
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalyzed decarboxylative C–S cross-coupling of carbonothioate: synthesis of allyl(aryl)sulfide†
Ren-Hua Zheng *a,
Hai-Chang Guoa,
Ting-Ting Chena,
Qing Huangb,
Guo-Bo Huanga and
Hua-Jiang Jiang*a
*a,
Hai-Chang Guoa,
Ting-Ting Chena,
Qing Huangb,
Guo-Bo Huanga and
Hua-Jiang Jiang*a
aSchool of Pharmaceutical and Chemical Engineering, Taizhou University, Taizhou 318000, China. E-mail: zhengrh@tzc.edu.cn; jhj@tzc.edu.cn
bCollege of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310014, China
First published on 12th July 2018
Abstract
A novel ruthenium-catalyzed decarboxylative cross-coupling of carbonothioate is disclosed. This method provides straightforward access to the corresponding allyl(aryl)sulfide derivatives in generally good to excellent yields under mild conditions and features a broad substrate scope, wide group tolerance and in particular, no need to use halocarbon precursors.
1. Introduction
Sulfur-containing molecules such as thioethers are commonly found in chemical biology, organic synthesis, and materials chemistry.1 The development of mild and general methods for C–S bond formation has received significant attention; among them, the transition metals have been applied in this field, and the palladium-catalyzed coupling of thiols with aryl halides is one of the most important methods in the synthesis of thioethers.2 Other metals have also been used for the same purpose.3 Despite the phenomenal growth in diverse synthetic methodologies, the synthesis of C–S bonds is generally limited to the condensation reaction between a metal thiolate and an organic halide.4 Organohalides have potential environmental and human health effects, and their wastes require costly remediation, particularly on an industrial scale.Recently, transition metal-catalyzed decarboxylative allylation reactions are a powerful method for the allylation of a wide variety of nucleophiles under neutral conditions.5 In particular, as one of the most efficient ways to capture the ketone enolates, palladium-catalyzed decarboxylative allylation of β-ketoesters6 has attracted considerable attention since the early discoveries reported by Tsuji7 and Saegusa.8 In contrast with C–H bond activation reactions, decarboxylative cross-coupling reactions through loss of CO2 generally do not need expensive organometallic reagents, while maintaining the advantage of regioselectivity offered by traditional cross-coupling reactions. Compared with a great wealth of studies on the decarboxylative allylation of C–C and C–N bond-forming transformations,9 the corresponding C–S bond-forming reactions were much less investigated and more challenging, mainly due to catalyst poisoning by the mercapto group. In 2009, Duan and co-workers reported the first decarboxylative coupling of ortho-substituted aryl carboxylic acids with thiols as an unprecedented synthetic entry to aryl sulfides (Scheme 1a).10 In a follow-up study, Ranjit and co-workers developed a versatile protocol, in which a CuI catalyst was used to initiate the decarboxylation of arylpropiolic acids, for the synthesis of vinyl sulfides (Scheme 1b)11 by the cross-coupling of the arylpropiolic acids with thiols. In 2018, Ichiishi12 and Ishitobi13 reported the decarbonylative C–S coupling of thioesters into thioethers respectively.
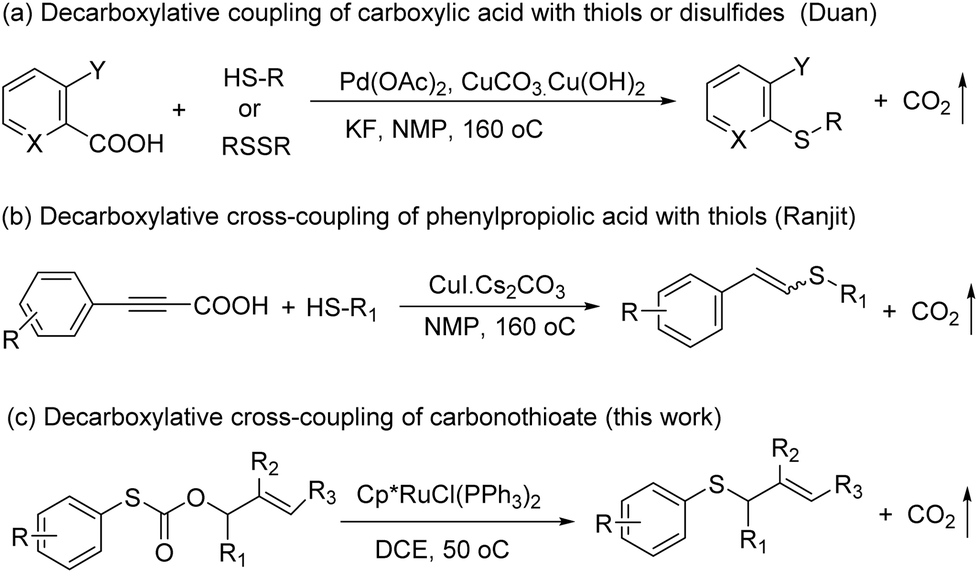 | ||
| Scheme 1 Synthesis of thioethers through the decarboxylative coupling reaction. | ||
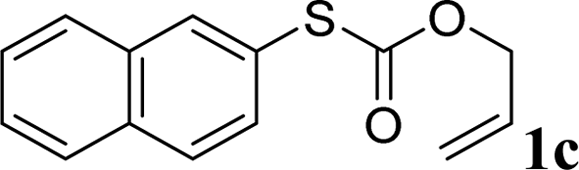
We reasoned that the direct decarboxylative C–S coupling of carbonothioate, if possible, would provide an alternative access to aryl sulfides without the need for halocarbon precursors (Scheme 1c). Herein, we describe the integration of these concepts into the transition-metal-catalyzed synthesis of a broad range of aryl sulfides.
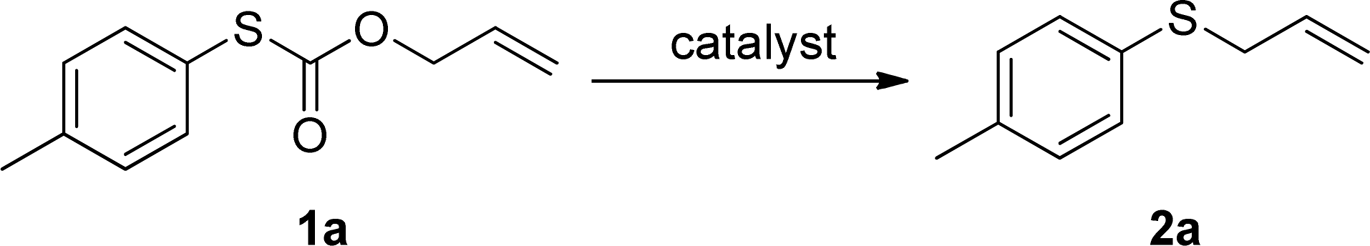
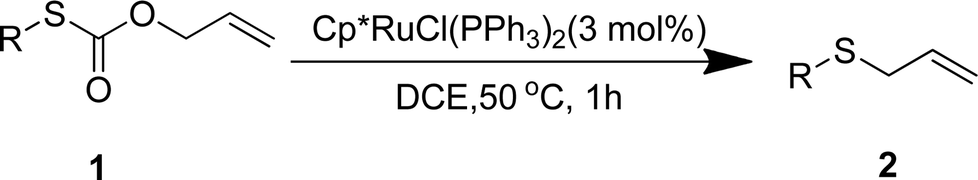
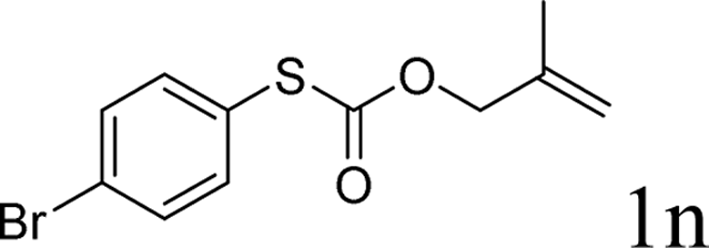
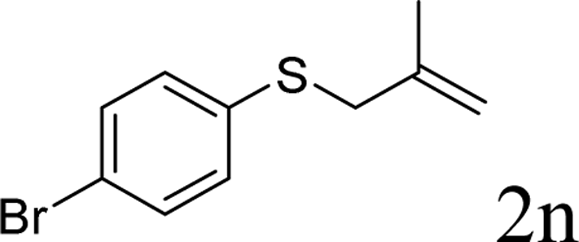
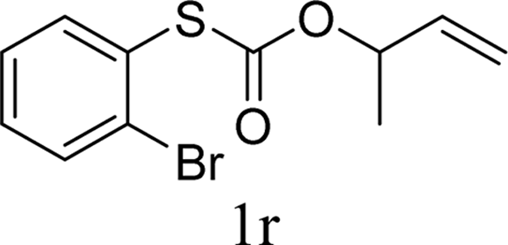
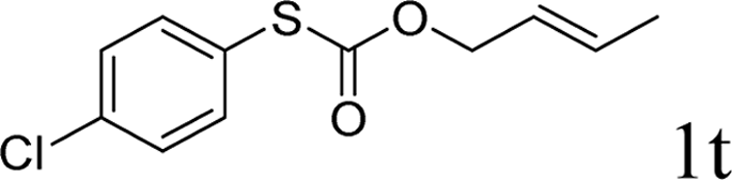
To test our hypothesis and also to identify an effective catalyst system, the decarboxylative allylation of O-allyl S-(p-tolyl) carbonothioate was selected as a model reaction and performed under different conditions (Table 1). To begin, we compared a variety of catalysts for their ability to effect the decarboxylative allylation of carbonothioate. Among them, nearly qualitative rate of decarboxylative allylation catalyzed by Pd and Ru catalysts (Table 1, entries 4–5), and that Fe, Ni and Au catalysts did not work with the reaction (entries 1–3). Different Ru catalysts was screened and the results showed that Cp*RuCl(PPh3)2 is the best catalyst (entries 5–7). A low yield was also resulted when the reaction was run at room temperature (entry 8). A series of solvents were examined (Table 1, entries 5, 9–11) and the desired product 2a was observed in 96% yield when DCE was utilized, whereas all the other solvents investigated afforded lower yields.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst/mol% | Solvent | T/°C | T/h | Yieldb (%) |
| a Reactions run in vials; [1a] = 0.05 M.b Estimated by 1H NMR using diethyl phthalate as an internal reference. | |||||
| 1 | Ni(acac)2(10)/PPh3(10) | DCE | 50 | 24 | nr |
| 2 | Fe(acac)3(10)/PPh3(10) | DCE | 50 | 24 | nr |
| 3 | Ph3PAuCl(5)/NaBArF4(10) | DCE | 50 | 24 | nr |
| 4 | Pd(PPh3)4(5) | DCE | 50 | 2 | 92 |
| 5 | Cp*RuCl(PPh3)2(3) | DCE | 50 | 1 | 96 |
| 6 | RuCl3(3) | DCE | 50 | 24 | nr |
| 7 | Ru(PPh3)3Cl2(3) | DCE | 50 | 5 | <5 |
| 8 | Cp*RuCl(PPh3)2(3) | DCE | rt | 12 | 56 |
| 9 | Cp*RuCl(PPh3)2(3) | THF | 50 | 1 | 87 |
| 10 | Cp*RuCl(PPh3)2(3) | CH3CN | 50 | 1 | 88 |
| 11 | Cp*RuCl(PPh3)2(3) | Toluene | 50 | 1 | 82 |
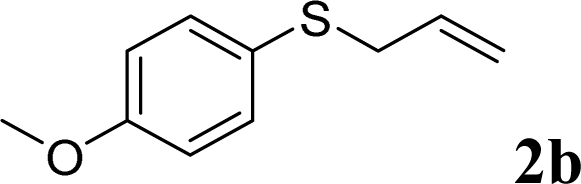
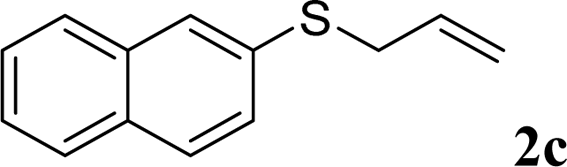
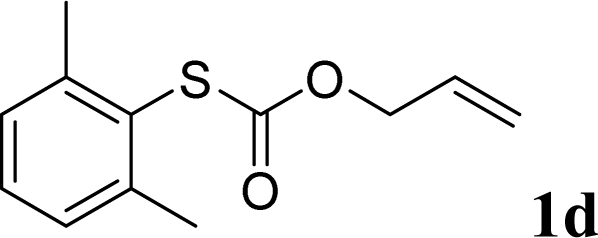
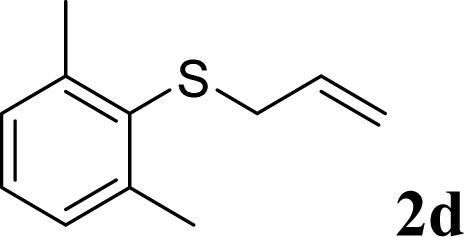
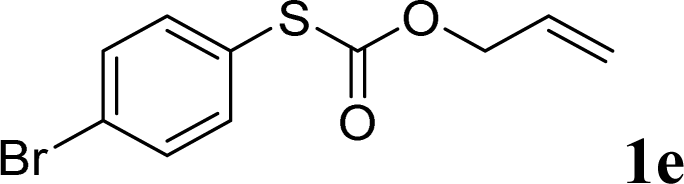
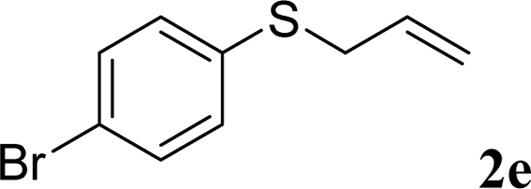
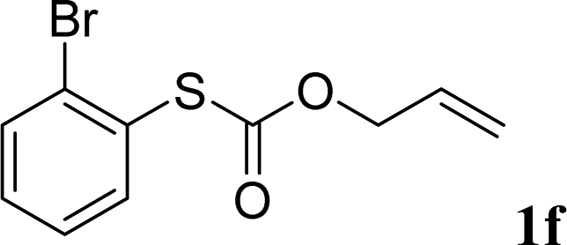
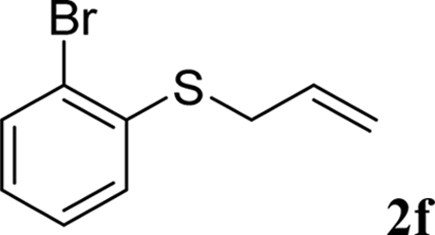
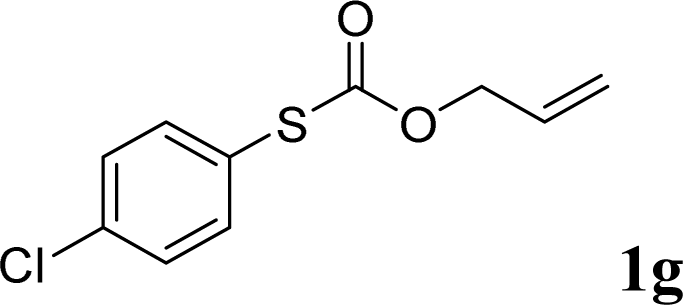
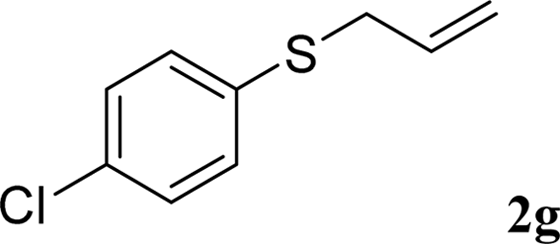
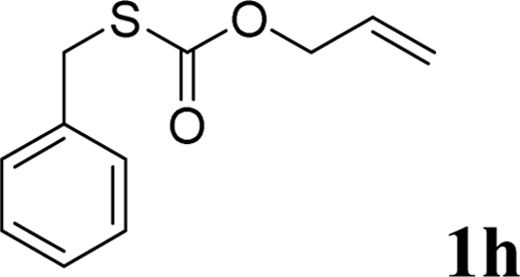
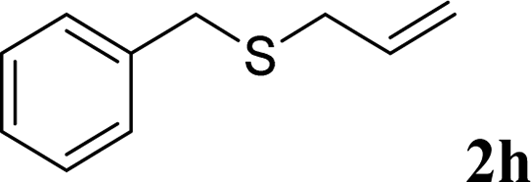
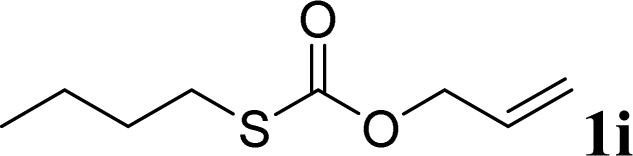
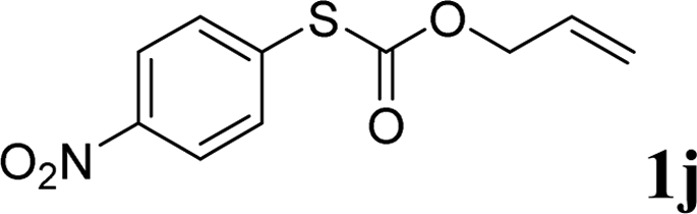
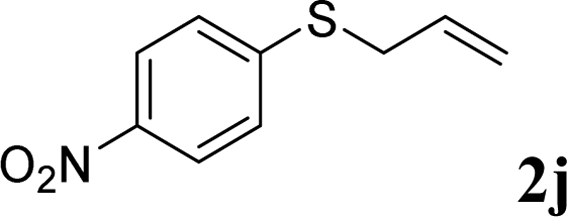
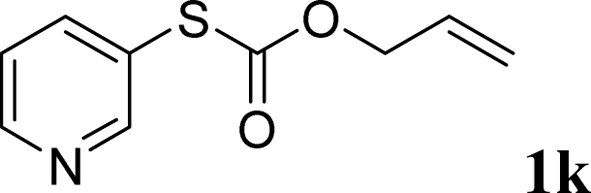
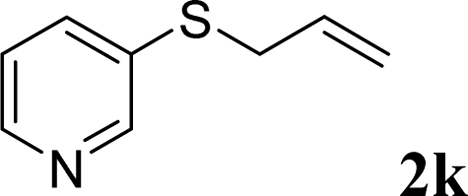
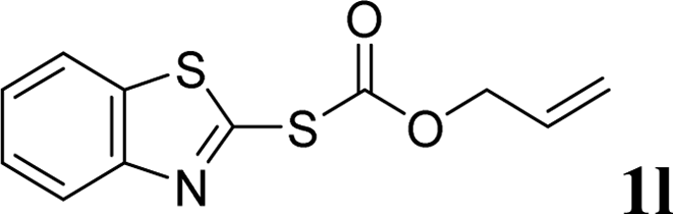
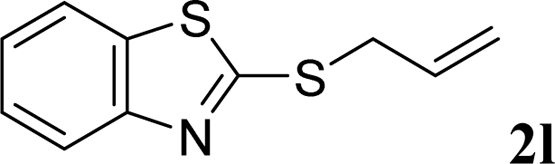
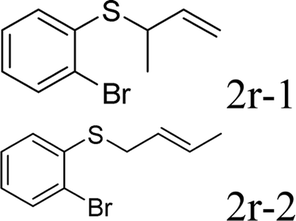
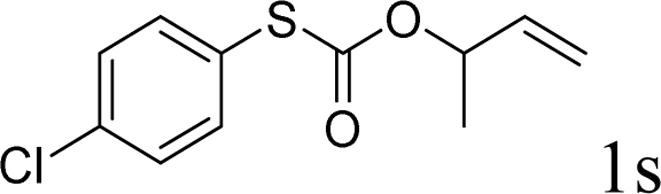
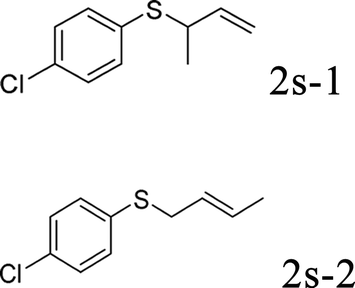
With the optimal reaction conditions in hand (Table 1, entry 5), the scope of the reaction was then examined. As summarized in Table 2, a variety of carbonothioate undergo decarboxylative allylation in high yield. In particular, the reaction is effective for nearly any substitution pattern about the carbonothioate; even sterically demanding ortho-substituted carbonothioates undergo allylation, albeit at a reduced rate (entries 3 and 5). Gratifyingly, halogen atoms (Cl and Br) could be tolerated well (entries 4–6), which have the potential to interfere with the analogous palladium-catalyzed reactions. Notably, a 4-NO2 group led to a significant decrease in the yield (entry 9). Replacing the phenyl group of 1 with a naphthyl (entry 2), a benzyl (entry 7), a butyl (entry 8), a pyridyl (entry 10), or a benzothiazolyl (entry 11) were all readily allowed and the corresponding products were isolated in good yields.
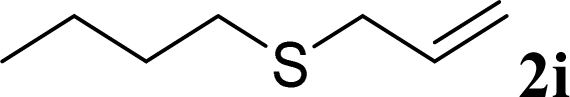
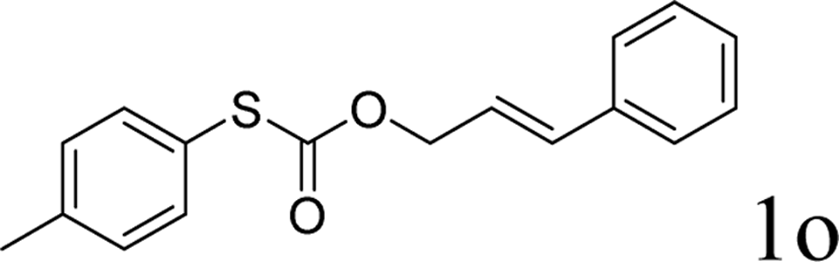
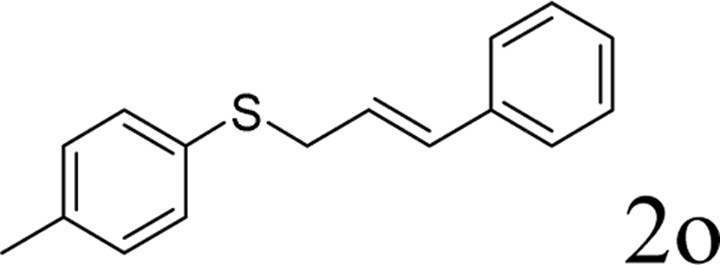
Next, the regioselectivity of the substituted allyl carbonothioates was investigated (Table 3). In all cases, the cinnamyl carbonothioates preferentially formed the linear allylic ethers in good yield (entries 3–5). As can be seen, cinnamyl carbonothioate substrates provided the linear product exclusively as judged by 1H NMR spectroscopy. Thus, the reaction with aryl-substituted allylic carbonothioates is regioselective. Next, we investigated the coupling of crotyl alcohol, which provided the branched and linear allylation product with the ratio nearly of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and high yield (entry 6–7). The isomeric branched carbonothioate also produced the branched and linear allylation product with the ratio nearly of 1:1 (entry 8). While the regiochemical outcome slightly depends on the regiochemistry of the starting allyl ester, the reaction is not strongly regiospecific. In general, ruthenium catalyst favors the branched products in allylic substitution,14 the higher reaction temperature maybe the main reason for the more stable linear product was obtained in our study.
:1 and high yield (entry 6–7). The isomeric branched carbonothioate also produced the branched and linear allylation product with the ratio nearly of 1:1 (entry 8). While the regiochemical outcome slightly depends on the regiochemistry of the starting allyl ester, the reaction is not strongly regiospecific. In general, ruthenium catalyst favors the branched products in allylic substitution,14 the higher reaction temperature maybe the main reason for the more stable linear product was obtained in our study.
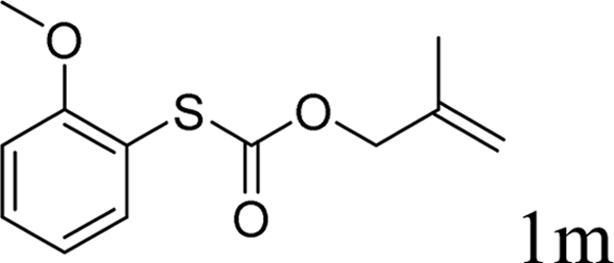
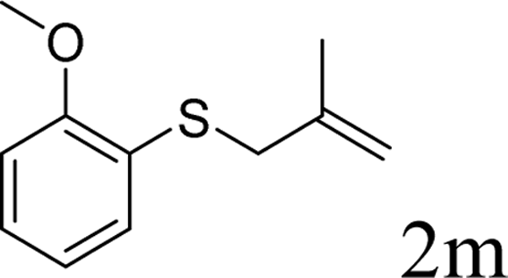
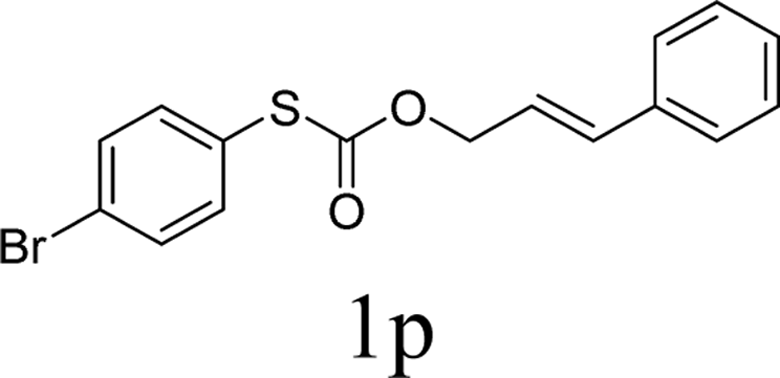
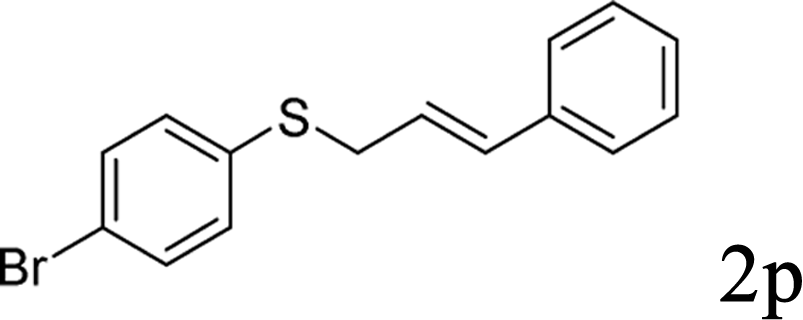
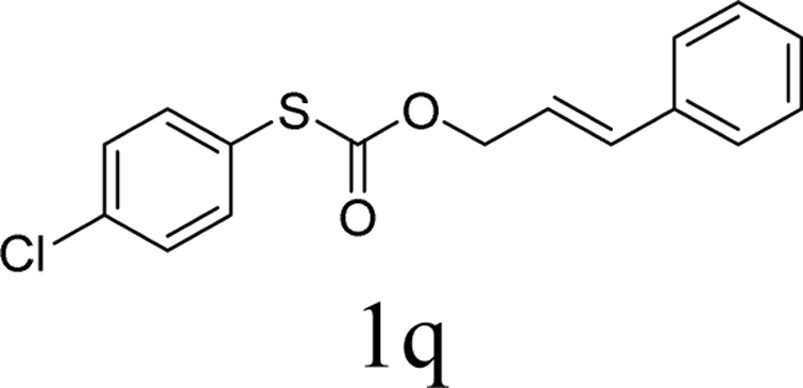
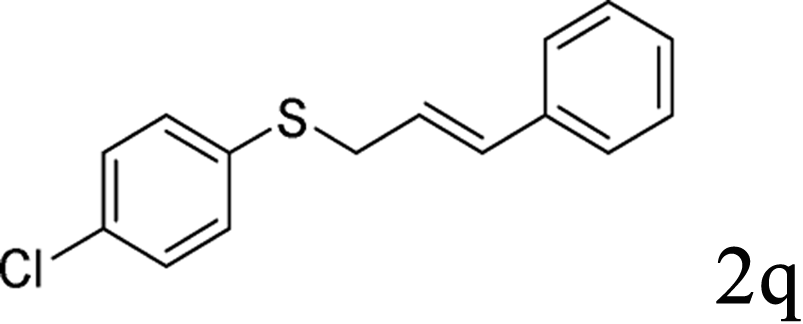
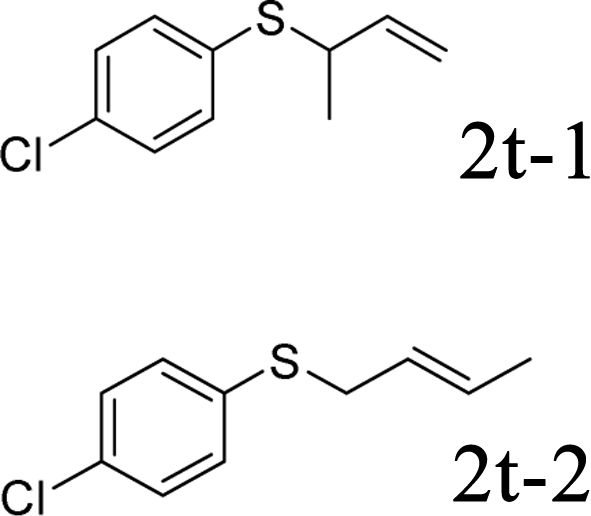
In conclusion, a ruthenium-catalyzed decarboxylative allylation of carbonothioates was developed. The yields of the reaction are generally high and the Ru catalyst often provides chemo- and regioselectivities that complement those of more standard palladium catalysts. This method is important not only for expanding our understanding of the decarboxylative reaction but also for providing a convenient synthetic pathway for facile synthesis of biologically or pharmaceutically relevant compounds. Further investigations of the substrate scope of this transformation and the reaction mechanism are currently in progress in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No. 21671146) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (2015-1098).Notes and references
- For recent reviews, see: (a) C. Shen, P. F. Zhang, Q. Sun, S. Bai, T. S. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (b) M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133 CrossRef PubMed; (c) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef PubMed; (d) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef PubMed.
- For selected examples, see: (a) S.-I. Murahashi, M. Yamamura, K.-I. Yanagisawa, N. Mita and K. Kondo, J. Org. Chem., 1979, 44, 2408 CrossRef; (b) T. Itoh and T. Mase, Org. Lett., 2004, 6, 4587 CrossRef PubMed; (c) M. A. Fernández-Rodríguez and J. F. Hartwig, J. Org. Chem., 2009, 74, 1663 CrossRef PubMed; (d) F.-L. Zeng and H. Alper, Org. Lett., 2011, 13, 2868 CrossRef PubMed; (e) Z.-J. Qiao, H. Liu, X. Xiao, Y. Fu, J. Wei, Y.-X. Li and X.-F. Jiang, Org. Lett., 2013, 15, 2594 CrossRef PubMed.
- For recent selected examples, see : (a) A.-X. Zhou, X.-Y. Liu, K. Yang, S.-C. Zhao and Y.-M. Liang, Org. Biomol. Chem., 2011, 9, 5456 RSC; (b) N. Park, Y. Heo, M. R. Kumar, Y. Kim, K.-H. Song and S.-W. Lee, Eur. J. Org. Chem., 2012, 1984 CrossRef; (c) H.-J. Xu, Y.-F. Liang, X.-F. Zhou and Y.-S. Feng, Org. Biomol. Chem., 2012, 10, 2562 RSC; (d) X.-B. Xu, J. Liu, J.-J. Zhang, Y.-W. Wang and Y. Peng, Org. Lett., 2013, 15, 550 CrossRef PubMed; (e) S. L. Buchwald and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5586 CrossRef PubMed; (f) H. Wang, L. Wang, J. Shang, X. Li, H. Wang, J. Gui and A. Lei, Chem. Commun., 2012, 48, 76 RSC; (g) G. Cahiez, A. Moyeux, J. Buendia and C. Duplais, J. Am. Chem. Soc., 2007, 129, 13788 CrossRef PubMed; (h) V. P. Reddy, A. V. Kumar, K. Swapna and K. R. Rao, Org. Lett., 2009, 11, 1697 CrossRef PubMed; (i) P. Malik and D. Chakraborty, Appl. Organomet. Chem., 2012, 26, 557 CrossRef.
- (a) E. Schaumann, Top. Curr. Chem., 2007, 274, 1 CrossRef; (b) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef PubMed; (c) E. Sperotto, G. P. M. van Klink, J. G. de Vries and G. van Koten, J. Org. Chem., 2008, 73, 5625 CrossRef PubMed.
- For recent reviews on catalytic decarboxylative coupling reaction, see: (a) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (b) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef PubMed; (c) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef PubMed; (d) Z. He, X. Qi, S. Li, Y. Zhao, G. Gao, Y. Lan, Y. Wu, J. Lan and J. You, Angew. Chem., Int. Ed., 2015, 54, 855 CrossRef PubMed.
- For recent selected examples, see: (a) N. Shibata, S. Suzuki, T. Furukawa, H. Kawai, E. Tokunaga, Z. Yuan and D. Cahard, Adv. Synth. Catal., 2011, 353, 2037 CrossRef; (b) Y. Ariyarathna and J. A. Tunge, Org. Biomol. Chem., 2014, 12, 8386 RSC; (c) A. N. Marziale, D. C. Duquette, R. A. Craig II, K. E. Kim, M. Liniger, Y. Numajiri and B. M. Stoltz, Adv. Synth. Catal., 2015, 357, 2238 CrossRef PubMed.
- I. Shimizu, T. Yamada and J. Tsuji, Tetrahedron Lett., 1980, 21, 3199 CrossRef.
- T. Tsuda, Y. Chujo, S. Nishi, K. Tawara and T. Saegusa, J. Am. Chem. Soc., 1980, 102, 6381 CrossRef.
- For selected examples, see: (a) W. Jia and N. Jiao, Org. Lett., 2010, 12, 2000 CrossRef PubMed; (b) P. Hu, Y. Shang and W. Su, Angew. Chem., Int. Ed., 2012, 51, 5945 CrossRef PubMed; (c) S. R. Mellegaard-Waetzig, D. K. Rayabarapu and J. A. Tunge, Synlett, 2005, 2759 Search PubMed; (d) C. Wang and J. A. Tunge, Org. Lett., 2006, 8, 3211 CrossRef PubMed; (e) C. Wang and J. A. Tunge, J. Am. Chem. Soc., 2008, 130, 8118 CrossRef PubMed; (f) R. Trivedi and J. A. Tunge, Org. Lett., 2009, 11, 5650 CrossRef PubMed.
- Z. Duan, S. Ranjit, P. Zhang and X. Liu, Chem.–Eur. J., 2009, 15, 3666 CrossRef PubMed.
- (a) S. Ranjit, Z. Duan, P. Zhang and X. Liu, Org. Lett., 2010, 12, 4134 CrossRef PubMed; (b) S. N. Riduan, J. Y. Ying and Y. Zhang, Org. Lett., 2012, 14, 1780 CrossRef PubMed.
- N. Ichiishi, C. A. Malapit, L. Woźniak and M. S. Sanford, Org. Lett., 2018, 20, 44 CrossRef PubMed.
- K. Ishitobi, R. Isshiki, K. K. Asahara, C. Lim, K. Muto and J. Yamaguchi, Chem. Lett., 2018, 47, 756 CrossRef.
- L. Egger, C. Tortoreto, T. Achard, D. Monge, A. Ros, R. Fernández, J. M. Lassaletta and J. Lacour, Adv. Synth. Catal., 2015, 357, 3325 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04783a |
| This journal is © The Royal Society of Chemistry 2018 |