
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProximity hybridization triggered strand displacement and DNAzyme assisted strand recycling for ATP fluorescence detection in vitro and imaging in living cells†
Fenglei Gao‡a,
Jing Wu‡
 a,
Yao Yaoa,
Yu Zhanga,
Xianjiu Liao*b,
Deqin Genga and
Daoquan Tang*a
a,
Yao Yaoa,
Yu Zhanga,
Xianjiu Liao*b,
Deqin Genga and
Daoquan Tang*a
aJiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, School of Pharmacy, Xuzhou Medical University, 221004, Xuzhou, China. E-mail: tdq993@hotmail.com; Fax: +86-516-83262138; Tel: +86-516-83262138
bSchool of Pharmacy, Youjiang Medical University for Nationalities, Baise, 533000, China. E-mail: lxj2006910@163.com; Fax: +86-516-83262138; Tel: +86-516-83262138
First published on 6th August 2018
Abstract
We developed a novel strategy for ATP detection in vitro and imaging in living cells based on integrating proximity hybridization-induced strand displacement and metal ion-dependent DNAzyme recycling amplification. Four DNA oligonucleotides were used in the sensing system including two aptamer probes, enzymatic sequences and FAM-linked substrate strands. Upon the addition of ATP, the proximity binding of two aptamers to ATP led to the release of the enzymatic sequences, which hybridized with the FAM-linked substrate strand on the graphene oxide (GO) surface to form the ion-dependent DNAzyme. Subsequent catalytic cleavage of the DNAzyme by the corresponding metal ions results in recycling of the enzymatic sequences and cyclic cleavage of the substrate strand, liberating many short FAM-linked oligonuleotide fragments separated from the GO surface, which results in fluorescence enhancement due to the weak affinity of the short FAM-linked oligonuleotide fragment to GO. The amount of produced short FAM-linked oligonuleotide fragments is positively related to the concentration of ATP. This means that one target binding could result in cleaving multiplex fluorophore labelled substrate strands, which provided effective signal amplification. The vivo studies suggested that the nanoprobe was efficiently delivered into living cells and worked for specific, high-contrast imaging of target ATP. More importantly, this target-responsive nanoscissor model is an important approach for intracellular amplified detection and imaging of various analytes by selecting appropriate affinity ligands.
1. Introduction
Adenosine 5′-triphosphate (ATP), one of the best known biological compounds, is a multifunctional molecule found in all living organisms.1,2 ATP is used as an intracellular precursor in kinase activity for proteins or lipids to provide a phosphate group, as well as in adenylate cyclase to produce the second messenger molecule cyclic adenosine monophosphate. The excess amount of ATP consumed by creatine kinase causes particular diseases.3,4 Therefore, the real-time monitoring of ATP levels is imperative for the study of multiple cellular mechanisms, and enzyme activity involving the production and consumption of ATP as well as clinical diagnosis.5,6Classic measurements of ATP are based on offline analysis methods, such as chromatographic, bioluminescence, and chemiluminescence. The chromatographic method is accurate but time-consuming; both bioluminescence and chemiluminescence methods involve chemical reactions among multiple components; in particular, the enzymes used in bioluminescence are costly and unstable.7–9 In recent years, the aptamer-based ATP sensing strategy has attracted tremendous interest because of the specific binding between selected aptamer and ATP including optical transduction, fluorescence, colorimetry, electrochemistry, and so on.10–16 Among these methods, fluorescence based detection has been widely used and continues to play an important role in future research because of its high sensitivity and easy operation.12 However, these methods could not monitor the dynamic expression and distribution of ATP in living cells. In situ fluorescence imaging technology provides an effective tool for visualizing intracellular ATP, which can offer valuable information for precise understanding of these biological processes. Employment of nanomaterials for detecting intracellular ATP in living cells has attracted intensive attention due to the unique quenching efficiency of such materials.17–20 For example, Wang group developed an ATP aptamer/GO platform to detect ATP in living cells. When ATP aptamer/GO entered cells, cellular ATP bound to ATP aptamers and released them from the surface of GO. However, in these assays, each ATP hybridizes to only a single complementary aptamer strand. Signal enhancement and the sensitivity of the assay are strictly limited because of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hybridization ratio. Thus, it is imperative to design an appropriate signal amplification strategy for intracellular biological molecules ATP detection and imaging.
:1 hybridization ratio. Thus, it is imperative to design an appropriate signal amplification strategy for intracellular biological molecules ATP detection and imaging.
Recently, metal ion-specific DNAzyme have been developed for detection of vital metal ions and even biomolecules.21,22 The metal ion-dependent DNAzymes exhibit high catalytic activity and excellent cleavage specificity toward the substrate sequence.23,24 And it showed broad prospects for in situ detection of intracellular analytes because no additional tool enzyme is required.25–27 Recently, a TAMRA-tagged substrate strands hybridized to DNAzyme strands functionalized gold nanoparticle probe was designed for in situ fluorescence imaging and detection of cytoplasmic ATP activity.25 Only the target molecule can activate the DNAzyme and then cleave and release the fluorophore-labeled substrate strands from the AuNP, resulting in fluorescence enhancement. However, the above-mentioned method has the disadvantages of lacking simplicity, suffering from time-consuming processes caused by immobilization of DNAzyme on gold nanoparticles, relatively poor salt stability of gold nanoparticles as signal transducer, which present some limitations for their practical implementation. Therefore, the development of robust methods for in situ tracking of intracellular ATP based on DNAzyme assistant probe recycling is still of great significance.
Inspired by the advantages of GO as the substrate for immobilization and the quencher of fluorescent dyes, several aptamers/GO based fluorescence “turn-on” biosensors have been developed for various targets based on the ssDNA and duplex with its target exhibit different affinity to GO. However, this strategy is not suitable for constructing DNAzyme/GO “turn-on” fluorescence biosensors, as a reversed conformation change from the duplex of DNAzyme with substrate to ssDNA (cleaved substrate) was involved in the DNAzyme/target reaction. To solve this problem, Zhang group developed a GO/DNAzyme based biosensor for fluorescence “turn-on” detection of Pb2+ based on the remarkable difference in affinity of GO with ssDNA containing a different number of bases in length.21 Inspired by this strategy, in this paper, we have developed a robust GO nanoprobe analysis platform based on proximity binding assay28–34 integrated with Pb2+-DNAzyme assistant probe recycling to achieve signal enhancement for detection of ATP activity and imaging of ATP in living cells (Scheme 1). The proximity binding of two aptamers to ATP leads to the release of the enzymatic sequences, which hybridize with the FAM-linked substrate strand on the graphene oxide (GO) surface to form the ion-dependent DNAzyme. Subsequent catalytic cleavage of the DNAzyme by the corresponding metal ions results in recycling of the enzymatic sequences and cyclic cleavage of the substrate strand, liberating many short FAM-linked oligonuleotide fragments separated from GO surface, which resulting in fluorescence enhancement due to the weak affinity of the short FAM-linked oligonuleotide fragment to GO. The amount of produced short FAM-linked oligonuleotide fragments are positively related to the concentrations of ATP. Therefore, this strategy has high sensitivity for providing amplified fluorescent signal in situ imaging of target ATP.
 | ||
| Scheme 1 Schematic illustration of the GO integrated proximity binding-induced strand displacement and metal ion-dependent DNAzyme recycling for ATP detection and imaging in living cells. | ||
2. Experimental
2.1 Materials and reagents
Oligomycin, ATP, cytidine triphosphate (CTP), guanosine triphosphate (GTP), and uridine triphosphate (UTP) were obtained from Sangon Biotech Co., Ltd. (Shanghai, China). MCF-7 cells and HepG2 cells were provided by KeyGEN BioTECH Corp., Ltd.DNA loading buffer was purchased from TaKaRa Bio Inc. (Dalian, China). SYBR was purchased from Invitrogen (USA). Hela cells were obtained from KeyGEN Biotech Co., Ltd. (Nanjing, China). Milli-Q water (resistance > 18 MΩ cm) was used in all experiments. Propidium iodide (PI), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), dimethylformamide (DMF), and dimethyl sulfoxide (DMSO) were purchased from Sigma Aldrich. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Annexin V-FITC/PI cell apoptosis kit were purchased from KeyGen Biotech. Co. Ltd. (Nanjing, China). Oligomycin was purchased from Sangon Biological Engineering Technology & Co. Ltd. (Shanghai, China). Graphene oxide (GO) was purchased from Xianfeng nanomaterials Co., LTD. (Nanjing, China). Before being used, GO (8 mg) was dissolved in 8 mL water and then strong sonicated for 1 h (60 W). Subsequently, large GO layers were removed by centrifugation (6000 rpm, 10 min) and stored in 4 °C. The HEPES buffer (pH 7.4) contained 10 mM HEPES, 137 mM NaCl, 2.7 mM KCl and 5 mM MgCl2 unless otherwise noted. HB (pH 7.4) buffer contained 10 mM Tris, 50 mM NaCl and 10 mM MgCl2·6H2O. TE (pH 8.0) buffer contained 10 mM Tris, 1 mM EDTA-2Na. All the oligonucleotides were synthesized and HPLC purified by Sangon Biotechnology Co., Ltd (Shanghai, China). The sequences of the oligonucleotides were described as follows:
S1: 5′-ACCTGGGGGAGTATTTTTTTGGGTTTCACTACGGCGGC-3′;
S2: 5′-GCCGCCGTAGTGAAACCCTTTTTTTGGAAGGAGGCGT-3′;
S3: 5′-AATCATCTCTGAAGTAGCGCCGCCGTAGTG-3′;
S4: 5′-FAM-CACTrAGGAAGAGATGATT-3′;
FAM-ATP aptamer: 5′-FAM-CACCTGGGGGAGTATTGCGGAGGAAGGTT-3′
Mismatched aptazyme S3: 5′-AATCATCTCTGAA![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) TAGGCCGCCGTAGTG-3′;
TAGGCCGCCGTAGTG-3′;
2.2 Apparatus
Zeta potential analysis was performed at room temperature on a Zetasizer (Nano-Z, 60 Malvern, UK). UV-vis-NIR spectra were measured on a UV-3600 spectrophotometer (Shimadzu, Japan). Atomic force microscopic (AFM) measurements were performed with ScanAsyst mode on Fastscan AFM (Bruker, Inc.) by directly casting sample dispersion onto mica sheet. The fluorescence spectra were obtained on a RF-5301 PC spectrofluorometer (Shimadzu, Japan) equipped with a xenon lamp. Flow cytometric analysis was performed on a FACSCalibur flow cytometer (Becton Dickinson, USA). Gel electrophoresis was performed on a DYCP-31 BN electrophoresis analyser (Liuyi Instrument Company, China) and imaged on the Bio-Rad ChemDoc XRS (USA). The cell images were observed by a Leica TCS SP5 confocal laser scanning microscope (CLSM, Germany). The MTT assay was performed using Varioskan Flash microplate reader (ThermoFisher Scientific) at 490 nm.2.3 Preparation of the DNA/GO nanocomplex
S1 (5 μL, 100 μM) and S3 (5 μL, 100 μM) were treated with 40 μL TE buffer at room temperature for 30 min. The GO (0.5 mg mL−1) was diluted to 0.1 mg mL−1 with HE buffer. To obtain the GO probe, the hybridized S1/S3 (5 μL), S2 (5 μL, 100 μM), S4 (5 μL, 100 μM) were added into GO (1 mL, 0.5 mg mL−1) for 1 h at 150 rpm and room temperature.2.4 Fluorescence response and selectivity of nanoprobe toward ATP
After the nanoprobe (150 μL, 0.1 mg mL−1) was incubated with different concentrations of ATP ranging in HEPES buffer (20 mM, pH 7.4, containing 15 μM Pb(AC)2) at room temperature for 80 min, the fluorescence measurement was carried out from 500 to 700 nm with the excitation wavelength at 496 nm at room temperature. To evaluate the selectivity, each type of nucleotide (1 mM ATP, 10 mM CTP, 10 mM UTP, 10 mM GTP) was incubated with the nanoprobe (150 μL, 0.1 mg mL−1) for 80 min to measure the fluorescence intensity at 523 nm, respectively.2.5 Cell culture
HeLa, MCF-7, and HepG2 cells were maintained as monolayer cultures in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37 °C in a CO2 incubator (Memmert, Germany. 5% CO2). The cells were pre-cultured until confluence was reached before each experiment.2.6 Cell imaging
For cell imaging experiments, the HeLa cells were seeded into 35 mm confocal dishes (Glass Bottom Dish) at 1 × 104 cells per dish and grown for 24 h at 37 °C. Then the cells were washed three times with HEPES buffer. Prior to the addition of the nanoprobes, the cells were incubated with or without 10 μg mL−1 oligomycin or 5 mM Ca2+ for 30 min. Afterward, the medium was replaced with 150 μL of nanoprobe (0.1 mg mL−1) and Pb(AC)2 (15 μM). 200 min later, the cells were rinsed three times with the HEPES buffer and imaged on the laser confocal microscope.2.7 Electrophoresis experiment
Electrophoresis experiments were performed using non-denaturing polyacrylamide gel electrophoresis (PAGE). The hybridized S1/S3 (7.5 μL, 20 μM), S3 (7.5 μL, 20 μM), S2 (7.5 μL, 20 μM), S4 (7.5 μL, 20 μM) and Sample 1, and Sample 2 were mixed with electrophoresis loading buffer (1.5 μL) and GelRed (0.5 μL) respectively; DNA ladder (4 μL, 1 μg μL−1) was added to TBE buffer (3.5 μL) diluted with the same method (DNA ladder (4 μL, 1 μg μL−1) diluted with TBE buffer (3.5 μL) was treated with same way). After centrifuging, it was added to the sample wells of the polyacrylamide gel. The buffer was tris–borate–EDTA (TBE) buffer, the electrophoresis conditions were 80 V, 80 min.Sample 1: S1/S3 (1.5 μL, 50 μM), S2 (1.5 μL, 100 μM), S4 (1.5 μL, 100 μM) were mixed with ATP (1.5 μL 10 mM) at 37 °C for 80 min.
Sample 2: S1/S3 (1.5 μL, 50 μM), S2 (1.5 μL, 100 μM), S4 (1.5 μL, 100 μM) were mixed with ATP (1.5 μL 10 mM) and Pb(AC)2 (0.5 μL 10 mM) at 37 °C for 80 min.
2.8 Cytotoxicity assay
After HeLa cells were cultivated in media each well of a 96-well plate for 12 h, the media were discarded. Either fresh medium alone or medium containing GO (150 μL, 0.1 mg mL−1) nanoprobes and Pb2+ (15 μM) was added to each well of the 96-well plate to cultivate for another 4 h. MTT (5 mg mL−1) was then added to each well. After incubation for 4 h, the medium was removed, and sodium dodecyl sulfate was added to each well to solubilize the formazan dye and vortexed. After 15 min, the absorbance of the GO (15 μg) and Pb2+ (15 μM) treated wells and the control wells that were not treated was measured using Varioskan Flash microplate reader (ThermoFisher Scientific) at 490 nm. The relative cell viability (%) was calculated by (A test/A control) × 100.2.9 Apoptosis experiment
The apoptosis experiments were carried out with PI staining. After the cells were seeded on glass cover slides, the culture media were replaced with media containing GO (150 μL, 0.1 mg mL−1) nanoprobes and Pb2+ (15 μM) for further 3 h. Afterwards, the cells were thoroughly washed and resuspended in fresh media to culture for another 24 h. The resulting cells were harvested, permeabilized with 96% ethanol and stained in binding buffer supplemented with PI for 10 min in dark. The HeLa cells were collected and analyzed by flow cytometry on FACSCalibur flow cytometer (Becton Dickinson, USA).3. Results and discussion
3.1 Characterization of the GO nanoprobe
The atomic force micrograph (AFM) and UV-vis spectra were used to demonstrate the binding of the DNA probe to GO. As shown in Fig. 1A, AFM of GO clearly showed its planar structure with a dimension of around 180 nm and a height of around 1.2 nm. Fig. 1B shows the typical AFM image of the DNA-GO complex, where the white areas on the GO surface might be due to the presence of DNA. The associated height profiles revealed the white areas was about 1.1 nm, which is matched with the diameter of single-stranded DNA. The UV-visible absorption spectra (Fig. 1C) of GO and DNA showed absorption peak at 230 and 260 nm. Upon addition of DNA to the GO solution, the absorption peaks showed remarkable hypochromicity, suggesting a static interaction between GO and DNA. Moreover, the zeta potential of the GO, DNA and DNA/GO were determined to be −30 mV, −14.1 mV and −40.4 mV (Fig. 1D), which suggested that DNA have been absorbed the GO surface and DNA/GO have great colloidal stability in aqueous media. These results verified the successful assembly of DNA on the surface of GO. | ||
| Fig. 1 (A) AFM height image of GO sheets deposited on mica substrates; (B) AFM height image of DNA-GO complex; (C) UV spectrum and (D) zeta-potential analysis of GO (a), DNA (b), DNA-GO probe (c). | ||
3.2 In vitro studies of ATP with the GO nanoprobe
In order to verify the feasibility of this design and the important role of Pb2+ during this sensing system, we next studied the fluorescence signal of the nanoprobe after the reaction of ATP with or without the treatment of Pb2+. As shown in Fig. 2A, in the absence of the target ATP with (curve a) and without (curve b) Pb2+, only the fluorophore-labeled substrate strand immobilized on GO surface showed relatively low fluorescent background. Because the displacement of S3 by S2 can be effectively avoided, and subsequent formation of the Pb2+-dependent DNAzyme are thus inhibited in the absence of the target ATP. The number of complementary bases of S1 and S3 are the same as the counterpart of S1 and S2. In the presence of target ATP without Pb2+, a little change in fluorescence intensity was observed (curve c). Binding of ATP to its aptamer sequences in S1 and S2 brought the two DNA probes together, resulting in the displacement of S3. Subsequently, the displaced S3 is hybridized with the S4 absorbed on the GO to form a Pb2+-dependent DNAzyme containing a large ssDNA loop to bind with the GO. However, Pb2+-dependent DNAzyme have weak affinity to GO compare with single strand DNA S4. Upon the addition of Pb2+, the fluorescence intensity sharply increased (curve d). This result can be contributed to the DNAzyme was activated and cleaved the substrate strand at the RNA site, releasing a short FAM-linked oligonuleotide fragment, and the S3 strand. The S3 strand can hybridize with another S4 substrate strand and then induce the second cycle of cleavage by binding Pb2+, providing an amplified detection signal for ATP. Moreover, PAGE analysis was also used to investigate the viability of the proximity binding-induced strand displacement and Pb2+-dependent DNAzyme catalytic cleavage. As shown in Fig. 2B, S2 (lane b), S3 (lane c), S4 (lane d) exhibited individual clear band, respectively. The S1 could hybridize with S3 for forming of dsDNA and produced a new band (lane a). Upon the addition of S1/S3 to the mixture of S2, S4 and ATP, a new band with a slowest migration speed appeared (lane e), demonstrating the formation of proximity complex S1/ATP/S2 and the other new band appear with a faster migration rate than S1/ATP/S2, which should be attributed to S3 hybridized with the S4 (S3/S4), which verifying the feasibility of proximity binding-induced strand displacement. After Pb2+ was added, a new band appeared (lane f), and showed the fastest migration rate, which should be contributed to the small DNA pieces produced in the cleavage of S4 by the Pb2+, verifying the feasibility of Pb2+-dependent DNAzyme catalytic cleavage. The other new band appear with a slower migration than S2, which should be attributed to S3 hybridized with the fragment of S4. The results further confirmed the feasibility of the target ATP triggered proximity binding-induced strand displacement and metal ion-dependent DNAzyme recycling. | ||
| Fig. 2 (A) Fluorescence intensity of GO nanoprobes incubated (a) without ATP and without Pb2+, (b) without ATP and with Pb2+, (c) with ATP and without Pb2+, (d) with ATP and with Pb2+ after 80 min; (B) PAGE image of (a) S1/S3, (b) S2, (c) S3, (d) S4, (e) mixture of S1/S3, S2, S4 and ATP after incubation for 80 min, (f) mixture of S1/S3, S2, S4, ATP and Pb2+ after incubation for 80 min. | ||
To achieve the best sensing performance, the corresponding experimental conditions including the reaction time and the concentration of Pb2+ were optimized (Fig. S1†). Under the optimal conditions, the fluorescence intensities of the mixtures of the nanoprobe and different concentrations of ATP were recorded. Fig. 3A depicts the typical fluorescence spectra of the assay towards ATP of varying concentrations. The fluorescence intensities gradually increased with the increasing concentration of ATP in the range from 2 μM to 1 mM. In the range from 2 μM to 90 μM, a linear dependency between FI and c (ATP) is obtained (Fig. 3B). The regression equation is FI = 18.63c + 102.3 (R = 0.9983) and the limit of detection was calculated to be 150 nM. The linear range of the nanocomplex met the need for the sensing of ATP in living cells, as the concentration of ATP in living cells is typically 0−10 mM.14 The proposed method is comparable to other reported aptasensors, the result was summed up in Table 1. The high sensitivity of this method could be attributed Pb2+-dependent DNAzyme assistant DNA recycling, demonstrated that the strategy could be used efficiently for ATP detection. To demonstrate the feasibility of the nanoprobe for ATP detection in complex biological environment, control experiments were performed by incubating the method with ATPs analogues such as CTP, UTP and GTP, respectively. The result is shown in Fig. 3C, even if the concentrations of CTP, GTP and UTP are 10 times higher than ATP, FI from ATP is very higher than those from the ATP analogues. The comparative results suggest the satisfactory ability of this biosensor to identify ATP, which proves that the ATP-binding aptamer sequence is very specificity to ATP.
 | ||
| Fig. 3 (A) Fluorescence spectra of nanoprobe in the presence of different concentrations of ATP (2 μM to 1 mM). (B) Plot of the fluorescence intensity of the system corresponding to the concentration of ATP. The inset showed a linear relationship in the concentration range from 2 μM to 90 μM for ATP. (C) FI of the nanoprobe in response to CTP, UTP, GTP (10 mM), and ATP (1 mM). (D) FI after mixing GO probe (0.1 mg mL−1) with (1) PBS buffer (pH 7.4); (2) 2 μg of DNase I (pH 7.4); (3) 2 μg of DNase I (pH 6.0); and (4) 10 μL ATP (1 mM) and 1.5 μL Pb2+ (15 μM). Inset: corresponding fluorescence spectra. | ||
3.3 Cytotoxicity of the GO nanoprobe
The in vitro response and efficient cytosolic delivery of the sensors provided us the possibility for fluorescent imaging of ATP in living cells. To realize the in situ target sensing in living cells, it is expected that the nanoprobe is with good biocompatibility and low toxicity. Prior to ATP imaging in the living cells, the cytotoxicity of GO and Pb2+ nanoprobe was by the standard MTT assay (Fig. 4A). After the HeLa cells were incubated with GO (15 μg) and Pb2+ (15 μM) nanoprobe for 4 h, higher than 83% cell survival rate was observed. The apoptosis of HeLa cells induced by the nanoprobe was monitored with flow cytometry analysis by propidium iodide staining. Compared to control group that was incubated in absence of the nanoprobe and presented an apoptotic ratio of 6.4% (Fig. 4B), the cells transfected with the nanoprobe displayed slightly higher apoptotic ratio 8.8% (Fig. 4C), which further suggested the nanoprobe could efficiently remain viability. To evaluate the nuclease stability of the nanoprobe, we carried out a series of experiments using enzyme deoxyribonuclease I (DNase I), a common endonuclease capable of effectively cutting both single and double-strand DNA. As shown in Fig. 3D, the nanoprobe treated with DNase I was not obviously degrade compared to the nanoprobe without DNase I at 80 min. These indicated that the probe possesses low background and high resistance to nuclease with the help of GO, and further demonstrated that the fluorescence recovery was indeed due to the target-induced activation–cleavage reaction instead of nuclease degradation. These results confirm that the nanoprobe has low toxicity and its stability is sufficient for application in cellular environments.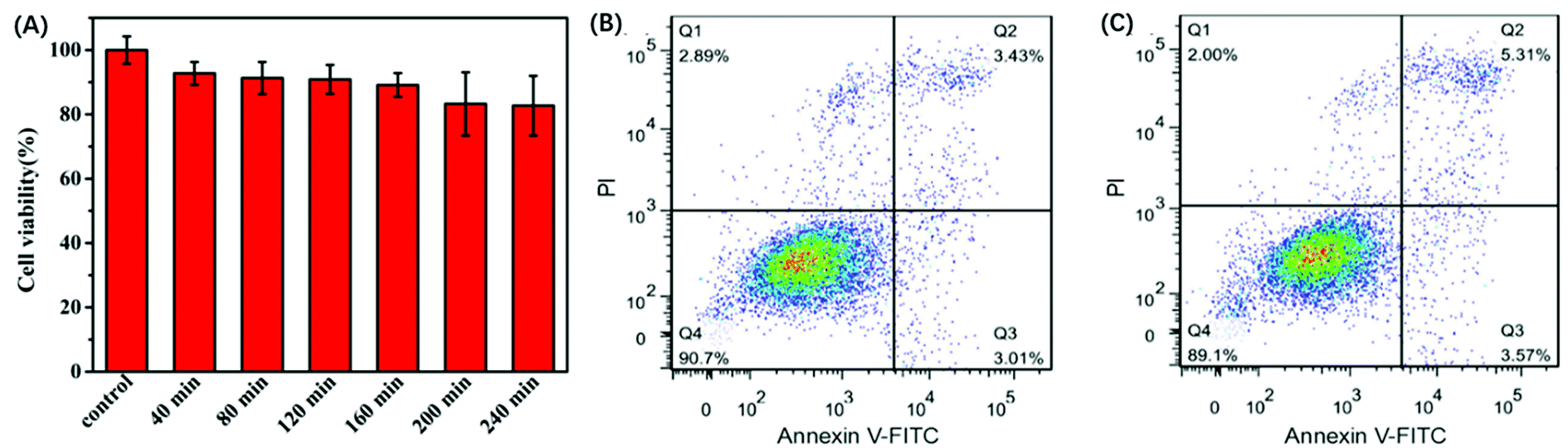 | ||
| Fig. 4 (A)Viability of HeLa cells after incubation with GO (15 μg) and Pb2+ (15 μM) nanoprobe for different time. Flow cytometric apoptotic analysis of (B) HeLa cells and (C) HeLa cells incubated with GO (15 μg) and Pb2+ (15 μM) nanoprobe. | ||
3.4 In situ monitoring of the dynamic changes of ATP
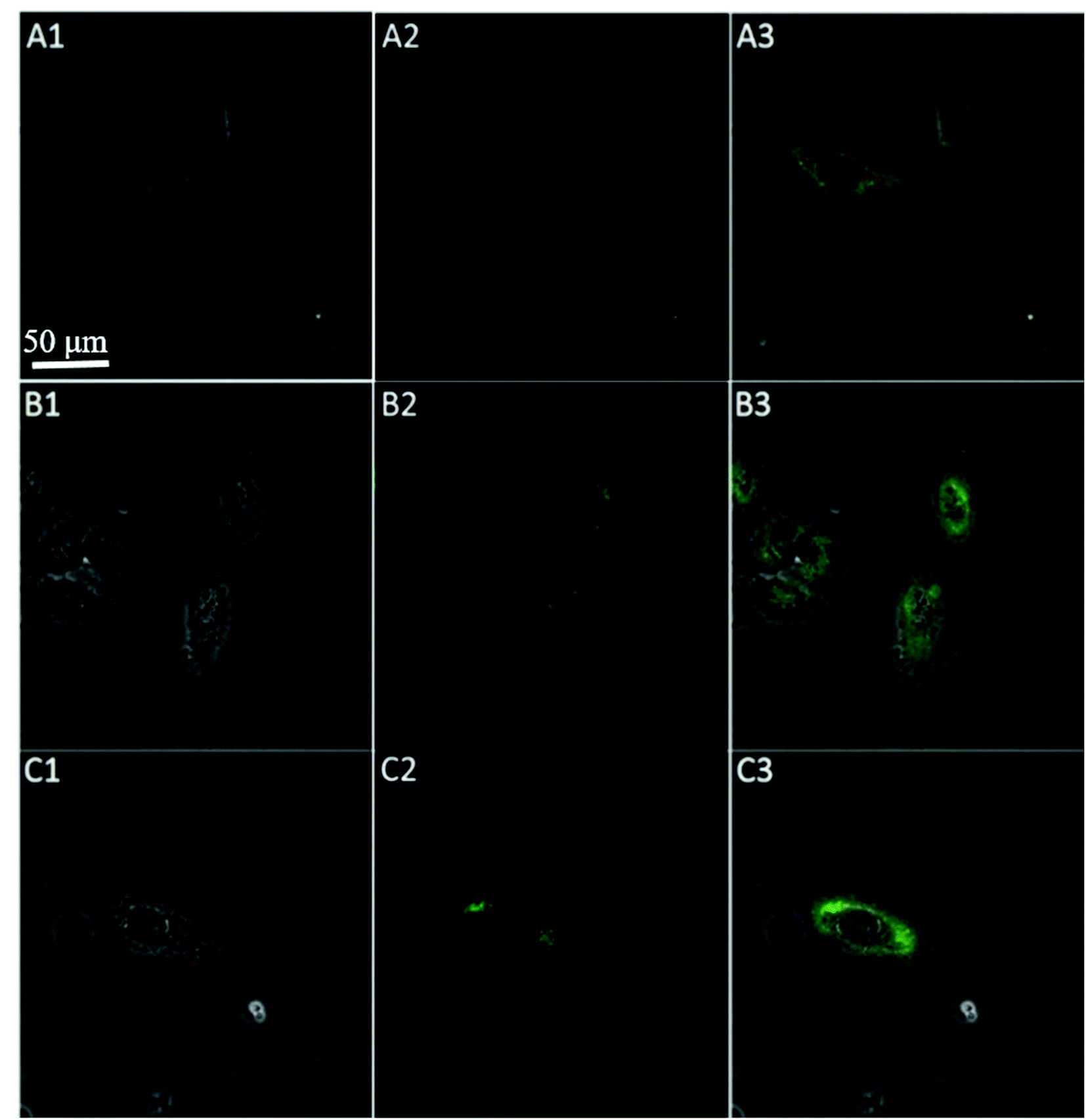
To further test the employment of this nanocomplex for intracellular ATP imaging study, HeLa cells were used to be incubated with nanoprobe (The stability of DNA–Go complex in 10% FBS buffer was proved in ESI†). After the HeLa cells were incubated with nanoprobe, a significant fluorescence signal was clearly observed from HeLa cells incubated with the nanoprobe and Pb2+ (Fig. 5A), with the increasing incubation time of HeLa cells in nanoprobe suspension (Fig. 6), the transfected cells showed increasing fluorescence signal, indicating the increased uptake of nanoprobe. While little fluorescence was obtained from HeLa cells incubated with nanoprobe and without Pb2+ (Fig. 5B), suggesting that the method must function under Pb2+, as Pb2+ is an important cofactor to aptazyme. To further manifest that the fluorescence observed was due to the target induced self-cleavage of DNAzyme strand, not only the target binding, we designed a control probe with mismatched bases in the DNAzyme catalytic domain, which possessed no catalytic activity even with target binding. In Fig. 5D, HeLa cells treated with such Mis-probes showed very less fluorescence as compared to those treated with the active probes. To confirm the sensitivity enhancement of DNAzyme-based amplification strategy, HeLa cells were incubated with the nanosystem that carries only the fluorescence self-quenching probe FAM-aptamer/GO, which could directly hybridize with target ATP and give a fluorescence signal in the cells. Fig. 5C shows low-contrast green fluorescence because of a lack of DNAzyme assistant recycling amplification. These results demonstrate that it is difficult to image low-level ATP using a direct probe with a 1:1 stoichiometric ratio, but the amplification strategy is advantageous for visualizing low levels of ATP in living cells. The feasibility of the method for in situ imaging of intracellular ATP was also investigated by dynamically observing the fluorescence of FAM upon the transfection of the nanoprobes (Fig. 6). In the beginning, no obvious fluorescence signal was observed within 60 min. After 1 h, weak fluorescence could be observed in the cytoplasm, and the intensity increased with the increasing incubation time for ATP-triggered desorb of GO. The green fluorescence was little stronger at 300 min than 200 min (fluorescence intensity histogram corresponding to each incubation time of cell was in ESI†). Consider saving time, we choose 200 min. For obtaining the best confocal images for monitoring intracellular ATP, the amount of the nanoprobe for HeLa cells were optimized to be 15 μg (Fig. 7) by confocal microscopy.
 | ||
| Fig. 5 Confocal microscopy bright field (1), fluorescence (2), and the overlay (3) images of HeLa cells after incubation with nanoprobe with Pb2+ (A), nanoprobe without Pb2+ (B), FAM-aptamer/GO (C), and Mis-probes (D) after 200 min. | ||
 | ||
| Fig. 6 Time course of confocal image (bright field (1), fluorescence (2), and the overlay (3)) of HeLa cells incubated with 150 μL GO probe (0.1 mg mL−1) (A) 0; (B) 40 min; (C) 80 min; (D) 120 min; (E) 160 min; (F) 200 min; (G) 240 min; (H) 300 min. | ||
 | ||
| Fig. 7 Imaging of confocal microscopy bright field (1), fluorescence (2), and the overlay (3) of HeLa cells after incubation with 50 (A), 80 (B), 100 (C), 150 (D) and 200 (E) μL GO nanoprobe (0.1 mg mL−1) for 200 min. | ||
To further confirm the fluorescence signal resulting from the endogenously produced ATP of the HeLa cells, we used the proposed method to measure intracellular ATP changes upon drug stimulation. Before incubating with the GO probe, the cells were treated with doses of oligomycin and 2-deoxy-D-glucose (a drug combination that is known to reduce ATP or with Ca2+ (a commonly used ATP inducer)) for 30 min. An untreated group served as a control. Fig. 8 showed that the fluorescent intensity was lower in the Hela cells treated with doses of oligomycin and 2-deoxy-D-glucose (A), while higher in the cells pre-incubated with Ca2+ (C), relative to that in the untreated cells (B). These results indicated that the sensor is capable of detecting changes in intracellular ATP levels. Moreover, ATP is the primary energy molecule in all living cells, to investigate whether this sensor could be used for intracellular sensing of ATP in other cells, MCF-7 and HepG2 cells were used to be incubated with the nanoprobes for 200 min. As shown in Fig. 9, the bright fluorescence signal was observed in two cells treated with nanoprobes, which indicated that the nanoprobes also be used for the intracellular sensing of ATP in other cells. These data clearly demonstrate that the method is a viable and reliable tool for high-contrast fluorescence imaging of small molecule ATP in living cells.
 | ||
| Fig. 8 Confocal microscopy bright field (1), fluorescence (2), and the overlay (3) images of HeLa cells after incubation with 10 μg mL−1 oligomycin (A), blank (B), and 5 mM Ca2+ (C) followed by incubation with nanoprobe and Pb2+. | ||
 | ||
| Fig. 9 Confocal image of MCF-7 (A) and HepG2 (B) cells incubated with nanoprobes and Pb2+ after 200 min (bright field (1), fluorescence (2), and the overlay (3)). | ||
4. Conclusions
In summary, we have developed a robust nanoprobe, named GO loaded split-DNAzyme-probe, for amplified detection of ATP and demonstrated its application for imaging of ATP in living cells. Taking advantage of proximity binding-induced DNA strand displacement and the Pb2+-dependent DNAzyme recycling amplification, the in vitro assays show that this strategy affords efficient signal amplification for fluorescence detection of ATP and exhibits high selectivity, good biocompatibility. The vivo studies suggested that the nanoprobe was efficiently delivered into living cells and worked for specific, high-contrast imaging of target ATP. More importantly, the strategy reported herein could provide a new platform to convert different aptazymes into intracellular sensors for amplified detecting and imaging of other specific molecules in living cells by selecting appropriate affinity ligands.Conflicts of interest
No.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21565002), Natural Science Foundation of Jiangsu Province (BK20171174), China Postdoctoral Science Foundation (2017M610355), Jiangsu Postdoctoral Science Foundation (1701045C), and Qing Lan Project.References
- Y. N. Jiang, N. N. Liu, W. Guo, F. Xia and L. Jiang, J. Am. Chem. Soc., 2012, 134, 15395–15401 CrossRef PubMed.
- J. J. Deng, K. Wang, M. Wang, P. Yu and L. Q. Mao, J. Am. Chem. Soc., 2017, 139, 5877–5882 CrossRef PubMed.
- Y. S. Guo, J. Liu, G. X. Yang, X. F. Sun, H. Y. Chen and J. J. Xu, Chem. Commun., 2015, 51, 862–864 RSC.
- L. Wang, L. Fang and S. F. Liu, Analyst, 2015, 140, 5877–5880 RSC.
- Y. T. Liu, J. P. Lei, Y. Huang and H. X. Ju, Anal. Chem., 2014, 86, 8735–8741 CrossRef PubMed.
- H. Xie, Y. Q. Chai, Y. L. Yuan and R. Yuan, Chem. Commun., 2017, 53, 8368–8371 RSC.
- L. L. Du, Y. Zhang, Y. Du, D. Z. Yang, F. L. Gao and D. Q. Tang, RSC Adv., 2015, 5, 100960–100967 RSC.
- T. Perez-ruiz, C. Martine-Lozano, V. Tomas and J. Martin, J. Anal. Bioanal. Chem., 2003, 377, 189–194 CrossRef PubMed.
- D. Compagnone and G. G. Guilbault, Anal. Chim. Acta, 1997, 340, 109–113 CrossRef.
- W. J. Chen, Y. P. Hu, J. S. Li, Y. H. Li, J. H. Bai, J. Zheng and R. H. Yang, Anal. Methods, 2014, 6, 33219–33222 Search PubMed.
- H. Xie, Y. Q. Chai, Y. L. Yuan and R. Yuan, Chem. Commun., 2017, 53, 8368–8371 RSC.
- X. Li, Y. Peng, Y. Q. Chai, R. Yuan and Y. Xiang, Chem. Commun., 2016, 52, 3673–3676 RSC.
- S. F. Liu, Y. Lin, T. Liu, C. B. Cheng, W. J. Wei, L. Wang and F. Li, Biosens. Bioelectron., 2014, 56, 12–18 CrossRef PubMed.
- P. Wang, Z. Y. Cheng, Q. Chen, L. L. Qu, X. M. Miao and Q. M. Feng, Sens. Actuators, B, 2018, 256, 931–937 CrossRef.
- X. H. Tan, T. Chen, X. L. Xiong, Y. Mao, G. Z. Zhu, E. Yasun, C. M. Li, Z. Zhu and W. H. Tan, Anal. Chem., 2012, 84, 8622–8627 CrossRef PubMed.
- X. L. Zuo, S. P. Song, J. Zhang, D. Pan, L. H. Wang and C. H. Fan, J. Am. Chem. Soc., 2007, 129, 11042–11043 CrossRef PubMed.
- L. Jia, L. Ding, J. W. Tian, L. Bao, Y. P. Hu, H. X. Ju and J. S. Yu, Nanoscale, 2015, 7, 115953–115961 Search PubMed.
- X. H. Tan, T. Chen, X. L. Xiong, Y. Mao, G. Z. Zhu, E. Yasun, C. M. Li, Z. Zhu and W. H. Tan, Anal. Chem., 2012, 84, 3568–3573 CrossRef PubMed.
- Y. Wang, Z. H. Li, D. H. Hu, C. T. Lin, J. H. Li and Y. H. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef PubMed.
- W. B. Qiang, H. T. Hu, L. Sun, H. Li and D. K. Xu, Anal. Chem., 2015, 87, 12190–12196 CrossRef PubMed.
- X. H. Zhao, R. M. Kong, X. B. Zhang, H. M. Meng, W. N. Liu, W. H. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2011, 83, 5062–5066 CrossRef PubMed.
- Y. J. Yang, J. Huang, X. H. Yang, X. X. He, K. Quan, N. L. Xie, M. Ou and K. M. Wang, Anal. Chem., 2017, 89, 5850–5856 CrossRef PubMed.
- L. B. Wang, H. Y. Zhou, B. Liu, C. Zhao, J. L. Fan, W. Wang and C. Y. Tong, Anal. Chem., 2017, 89, 111014–111020 Search PubMed.
- X. Li, J. Q. Xie, B. Y. Jiang, R. Yuan and Y. Xiang, ACS Appl. Mater. Interfaces, 2017, 9, 5733–5738 CrossRef PubMed.
- Y. J. Yang, J. Huang, X. H. Yang, K. Quan, H. Wang, L. Ying, N. L. Xie, M. Ou and K. M. Wang, Anal. Chem., 2016, 88, 5981–5987 CrossRef PubMed.
- L. Li, J. Feng, Y. Y. Fan and B. Tang, Anal. Chem., 2015, 87, 44829–44835 Search PubMed.
- X. H. Zhao, L. Gong, X. B. Zhang, B. Yang, T. Fu, R. Hu, W. H. Tan and R. Q. Yu, Anal. Chem., 2013, 85, 3614–3620 CrossRef PubMed.
- F. Li, Y. W. Lin and X. C. Le, Anal. Chem., 2013, 85, 10835–10841 CrossRef PubMed.
- J. M. Yang, B. T. Dou, R. Yuan and Y. Xiang, Anal. Chem., 2016, 88, 8218–8223 CrossRef PubMed.
- F. Li, Y. N. Tang, S. M. Traynor, X. F. Li and X. C. Le, Anal. Chem., 2016, 88, 8152–8157 CrossRef PubMed.
- F. Li, H. Q. Zhang, C. Lai, X. F. Li and X. C. Le, Angew. Chem., 2012, 124, 9451–9454 CrossRef.
- F. Li, H. Q. Zhang, Z. X. Wang, X. K. Li, X. F. Li and X. C. Le, J. Am. Chem. Soc., 2013, 135, 2443–2446 CrossRef PubMed.
- L. Zhang, K. X. Zhang, G. C. Liu, M. J. Liu, Y. Liu and J. H. Li, Anal. Chem., 2015, 87, 5677–5682 CrossRef PubMed.
- H. Imamura, K. P. Huynh Nhat, H. Togawa, K. Saito, R. Iino, Y. Kato-Yamada, T. Nagai and H. Noji, Proc. Natl. Acad. Sci., 2009, 106, 15651–15656 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05193c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |