mL) were added into a Schlenk flask. Subsequently, racemic catalyst C1 (ref. 18) (13.1 mg, 0.027 mmol) was added and the yellow solution was stirred at the stipulated temperature for 5 min. Following that, the respective ferrocenyl enone (0.27 mmol) and NEt3 (27.3 mg, 37.6 μL, 0.27 mmol), diluted with degassed MeOH (2 mL), were added. The resulting purple mixture was allowed to stir at room temperature for 24 h. Thereafter, NMR structural confirmation, high resolution mass spectrometry (HRMS) characterization and melting-point and purity analysis of the compounds were performed. Detailed characterization parameters and NMR spectra of each compound are provided as ESI.† All the compounds are soluble in polar organic solvents, including dimethyl sulfoxide (DMSO), and are bench stable. These compounds could be purified via silica gel column chromatography under ambient conditions. | ||
| Fig. 1 Scheme for the hydrophosphination of ferrocenyl enones and structures of ferrocenyl–phosphine derivatives FD1 to FD10. | ||
Patient-derived hematolymphoid cell lines and culture
We used four different types of CTCL cell lines – (1) HuT78, derived from the peripheral blood of a 53 years old Caucasian male suffering from Sézary syndrome; (2) MyLa, derived from a patient suffering from non-Hodgkin CTCL; (3) MJ, derived from a plaque biopsy of a 50 years old Caucasian male suffering from mycosis fungoides; (4) HH, derived from a tumor nodule of a 61 years old Caucasian male suffering from aggressive mycosis fungoides. HuT78, MJ and HH cell lines were obtained from the American Type Culture Collection (ATCC) and the MyLa cell line was procured from Sigma-Aldrich (now Merck). HuT78 and HH cells were cultured in Gibco® RPMI-1640 supplemented with 10% fetal bovine serum (FBS). MJ cells were cultured in Gibco® IMDM supplemented with 20% FBS. MyLa cells were cultured in Gibco® RPMI-1640 supplemented with 10% human serum and 4 ng mL−1 each of IL-2 and IL-4. Gibco® penicillin–streptomycin (100 U mL−1) was added to all the culture media.Human primary peripheral blood mononuclear cells (PBMCs) were isolated from buffy-coat using Lymphoprep™ (Axis Shield) density gradient centrifugation. Buffy-coat blood packs obtained from anonymous healthy volunteers at the Blood Services Group, Health Sciences Authority (HSA) Singapore were used. Protocols were approved by the Nanyang Technological University Singapore Institutional Review Board (IRB-2014-09-007). Cells were cultured in Gibco® RPMI-1640 supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 10% FBS and Gibco® penicillin–streptomycin as described previously.19
MTS-based cell viability assay
The cytotoxic effect of compounds was determined using CellTiter 96® AQueous One Solution Cell Proliferation Assay kit according to manufacturer's instructions (Promega Corporation, Madison, WI, USA). Compounds were diluted in DMSO to obtain 20 mM stock solutions. Appropriate aliquots of these stock solutions were further diluted in the cell culture medium to obtain final working concentrations. Cells (HuT78, MyLa, MJ, HH and PBMCs) were seeded at 5 × 104 cells per well in 96-well tissue culture plates (Nunc). Cells were treated with the various compounds at concentrations ranging from 1 to 100 μM for 24 h in triplicates. After the treatment period, cells were incubated with 20 μL MTS reagent (provided with the assay kit) for 2 h at 37 °C. Absorbance was recorded at 490 nm using a microplate reader (BioTek) and IC50 values for each compound were determined using GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA).Flow-cytometric analysis for cell cycle and apoptosis
For cell cycle analysis, hematolymphoid cells after treatment (as indicated in the corresponding figure legends) were fixed with 70% ethanol and then stained with 50 μg mL−1 propidium iodide (PI) containing 0.25 mg mL−1 RNaseA at room temperature for 30 min. For apoptosis analysis, treated cells were washed with ice-cold phosphate-buffered saline and incubated with a combination of 5 μL each of FITC-Annexin-V and PI (Apoptosis detection kit, Thermo Fisher Scientific, USA) at room temperature in the dark for 15 min. Flow-cytometry analysis was performed to determine cell cycle distribution and apoptosis using a BD FACSCalibur™ flow-cytometer (BD Biosciences).Western-immunoblot analysis
Following treatment as described above, cells were washed with ice-cold phosphate-buffered saline and lysed in the lysis buffer as described.20 Equal amounts of cell lysates (20 μg each, as determined by Bio-Rad protein estimation kit) were resolved on SDS-PAGE gel and subsequently transferred onto the polyvinylidene difluoride (PVDF) membrane. After blocking in 5% bovine serum albumin for 1 h at room temperature, membranes were washed three times in tris-buffered saline containing Tween 20. The membranes were then incubated overnight at 4 °C with primary antibodies (diluted in 5% bovine serum albumin in tris-buffered saline-Tween 20 buffer) with gentle rocking. After three washes in tris-buffered saline-Tween 20, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. Primary antibodies: cleaved PARP, cleaved caspase-3 (Cell Signaling Technology) and β-actin (Santa Cruz Biotechnology). Secondary antibodies: horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG (Cell Signaling Technology). The immunoreactive bands on PVDF membrane were reacted to the ECL detection reagents and visualised by exposure to X-ray films (Kodak, Rochester, NY, USA). Densitometric analysis of the blots was performed by ImageJ software.Statistical analysis
Where applicable, all the data were expressed as mean ± SEM. GraphPad prism 4.0 software (GraphPad software) was used for statistical analysis. Student's t-test was performed and used to compare the difference between variables and p < 0.05 was considered statistically significant.Results and discussion
Chemistry
A panel of 10 ferrocenyl–phosphine derivatives (FD1 to FD10) was designed and synthesised via the palladacycle-catalysed hydrophosphination reaction followed by subsequent derivatization steps involving conversion of the air sensitive tertiary phosphine moiety to stable oxide/sulphide derivatives or by coordination to gold (Fig. 1). For the synthesis of FD1–FD7 compounds, hydrophosphination was followed by protection of the tertiary phosphine as oxide or sulphide. The tertiary phosphine adduct compounds FD8–FD10 were subsequently coordinated directly to dimethylsulfide gold(I)chloride salt. 1H-NMR analysis confirmed the reduction of the conjugate linker of the parent ferrocenyl chalcone and installation of a phosphine group at the intended position adjacent to the ferrocene unit via the Michael addition protocol described earlier.21Traditionally, ferrocene-based molecules are synthesized by incorporating a conjugated linker that lowers the oxidation potential of ferrocene moiety as well as introducing an amino acid or peptide moiety that can interact with other molecules via hydrogen-bonding.11,22–24 Additionally, the extent of the generation of free radicals by the electroactive molecule is primarily influenced by its physicochemical properties (e.g. lipophilicity, polarity and planarity).25,26 Our primary design rationale for the ferrocene–phosphine derivatives diverges from the traditional method in which we applied a catalytic chemical reduction of the conjugate linker by utilizing an efficient metallacycle catalyzed hydrophosphination protocol. The generic idea stems from previous studies in which the reduced conjugate linkers (of chalcones) have shown markedly differing extent of biological activity from its parent compound.27–29 Adoption of this methodology also provides lipophilic modularity of the target compound and installs a functional phosphine handle which can be derivatized by either simple oxidation or coordination to transition metals of interest. The structural framework of these compounds are categorically made up of 3 synthetically customizable pharmacophores: (i) metallocene, (ii) phosphine, and (iii) acetylketone. In this regard, differing biological activities are expected with various combination of each component's potential pharmacological property.
Anti-proliferative activities of ferrocenyl–phosphine derivatives against various hematolymphoid cell lines
We initially screened the above panel of 10 ferrocenyl–phosphine derivatives (FD1 to FD10) for their anti-proliferative activities on 4 different CTCL cell lines (HuT78, HH, MJ and MyLa). Cells (5 × 104 cells per well in 96-well plate) were treated with increasing concentrations of compounds ranging from 1 μM to 100 μM for 24 h and cell viability was quantified by MTS-based assay. The concentrations that inhibited cell growth by 50% (IC50) were determined and results are presented in Table 1. Notably, the gold-sandwiched compound FD10 demonstrated a significant anti-proliferative activity in all the four hematolymphoid cell lines HuT78, HH, MJ and MyLa with IC50 values 5.55 ± 0.20 μM, 7.80 ± 0.09 μM, 3.16 ± 0.10 μM and 6.46 ± 0.24 μM respectively (Table 1). Whereas, the IC50 of FD10 in human primary PBMCs isolated from healthy volunteers was recorded to be 9.72 ± 0.24 μM (Fig. S1, ESI†), suggesting cancer cell selectivity of FD10 to a certain extent.| Compound | Cell Type | |||
|---|---|---|---|---|
| IC50 (μM) | ||||
| HuT78 | HH | MJ | MyLa | |
| FD1 | >50 | 14.99 ± 1.04 | >50 | >50 |
| FD2 | 19.20 ± 0.82 | 12.37 ± 0.95 | 30.20 ± 0.32 | 30.02 ± 1.07 |
| FD3 | 11.22 ± 0.47 | 24.07 ± 0.69 | >50 | >50 |
| FD4 | >50 | >50 | >50 | >50 |
| FD5 | >50 | >50 | >50 | >50 |
| FD6 | >50 | >50 | >50 | >50 |
| FD7 | 19.62 ± 1.06 | 14.78 ± 1.12 | >50 | >50 |
| FD8 | 20.82 ± 6.00 | 16.10 ± 1.09 | >50 | >50 |
| FD9 | >50 | 39.54 ± 8.05 | >50 | >50 |
| FD10 | 5.55 ± 0.20 | 7.80 ± 0.09 | 3.16 ± 0.10 | 6.46 ± 0.24 |
| SAHA | 2.10 ± 0.25 | 0.50 ± 0.15 | 3.01 ± 0.40 | 1.75 ± 0.20 |
We speculate that the anti-proliferative activity of the ferrocenyl–phosphine compounds lacking the generally desired conjugate linker is due to the augmentation of spatial morphology of the parent ferrocenyl compound while still providing the dual hydrophilicity (when Fc is oxidized to Fc+) and hydrophobicity attributed to the organic fragment. The prominent anticancer activity of FD10 could be due to the presence of a novel gold addition PPh2-AuCl along with an aromatic furan side chain. Up to some extent, fair success to FD8 could be attributed to the presence of PPh2-AuCl functional group and an aromatic methylbenzene side chain. A significant anticancer activity of mono ferrocenyl, chloride-containing gold complex has previously been reported.13 Based on the cytotoxicity screening data showing the robust and consistent anti-proliferative activity of FD10, this compound was chosen for further biological studies to investigate the mechanism of action in CTCL cells.
FD10 induces apoptosis in CTCL cells
Apoptosis or programmed cell death is an essential physiological process for proper homeostatic maintenance by removing injured or mutated cells. Any dysregulation in apoptosis results in high or decreased cell death, which supports the malignancy progression.30,31 Thus, anticancer therapeutics agents are designed to favour the apoptotic cells death.The generation of reactive oxygen species (ROS) is one of the key factors that induce apoptosis under physiologic as well as pathologic conditions32 and therefore targeting ROS response pathway is a viable strategy for anticancer development. Since, ferrocene complexes have been found to elevate ROS within cancer cells,33,34 we speculate that FD10 targets cellular antioxidant systems leading to intracellular ROS generation resulting in cell death via apoptosis.
In order to characterize the cellular basis for the loss of cell viability in FD10-treated cells, we investigated the potential ability of this compound to induce cell death via apoptosis. For this purpose, Annexin V-FITC/PI binding assay was performed using flow-cytometry. This assay evaluates translocation of phosphatidylserine from the cell's inner membrane to the outer lipid layer of the plasma membrane, an important hallmark of apoptosis induction.35 Flow-cytometric analysis of Annexin V-FITC/PI stained cells can distinguish cells into four groups: (1) viable i.e. Annexin V−/PI−, (2) early apoptosis i.e. Annexin V+/PI−, (3) late apoptosis i.e. Annexin V+/PI+, and (4) necrotic i.e. Annexin V−/PI+. Treatment of CTCL cells (HuT78, MJ and MyLa) with IC50 of FD10 for 24 h resulted in significantly high proportion of cells (approximately 50–60%) undergoing early or late apoptosis (Fig. 2). The observed apoptosis induction by FD10 was consistent in all the three CTCL cell types – HuT78, MJ and MyLa (Fig. 2).
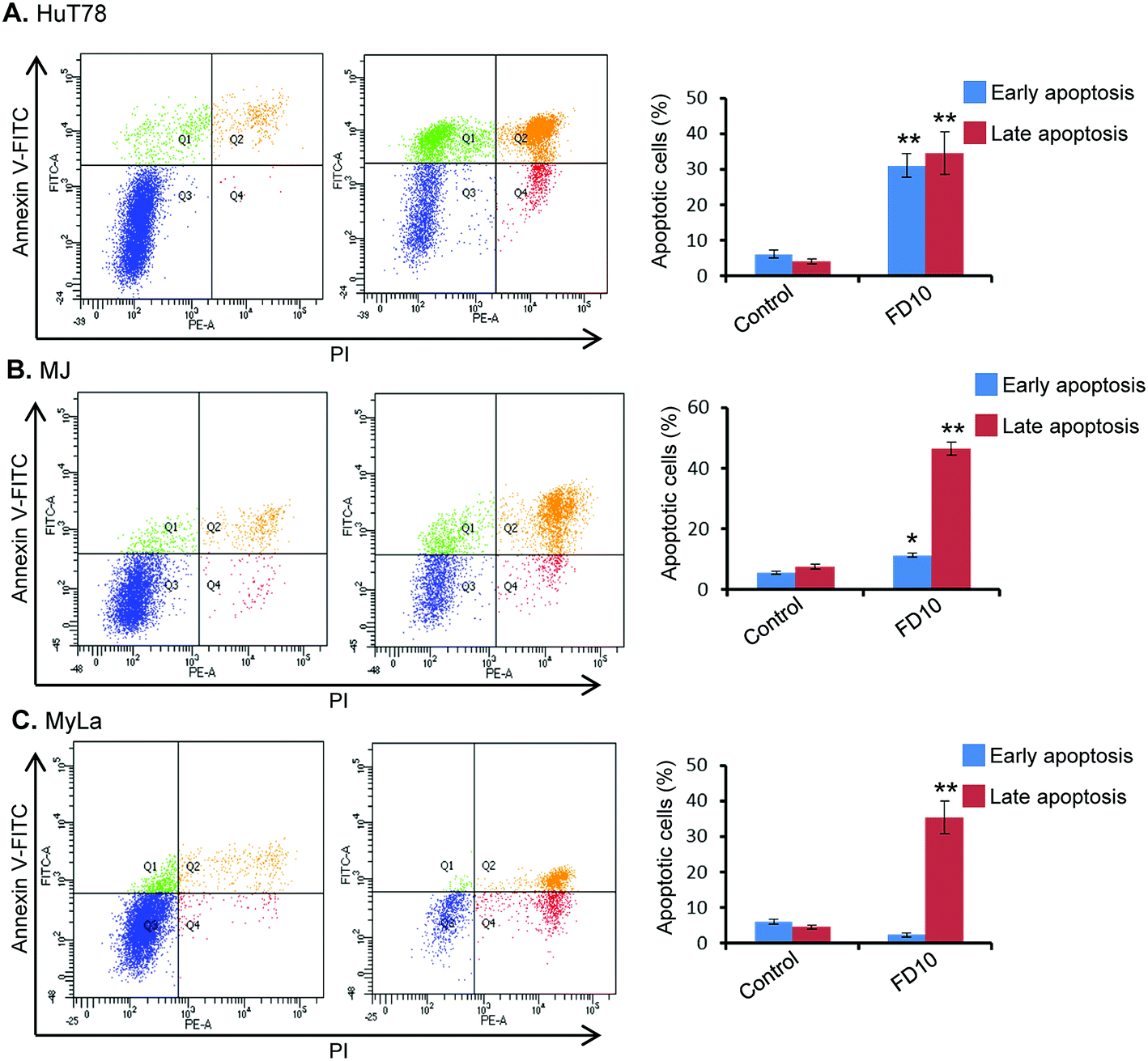 | ||
| Fig. 2 FD10 induces apoptosis in human hematolymphoid cells. Flow-cytometry analysis of HuT78 (A), MJ (B) and MyLa (C) cells treated with FD10 IC50 for 24 h. Representative figures showing population of viable (Q3, Annexin V−/PI−), early apoptotic (Q4, Annexin V+/PI−), late apoptotic (Q2, Annexin V+/PI+) and necrotic (Q1, Annexin V−/PI+) cells. Bar chart showing increased proportion of early and late apoptotic cells due to FD10 treatment (mean ± SEM of three independent experiments, *P < 0.05, **P < 0.01). | ||
FD10 induces caspase-3 activation and PARP cleavage in CTCL cells
Induction of apoptosis is a complex process that mobilizes a number of molecules and can be classified into caspase-dependent or caspase-independent mechanisms.35,36 Caspase-mediated apoptotic cell death is facilitated through the cleavage of several essential proteins required for cellular functioning and survival. Among all the caspase proteins, caspase-3 is perhaps the best understood concerning its specificity and roles in apoptosis. Caspase-3 is present in an inactive state and gets activated by receiving apoptotic stimuli from both, by extrinsic (death ligand) and intrinsic (mitochondrial) pathways.36–38 Cleavage of poly(ADP-ribose)polymerase (PARP) by caspase-3 is an important downstream signaling event and is considered as a hallmark of the apoptosis.39,40 PARP-1 is a nuclear protein with a wide range of physiological as well as pathological functions.41 PARP-1 cleavage by caspase-3 results in formation of two peptides 24 and 89 kDa and resulting in abolition of catalytic properties of PARP.42,43To examine whether caspase-3 and the downstream PARP molecule were involved in FD10-induced apoptosis in hematolymphoid cells, we performed Western immunoblot analysis using cell lysates of untreated/FD10-treated CTCL cells (HuT78, MyLa and MJ, 24 h treatment). We included suberoylanilide hydroxamic acid (SAHA)-treated CTCL cell lysates as a positive control. Results clearly showed that FD10 treatment caused significant activation of caspase-3 in all the three CTCL cell lines – HuT78, MyLa and MJ (Fig. 3A–C). In addition, PARP cleavage was also detected in FD10-treated CTCL cells (Fig. 3A–C), suggesting involvement of caspase-3 and PARP inactivation in FD10-induced apoptosis.
 | ||
| Fig. 3 FD10 induces apoptosis through intrinsic caspase-3/PARP-1 pathway. Western immunoblots showing the expression levels of cleaved PARP-1 and cleaved caspase-3 in HuT78 (A), MyLa (B) and MJ (C) cells treated with DMSO (control) or FD10 IC50 for 24 h. Cells were treated with SAHA as a positive control. β-actin served as a loading control. Graph shows densitometry values of cleaved PARP-1 and caspase-3 (mean ± SEM). Data represent at least three independent experiments; *P < 0.05. | ||
FD10 inhiits the expression and activity of STAT3 in CTCL cells
CTCL cells possess constitutively phosphorylated and relatively high levels of the STAT3 protein and are critically dependent of STAT3 for their survival.44 Indeed inhibition of STAT3 has been found to inhibit the growth of CTCL cells.20 In view of the anti-proliferative properties of FD10, we examined whether this compound can inhibit the constitutive STAT3 activation in CTCL cells. HuT78 cells were treated with increasing FD10 concentrations for 24 h and subjected to Western immunoblot analysis to assess changes in the expression and phosphorylation of STAT3. FD10 treatment significantly and dose-dependently reduced the expression as well as Tyr705 phosphorylation of STAT3 (Fig. 4A). To further confirm the effect of FD10-mediated inhibition of STAT3 on other CTCL cell types, we treated HuT78, MyLa and MJ cells with FD10 IC50 for 24 h. Western immunoblot analysis showed that FD10 significantly inhibited the expression and phosphorylation of STAT3 in all the three CTCL cell lines (Fig. 4B–D). | ||
| Fig. 4 FD10 inhibits STAT3 expression and phosphorylation in human hematolymphoid cells. Western immunoblots showing the expression levels of Y705 phosphorylated STAT3 (pSTAT3) and total STAT3 protein in HuT78 (A and B), MyLa (C) and MJ (D) cells treated with DMSO (control) or FD10 IC50 for 24 h. Cells were treated with SAHA as a positive control. Blots were re-probed with GAPDH as a loading control. Graph shows densitometry values of pSTAT3 and STAT3 (mean ± SEM). Data represents at least three independent experiments; *P < 0.05. | ||
FD10 induces G0/G1 phase cell cycle arrest in hematolymphoid cells
In addition to programmed cell death, cell cycle is another main biological process implicated in the pathogenesis and progression of cancers.45 Cell cycle depends upon the several checkpoints, which synchronise in different phases. Blockade of cell cycle at specific phase causes an accumulation of cells at that phase resulting into the induction of apoptosis.46,47To further elucidate the mechanisms underlying the anti-proliferative activity of FD10, we examined the cell cycle distribution by staining FD10-treated CTCL cells with PI and analysed the percentages of G0/G1, S, G2/M cell population using flow-cytometry. The results demonstrated that treatment of HuT78 cells with FD10 IC50 significantly increased the proportion of cells in the G0/G1 cell cycle phase (48%) compared to 32% in control cells (Fig. 5A). Similarly, MJ cells treated with IC50 concentration of FD10 showed significantly higher G0/G1 population (44%) compared with 34% in the control (Fig. 5B). In addition, FD10 treatment caused a concomitant decrease in the proportion of cells in G2/M phase of the cell cycle from control (12%) to treated HuT78 cells (7%), and from control (13%) to treated MJ cells (8%) (Fig. 5A and B). We next examined the effect of FD10 on the expression and phosphorylation levels of proteins that regulate cell cycle progression – Akt and c-Myc. Western immunoblot analysis revealed that FD10 IC50 treatment significantly decreased the S473 phosphorylation of Akt and the expression of c-Myc in HuT78 CTCL cells (Fig. 5C). These data indicate that FD10 is able to induce cell cycle arrest at the G0/G1 phase.
 | ||
| Fig. 5 FD10 induces cell cycle arrest at G0/G1 stage. HuT78 (A) and MJ (B) cells were treated with FD10 IC50 for 24 h. Cells were stained with PI and changes in cell cycle were analysed by flow-cytometry. Graphs show accumulation of FD10-treated cells in G0/G1, S or G2/M stage (mean ± SEM of three independent experiments, *P < 0.05). (C) HuT78 cells were treated with DMSO (control) or FD10 IC50 or SAHA (as a positive control) for 24 h and lysed. Cellular lysates were analysed by Western immunoblotting for the expression levels of S473 phosphorylated Akt, (pAkt), total Akt, c-Myc proteins. Blots were re-probed with GAPDH as a loading control. Data represent at least three independent experiments. | ||
Conclusions
In conclusion, this study identified the gold-coordinated ferrocenyl–phosphine complex FD10 as a potential anticancer compound. The introduction of the gold centre was in view of the proven ability of gold to amplify biological activity in many classes of anticancer drugs, including several instances involving phosphine-based drugs.48–50 FD10 consistently blocked cell growth, inhibited STAT3, Akt and c-Myc, induced apoptosis and arrested the cell-cycle in hematolymphoid cells. The structural framework of this compound provides high modularity. To further improve the characteristics of FD10 and its potency, future studies would focus on the (i) substitution of the chloride on gold(I) with glucose/dithiocarbamate derivatives, (ii) generation of binuclear gold(I)–phosphine derivatives or incorporation of hydroxyalkyl functionality on the ferrocene scaffold, (iii) improvement in the overall structural rigidity of the compound with bulkier cyclopentadiene groups, and (iv) an elaborate analysis of structure activity relationship. Furthermore, it would be imperative to understand cellular uptake and distribution of the compound and investigate possible targeting of ROS response pathways by FD10 and its functional derivatives. Although further refinement of FD10, development of its functionalized derivatives, elucidation of the detailed mechanism of action and their assessment in in vivo models are needed, our results suggest that the gold-coordinated ferrocenyl–phosphine complex FD10 is a new class of organometallic anticancer compound with promising therapeutic potential.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr M. H. U. T Fazil for assisting in flow-cytometry analysis. We also thank Mr Jing Yew Jeremy Lim, Ms Ching Yee Ivory Yeo and Ms Praseetha P. for technical assistance. This work was supported in part by the Lee Kong Chian School of Medicine, Nanyang Technological University Singapore Start-Up Grant to N. K. V. and the Singapore Ministry of Education under its Singapore Ministry of Education Academic Research Fund Tier 1 (2017-T1-001-175) to S. A. P. Researchers A. S., A. K. and Q. Y. A. C. were supported by Nanyang Technological University Singapore, Lee Kong Chian School of Medicine and IDR-URECA project, respectively.References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef.
- G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef.
- G. Wilkinson, J. Organomet. Chem., 1975, 100, 273–278 CrossRef.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef.
- C. Ornelas, New J. Chem., 2011, 35, 1973–1985 RSC.
- V. J. Fiorina, R. J. Dubois and S. Brynes, J. Med. Chem., 1978, 21, 393–395 CrossRef PubMed.
- J. A. Carmona-Negron, M. M. Flores-Rivera, Z. Diaz-Reyes, C. E. Moore, A. L. Rheigold and E. Melendez, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 868–871 CrossRef PubMed.
- H. Atmaca, A. N. Ozkan and M. Zora, Chem.-Biol. Interact., 2017, 263, 28–35 CrossRef PubMed.
- S. Top, B. Dauer, J. Vaissermann and G. Jaouen, J. Organomet. Chem., 1997, 541, 355–361 CrossRef.
- J. Cazares Marinero Jde, M. Lapierre, V. Cavailles, R. Saint-Fort, A. Vessieres, S. Top and G. Jaouen, Dalton Trans., 2013, 42, 15489–15501 RSC.
- G. Jaouen, A. Vessieres and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC.
- M. M. Santos, P. Bastos, I. Catela, K. Zalewska and L. C. Branco, Mini-Rev. Med. Chem., 2017, 17, 771–784 CrossRef PubMed.
- E. Fourie, E. Erasmus, J. C. Swarts, A. Jakob, H. Lang, G. K. Joone and C. E. Van Rensburg, Anticancer Res., 2011, 31, 825–829 Search PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- A. Miranda-Filho, M. Piñeros, J. Ferlay, I. Soerjomataram, A. Monnereau and F. Bray, Lancet Haematol., 2018, 5, e14–e24 CrossRef PubMed.
- S. A. Pullarkat, Synthesis, 2016, 48, 493–503 CrossRef.
- S. A. Pullarkat and P.-H. Leung, in, Hydrofunctionalization, Springer, 2011, pp.145–166 Search PubMed.
- P.-H. Leung, G. M. McLaughlin, J. W. Martin and S. B. Wild, Inorg. Chem., 1986, 25, 3392–3395 CrossRef.
- S. T. Ong, M. L. S. Chalasani, M. H. U. T. Fazil, P. Prasannan, A. Kizhakeyil, G. D. Wright, D. Kelleher and N. K. Verma, Front. Immunol., 2018, 9, 397 CrossRef PubMed.
- N. K. Verma, A. M. Davies, A. Long, D. Kelleher and Y. Volkov, Cell. Mol. Biol. Lett., 2010, 15, 342–355 Search PubMed.
- K. Gan, A. Sadeer, C. Xu, Y. Li and S. A. Pullarkat, Organometallics, 2014, 33, 5074–5076 CrossRef.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef PubMed.
- V. Janka, D. Žatko, V. Ladislav, P. Pál, P. Janka and M. Gabriela, In Vitro Cell. Dev. Biol.: Anim., 2015, 51, 964–974 CrossRef PubMed.
- A. Podolski-Renic, S. Bősze, J. Dinic, L. L. Kocsis, F. Hudecz, A. Csámpai and M. Pesic, Metallomics, 2017, 9, 1132–1141 RSC.
- E. Hillard, A. Vessières, L. Thouin, G. Jaouen and C. Amatore, Angew. Chem., Int. Ed. Engl., 2006, 45, 285–290 CrossRef PubMed.
- X. Wu, E. R. Tiekink, I. Kostetski, N. Kocherginsky, A. L. Tan, S. B. Khoo, P. Wilairat and M.-L. Go, Eur. J. Pharm. Sci., 2006, 27, 175–187 CrossRef PubMed.
- D. Elias, M. Beazely and N. Kandepu, Curr. Med. Chem., 1999, 6, 1125–1149 Search PubMed.
- N. Tadigoppula, V. Korthikunta, S. Gupta, P. Kancharla, T. Khaliq and A. Soni, et al., J. Med. Chem., 2012, 56, 31–45 CrossRef PubMed.
- H. Wei, J. Ruan and X. Zhang, RSC Adv., 2016, 6, 10846–10860 RSC.
- G. Evan and T. Littlewood, Science, 1998, 281, 1317–1322 CrossRef PubMed.
- Y. Ishizaki, L. Cheng, A. W. Mudge and M. C. Raff, Mol. Biol. Cell, 1995, 6, 1443–1458 CrossRef PubMed.
- M. Redza-Dutordoir and D. A. Averill-Bates, Biochim. Biophys. Acta, 2016, 1863, 2977–2992 CrossRef PubMed.
- J. F. Arambula, R. McCall, K. J. Sidoran, D. Magda, N. A. Mitchell, C. W. Bielawski, V. M. Lynch, J. L. Sessler and K. Arumugam, Chem. Sci., 2016, 7, 1245–1256 RSC.
- X. Peng and V. Gandhi, Ther. Delivery, 2012, 3, 823–833 CrossRef.
- A. Ashkenazi and V. M. Dixit, Science, 1998, 281, 1305–1308 CrossRef PubMed.
- H. Hirata, A. Takahashi, S. Kobayashi, S. Yonehara, H. Sawai, T. Okazaki, K. Yamamoto and M. Sasada, J. Exp. Med., 1998, 187, 587–600 CrossRef PubMed.
- S. Fulda, Semin. Cancer Biol., 2015, 31, 84–88 CrossRef PubMed.
- J. Walters, C. Pop, F. L. Scott, M. Drag, P. Swartz, C. Mattos, S. G. Salvesen and A. C. Clark, Biochem. J., 2009, 424, 335–345 CrossRef PubMed.
- R. U. Janicke, M. L. Sprengart, M. R. Wati and A. G. Porter, J. Biol. Chem., 1998, 273, 9357–9360 CrossRef PubMed.
- S. H. Kaufmann, Cancer Res., 1989, 49, 5870–5878 Search PubMed.
- T. Lindahl, M. S. Satoh, G. G. Poirier and A. Klungland, Trends Biochem. Sci., 1995, 20, 405–411 CrossRef PubMed.
- N. Margolin, S. A. Raybuck, K. P. Wilson, W. Chen, T. Fox, Y. Gu and D. J. Livingston, J. Biol. Chem., 1997, 272, 7223–7228 CrossRef PubMed.
- M. Tewari, L. T. Quan, K. O'Rourke, S. Desnoyers, Z. Zeng, D. R. Beidler, G. G. Poirier, G. S. Salvesen and V. M. Dixit, Cell, 1995, 81, 801–809 CrossRef PubMed.
- V. H. Sommer, O. J. Clemmensen, O. Nielsen, M. Wasik, P. Lovato, C. Brender, K. W. Eriksen, A. Woetmann, C. G. Kaestel, M. H. Nissen, C. Ropke, S. Skov and N. Ødum, Leukemia, 2014, 18, 1288–1295 CrossRef PubMed.
- Y. A. Fouad and C. Aanei, Am. J. Cancer Res., 2017, 7, 1016–1036 Search PubMed.
- G. H. Williams and K. Stoeber, J. Pathol., 2012, 226, 352–364 CrossRef PubMed.
- M. Malumbres, Clin. Transl. Oncol., 2006, 8, 399–408 CrossRef PubMed.
- B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209–4219 RSC.
- C. Nardon, G. Boscutti and D. Fregona, Anticancer Res., 2014, 34, 487–492 Search PubMed.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Ferrocenyl–phosphine derivatives and their characterization. See DOI: 10.1039/c8ra05224g |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |