DOI:
10.1039/C8RA04719G
(Paper)
RSC Adv., 2018,
8, 26356-26363
Characterization and electrochemical properties of iron-doped tetrahedral amorphous carbon (ta-C) thin films†
Received
2nd June 2018
, Accepted 13th July 2018
First published on 24th July 2018
Abstract
Iron-doped tetrahedral amorphous carbon thin films (Fe/ta-C) were deposited with varying iron content using a pulsed filtered cathodic vacuum arc system (p-FCVA). The aim of this study was to understand effects of iron on both the physical and electrochemical properties of the otherwise inert sp3-rich ta-C matrix. As indicated by X-ray photoelectron spectroscopy (XPS), even ∼0.4 at% surface iron had a profound electrochemical impact on both the potential window of ta-C in H2SO4 and KOH, as well as pseudocapacitance. It also substantially enhanced the electron transport and re-enabled facile outer sphere redox reaction kinetics in comparison to un-doped ta-C, as measured with electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) using outer-sphere probes Ru(NH3)6, IrCl6, and FcMeOH. These increases in surface iron loading were linked to increased surface oxygen content and iron oxides. Unlike few other metals, an iron content even up to 10 at% was not found to result in the formation of sp2-rich amorphous carbon films as investigated by Raman spectroscopy. Atomic force microscopy (AFM) and transmission electron microscopy (TEM) investigations found all films to be amorphous and ultrasmooth with Rq values always in the range of 0.1–0.2 nm. As even very small amounts of Fe were shown to dominate the electrochemistry of ta-C, implications of this study are very useful e.g. in carbon nanostructure synthesis, where irregular traces of iron can be readily incorporated into the final structures.
Introduction
Iron is a ubiquitous non-precious metal catalyst found in numerous applications of electrocatalysis1,2 electrochemistry,3 and for example in carbon nanomaterial synthesis.4,5 Yet, in many of these high-performance applications, such as carbon nanotubes, the origin of electrochemical performance can be often attributed to both the iron impurities and the supporting carbon nanostructure framework.6–8 If such metal impurities exhibit dominating electrochemical effects, the catalyst metal used in synthesis should be selected based on its desired electrochemical role and the end application.9 Consequently, it is of interest to investigate the evolution of both electrochemical as well as electrical properties of an inert carbon framework as iron is gradually introduced into the matrix. Since surface area and morphology can also impact carbon electrochemistry,10 the geometry of any carbon framework would have to be strictly controlled. One such relatively inert carbon framework is tetrahedral amorphous carbon thin films, or ta-C. Due to the high fraction of >60% sp3-bonded carbon,11 pure ta-C is a very hard, ultrasmooth,12 protective diamond-like carbon (DLC) coating, which is chemically reasonably inert, has a wide water window, is biocompatible, and is resistant against biofouling.13–15 Furthermore, the physicochemical properties of ta-C thin films can be tuned by doping it with metals or gases,16,17 as well as adjusting the sp3/sp2 ratio.18,19 With pure ta-C, the overall kinetics of outer sphere redox probe electron transfer can be tuned by adjusting the thickness of the film i.e. the electron transport through the film.20 Such adjustability makes DLC and ta-C films applicable to many demanding electrochemical environments in for example biomedical applications,21 fuel cell bipolar plates,22 lithium air batteries,23 and ozone generation17 as well as wastewater treatment.24,25 Other tunable properties include superhydrophobicity/superhydrophilicity26 and superoleophilicity27 for oil-water separation. DLC and ta-C films have been used in a broad range of analyte detection applications, such as enzymatic hydrogen peroxide sensing,14 detection of drug molecules16 and neurotransmitters,28,29 as well as gene expression profiling.30
In this work, the addition of iron into a pure ta-C matrix (Fe/ta-C) is investigated to enable the adjustment of both electrical and electrochemical properties, while retaining other properties, such as the wide water window. Although facile outer sphere electron transfer has been achieved for 7 nm thick ta-C electrodes,20,31 such ultrathin films commonly lack uniformity and are not feasible for application on non-planar surfaces. Thicker films on the other hand, such as the 30 nm thick reference ta-C in this study, become too resistive for most applications. p-FCVA deposited Fe/ta-C films are characterized in this study by correlating changes in physical structure and surface chemistry to electrochemical properties. Physical structure is characterized by X-ray reflectivity (XRR), atomic force microscopy (AFM), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), four-point probe, and transmission electron microscopy (TEM). These results are correlated to electrochemical properties obtained from cyclic voltammetry (CV) measurements of potential windows (KOH, H2SO4) and outer sphere redox probes (Ru(NH3)6, FcMeOH, IrCl6), as well as electrochemical impedance spectroscopy (EIS) of (Ru(NH3)6).
Experimental
Deposition
Iron doped tetrahedral amorphous carbon (Fe/ta-C) thin films were deposited with a pulsed filtered cathodic vacuum arc system (p-FCVA). The vacuum chamber was evacuated to pressures below 5 × 10−7 torr pressure using a CTI cryogenics CTI 8F cryopump prior to deposition. The p-FCVA system uses a toroidal 45° bent electromagnetic filter, with a pulse forming network unit of 2.6 mF charged to −400 V (pulse current 650 A) to initiate a triggerless arc between the cathode and the surrounding anode body. The cathode assembly consists of two separate cathodes of different materials, carbon and iron, surrounded by a shared anode body. A self-designed Labview based software running on National Instruments hardware controls the triggering of the pulse-forming network with a frequency of 1.0 Hz and triggers one cathode after the other as defined by the chosen Fe/C pulse ratios. X-ray reflectivity (XRR) was used to determine the deposition rates for both carbon and iron p-FCVA sources. These thickness and density values were used to calculate the respective molar deposition rates, which were then used to approximate sample iron atomic percentages (Fe at%) in Fe/ta-C (ESI†). Based on previous metal doped DLC studies,32–35 Fe/ta-C thin films with approximate iron concentrations of 2, 5, and 10 at% were deposited in 30 nm thickness. A reference ta-C sample was also deposited for comparison. Boron doped p-type silicon (100) prime wafers (<0.005 Ωcm, Siegert Wafer), microscope glass slides (Menzel), and NaCl single crystals (TedPella), were placed 110 mm away from the end of filter coil at floating potential and room temperature. The substrate holder was rotated at 17 rpm to ensure uniform film thickness. The source material for carbon plasma is a graphite rod of 6.35 mm diameter and purity of 99.95% (Graphitestore). The source material for iron plasma is an iron rod of similar diameter and purity 99.99% (Goodfellow).
Physical characterization
The film deposited on the salt crystals was prepared into a Transmission Electron Microscopy (TEM) specimen by floating off the film on top of DI water. By inserting the crystal into DI water bath at a shallow angle a suitably small piece of film was broken off and fished onto an M75 copper-only TEM grid (Agar). TEM samples were heated on a hotplate at 60 °C for 10 minutes to remove excess water. TEM analysis was performed by a FEI Tecnai F-20 TEM at 200 kV acceleration voltage. X-ray reflectivity (XRR) measurements were performed in parallel beam mode using a Rigaku SmartLab diffractometer equipped with a 9 kW rotating Cu anode (0.154 nm), HyPix-3000 2D single photon counting detector in 0D mode, Kα1 monochromator, 5 mm incident horizontal slit, and 5° Soller slits at both incident and detector ends. X-ray photoelectron spectroscopy (XPS) was performed using an Axis Ultra electron spectrometer (Kratos Analytical) with monochromatic Al Kα irradiation at 100 W under neutralization. Before analysis, pre-evacuation was carried out overnight. High-resolution spectra of C 1s, O 1s, and Fe 2p as well as survey spectra of 3–4 locations were recorded for each sample. The data were fitted with CasaXPS software, assuming Gaussian line shapes. 100% filter paper (Whatman) was used as an in situ reference for charge correction.36 A Veeco Dimension 5000 Atomic Force Microscope (AFM) was used in tapping mode with Mikromasch HQ:NSC14/Al BS tips of 8 nm typical tip radius and 5 N m−1 force constant. Data on 1 × 1 μm (2048 × 2048) areas were collected for height topography. Images were processed using Gwyddion 2.47 software for artifact correction and calculating the root mean squared surface roughness (Rq). Four-point probe measurements were carried out for glass substrate samples on a Hewlett Packard 3458A multimeter attached to a Jandel RM3000 multi-height probe. Visible-Raman spectroscopy was performed on a Horiba Jobin-Yvon Labram HR confocal Raman system with a 488 nm argon laser with 10 mW power on sample. Spot size of 1 μm was used with an Olympus 100× objective. Raman spectra were acquired in the range of 50 to 3000 cm−1 with a 600 lines per inch diffraction grating, exposure time of 15 s, and accumulation averaging count of two. Spectroscopic calibration was performed on intrinsic Si wafer (Ultrasil). Raman spectra were obtained from three places and fitted by two Gaussian peaks for D and G regions of amorphous carbon to obtain the ID/IG peak intensity ratios, as explained in literature.37,38
Electrochemical characterization
All electrochemical measurements were carried out on a Gamry Reference 600 potentiostat and analysed using Gamry Echem Analyst software. Both cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were carried out in a cell with a three-electrode configuration and an Ag/AgCl (+0.199 V vs. SHE, Radiometer Analytical) reference electrode and a platinum wire counter electrode. Aqueous solutions were prepared from distilled water and were de-aerated for at least 15 minutes with N2 (g) before measurements. In addition, during the measurements the cell was blanketed in a continuous flow of nitrogen. Before all measurement sets, preliminary scans were used to determine the appropriate potential windows for the unknown samples. All electrodes were prepared by masking the samples with PTFE tape (Irpola) onto FR-4 copper laminate sheets (MG Chemicals). Electrode preparation quality was evaluated with uncompensated resistance Ru approximations in the supporting electrolyte. Hence, when measuring low currents of around 10 μA, the ohmic drop is only a few millivolts and can be disregarded. For all cyclic voltammetry measurements, the following data were computed: oxidation and reduction peak currents (Ipa, Ipc), potentials (Ep), peak potential separation (ΔEp), peak current ratios (Ipa/Ipc), and respective background corrections. The active electrode surface area was 3 mm in diameter equating to 0.07 cm2. The potential windows were determined in 0.15 M H2SO4 (Merck Suprapur) and 1 M KOH (Honeywell) by cycling the samples at a scan rate of 400 mV s−1 starting from the cathodic end, so that the absolute value of both anodic and cathodic current was about 200 μA. Cyclic voltammetry measurements in 1 mM Ru(NH3)6 (hexaammineruthenium(III) chloride, Sigma-Aldrich), 1 mM IrCl6 (potassium hexachloroiridate(IV), Sigma-Aldrich) and 1 mM FcMeOH (ferrocenemethanol, Sigma-Aldrich) dissolved in 1 M KCl (Sigma-Aldrich) were performed at scanning rates of 25, 50, 100, 200, 300, 400, and 500 mV s−1, of which only the 100 mV s−1 is shown here. Redox peak currents were plotted as a function of the square root of scan rate. EIS measurements were conducted in 5 mM Ru(NH3)6 dissolved in 1 M KCl to investigate the effect of added iron on film apparent charge transfer resistance (Rct) and double-layer capacitance (Cdl). An AC signal of 15 mV amplitude was used in the frequency range from 200 kHz to 100 mHz. CV measurements at 400 mV s−1 were used to determine the formal potentials of the Ru(NH3)63+/2+ redox system, which were set as the DC potentials for EIS. Obtained EIS spectra were fitted and analyzed using Gamry Echem Analyst software. The equivalent circuit used for fitting the Nyquist plots was a modified Randles circuit20 with a solution resistance (Rs) in series with parallel circuit of a charge transfer resistance (Rct), a Warburg element, and a constant phase element (CPE) for the double-layer capacitance (Cdl). The Rct element comprises of two resistance elements: electron transfer to the film surface, and electron transport through the film. For the Cdl, ideal capacitor parameter (a = 1 for ideal capacitor), and apparent heterogeneous rate constant (k0), were calculated as described by Hsu & Mansfeld.39
Results and discussion
Physical characteristics
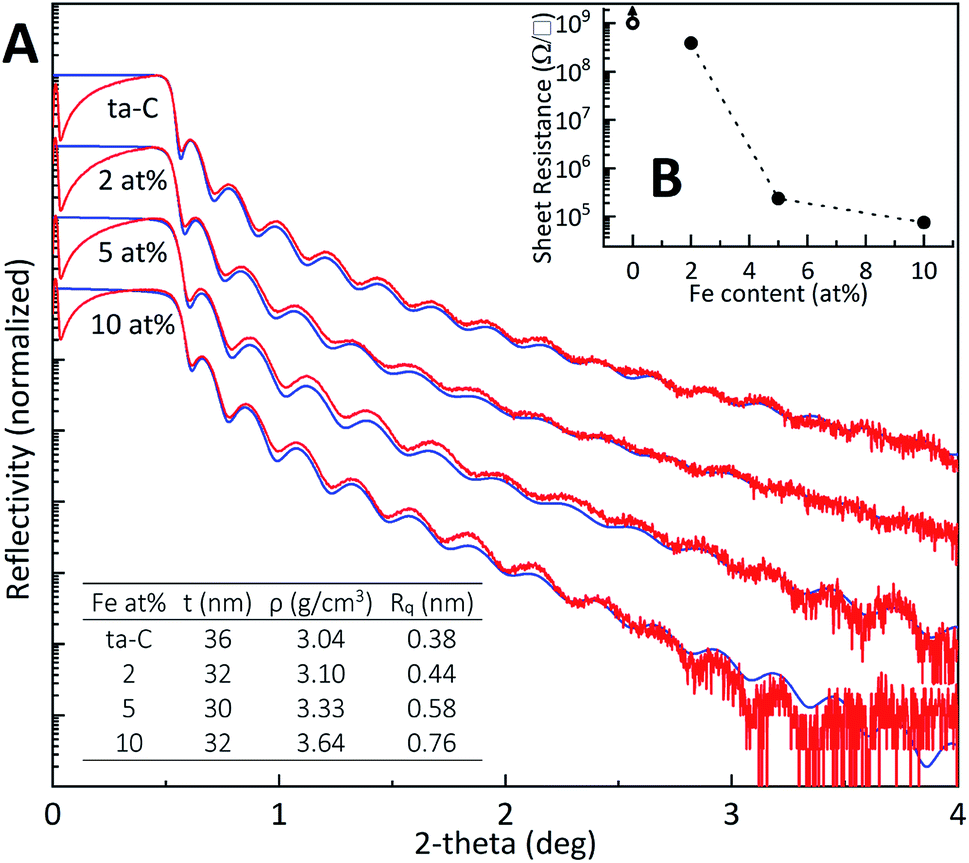
XRR. Film thickness for all samples was measured to be around 30 nm using X-ray reflectivity (XRR). All Fe/ta-C films followed a fit comprising a ∼1 nm native silicon oxide layer with a single Fe/ta-C layer of varying density (Table S4†). The XRR density of 3.04 g cm−3 obtained for the reference ta-C in Fig. 1 (A) corresponds to an sp3-fraction of about 80%, according to Robertson,15 which is a typical value for unbiased p-FCVA process. Successive additions of iron increased the density up to 3.64 g cm−3 for the 10 at% sample, indicating incorporation of iron into the carbon matrix. These additions are also directly evident from X-ray photoelectron spectroscopy wide spectrums shown in Fig. 2(B). Complementary to the resistive sp3-rich bulk of ta-C, the surface of the film is characteristically sp2-rich due to “ion peening”-growth model, as proposed by Caro et al.40 in a recent computational study utilizing machine-learned interatomic potential trained from ab initio data. Several other computational31,41,42 as well as XAS, XPS, and EELS studies18,43 have investigated the nature of ta-C and its sp2-rich surface structures. For example, Caro et al.42 showed that the surface of ta-C films consisted of clustered olefinic chains, with a lower ratio of ringed carbon structures.
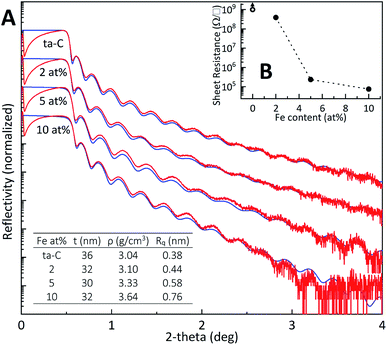 |
| | Fig. 1 (A) X-ray reflectivity scans showing experimental and simulation data in red and blue, respectively. Fitted parameters thickness (t), density (ρ), and roughness (Rq) presented in the inset table were calculated from the simulated curve. (B) Four-point probe average sheet resistances and standard deviations (N = 10) plotted as a function of Fe at%. Error bars are smaller than their respective dots. The reference ta-C, represented with an open circle, was too resistive to be measured. | |
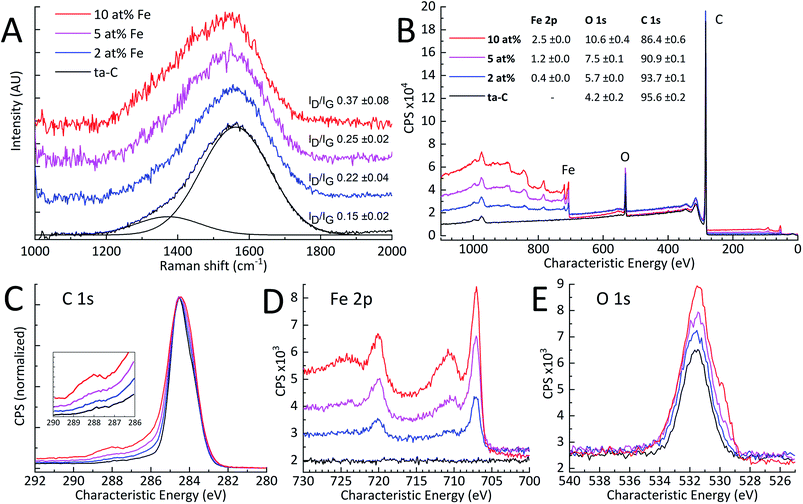 |
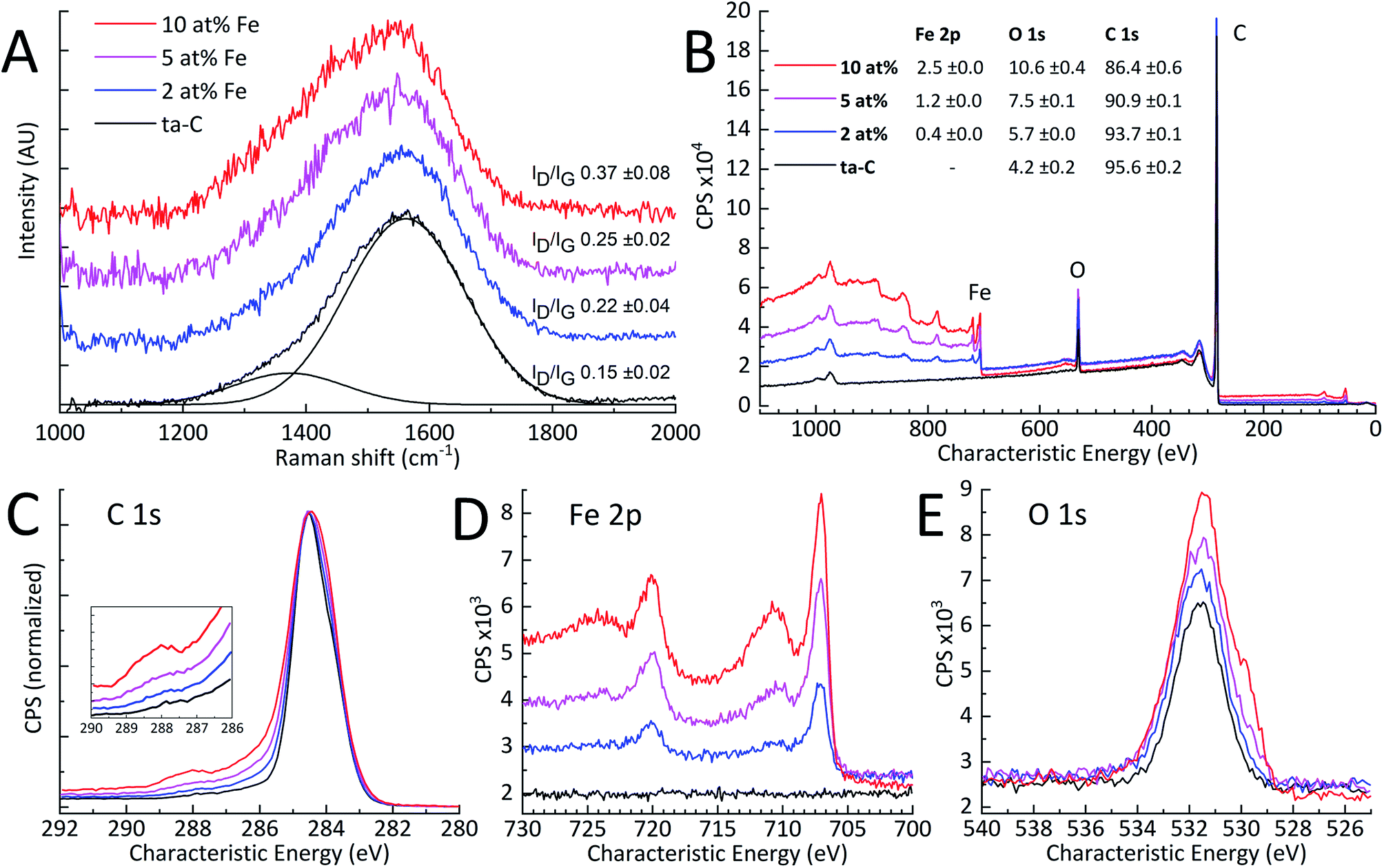
| | Fig. 2 (A) Obtained Raman spectra with calculated ID/IG ratios (N = 3), (B) XPS survey spectra with fitted atomic concentrations in the inset table (N = 3). High-resolution XPS spectra of (C) normalized C 1s, (D) Fe 2p, and (E) O 1s. | |
Sheet resistance. The incorporation of iron into the ta-C matrix is found to have a substantial effect in 4-point probe measurements in Fig. 1(B), where even the smallest addition of 2 at% Fe makes the film resistivity measurable compared to the more dielectric reference ta-C, which is too resistive for measurement. High density (>3.0 g cm−3) and high sp3-fraction ta-C films can have dielectric constants around ∼6.5, which is higher than that of diamond at 5.7.44 A further iron addition to 5 at% decreases sheet resistance by three orders of magnitude. Very similar trends have been observed e.g. for Zn, Mo and W/DLC composite films.34,35,45
XPS. As indicated by the high-resolution Fe 2p and O 1s spectra in Fig. 2(D) and (E), added amounts of iron in ta-C resulted in a clear increase of surface iron and oxygen loading. Also, the Fe 2p spectra indicates successive increases in iron-oxide content. High-resolution C 1s on the other hand, apart from the increasing shoulder indicating increased surface oxygen functionalities,46,47 shows no other major changes as a function of Fe at%. Overall, increased iron loading can be correlated with the increased surface oxygen content. In addition, the relative ratios of surface iron correspond to expected Fe atomic percentages of 2, 5 and 10 at%.
Raman spectroscopy. To examine iron-induced changes in carbon-to-carbon bonding, Raman spectroscopy investigations were carried out. Raman spectra shown in Fig. 2(A), fitted according to Ferrari & Robertson,37,38 show two Gaussian peaks for D and G regions of amorphous carbon. The ID/IG peak intensity ratios, representative of the sp2 carbon clustering, are also shown in Fig. 2(A). These results indicate an increase in sp2-clustering with increased Fe-content. Oxidized surface iron, as indicated by XPS, may exhibit sharp Raman excitation close to the D and G regions.48 There is, however, no apparent evidence of this in Fig. 2(A) since the amount of iron oxide is low compared to the excitation of sp2 carbon. The 2 at% Fe sample has a ID/IG of 0.22 ± 0.04, which is typical for a ta-C thin film of this thickness.20 The ID/IG of 0.37 ± 0.08 measured for the 10 at% Fe sample, is over twice than the 0.15 ± 0.02 measured for the reference ta-C, but still comparable to ID/IG values reported for ta-C films in literature.20,31,49 The higher ID/IG with increased Fe content points to an increased amount of sp2-clustering or total sp2-fraction in the Fe/ta-C matrix.37 Therefore, added iron appears to have an effect on the carbon–carbon bonding in ta-C. Such effects can be much larger in metal/ta-C composites: a very similar p-FCVA study by Pasaja et al.35 found that even the smallest addition of molybdenum increased the ID/IG to about 0.97 compared to their similar reference ta-C value of 0.15. This can be expected based on the higher solubility of carbon to iron in comparison to that of to molybdenum.50,51
AFM. Films deposited by p-FCVA have typically ultrasmooth surfaces with RMS roughness (Rq) values as low as 0.1 nm.12,31 This smoothness is also evident from AFM rasters shown in Fig. 3, where all samples were invariably smooth with Rq values in the 0.1–0.2 nm range. There is also no indication of differentiating surface features or cluster formation between the samples, which is in agreement with transmission electron microscopy (TEM) results, where all films were found to be amorphous with no particle formation (Fig. S4–S6†).
 |
| | Fig. 3 Atomic force microscopy height topography images. False color scale in all images is 2 nm. Root mean squared surface roughness (Rq) is calculated from particle-free areas. | |
Summary. In summary (Table 1), reference ta-C films were high in sp3-content, highly resistive, and amorphous with some oxygen content on the ultrasmooth carbon surface. The following changes were induced as a function of added iron: considerably decreased sheet resistance, slightly increased sp2-clustering, and increased surface loading of iron, iron oxide, and oxygen. There was no visible formation of particles or changes in surface smoothness.
Table 1 Summary of measured physical characteristics: Raman spectroscopy ID/IG-ratio, four-point probe sheet resistance (Ω □−1), and XPS wide scan atomic concentrations (at%)
| Fe at% |
Raman ID/IG |
Sheet resistance |
XPS wide at% |
| Fe 2p |
O 1s |
C 1s |
| ta-C |
0.15 ± 0.08 |
— |
— |
4.2 ± 0.2 |
95.6 ± 0.2 |
| 2 |
0.22 ± 0.02 |
3.9 × 108 |
0.4 ± 0.0 |
5.7 ± 0.0 |
93.7 ± 0.1 |
| 5 |
0.25 ± 0.04 |
2.4 × 105 |
1.2 ± 0.0 |
7.5 ± 0.1 |
90.9 ± 0.1 |
| 10 |
0.37 ± 0.02 |
7.6 × 104 |
2.5 ± 0.0 |
10.6 ± 0.4 |
86.4 ± 0.6 |
Electrochemical Characteristics
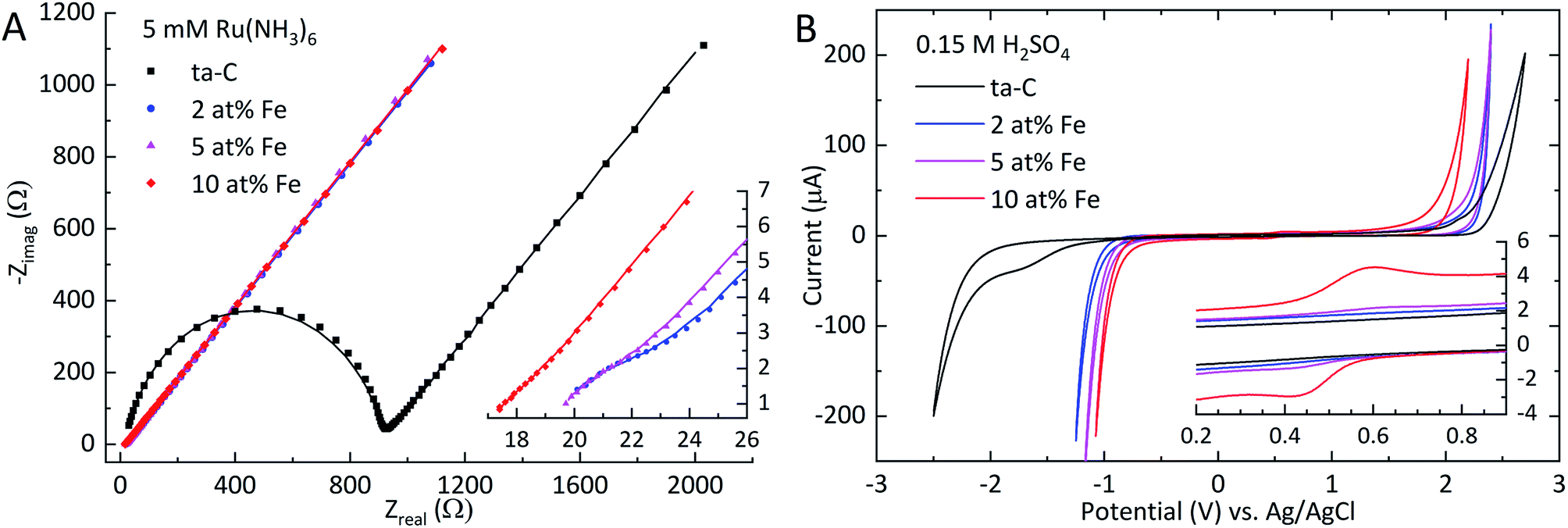
Electrochemical impedance spectroscopy. EIS measurements were carried out to probe the electrochemical properties of Fe/ta-C films. The Nyquist plot in Fig. 4(A) was fitted to obtain the exponent a, solution resistance Rs, charge transfer resistance Rct and double layer capacitance Cdl, as displayed in Table 2. The apparent heterogeneous electron transfer rate constant k0 was calculated according to Hsu & Mansfeld.39
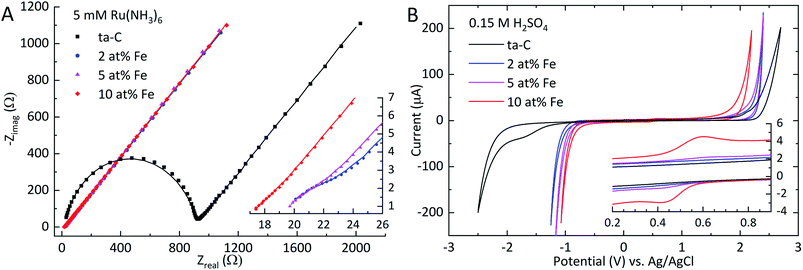 |
| | Fig. 4 (A) Nyquist plots of EIS measurements in 5 mM Ru(NH3)6 in 1 M KCl showing both experimental data (markers) and the fitted simulation (solid line). The inset in (A) shows a magnification of the high-frequency range to show the low Rct values of Fe/ta-C electrodes. (B) Cyclic voltammogram of 0.15 M H2SO4 in 400 mV s−1 for a 200 μA threshold. The inset in (B) shows a redox couple attributable to iron species. | |
Table 2 EIS results calculated from fitting the Nyquist plots in Fig. 4(A) with the modified Randles circuit.20 Values and errors are averages and standard deviations of three measurements
| Fe at% |
a |
Rs (Ω) |
Rct (Ω) |
Cdl (μF cm−2) |
k0 (cm s−1) |
| ta-C |
0.89 ± 0.00 |
16 ± 2 |
841 ± 160 |
0.3 ± 0.0 |
0.002 ±![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 × 10−4 3 × 10−4 |
| 2 |
0.76 ± 0.07 |
18 ± 1 |
4.0 ± 0.6 |
5.0 ± 0.2 |
0.37 ± 0.05 |
| 5 |
0.82 ± 0.05 |
18 ± 1 |
2.7 ± 0.5 |
5.9 ± 1.0 |
0.54 ± 0.11 |
| 10 |
0.84 ± 0.05 |
22 ± 8 |
1.6 ± 0.3 |
7.0 ± 0.4 |
0.94 ± 0.16 |
Analogous to the significant decrease in sheet resistance, also the apparent charge transfer resistance Rct exhibits a decrease of over two magnitudes from 840 Ω for ta-C to 4 Ω with 2 at% Fe. Further additions show a consistent trend of decreasing Rct. This trend is also directly reflected in the increased apparent heterogeneous electron transfer rate constant k0. This result is as expected, since the overall kinetics of outer sphere redox probe electron transfer on thicker ta-C films has been shown to be largely dependent on the electron transport through the electrode structure.13,20 The solution resistance Rs remained relatively constant throughout the series. The EIS measurements in Table 2 indicate over an order of magnitude increase in double-layer capacitance Cdl even with the smallest addition of iron (2 at% Fe). Such a large increase in Cdl is noteworthy given the unchanged surface roughness Rq. Additional iron further increases Cdl. As outlined by Frackowiak & Béguin,10 such surface-area independent enhancements in pseudocapacitance on carbon materials can be linked to increased electroactive surface oxygen content and the presence of transition metal oxides. The exponent a, describing the ideality of the capacitive characteristics of the electrode, shows some variation as a function of iron content, but the changes are too inconsistent to show a clear trend.
Potential windows. The effect of added iron on the water window of ta-C in 0.15 M H2SO4 and 1 M KOH was investigated to evaluate the applicability of Fe/ta-C in different electrochemical environments. In 0.15 M sulfuric acid, the addition of iron was found to have a profound electrochemical effect on both the cathodic and anodic branches of the potential window. The 5.2 V wide potential window for reference ta-C, shown in Fig. 4(B) and Table 3, is typical for a pure high sp3-fraction ta-C matrix.13,31 With an addition of 2 at% Fe, the anodic branch exhibits a negative shift from +2.7 V to +2.4 V, while the cathodic branch experiences a more noticeable shift of +1.3 V from −2.5 V to −1.2 V. In total, the cathodic and anodic branches shift by +1.4 V and −0.5 V, respectively to a potential window of 3.3 V for the 10 at% Fe sample. The peak current of a redox couple shown in the inset of Fig. 4(B) is in direct correlation with increased iron content. These peaks can be attributed to the oxidation and reduction of different iron species, as iron tends to dissolve in aqueous solutions at anodic potentials.52,53 However, it is not possible to straightforwardly associate these peaks with specific Fe-redox reactions, as the corrosion behavior of iron is known to be very sensitive to the presence of impurities, such as carbon or other metals.52,54 Nevertheless, together with the XPS results, these observations clearly indicate the presence of oxidized iron species on the surface, which is consistent with the decreased anodic and cathodic branches.52
Table 3 Cyclic voltammetry potential window results in volts (V) at 400 mV s−1 scan speed in 0.15 M H2SO4 and 1 M KOH at 200 μA threshold
| Fe at% |
0.15 M H2SO4 |
1 M KOH |
| Epa |
Epc |
Pot. window |
Epa |
Epc |
Pot. window |
| ta-C |
2.7 |
−2.5 |
5.2 |
1.8 |
−2.3 |
4.0 |
| 2 |
2.4 |
−1.2 |
3.6 |
1.4 |
−1.8 |
3.1 |
| 5 |
2.4 |
−1.1 |
3.5 |
1.1 |
−1.8 |
2.8 |
| 10 |
2.2 |
−1.1 |
3.3 |
1.0 |
−1.7 |
2.7 |
In 1 M KOH, the effect of iron is much more pronounced in the negative shift of the anodic branch, as displayed in Fig. S7† and Table 3. The initial potential window of 4.0 V for ta-C is considerably wide, similar to that in H2SO4 shown in Fig. 4(B). With each subsequent addition of iron, the anodic branch of ta-C consequently decreases from +1.8 V to +1.0 V for the 10 at% Fe. The cathodic branch in contrast experiences a single, much larger positive shift of +0.5 V from −2.3 V to −1.8 V for the 2 at% Fe, but then remains practically unchanged. The potential window for 10 at% Fe is still somewhat wider than that of polycrystalline iron, which has typically a window from −1.6 V to +0.8 V.55 Similar to the inset of Fig. 4(B) the cyclic voltammogram in 1 M KOH (Fig. S7†) shows the presence of a redox couple which can be also attributed to surface iron species, however this time at a more cathodic potential due to the higher pH of 1 M KOH.55,56 Although the water windows are notably decreased by the addition of iron in both H2SO4 and KOH, they remain flat, stable and relatively wide.
Outer sphere redox couples. Electron transfer kinetics of Fe/ta-C films were investigated by using cyclic voltammetry and three outer sphere redox probes: rutheniumhexaammine (Ru(NH3)6), ferrocenemethanol (FcMeOH or C11H12FeO), and iridium chloride (IrCl6). Iridium chloride and rutheniumhexaammine are known to be true outer sphere redox probes insensitive to changes in surface chemistry57 whereas for FcMeOH there are some indications that it may adsorb on carbon surfaces.58,59 However, all three probes exhibited identical behavior as a function of added iron, as observed in Table 4 and Fig. S8–S10:† electron transfer for reference ta-C was slow and irreversible, while even the slightest addition of iron to 2 at% Fe rendered the electron transfer reversible or close to reversible, as reflected by the ΔEp and Ipa/Ipc values listed in Table 4. This is consistent with the results from EIS and sheet resistance measurements indicating substantially decreased electron transport resistance through the Fe/ta-C films. Also, all outer sphere redox reactions were diffusion limited (Fig. S11–S13†). For comparison to EIS data, k0 values were calculated from the CV data using Nicholson's method60 for the measured outer sphere redox probes (Fig. S14†).
Table 4 Cyclic voltammetry redox peak potential separations ΔEp (mV) and peak current ratios Ipa/Ipc at 100 mV s−1 scan speed of the three redox probes: rutheniumhexaammine, ferrocenemethanol, and iridium chloride
| Fe at% |
1 mM Ru(NH3)6 |
1 mM FcMeOH |
1 mM IrCl6 |
| ΔEp |
Ipa/Ipc |
ΔEp |
Ipa/Ipc |
ΔEp |
Ipa/Ipc |
| ta-C |
101 |
0.856 |
88 |
0.955 |
122 |
0.950 |
| 2 |
59 |
0.920 |
61 |
0.986 |
63 |
0.980 |
| 5 |
61 |
0.916 |
61 |
0.983 |
68 |
0.986 |
| 10 |
61 |
0.907 |
61 |
0.986 |
59 |
0.990 |
Conclusions
Iron was added into pure ∼80% sp3-rich ta-C thin films for the tailoring of electrical and electrochemical properties. Fe/ta-C thin films were fabricated with three different iron contents, approximately 2, 5, and 10 at%, using a room temperature p-FCVA process. TEM and AFM found all films amorphous with ultrasmooth surfaces with the RMS roughness being in the range of 0.1–0.2 nm. Unlike metals with low carbon solubility, such as molybdenum, iron contents even up to 10 at% in ta-C were not found to result into the formation of sp2-rich amorphous carbon as investigated by Raman spectroscopy. As observed by the sheet resistance measurements, EIS, and several outer-sphere probes (Ru(NH3)6, IrCl6, FcMeOH), even the smallest iron addition of 2 at% was found to substantially enhance electron transport properties of the films and re-enable facile outer sphere redox reactions on the otherwise highly resistive and dielectric 30 nm thick ta-C film. XPS results indicated increases in surface iron loading, that could be connected with the increased surface oxygen content and subsequent formation of iron oxides. These iron-induced changes in surface chemistry were associated to (i) an order of magnitude increase in pseudocapacitance, and (ii) considerable decreases in the width of the potential windows measured in 0.15 M H2SO4 and 1 M KOH. In both cases 0.4 at% surface iron (XPS) produced effects comparable to 2.5 at% surface iron. It can therefore be expected that even iron impurity levels of 0.5 at% and below on inert carbon surfaces can have a profound electrochemical role. It is to be noted that these impurity levels are already at the limit of resolution of XPS that is commonly used to verify the purity of carbon nanostructures. This notion can be critical e.g. in carbon nanostructure synthesis, where irregular trace amounts of iron impurities can contaminate the process.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Finnish Funding Agency for Technology and Innovation (FEDOC project, grant number 211637). The authors acknowledge the provision of facilities by Aalto University Bioeconomy and OtaNano – Nanomicroscopy Center (Aalto-NMC).
References
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef.
- M. Tavakkoli, T. Kallio, O. Reynaud, A. G. Nasibulin, J. Sainio, H. Jiang, E. I. Kauppinen and K. Laasonen, J. Mater. Chem. A, 2016, 4, 5216–5222 RSC.
- D. Huber, Small, 2005, 1, 482–501 CrossRef PubMed.
- A. Moisala, A. G. Nasibulin and E. I. Kauppinen, J. Phys.: Condens. Matter, 2003, 15, 3011–3035 CrossRef.
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef PubMed.
- K. Jurkschat, X. Ji, A. Crossley, R. G. Compton and C. E. Banks, Analyst, 2007, 132, 21–23 RSC.
- C. E. Banks, A. Crossley, C. Salter, S. J. Wilkins and R. G. Compton, Angew. Chem., Int. Ed., 2006, 45, 2533–2537 CrossRef PubMed.
- J. Kruusma, N. Mould, K. Jurkschat, A. Crossley and C. E. Banks, Electrochem. Commun., 2007, 9, 2330–2333 CrossRef.
- T. Laurila, S. Sainio, H. Jiang, N. Isoaho, J. E. Koehne, J. Etula, J. Koskinen and M. Meyyappan, ACS Omega, 2017, 2, 496–507 CrossRef PubMed.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef.
- C. Casiraghi, J. Robertson and A. C. Ferrari, Mater. Today, 2007, 10, 44–53 CrossRef.
- M. Moseler, Science, 2005, 309, 1545–1548 CrossRef PubMed.
- T. Laurila, S. Sainio and M. A. Caro, Prog. Mater. Sci., 2017, 88, 499–594 CrossRef.
- N. Tujunen, E. Kaivosoja, V. Protopopova, J. J. Valle-Delgado, M. Österberg, J. Koskinen and T. Laurila, Mater. Sci. Eng., C, 2015, 55, 70–78 CrossRef PubMed.
- J. Robertson, Mater. Sci. Eng., R, 2002, 37, 129–281 CrossRef.
- N. Wester, J. Etula, T. Lilius, S. Sainio, T. Laurila and J. Koskinen, Electrochem. Commun., 2018, 86, 166–170 CrossRef.
- L.-C. Cheng, T.-F. Hung, P.-H. Lee, I.-C. Lin, H.-L. Wen, L.-H. Lu, C.-L. Chiu, S.-C. Chen, J. C. Sung, B.-J. Weng and R.-S. Liu, RSC Adv., 2013, 3, 5917 RSC.
- S. Sainio, D. Nordlund, M. A. Caro, R. Gandhiraman, J. Koehne, N. Wester, J. Koskinen, M. Meyyappan and T. Laurila, J. Phys. Chem. C, 2016, 120, 8298–8304 CrossRef.
- W. Volksen, R. D. Miller and G. Dubois, Chem. Rev., 2010, 110, 56–110 CrossRef PubMed.
- T. Palomäki, N. Wester, M. A. Caro, S. Sainio, V. Protopopova, J. Koskinen and T. Laurila, Electrochim. Acta, 2017, 225, 1–10 CrossRef.
- R. K. Roy and K.-R. Lee, J. Biomed. Mater. Res., Part B, 2007, 83B, 72–84 CrossRef PubMed.
- R. L. Borup and N. E. Vanderborgh, MRS Proceedings, 1995, 393, 151 CrossRef.
- Y. Yang, Q. Sun, Y.-S. Li, H. Li and Z.-W. Fu, J. Power Sources, 2013, 223, 312–318 CrossRef.
- S. Miyagawa, S. Nakao, J. Choi, M. Ikeyama and Y. Miyagawa, New Diamond Front. Carbon Technol., 2006, 16, 33–38 Search PubMed.
- M. Janotta, F. Vogt, H.-S. Voraberger, W. Waldhauser, J. M. Lackner, C. Stotter, M. Beutl and B. Mizaikoff, Anal. Chem., 2004, 76, 384–391 CrossRef PubMed.
- D. Caschera, B. Cortese, A. Mezzi, M. Brucale, G. M. Ingo, G. Gigli and G. Padeletti, Langmuir, 2013, 29, 2775–2783 CrossRef PubMed.
- B. Cortese, D. Caschera, F. Federici, G. M. Ingo and G. Gigli, J. Mater. Chem. A, 2014, 2, 6781–6789 RSC.
- H. Dong, S. Wang, A. Liu, J. J. Galligan and G. M. Swain, J. Electroanal. Chem., 2009, 632, 20–29 CrossRef.
- E. Kaivosoja, E. Berg, A. Rautiainen, T. Palomaki, J. Koskinen, M. Paulasto-Krockel and T. Laurila, in 2013 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), IEEE, 2013, vol. 2013, pp. 631–634 Search PubMed.
- Tuoya, K. Hirayama, T. Nagaoka, D. Yu, T. Fukuda, H. Tada, H. Yamada and M. Seno, Biochem. Biophys. Res. Commun., 2005, 334, 263–268 CrossRef PubMed.
- V. S. Protopopova, N. Wester, M. A. Caro, P. G. Gabdullin, T. Palomäki, T. Laurila and J. Koskinen, Phys. Chem. Chem. Phys., 2015, 17, 9020–9031 RSC.
- Š. Meškinis, A. Vasiliauskas, K. Šlapikas, G. Niaura, R. Juškėnas, M. Andrulevičius and S. Tamulevičius, Diamond Relat. Mater., 2013, 40, 32–37 CrossRef.
- Š. Meškinis, A. Čiegis, A. Vasiliauskas, K. Šlapikas, R. Gudaitis, I. Yaremchuk, V. Fitio, Y. Bobitski and S. Tamulevičius, Nanoscale Res. Lett., 2016, 11, 146 CrossRef PubMed.
- H. Wong, Y. M. Foong and D. H. C. Chua, Appl. Surf. Sci., 2011, 257, 9616–9620 CrossRef.
- N. Pasaja, S. Sansongsiri, S. Intarasiri, T. Vilaithong and A. Anders, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 259, 867–870 CrossRef.
- L.-S. Johansson, J. M. Campbell, P. Fardim, A. H. Hultén, J.-P. Boisvert and M. Ernstsson, Surf. Sci., 2005, 584, 126–132 CrossRef.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 075414 CrossRef.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef.
- C. H. Hsu and F. Mansfeld, Corrosion, 2001, 57, 747–748 CrossRef.
- M. A. Caro, V. L. Deringer, J. Koskinen, T. Laurila and G. Csányi, Phys. Rev. Lett., 2018, 120, 166101 CrossRef PubMed.
- M. A. Caro, R. Zoubkoff, O. Lopez-Acevedo and T. Laurila, Carbon, 2014, 77, 1168–1182 CrossRef.
- M. A. Caro, J. Määttä, O. Lopez-Acevedo and T. Laurila, J. Appl. Phys., 2015, 117, 034502 CrossRef.
- C. A. Davis, G. A. J. Amaratunga and K. M. Knowles, Phys. Rev. Lett., 1998, 80, 3280–3283 CrossRef.
- J. P. Sullivan, T. A. Friedmann, C. A. Apblett, M. P. Siegal, N. Missert, M. L. Lovejoy, P. B. Mirkarimi and K. F. McCarty, MRS Proceedings, 1995, 381, 273 CrossRef.
- K. Baba, R. Hatada and Y. Tanaka, Surf. Coat. Technol., 2007, 201, 8362–8365 CrossRef.
- N. Dwivedi, R. J. Yeo, N. Satyanarayana, S. Kundu, S. Tripathy and C. S. Bhatia, Sci. Rep., 2015, 5, 7772 CrossRef PubMed.
- K. Song, Y. Lee, M. R. Jo, K. M. Nam and Y.-M. Kang, Nanotechnology, 2012, 23, 505401 CrossRef PubMed.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef.
- R. McCann, S. S. Roy, P. Papakonstantinou, G. Abbas and J. A. McLaughlin, Diamond Relat. Mater., 2005, 14, 983–988 CrossRef.
- W. R. Thomas and G. M. Leak, London, Edinburgh Dublin Philos. Mag. J. Sci., 1954, 45, 986–987 CrossRef.
- J. Imai, O. Taguchi, G. P. Tiwari and Y. Iijima, Mater. Trans., 2014, 55, 1786–1791 CrossRef.
- A. C. Makrides, J. Electrochem. Soc., 1960, 107, 869 CrossRef.
- C. Wang, S. Chen and X. Yu, Electrochim. Acta, 1994, 39, 577–580 CrossRef.
- A. C. Makrides and M. Stern, J. Electrochem. Soc., 1960, 107, 877 CrossRef.
- G. Kreysa and B. Håkansson, J. Electroanal. Chem. Interfacial Electrochem., 1986, 201, 61–83 CrossRef.
- H. Alemu and K. Jüttner, Electrochim. Acta, 1988, 33, 1101–1109 CrossRef.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef PubMed.
- A. S. Cuharuc, G. Zhang and P. R. Unwin, Phys. Chem. Chem. Phys., 2016, 18, 4966–4977 RSC.
- A. M. Bond, E. A. McLennan, R. S. Stojanovic and F. G. Thomas, Anal. Chem., 1987, 59, 2853–2860 CrossRef.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Deposition method calibration data, XRR details, additional XPS data, TEM images, CV curves, calculated CV rate constants. See DOI: 10.1039/c8ra04719g |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Niklas Westera,
Sami Sainioa,
Tomi Laurilab and
Jari Koskinen
*a,
Niklas Westera,
Sami Sainioa,
Tomi Laurilab and
Jari Koskinen