DOI:
10.1039/C8RA04814B
(Paper)
RSC Adv., 2018,
8, 30387-30395
The influence of composition on the functionality of hybrid CuO–ZnO–Al2O3/HZSM-5 for the synthesis of DME from CO2 hydrogenation
Received
6th June 2018
, Accepted 18th August 2018
First published on 28th August 2018
Abstract
A series of CuO–ZnO–Al2O3/HZSM-5 hybrid catalysts with different Cu/Zn ratios and disparate Al2O3 doping were prepared and characterized by XRD, BET, H2-TPR, NH3-TPD and XPS techniques. The optimal Cu/Zn ratio is 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, and the introduction of a suitable amount of Al2O3 to form hybrid catalysts increased the BET specific area and micropore volume, facilitated the CuO dispersion, decreased the CuO crystallite size, increased the interaction between CuO and ZnO, enhanced the number of weak acid sites, altered the copper chemical state and improved the catalytic performance consequently. The highest CO2 conversion, DME selectivity and DME yield of 27.3%, 67.1% and 18.3%, respectively, were observed over the CZA7H catalyst. The suitable temperature of 260 °C and the appropriate space velocity of 1500 h−1 for one-step synthesis of dimethyl ether (DME) from carbon dioxide (CO2) hydrogenation were also investigated. The 50 h stability of the CZA7H catalyst was also tested.
:3, and the introduction of a suitable amount of Al2O3 to form hybrid catalysts increased the BET specific area and micropore volume, facilitated the CuO dispersion, decreased the CuO crystallite size, increased the interaction between CuO and ZnO, enhanced the number of weak acid sites, altered the copper chemical state and improved the catalytic performance consequently. The highest CO2 conversion, DME selectivity and DME yield of 27.3%, 67.1% and 18.3%, respectively, were observed over the CZA7H catalyst. The suitable temperature of 260 °C and the appropriate space velocity of 1500 h−1 for one-step synthesis of dimethyl ether (DME) from carbon dioxide (CO2) hydrogenation were also investigated. The 50 h stability of the CZA7H catalyst was also tested.
1. Introduction
CO2 emission is increasing at an unparalleled rate due to fossil fuel combustion in recent years.1 As a consequence, excessive CO2 release leads to a series of problems such as global warming and environmental pollution.2,3 Therefore, CO2 recycling and utilization are very imperative.4,5 CO2 hydrogenation to dimethyl ether (DME) is an excellent option.6
DME is attracting more and more interest as a green fuel because it produces less COx, NOx, and SOx particulates when combusted.7 Therefore, DME is considered as a potential environmentally friendly alternative option to petrol diesel for diesel engines.8 Meanwhile, DME, as a vital chemical intermediate, can also be used to prepare diversified valuable chemical products especially esters and lower olefins.9 Furthermore, it can be utilized in several fields, for example, in fuel additives of diesel, home heating and cooking gas, and pro-environmental refrigerants.10,11
The hydrogenation of CO2 to DME over a hybrid catalyst for DME synthesis is principally composed of three independent reactions, methanol synthesis from CO2 hydrogenation (CO2 + 3H2 ↔ CH3OH + H2O reaction 1), methanol dehydration to form DME (2CH3OH ↔ CH3OCH3 + H2O reaction 2), and a reverse water gas shift reaction (RWGS) (CO2 + H2 ↔ CO + H2O reaction 3). DME can be produced via a two-step process including reaction 1 on a copper based catalyst and subsequent reaction 2 over a solid acid catalyst. Furthermore, one-step process (performing the two steps simultaneously over a hybrid catalyst) has a better performance as a thermodynamically favorable course because the equilibrium limitation of reaction 1 can be readily overcame by reaction 2.12,13
Currently, hybrid catalysts of methanol synthesis component and methanol dehydration component are applied to the one-step direct DME synthesis from CO2.14,15 The methanol dehydration components are generally solid acid catalyst such as γ-Al2O3, but the active sites of the γ-Al2O3 is Lewis acid sites which are favored for water adsorption, resulting in a significant reduction of the amount of acid sites and then a decrease of catalytic activity. Another intriguing solid acid catalyst is WOx/ZrO2, but the catalytic activity of WOx/ZrO2 is lower due to its weaker acid strength present.16 Besides, SAPO-18 has been used as acid function owing to its uniform microporous structure and a high stability, but SAPO-18 possesses a large number of moderate strength acid sites, which cause catalyst deactivation as well as undesired products by coke deposition.17 By contrast, HZSM-5 is the most suitable acidic component due to its more hydrophobic character and advantage of Brønsted acidity, which significantly reduces the impact of water on HZSM-5.18–23 Furthermore, HZSM-5 possesses weak acid sites which are in favor of methanol dehydration to DME at low temperatures and exhibits high catalytic activity.24 However, the methanol synthesis components of the hybrid catalysts are various, and the Cu–Zn based catalyst is regarded as the most promising binary catalyst among them. Xiao et al. reported the deposition of Zn on the surface of copper can increase surface area and can form a unique site at the Cu–ZnO interface to improve the adsorption of CO2 and catalyst activity.25,26 To further improve the performance of the Cu–Zn bimetallic compound catalyst, adding an amphoteric metal oxide species as additive to Cu–Zn based catalyst is an efficient method. Al2O3 is a prospective co-catalyst of sufficient attraction due to its mechanical and thermal stability, its excellent dispersion feature of copper and zinc. Nevertheless, only a few literatures have reported Al2O3 as a single additive of the Cu–Zn based catalyst for one-step direct DME synthesis from CO2 hydrogenation at present. Lei et al. studied hydrogenation of CO2 to CH3OH over CuO–ZnO–Al2O3 catalysts prepared via a solvent-free route, they found that the combustion processes and physicochemical properties of catalysts depend strongly on the type and amount of fuel.27 Allahyari et al. reported CuO–ZnO–Al2O3/HZSM-5 nanocatalyst for direct synthesis DME as a green fuel from syngas, the catalysts were prepared by ultrasound-assisted co-precipitation method at different irradiation times, and the result revealed the relationship between irradiation time and catalytic performance.28 Bowker et al. studied the mechanism of methanol synthesis on copper/zinc oxide/aluminum catalyst, and found that the energetics of formate hydrogenation/hydrogenolysis on the copper component of the catalyst was unaffected by the intimate mixing of copper and zinc oxide in the catalyst.29 Liu reported preparation of HZSM-5 membrane packed CuO–ZnO–Al2O3 nanoparticles for catalysing carbon dioxide hydrogenation to DME, they found zeolite capsule catalysts exhibited an excellent DME selectivity compared with the conventional hybrid catalyst because the further dehydration of DME was restrained.10 Unfortunately, those above mentioned literature of CO2 hydrogenation over CuO–ZnO–Al2O3 catalyst were mainly related to the influence of catalyst preparation methods to the catalyst activity as well as the mechanism of CO2 hydrogenation. On the contrary, the effect of the amount of the additive Al2O3 of CuO–ZnO–Al2O3 catalyst on catalyst activity was rarely investigated. Meanwhile, the Cu/Zn ratio of the CuO–ZnO–Al2O3 catalyst currently still derived from methanol synthesis from CO2 hydrogenation or from syngas hydrogenation.30
Here, we prepared a series of CuO–ZnO–Al2O3/HZSM-5 hybrid catalysts of different Al2O3 contents by the oxalate co-precipitation method, on the basis of optimizing Cu/Zn ratio of catalyst for DME synthesis from CO2 hydrogenation. The effect of Al2O3 content on catalytic properties especially CO2 conversion and DME selectivity of the hybrid catalysts was investigated. Moreover, the influence of reaction temperature and velocity on the catalyst activity of the hybrid catalyst with optimum Al2O3 content was also investigated.
2. Experimental
2.1 Preparation of CuO–ZnO/HZSM-5 catalysts
A series of CuO–ZnO/HZSM-5 catalysts of diverse Cu/Zn molar ratio were prepared, aiming at confirming optimum composition. Cu(NO3)2·3H2O, Zn(NO3)2·6H2O (Cu/Zn molar ratio ranging from 2:8 to 8:2) were dissolved in ethanol to gain the metal nitrates solution and coprecipitated by oxalate under vigorous stirring at 60 °C in a solution containing the HZSM-5 finely dispersed, with a final CuO–ZnO:HZSM-5 weight ratio of 2:1, for convenience of comparison with previous work.7,31,32 The HZSM-5 with a SiO2/Al2O3 ratio of 38 was purchased from Catalyst Plant of Nankai University (China). The slurry was heated to 80 °C and was aged for 2 h under stirring. Subsequently, the slurry was evaporated at the same temperature. Then the obtained product was dried in muffler furnace at 120 °C for 12 h and calcined in air at 400 °C for 4 h. The final product was hybrid catalyst named as CxZ10−xH (the x express the molar ratio of CuO:CuO–ZnO) and was pressed, crushed and griddled to get the granules (20–40 mesh).
2.2 Preparation of CuO–ZnO–Al2O3/HZSM-5 hybrid catalysts
Thereafter, a series of CuO–ZnO–Al2O3/HZSM-5 hybrid catalysts of different Al2O3 contents were prepared. Cu(NO3)2·3H2O, Zn(NO3)2·6H2O were weighed according to the optimal Cu/Zn mole ratio which was determined in the preliminary experiments. The mixture was dissolved in ethanol followed by the addition of Al(NO3)2·9H2O. The amount of the Al(NO3)2·9H2O used depended on the required Al2O3 contents of 0, 1.0, 3.0, 5.0, 7.0, 10.0 wt% in CuO–ZnO–Al2O3. Then, the solution of precursors was added to a vigorously stirred ethanol solution containing the oxalic acid and the powdered HZSM-5 in a constant and slow manner at 60 °C. The weight ratio of the final oxide of CuO–ZnO–Al2O3 to HZSM-5 is 2:1. The slurry was dealt with the same procedure as the CxZ10−xH and the final product was abbreviated as CZAyH, where the y represents theoretical Al2O3:CuO–ZnO–Al2O3 wt%.
2.3 Catalyst testing
The hybrid catalyst activity was measured in a continuous-flow fixed-bed reactor made of stainless steel (10 mm inner diameter). 1 mL of the hybrid catalyst was diluted with 0.5 mL of the quartz sand (both in 20–40 mesh). Prior to the test, the catalyst was reduced in situ in a stream of 10% H2/N2 at 300 °C for 3 h under atmospheric pressure. After the reduction, the reactor was cooled to room temperature under flowing N2 and reactant gas flow (H2/CO2/N2 = 3/9/1, molar) was introduced, subsequently raising the pressure to 3.0 MPa and the reaction temperature was raised to 260 °C. Analysis of the products were performed applied a gas chromatograph (SP2100A) equipped with both a TCD (for N2, CO and CO2, GDX-101 connected with Porapak T column) and a FID (for CH4, CH3OH and DME, Porapak Q column). The CO2 conversion as well as the selectivity toward CO, CH3OH and DME are calculated by internal standard method.32,33 XCO2, SPi and YDME represent the conversion of CO2, the selectivity of the product (DME, MeOH, CO) and the yield of DME, respectively. Each experimental data was corresponds to an average of three independent measurements, with error of ±2%.| |
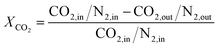 | (1) |
| |
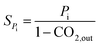 | (2) |
where Pi stands for the concentration of a specific i product (DME, MeOH, CO).
2.4 Catalyst characterization
X-ray powder diffraction (XRD) patterns of all samples were performed on a Rigaku D/max 2500 pc X-ray diffractometer with Cu-Kα radiation (λ = 1.54156 Å) at a scan rate of 4° min−1 at 50 kV and 250 mA.
The BET surface area of the samples were conducted by N2 adsorption at −196 °C using a Quantachrome Autosorb 1-C. Before the absorption–desorption measurements, samples were degassed under vacuum at 300 °C for 3 h. The specific BET (SBET) was estimated from the linear part of the Brunauer–Emmett–Teller (BET) plot.
The X-ray photoelectron spectroscopy (XPS) and X-ray-induced Auger electron spectroscopy were recorded on an ESCALA 250 Xi spectrometer, using a standard Al-Kα X-ray source (1486.6 eV). The binding energy (BE) values were referenced to the adventitious C 1s peak (284.6 eV). Quantification of the surface atomic concentrations was carried out using the sensitivity factors supplied for the XPS instrument.
H2-TPR of catalysts were performed on chemisorptions (ChemBET 3000). Before reduction, 0.02 g of sample was preheated with flowing He at 400 °C for 60 min, then cooled down room temperature. Subsequently, the temperature was raised in 10% H2/Ar (50 mL min−1) at a ramp rate of 10 °C min to 400 °C. H2 consumption was detected by TCD.
NH3-TPD of the samples were measured on the same apparatus for H2-TPR measurements. 0.1 g sample was heated to 400 °C and maintained for 30 min, then the temperature was cooled to 100 °C. After that, a 6 vol% NH3/Ar mixture stream was introduced to the sample for 60 min. Then, the examined sample was flushed with helium stream (30 mL min−1) for 60 min to remove the weak adsorbed NH3. The temperature was raised from 100 °C to 800 °C at a rate of 10 °C min.
3. Results and discussion
3.1 The effect of the Cu/Zn molar ratio on the catalytic performance of CxZ10−xH catalysts
In order to investigate the optimal Cu/Zn molar ratio of the hybrid catalysts, a series of CxZ10−xH catalysts were prepared. The catalytic performances of catalysts with different Cu/Zn molar ratio were summarized in Table 1. The CO2 conversion, DME selectivity and the yield of DME increased with the Cu/Zn increased from 2:8 to 7:3, and decreased if continue raising the Cu/Zn to 8:2. The C7Z3H exhibited the maximum CO2 conversion, DME selectivity and DME yield values of 19.2%, 47.8% and 9.2%, respectively. The result revealed the Cu/Zn molar ratio can affect the catalytic performance and the optimal Cu/Zn molar ratio of the hybrid catalysts was 7:3.
Table 1 Catalytic performance of the CxZ10−xH Hybrid catalystsa
| Catalyst |
Conversion of CO2/% |
Selectivity/% |
Yield/% DME |
| DME |
CH3OH |
CO |
Hydrocarbons |
| Reaction conditions: T = 260 °C; P = 3.0 MPa; CO2:H2:N2 = 3:9:1; GHSV = 1500 h−1. |
| C2Z8H |
16.06 |
25.41 |
5.34 |
67.35 |
1.90 |
4.08 |
| C3Z7H |
16.44 |
27.04 |
8.03 |
63.14 |
1.79 |
4.45 |
| C4Z6H |
17.71 |
36.71 |
5.23 |
56.16 |
1.90 |
6.50 |
| C5Z5H |
17.46 |
37.73 |
5.22 |
55.22 |
1.83 |
6.59 |
| C6Z4H |
18.06 |
38.42 |
6.01 |
53.70 |
1.87 |
6.94 |
| C7Z3H |
19.20 |
47.80 |
1.70 |
47.80 |
2.70 |
9.20 |
| C8Z2H |
18.16 |
39.35 |
5.15 |
53.49 |
2.01 |
7.15 |
3.2 Catalytic performance of the CZAyH catalysts
The catalytic performances of the CZAyH catalysts with different Al2O3 contents were listed in Table 2. DME was the major product and the by products included CO and methanol under the reaction temperature of 260 °C, pressure of 3.0 MPa and GHSV of 1500 h−1. Moreover, several hydrocarbons such as CH4 were also detected. The catalytic performances of the Al2O3-modified hybrid catalysts were enhanced enormously compared with the Al2O3-free hybrid catalyst. With the Al2O3 contents increase in the catalysts (range from CZA0H to CZA7H), the CO2 conversion, DME selectivity and the DME yield increased significantly. Further increasing the contents of Al2O3 (CZA10H) decreased the catalysts activity. The CZA7H catalyst exhibited the highest catalyst performance and a maximum CO2 conversion of 27.3% with a DME selectivity of 67.1% and a yield of DME of 18.3%, as shown in Table 2. The DME yield and the DME selectivity of CZA7H catalyst were appreciable in comparison of nearly 13.5% and 63.4%, respectively, obtained over CuO–ZnO–ZrO2/HZSM-5 catalyst reported by Li et al.34 Meanwhile, the DME selectivity of 67.1% for CZA7H catalyst was significantly higher than 49.2% observed over CuO–ZnO–Al2O3/HZSM-5 catalyst studied by Zhang et al.31 These results indicated suitable Al2O3 additive can improve the catalyst performance significantly.
Table 2 Catalytic performance of the CZAyH hybrid catalystsa
| Catalyst |
Conversion of CO2/% |
Selectivity/% |
Yield/% DME |
| DME |
CH3OH |
CO |
Hydrocarbons |
| Reaction conditions: T = 260 °C; P = 3.0 MPa; CO2:H2:N2 = 3:9:1; GHSV = 1500 h−1. |
| CZA0H |
19.2 |
47.8 |
1.7 |
47.8 |
2.7 |
9.2 |
| CZA1H |
23.6 |
60.6 |
2.0 |
34.6 |
2.8 |
14.3 |
| CZA3H |
22.9 |
61.6 |
2.6 |
33.1 |
2.7 |
14.1 |
| CZA5H |
23.0 |
64.3 |
5.5 |
26.5 |
3.7 |
14.8 |
| CZA7H |
27.3 |
67.1 |
3.9 |
25.5 |
3.5 |
18.3 |
| CZA10H |
22.6 |
60.9 |
3.6 |
32.7 |
2.8 |
13.8 |
Fig. 1 shows the effect of reaction temperature on the catalytic performance of CZA7H catalyst. It can be found that volcanic shape change trends of the CO2 conversion, DME selectivity and the yield of DME versus the variation of the temperature. With the temperature rising from 220 °C to 260 °C, the conversion of CO2, the selectivity of the DME and the DME yield increased obviously 12.7%, 2.7% and 8.9%, respectively, and then reduced sharply when the reaction temperature reached 280 °C. Comparing the catalytic performance data at several reaction temperatures, the optimal catalytic performance for the CZA7H catalyst were obtained at 260 °C. These results can be explained by the hydrogenation of CO2 for synthesis of DME was a reversible exothermic reaction, the reverse reaction favored with an excessive temperature. Meanwhile, the main side reaction RWGS was an endothermic reaction thus the CO selectivity increased and the selectivity and yield of DME decreased with the reaction temperature higher than 260 °C.5 In addition, the CZA7H catalyst activity may also decrease due to the Cu sintered and crystallized gradually with the reaction temperature increased.18
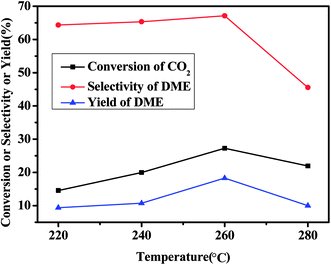 |
| | Fig. 1 Effect of temperature on catalytic CO2 hydrogenation to DME for CZA7H hybrid catalysts. | |
Fig. 2 illustrates the influence of the space velocity of on the catalytic performance of the CZA7H catalyst. It can be found that the CO2 conversion, the DME selectivity and the DME yield decreased continuously from 27.3% to 20.5%, 67.1% to 52.3% and 18.3% to 10.7%, respectively, as the space velocity increased from 1500 to 3000 h−1. With the space velocity increasing, the contact time between reactants and the catalyst becomes short, consequently both the CO2 hydrogenation to methanol and methanol dehydration to DME reactions could not proceed sufficiently.18 Therefore, the conversion of CO2 and the selectivity of the DME reduced distinctly indicating the efficient effect the space velocity on the catalytic performance.
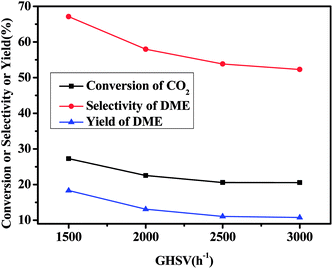 |
| | Fig. 2 Effect of space velocity on catalytic CO2 hydrogenation to DME for CZA7H hybrid catalysts. | |
3.3 Stability of the CZA7H catalyst
The stability of CZA7H catalyst was studied for the reaction of DME synthesis via CO2 hydrogenation at 260 °C and 3.0 MPa with GHSV = 1500 h−1. The stability results were recorded every 2 h, as shown in Fig. 3. It can be found that the CO2 conversion of and the DME selectivity decrease slightly from 27.3% and 67.1% to 25.3% and 62.1% during the continuous 50 h reaction process, respectively. The result indicated that the CZA7H catalyst possessed better catalytic performance and stability.
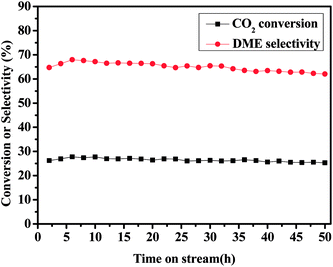 |
| | Fig. 3 Effects of the conversion of CO2 and the selectivity of DME with CZA7H hybrid catalysts with time on stream. | |
3.4 The structure of the catalysts
Fig. 4 illustrates the XRD patterns of a series of CZAyH catalysts with different Al2O3 contents before H2 reduction. For all catalysts, the diffraction peaks at 2θ = 35.5°, 38.7°, 48.7°, 53.4°, 58.2°, 61.5°, 66.2°, 72.3° and 74.9° can be ascribed to CuO phase (JCPDS no. 48-1548), the peaks at 2θ = 31.7°, 34.3°, 36.1°, 47.4°, 56.5°, 62.8° and 67.8° can be attributed to ZnO phase (JCPDS no. 65-3411), and the peaks appear in the 2θ range of 21–25° can be matched to HZSM-5 (JCPDS no. 44-0003). The peaks corresponding to Al2O3 phase cannot be observed, indicating that the presence of Al2O3 were amorphous phase or highly dispersed in the catalyst body phase. Compared with the peaks of catalyst CZA0H, the peaks of catalysts with Al2O3 become weaker, indicating the CuO–ZnO oxides possess less intensive crystallinity. The diffraction peaks of CuO and ZnO weakened and broadened gradually with the increasing of Al2O3 contents, even some diffraction peaks of CuO and ZnO phase disappeared, suggesting the decrease of grain size. The grain sizes of CuO (dCuO) and ZnO (dZnO) for CZAxH catalysts calculated by Scherrer's equation are listed in Table 3. These results indicated obviously that the additive of Al2O3 can improve the CuO dispersion and can decrease the grain size of CuO. For all catalysts, the grain size of ZnO phase is greater than that of CuO phase, indicating ZnO phase closely relates to CuO phase. Shimokawabe et al. considered the grain size of ZnO is larger than CuO due to CuO can be well dissolved on the ZnO particles.35 As a consequence, the active interface increased with the improving the dispersion of CuO.35,36 Fig. 5 shows the XRD patterns of the reduced CZA0H and CZA7H catalysts. For these two catalysts, the diffraction peaks at 2θ = 43.3°, 50.4° and 74.1° can be ascribed to metallic copper phase (JCPDS no. 04-0836). No diffraction peaks of CuO can be detected, indicating all CuO in the catalysts reduced to Cu. The intensity of diffraction peaks of Cu for reduced CZA7H catalyst decrease significantly, compared to the corresponding peaks for CZA0H catalyst. Furthermore, the grain size of Cu decreased significantly for CZA7H catalyst. As shown in Table 3, and the grain sizes of Cu (dCu) which calculated by Scherrer's equation for reduced CZA0H and CZA7H are 21.4 nm and 9.7 nm, respectively. These results suggest that the addition of Al2O3 can effectively inhibit the Cu crystals growth.
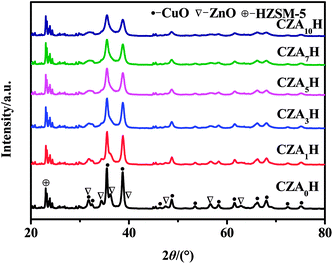 |
| | Fig. 4 XRD patterns of the CZAyH hybrid catalysts. | |
Table 3 Physicochemical properties of the CZAyH hybrid catalysts
| Catalyst |
SBET/(m2 g−1) |
V1 (Micropore volume)/(cm3 g−1) |
D (Average pore diameter)/nm |
dCuOa/nm |
dCua/nm |
dZnOa/nm |
| Determined by Scherrer's equation. |
| CZA0H |
131.7 |
0.032 |
8.0 |
20.4 |
21.4 |
29.1 |
| CZA1H |
132.1 |
0.024 |
8.7 |
15.4 |
— |
17.6 |
| CZA3H |
133.4 |
0.028 |
7.7 |
14.4 |
— |
16.5 |
| CZA5H |
134.1 |
0.031 |
7.6 |
11.4 |
— |
14.1 |
| CZA7H |
161.2 |
0.037 |
6.8 |
10.9 |
9.7 |
13.4 |
| CZA10H |
143.5 |
0.030 |
7.6 |
10.6 |
— |
12.3 |
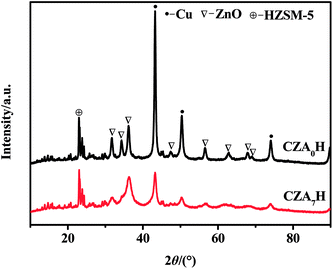 |
| | Fig. 5 XRD patterns of the reduced CZA0H and CZA7H hybrid catalysts. | |
The BET Surface areas (SBET) and the calculated average pore diameter of the catalysts are also listed in Table 3. The SBET of the catalysts approximately increases from 131.7 m2 g−1 to 161.2 m2 g−1 with the increasing of Al2O3 contents (from CZA0H to CZA7H), then sharply decreases to 143.5 m2 g−1 when the Al2O3 contents continuing increasing (CZA10H). The variation trend of the micropore volumes is the same as the SBET while the trend of average pore diameter of the catalysts is contrary to the SBET generally. These results illustrate that the pore structure of the hybrid catalysts can be changed by the introduction of Al2O3. Meanwhile, a maximum SBET of 161.2 m2 g−1 and a minimum average pore diameter of 6.8 nm are detected for the CZA7H catalyst which possesses the smallest grain sizes of Cu (dCu), as determined from XRD. The result revealed that why the catalyst of CZA7H exhibited the maximum of the CO2 conversion, the DME selectivity and the DME yield simultaneously.
3.5 The reducibility of the catalysts
TPR measurements were carried out in order to evaluate the reduction behavior of all the catalysts. Fig. 6 shows the H2-TPR profiles of the CZAxH catalysts with different Al2O3 contents. It can be found only a single reduction peak with any satellite peaks for all the catalysts. The result illustrate the opinion the copper oxides on the CZAxH catalysts are easy to be reduced.37 Since ZnO and Al2O3 cannot be reduced under the experimental condition, the single high temperature reduction peak can be ascribed to the reduction of CuO species with strongly interaction with ZnO and Al2O3.38 With Al2O3 content increasing, the reduction peak maximum firstly shifted towards higher temperature from CZA0H to CZA7H catalysts, and then shifted towards lower temperature for CZA10H, as shown in Fig. 6. The CZA7H catalyst possessed the highest reduction temperature at 373 °C, which indicated the strongest interaction between the heterogeneous CuO particles with metal oxides. The CZA7H catalyst exhibited the best catalytic performance which might be explained that strongly interaction between CuO and oxides was beneficial to CO2 hydrogenation to DME.39 Meanwhile, the CZA7H catalyst possessed the largest area among the reducing peaks, which revealed that the hydrogen consumption of the catalyst was enhanced significantly, as shown in Table 4. The result implied the largest amount of easily reducible well-dispersed copper oxide existed, which might also be ascribed to the improving dispersion of the CuO particles.40 This may be one reason why when increase Al2O3 contents in the catalysts from CZA0H to CZA7H, the conversion and selectivity significantly increased and a further increase in Al2O3 contents adversely influenced conversion and selectivity simultaneously.
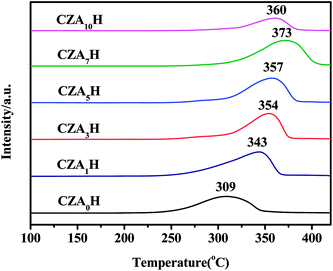 |
| | Fig. 6 H2-TPR profiles of the CZAyH hybrid catalysts. | |
Table 4 The reduction peak areas of the CZAyH hybrid catalysts
| Catalyst |
CZA0H |
CZA1H |
CZA3H |
CZA5H |
CZA7H |
CZA10H |
| Peak area |
25065 |
30247 |
26320 |
32048 |
38894 |
15425 |
3.6 Surface acidity of the CuO–ZnO–Al2O3/HZSM-5 catalysts
Fig. 7 shows NH3-TPD profiles of the CZAxH catalysts with different Al2O3 contents. It can be found three NH3 desorption peaks which located in the range of 50–200 °C (denoted as peak α), 200–350 °C (denoted as peak β), and 350–600 °C (denoted as peak γ), respectively. The α, β and γ peaks were attributed to weak, medium and strong acid sites, respectively, indicating the presence of three strength acid sites on the hybrid catalysts.33 The intensity of the low-temperature peak α of weak acid sites firstly increased with increasing Al2O3 contents (from CZA0H to CZA7H) and then declined with further increasing Al2O3 additive, which was agreement with the trend of SBET and H2-TPR. The CZA7H catalyst of the highest peak intensity possessed the largest amount of weak acid sites which were beneficial for methanol dehydration to DME at low temperatures.24 This might be ascribed to the fact Al2O3 owned weak acidic characters. In addition, the intensity and the position of the peaks β exhibited unconspicuous changes with doping different Al2O3 contents, implying that the strength and amount of medium acid sites cannot be influenced by Al2O3 additive substantially. The position of peaks γ shifted towards higher temperature gradually, indicating the intensity of strong acid sites increased with increasing Al2O3 doping, leading to the increase of hydrocarbons in the products. This might be attributed to the fact that the HZSM-5 with possessed strong acid sites could be modified by more oxalic acid with the addition of the more Al2O3 contents, during the preparation of hybrid catalysts by the co-precipitation method.11,34 From the above, the amount of weak acid sites enhanced with optimal Al2O3 modified, which was in favor of obtaining the highest catalytic performance for one-step DME synthesis reaction.24
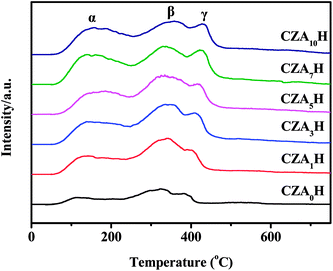 |
| | Fig. 7 NH3-TPD profiles of the CZAyH hybrid catalysts. | |
3.7 XPS analysis
The XPS spectra of reduced catalysts are shown in Fig. 8. Both of the spectra contained two peaks at about 1022 and 1045 eV (Fig. 8a), which were assigned to Zn 2p3/2 and Zn 2p1/2 peaks of ZnO, respectively, with a spin energy separation of 23 eV.41 This illustrates that the Zn atoms are in a completely oxidized state and were not reduced.42 It can be observed a single peak centered at near 74.2 eV in Fig. 8b, which was attributed to Al 2p3/2 of Al2O3, therefore, the form of Al was Al2O3. Fig. 8c showed the XPS spectra of the Cu 2p for the reduced CZA0H and CZA7H catalysts. Two reduced catalysts displayed Cu 2p3/2 and Cu 2p1/2 main characteristics peaks with binding energy (BE) values of approximately 932.6 eV and 952.6 eV, respectively, with a Cu 2p core level split spin–orbit components (ΔCu 2p) of 20.0 eV. The shake-up satellite peaks between 940 eV and 945 eV were not detected, which illustrates the absence of Cu2+ species in both CZA0H and CZA7H catalysts can be reduced to Cu+ and/or Cu0 species completely.43,44 Meanwhile, the binding energy (BE) of Cu 2p3/2 and Cu 2p1/2 for reduced CZA7H can be observed slightly shifted towards lower BE values, which revealed an enhancement on the interaction between Cu and ZnO with the increasing Al2O3 contents.45 The result indicated an influence on Cu chemical combination state due to the increase of the outer-shell electron density of Cu as a consequence of the introduction of additive Al2O3. Liu et al. thought that the chemical state of Cu was tightly related to the catalysts performance for CO2 hydrogenation to DME.46 This point might be one reason answerable for doping Al2O3 affect the activity of the hybrid catalysts. To distinguish Cu+ and Cu0 species, the kinetic energies (KE) of the Cu LMM X-ray auger electron spectroscopy (XAES) spectrum are measured. As shown in Fig. 8d, a broad and asymmetric peak was divided into two peaks, which centered at near 916.5 and 918.7 eV for both reduced CZA0H and CZA7H catalysts. The two symmetrical peaks are corresponding to the Cu+ and Cu0 species, respectively. The line position in the Cu LMM Auger electron spectrum of the two reduced catalysts illustrated that the Cu0 was the predominant copper species on the CZA0H and CZA7H catalysts surface. Furthermore, the Cu0/Cu+ area ratios of the two reduced catalysts were calculated based on the corresponding Cu LMM peaks. The area ratio of the reduced CZA7H catalyst is a bit higher than that of the CZA0H catalyst, which indicated that Cu0 species but not Cu+ species might in charge of the activity of these catalysts.47 The result might be another reason responsible for the improvement in catalytic performance for the Al2O3-modified hybrid catalysts.48,49
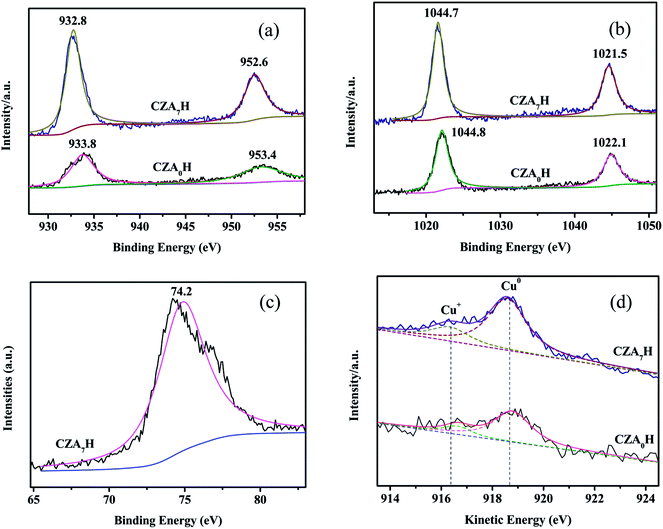 |
| | Fig. 8 Spectra of the reduced CZA0H and CZA7H hybrid catalysts: (a) Cu 2p; (b) Zn 2p; (c) Al 2p; (d) Cu (LMM) Auger. | |
4. Conclusions
The work discussed the effect of the amount of Al2O3 additive of the CuO–ZnO–Al2O3/HZSM-5 catalysts on the physicochemical properties and catalytic performance of the hybrid catalysts based on the optimizing Cu/Zn ratio. The hybrid catalysts for the reaction of CO2 directly hydrogenation to DME improved the CuO dispersion, reduced the CuO crystallite size, decreased the grain size of the Cu, enhanced the BET surface areas, increased the interaction between the CuO and ZnO, enhanced the amount of weak acid sites, changed the copper chemical state on the catalysts surface and enhanced the catalytic activity and stability with the introduction of the Al2O3 contents. In addition, the influence of the reaction temperature and the space velocity on the catalytic performance of the catalysts modified by Al2O3 was also investigated. The optimal Al2O3 contents (Al2O3:CuO–ZnO–Al2O3 wt%) of the CuO–ZnO–Al2O3/HZSM-5 catalysts was 7.0 wt% under the proper Cu/Zn ratio of 7:3. The CZA7H catalyst for CO2 hydrogenation to DME showed the maximum CO2 conversion, DME selectivity and DME yield of 27.3%, 67.1% and 18.3%, respectively, under the reaction conditions of the optimal reaction temperature 260 °C, the appropriate space velocity of GHSV 1500 h−1 and the conventional pressure of 3.0 MPa.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has been supported by National Nature Science Foundation of China (51301114, 21203125, 51602206), Natural Science Foundation of Liaoning Province (201602598, 2015020649), Science research Foundation of Education Department of Liaoning Province (LQ2017011, L2016003).
References
- T. Witoon, T. Permsirivanich, N. Kanjanasoontorn, C. Akkaraphataworn, A. Seubsai, K. Faungnawakij, C. Warakulwit, M. Chareonpanich and J. Limtrakul, Catal. Sci. Technol., 2015, 5, 2347–2357 RSC
 .
. - T. Phongamwong, U. Chantaprasertporn, T. Witoon, T. Numpilai, Y. Poo-arporn, W. Limphirat, W. Donphai, P. Dittanet, M. Chareonpanich and J. Limtraku, Chem. Eng. J., 2017, 316, 692–703 CrossRef .
- T. Witoon, N. Kachaban, W. Donphai, P. Kidkhunthod, K. Faungnawakij, M. Chareonpanich and J. Limtraku, Energy Convers. Manage., 2016, 118, 21–31 CrossRef .
- T. Witoon, S. Bumrungsalee, M. Chareonpanich and J. Limtraku, Energy Convers. Manage., 2015, 103, 886–894 CrossRef .
- K. H. Sun, Z. G. Fan, J. Y. Ye, J. M. Yan, Q. F. Ge, Y. N. Li, W. J. He, W. M. Yang and C. J. Liu, Journal of CO2 Utilization, 2015, 12, 1–6 CrossRef .
- R. J. da Silva, A. F. Pimentel, R. S. Monteiro and C. J. A. Mota, Journal of CO2 Utilization, 2016, 15, 83–88 CrossRef .
- G. Bonura, C. Cannilla, L. Frusteri, A. Mezzapica and F. Frusteri, Catal. Today, 2017, 281, 337–344 CrossRef .
- Y. Suwannapichat, T. Numpilai, N. Chanlek, K. Faungnawakij, M. Chareonpanich, J. Limtraku and T. Witoon, Energy Convers. Manage., 2018, 159, 20–29 CrossRef .
- Y. X. Hua, X. M. Guo, D. S. Mao, G. Z. Lu, G. L. Rempel and F. T. T. Ng, Appl. Catal., A, 2017, 540, 68–74 CrossRef .
- R. Liu, H. F. Tian, A. M. Yang, F. Zha, J. Ding and Y. Chang, Appl. Surf. Sci., 2015, 345, 1–9 CrossRef .
- D. S. Mao, J. C. Xia, B. Zhang and G. Z. Lu, Energy Convers. Manage., 2010, 51, 1134–1139 CrossRef .
- G. X. Jia, Y. S. Tan and Y. Z. Han, Ind. Eng. Chem. Res., 2006, 45, 1152–1159 CrossRef .
- R. Vakili, H. Rahmanifard, P. Maroufi, R. Eslamloueyan and M. R. Rahimpour, Int. J. Hydrogen Energy, 2011, 36, 4354–4365 CrossRef .
- W. G. Gao, H. Wang, Y. H. Wang, W. Guo and M. Y. Jia, J. Rare Earths, 2013, 31, 470–476 CrossRef .
- A. García-Trenco and A. Martínez, Appl. Catal., A, 2015, 493, 40–49 CrossRef .
- T. Witoon, P. Kidkhunthod, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2018, 348, 713–722 CrossRef .
- A. Ateka, I. Sierra, J. Ereña, J. Bilbao and A. T. Aguayo, Fuel Process. Technol., 2016, 152, 34–45 CrossRef .
- X. H. Zhou, T. M. Su, Y. X. Jiang, Z. Z. Qin, H. B. Ji and Z. H. Guo, Chem. Eng. Sci., 2016, 153, 10–20 CrossRef .
- A. García-Trenco and A. Martínez, Appl. Catal., A, 2012, 411–412, 170–179 CrossRef .
- S. Wang, D. S. Mao, X. M. Guo, G. S. Wu and G. Z. Lu, Catal. Commun., 2009, 10, 1367–1370 CrossRef .
- T. Takeguchi, K. Yanagisawa, T. Inui and M. Inoue, Appl. Catal., A, 2000, 192, 201–209 CrossRef .
- V. Vishwanathan, K. W. Jun, J. W. Kim and H. S. Roh, Appl. Catal., A, 2004, 276, 251–255 CrossRef .
- M. Xu, J. H. Lunsford, D. W. Goodman and A. Bhattacharyya, Appl. Catal., A, 1997, 149, 289–301 CrossRef .
- Y. J. Lee, M. H. Jung, J. B. Lee, K. E. Jeong, H. S. Roh, Y. W. Suh and J. W. Bae, Catal. Today, 2013, 228, 175–182 CrossRef .
- J. Xiao, D. S. Mao, X. M. Guo and J. Yu, Appl. Surf. Sci., 2015, 338, 146–153 CrossRef .
- J. Toyira, M. Saitob, I. Yamauchic, S. Luoa, J. Wua, I. Takaharab and M. Takeuchid, Catal. Today, 1998, 45, 245–250 CrossRef .
- H. Lei, Z. Y. Hou and J. W. Xie, Fuel, 2016, 164, 191–198 CrossRef .
- S. Allahyari, M. Haghighi, A. Ebadi and H. Saeedi, J. Power Sources, 2014, 272, 929–939 CrossRef .
- M. Bowker, R. A. Hadden, H. Houghton, J. N. K. Hyland and K. C. Waugh, J. Catal., 1988, 109, 263–273 CrossRef .
- F. Arena, G. Italiano, K. Barbera, S. Bordiga, G. Bonura, L. Spadaro and F. Frusteri, Appl. Catal., A, 2008, 350, 16–23 CrossRef .
- Y. J. Zhang, D. B. Li, S. J. Zhang, K. J. Wang and J. Wu, RSC Adv., 2014, 4, 16391–16396 RSC .
- Y. J. Zhang, Y. Zhang, F. Ding, K. J. Wang, X. L. Wang, B. J. Ren and J. Wu, Chem. Ind. Chem. Eng. Q., 2017, 23, 49–56 CrossRef .
- G. Bonura, M. Cordaro, L. Spadaro, C. Cannilla, F. Arena and F. Frusteri, Appl. Catal., B, 2013, 140–141, 16–24 CrossRef .
- L. Y. Li, D. S. Mao, J. Xiao, L. Li, X. M. Guo and J. Yu, Chem. Eng. Res. Des., 2016, 111, 100–108 CrossRef .
- M. Shimokawabe, H. Asakawa and N. Takezawa, Appl. Catal., 1990, 59, 45–58 CrossRef .
- L. Shi, C. Y. Zeng, T. J. Wang and N. Tubaki, Catal. Sci. Technol., 2012, 2, 2569–2577 RSC .
- P. S. Sai Prasad, J. W. Bae, S. H. Kang, Y. J. Lee and K. W. Jun, Fuel Process. Technol., 2008, 89, 1281–1286 CrossRef .
- X. M. Guo, D. S. Mao, G. Z. Lu, S. Wang and G. S. Wu, J. Catal., 2010, 271, 178–185 CrossRef .
- L. P. Cui, T. Zhou, N. Li, Z. H. Gao, Z. J. Zuo and W. Huang, Catal. Lett., 2017, 147, 2023–2027 CrossRef .
- Y. J. Zhang, D. B. Li, Y. Zhang, Y. Cao, S. J. Zhang, K. J. Wang, F. Ding and J. Wu, Catal. Commun., 2014, 55, 49–52 CrossRef .
- O. Lupan, G. A. Emelchenko, V. V. Ursaki, G. Chai, A. N. Redkin, A. N. Gruzintsev, I. M. Tiginyanu, L. Chow, L. K. Ono, B. Roldan Cuenya, H. Heinrich and E. E. Yakimov, Mater. Res. Bull., 2010, 45, 1026–1032 CrossRef .
- R. Al-Gaashani, S. Radiman, A. R. Daud, N. Tabet and Y. Al-Douri, Ceram. Int., 2013, 39, 2283–2292 CrossRef .
- Y. H. Wang, W. G. Gao, H. Wang, Y. E. Zheng, W. Na and K. Z. Li, RSC Adv., 2017, 7, 8709–8717 RSC .
- Z. Q. Wang, Z. N. Xu, S. Y. Peng, M. J. Zhang, G. Lu, Q. S. Chen, Y. M. Chen and G. C. Guo, ACS Catal., 2015, 5, 4255–4259 CrossRef .
- Y. J. Zhao, B. Shan, Y. Wang, J. H. Zhou, S. P. Wang and X. B. Ma, Ind. Eng. Chem. Res., 2018, 57, 4526–4534 CrossRef .
- R. W. Liu, Z. Z. Qin, H. B. Ji and T. M. Su, Ind. Eng. Chem. Res., 2013, 52, 16648–16655 CrossRef .
- P. Gao, L. S. Zhong, L. N. Zhang, H. Wang, N. Zhao, W. Wei and Y. H. Sun, Catal. Sci. Technol., 2015, 5, 4365–4377 RSC .
- B. Xue, L. Hui, H. Q. Yang, Y. L. Zhao, L. X. Hou and W. Li, Ind. Eng. Chem. Res., 2017, 56, 135–142 CrossRef .
- Y. J. Zhang, N. Zheng, K. J. Wang, S. J. Zhang and J. Wu, J. Nanomater., 2013, 7, 246–247 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Jie Du,
Chunyan Li,
Kangjun Wang*,
Lidong Liu,
Xinrui Yu,
Kai Wang and
Nan Liu
*,
Jie Du,
Chunyan Li,
Kangjun Wang*,
Lidong Liu,
Xinrui Yu,
Kai Wang and
Nan Liu









