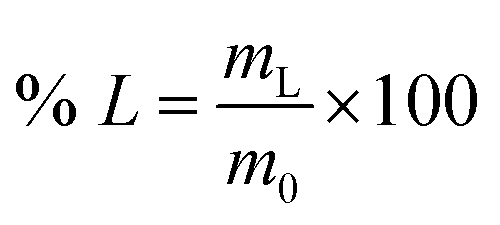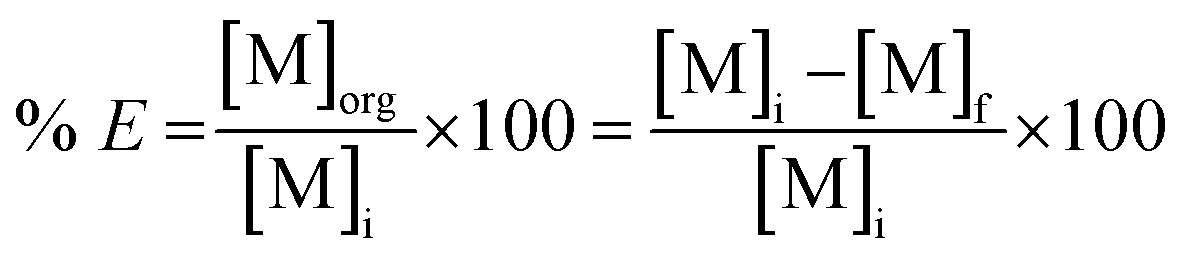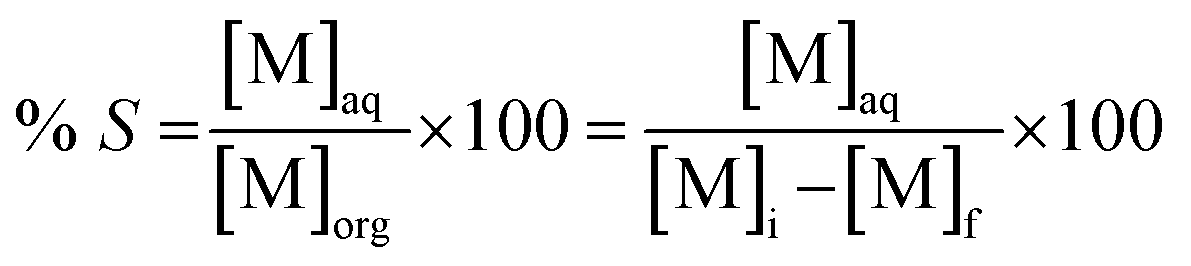DOI:
10.1039/C8RA04532A
(Paper)
RSC Adv., 2018,
8, 26349-26355
Recovery of rare earths from the green lamp phosphor LaPO4:Ce3+,Tb3+ (LAP) by dissolution in concentrated methanesulphonic acid
Received
28th May 2018
, Accepted 17th July 2018
First published on 24th July 2018
Abstract
A process was developed for the recovery of rare earths from terbium-rich lamp phosphor waste. The process consists of a solvometallurgical leaching step with concentrated methanesulphonic acid (MSA) at temperatures between 433 K to 473 K, followed by solvent extraction with the acidic extractant di-(2-ethylhexyl)phosphoric acid (D2EHPA). Preliminary tests were performed on a synthetic lamp phosphor (LaPO4:Ce3+,Tb3+, LAP). The optimised conditions were afterwards applied to a real lamp phosphor waste residue, that was obtained after removal of yttrium and europium from lamp phosphor waste powder by a hydrometallurgical process. The leaching can be carried out at lower temperatures than digestion in concentrated sulphuric acid or fused alkali. The process takes advantage of the much higher solubility of the rare-earth methanesulphonates compared to the corresponding sulphates, so that solvent extraction can be performed directly on the leachate after dilution, without the need of several additional steps to convert the rare-earth sulphates into chlorides or nitrates.
Introduction
Lamp phosphor waste recovered from end-of-life fluorescent lamps is a potential valuable secondary source of rare-earth elements (REEs).1–4 Prior to recovery of valuable metals from the lamp phosphor waste powder, the crushed lamps are sieved in order to remove the coarse glass particles. The mercury present in the lamps can be removed in a separate step. The lamp phosphor waste powder fraction is a complex mixture of inorganic compounds. The largest fraction is the white phosphor (Sr,Ca)10(PO4)6(Cl,F)2:Sb3+,Mn2+ (HALO) which does not contain REEs, but which is relatively rich in antimony.5 The other phosphors are the red phosphor Y2O3:Eu3+ (YOX), the green phosphors LaPO4:Ce3+,Tb3+ (LAP), CeMgAl11O19:Tb3+ (CAT), (Ce,Gd)MgB5O10:Tb3+ (CBT), and the blue phosphor BaMgAl10O17:Eu2+ (BAM). Additionally, the waste powder contains fine glass particles and Al2O3 from the binder. Most of the previous research activities have focused on the recovery of rare earths from YOX phosphor, because this phosphor has the highest economic value.6–12 YOX consists almost entirely of the two critical REEs yttrium and europium. In a typical recycling process, the lamp phosphor waste is treated with a dilute acid solution to dissolve first the HALO phosphor, followed by dissolution of YOX in a more concentrated acid solution. After dissolution of YOX, the REEs are recovered from the solution by precipitation with oxalic acid or the solution is further purified by solvent extraction. For example, in the HydroWEEE process, the waste powder is fed into a leaching reactor where leaching with 4 N H2SO4 is carried out (20% of pulp density, T = 343 K, t = 2–3 h).9 The leachate is separated from the residue by filtration and is further treated by precipitation with oxalic acid to recover yttrium and europium as a mixed oxalate. An ionic liquid process allows the selective recovery of the YOX, without the need for prior dissolution of the HALO phosphor.13 The other phosphors in the lamp waste fraction are much more difficult to dissolve than HALO or YOX. The residue after dissolution of YOX is often just landfilled, although it contains large concentrations of the valuable REE terbium. Lamp phosphor recycling processes are presently economically feasible if not only yttrium and europium is recovered, but terbium as well. LAP is the most interesting terbium-containing compound in the lamp phosphor waste powder, because of its high terbium content and because it is easier to dissolve than CAT and CBT. However, very long leaching times with concentrated acids are required to dissolve LAP.14 To dissolve LAP in a short time, harsh conditions must be applied, similarly to those used to treat monazite ore. Typically, the LAP is treated with concentrated H2SO4 at temperatures above 523 K or by alkali fusion.15–18 A new evolution is the use of mechanical activation by ball-milling to activate LAP for easier dissolution, but processes making use of a planetary ball mill are difficult to upscale.19–22
In this paper we present a milder approach for dissolving the LAP phosphor, by making use of concentrated methane-sulphonic acid (MSA, CH3SO3H). The process is applied on a residue obtained after leaching lamp phosphor waste with a H2SO4 solution. This work was inspired by the use of trifluoromethanesulphonic acid (triflic acid, CF3SO3H) to dissolve thoria.23 However, MSA is much safer to work with than triflic acid and it is even considered to be a green acid.24 It has a high boiling point and is thermally stable so that it can be used at temperatures up to 473 K. This new process can be classified as solvometallurgy, because the dissolution of LAP occurs in anhydrous conditions.25 The leaching step is followed by dilution of the leachate and solvent extraction with the acidic extractant di-(2-ethylhexyl)phosphoric acid (D2EHPA).
Experimental
Chemicals
The LAP phosphor (LaPO4:Ce3+,Tb3+) (99% purity) was obtained from Nichia (Japan) with 99% purity. It contained 34.1 wt% La, 16.0 wt% Ce and 8.2 wt% Tb. The lamp phosphor residue was provided by Relight Srl (Rho, Italy). Methanesulphonic acid (99.5%) was purchased from Carl Roth (Karlsruhe, Germany), di-(2-ethylhexyl)phosphoric acid (95%), xylene ≥98.5% (mixture of isomers) and Triton X-100 were purchased from Acros Organics (Geel, Belgium). HCl (37%) and toluene (>99.5%) were obtained from VWR Chemicals (Leuven, Belgium). Bis(2,4,4-trimethylpentyl)phosphinic acid (Cyanex® 272) was provided by Cytec Industries (Canada), tri-n-butyl phosphate (>99%) was obtained from Chem-Lab (Belgium), n-dodecane (≥99.0%) and the standard solutions (1000 ppm Y, La, Ce, Pr, Eu, Gd, Tb, Lu) were obtained from Merck (Overijse, Belgium). LiBO2 (≥99.99%) was purchased from Alfa Aesar (Karlsruhe, Germany), the silicone solution in isopropanol from SERVA Electrophoresis GmbH (Heidelberg, Germany) and triflic acid (99%) from Iolitec (Heilbronn, Germany). Water was always of ultrapure quality, deionized to a resistivity of 18.2 MΩ cm with a Sartorius Arium® Pro ultrapure water system. All chemicals were used as received without any further purification.
Borate fusion procedure
The composition of the real lamp phosphor residue was determined after borate fusion. A sample of 100 mg was mixed with 500 mg of LiBO2 and the mixture was placed in a graphite crucible. The crucibles were then located in a furnace preheated at 1273 K. After 10 minutes, the molten mixture was poured in an acidic solution (2.5 mol dm−3 HCl), while stirring, and the resulting solution was analysed by ICP-OES.
Analytical techniques
The concentration of metals in solution was determined by total reflection X-ray fluorescence spectrometry (TXRF) with a Bruker S2 Picofox TXRF spectrometer equipped with a molybdenum source.26 Polypropylene microtubes were filled with a certain amount of the sample; an appropriate internal standard was then added, with the X-ray fluorescence energy as close as possible to that of the element to be determined in order to reduce the effects caused by absorption of secondary X-rays. Samples were finally diluted to 1 mL with a Triton solution (5% v/v).27 After shaking the samples on a vibrating plate (IKA MS 3 basic), a small droplet (1.5 μL) was put on a quartz glass carrier, previously treated with a silicone–isopropanol solution to avoid spreading of the droplet. The quartz carriers were then dried for 30 min at 333 K prior to analysis. Each sample was measured for 500 s.
The composition of the residue was determined by using a Perkin Elmer Optima 8300 inductively coupled plasma optical emission spectrometer (ICP-OES) using a Scott injector. Standard solutions were prepared containing 10 ppb to 20 ppm La, Ce, Eu, Gd, Tb, Y in 2% v/v HNO3. Lu was selected as internal standard. Measurements were performed in radial mode.
Methods
Unless otherwise specified, leaching tests were performed by contacting 100 mg of LAP phosphor with the selected leaching agent in glass vials (4 cm3) inserted in a copper block located on a stirring plate. Temperature control was performed via a temperature probe. After the leaching step, the leachate was separated from the solid residue by centrifugation (5300 rpm, 30 min). For the experiments on the LAP phosphors, the leaching efficiency was determined by weighing the sample before and after the leaching step. For the experiments on the real lamp phosphor residue, the leachate was analysed by TXRF to determine the leaching efficiency of the individual rare earths (% L), which was calculated according to eqn (1):| |
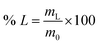 | (1) |
where mL is the mass of the dissolved metal (mg) and m0 is the mass of metal in the initial sample (mg).
The leachate was diluted with ultrapure water prior to extraction; several dilution factors were tested (10, 30, 40, 50). Solvent extraction tests were performed with tri-n-butyl phosphate (TBP), bis(2,4,4-trimethylpentyl)phosphinic acid (Cyanex 272) and di-(2-ethylhexyl)phosphoric acid (D2EHPA) as extractants. Toluene, xylene and n-dodecane were tested as diluents. These tests were performed by contacting the leachate with the selected extractant in glass vials located on a mechanical shaker (TMS-200 Turbo Thermoshaker), by varying several operative parameters, such as the dilution factor of the feed, the phase ratio (D2EHPA phase/MSA phase) and the number of extraction steps. The agitation was kept constant at 2500 rpm in all the experiments. The same procedure was followed for the solvent extraction tests performed on the raffinate of the first solvent extraction step. After the test, the aqueous phase was separated from the organic phase by centrifugation and analysed by TXRF to determine the percentage extraction % E which can be expressed, for phase ratio equal to 1, according to eqn (2):
| |
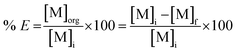 | (2) |
where [M]
i is the metal concentration in the feed, [M]
org is the metal concentration in the organic phase and [M]
f is the metal concentration in the aqueous phase after extraction. Metal recovery from the organic phase was investigated through precipitation-stripping. These tests were performed by contacting the loaded organic phase with the stripping solution in glass vials located on a mechanical shaker. Oxalic acid and hydrochloric acid were selected as stripping agents. The precipitation efficiency %
S was calculated according to
eqn (3):
| |
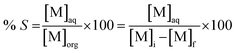 | (3) |
where [M]
aq is the metal concentration in the aqueous phase after stripping. The obtained oxalates were separated from the aqueous phase by centrifugation and calcined in a muffle furnace at a temperature 973 K for 4 hours in order form the corresponding oxides. Since the amount of oxides obtained from these tests was not sufficient for analysis
via ICP-OES, their composition was calculated knowing the amount of rare earths precipitated as oxalates and converting them into oxides.
Results and discussion
Dissolution of LAP phosphor
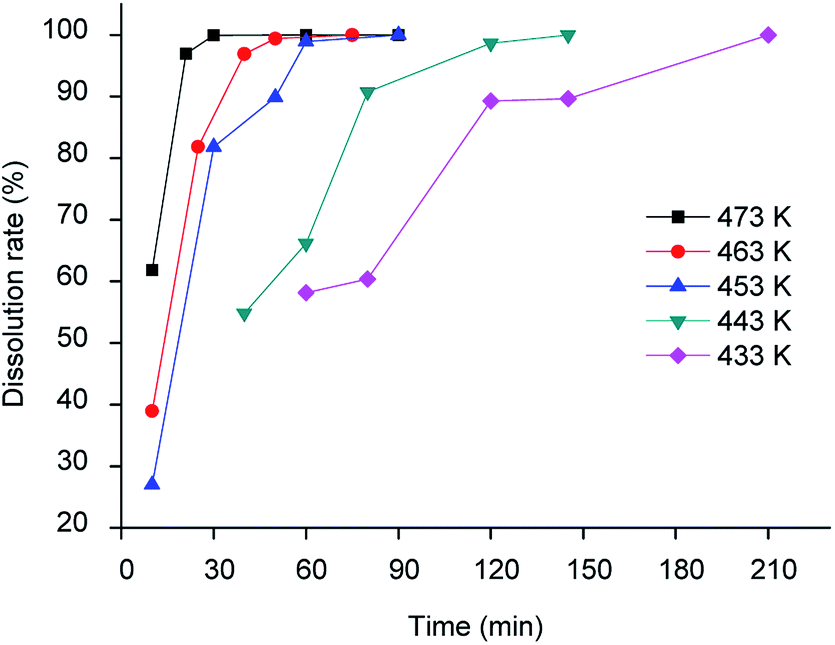
The dissolution of LAP phosphor (containing 34.1 wt% La, 16.0 wt% Ce and 8.2 wt% Tb) was tested first in methanesulphonic acid (MSA, 13 mol dm−3) and triflic acid (9 mol dm−3), at 433 K using a liquid-to-solid (L/S) ratio of 60 cm3 g−1. More than 80% of the sample was dissolved after 1 hour. However, a gel in which cerium and lanthanum aggregated was formed in both solvents. Gel formation could be prevented by using concentrated MSA (15.4 mol dm−3). Quantitative dissolution of the LAP phosphor was observed after heating for 3 h at 433 K and a L/S of 60 cm3 g−1. Therefore, concentrated MSA was selected as the lixiviant for further experiments, which were performed to optimise both the temperature and the L/S ratio. As expected, higher temperatures resulted in faster dissolution rates. However, to prevent thermal decomposition of MSA, the maximum temperature should not exceed 473 K. Quantitative dissolution could be achieved in 40 min at 473 K, but 1 h was selected as the optimised time to ensure full dissolution (Fig. 1).
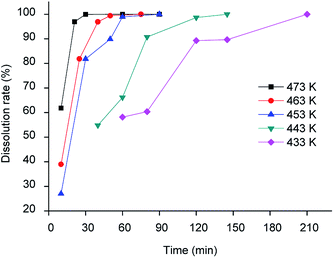 |
| | Fig. 1 Dissolution rate of LAP phosphor with concentrated MSA as a function of temperature (L/S = 15 cm3 g−1). | |
In this test, the dissolution efficiency was determined by weighing the LAP phosphor before and after the leaching step. Since terbium, cerium and lanthanum are in the same host matrix, the dissolution efficiency of the individual elements separately is equal to the total dissolution efficiency. The optimal L/S ratio was determined to be 15 cm3 g−1. Smaller ratios (i.e. using less liquid) resulted again in gel formation, thus lowering the leaching yield. The reaction between the LAP phosphor and methanesulphonic acid can be represented as follows for the main component LaPO4:
| | |
LaPO4 + 3CH3SO3H → La(CH3SO3)3 + H3PO4
| (4) |
A main advantage of the rare-earth methanesulphonates is their high solubility in aqueous solutions; for instance, the solubility of La(CH3SO3)3 in water is 1110 g dm−3 at 296 K.28
After the MSA leachate was allowed to cool to room temperature, the recovery of the REEs was first tested by addition of oxalic acid as precipitating agent. Since the REE oxalates could not be precipitated from concentrated MSA, the leachate was diluted first by addition of water. However, the dilution factor of the leachate and the excess of oxalic acid required for quantitative precipitation were too high to be of any practical value. Therefore, solvent extraction was considered as an alternative.
Solvent extraction
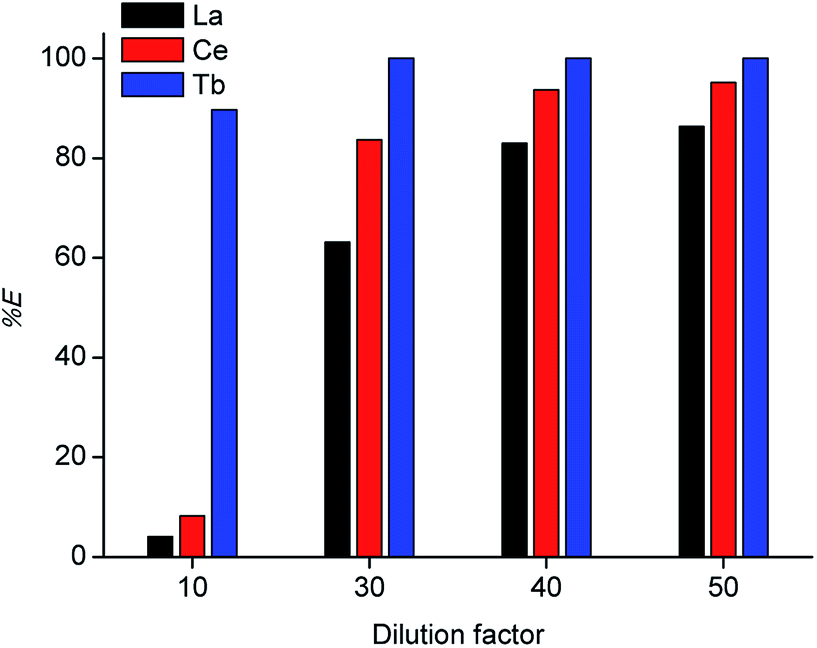
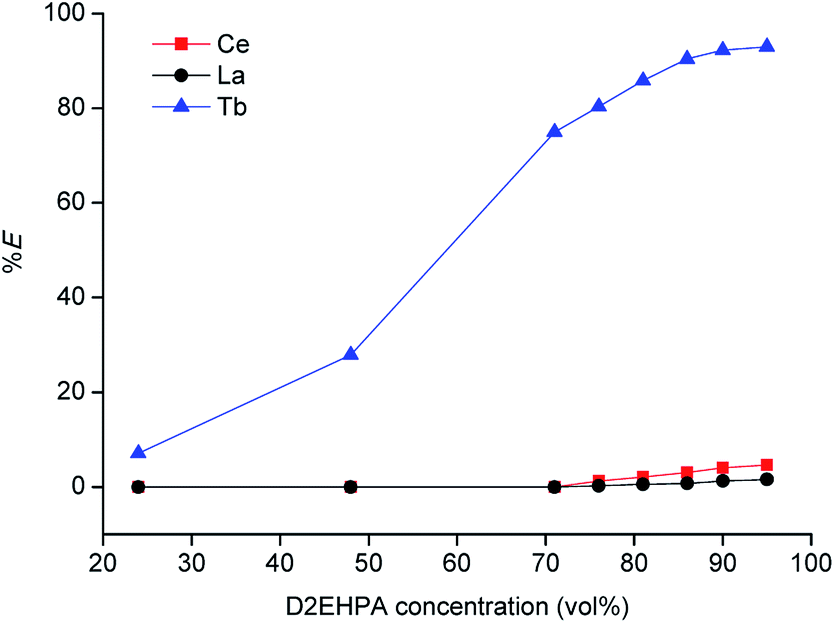
Tri-n-butyl phosphate (TBP), bis(2,4,4-trimethylpentyl)phosphinic acid (Cyanex 272) and di-(2-ethylhexyl)phosphoric acid (D2EHPA) were tested as extractants, with toluene, xylene and n-dodecane as diluents. The results are shown in Tables 1 and 2. The best results were obtained for undiluted D2EHPA (highest extraction of La and Ce) and for D2EHPA diluted in xylene (selectivity for Tb). These results confirm the strong effect of the type of diluent on the extraction REEs by D2EHPA, as recently described by our research group.29 Aliphatic diluents give much higher extraction efficiency for all the REEs than aromatic diluents (with the highest efficiency for the undiluted system), but the separation factors can be improved by selecting an aromatic diluent. An aromatic diluent suppresses the extraction of the light REEs (La and Ce), while the extraction efficiencies for the heavy REEs (e.g. Tb) remains high. An additional advantage of aromatic diluents is that gel formation is suppressed for the extraction of REEs by D2EHPA.29 In Fig. 2 solvent extraction tests with undiluted D2EHPA are reported for different dilution factors of the concentrated MSA leachate by water before solvent extraction. By selecting a low dilution factor of 10, selective extraction of terbium over cerium/lanthanum could be achieved. This effect could be further enhanced by lowering the extractant concentration: by using ≤70 vol% D2EHPA in xylene, co-extraction of lanthanum and cerium was completely suppressed (Fig. 3), although the extraction efficiency of terbium also decreased compared to extractions from solutions that were diluted 30 to 50 times. Lower phase ratios resulted in lower terbium extraction efficiencies, so that a phase ratio of 1 was chosen as the optimal one. Based on these observations, a three-stage cross-current extraction was performed from 10 times diluted MSA leachate containing La, Ce and Tb, 70 vol% D2EHPA in xylene, phase ratio = 1, T = 298 K and t = 15 min. Only terbium was extracted, with a cumulative percentage extraction of 74.9, 87.0 and 97.5% obtained in the three steps. Co-extraction of lanthanum and cerium was not observed at all (0% extraction). Hence, a pure terbium phase was obtained in a high yield.
Table 1 Percentage extraction (%) from 40 times diluted MSA leachate with several extraction systems (phase ratio = 1, T = 298 K, t = 1 h)
| Extraction system |
La |
Ce |
Tb |
| 50% Cyanex 272 in xylene |
0 |
0 |
0 |
| 50% Cyanex 272 in n-dodecane |
0 |
0 |
0 |
| Undiluted Cyanex 272 |
0 |
0 |
0 |
| 50% D2EHPA in xylene |
3.6 |
14.0 |
100 |
| 50% D2EHPA in n-dodecane |
47.9 |
79.7 |
100 |
| Undiluted D2EHPA |
77.1 |
94.9 |
100 |
Table 2 Percentage extraction (%) from 10 times diluted MSA leachate with several extraction systems (phase ratio = 1, T = 298 K, t = 1 h)
| Extraction system |
La |
Ce |
Tb |
| 30% TBP in toluene |
0 |
0 |
0 |
| 60% TBP in toluene |
0 |
0 |
0 |
| Undiluted TBP |
0 |
0 |
0 |
| 29% D2EHPA in toluene |
0 |
0 |
5.5 |
| 57% D2EHPA in toluene |
0 |
0 |
47.9 |
| Undiluted D2EHPA |
4.1 |
8.2 |
89.7 |
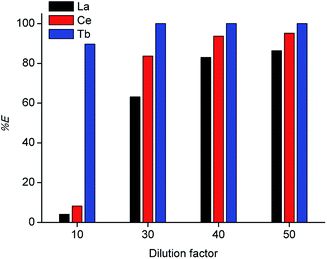 |
| | Fig. 2 Percentage extraction (% E) for solvent extraction from diluted MSA leachate with undiluted D2EHPA extractant, for different dilution factors (phase ratio = 1, T = 298 K, t = 1 h). | |
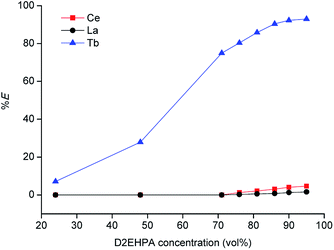 |
| | Fig. 3 Percentage extraction (% E) of La, Ce and Tb for solvent extraction from 10 times diluted MSA leachate by D2EHPA extractant in xylene diluent (phase ratio = 1, T = 298 K, t = 15 min), first extraction step. | |
Cerium and lanthanum could be extracted after removal of terbium, by further diluting the raffinate to a total dilution factor of 40. By contacting this diluted raffinate with undiluted D2EHPA (at a phase ratio of 1), cerium and lanthanum could be extracted with an efficiency of 93.1% and 78.1%, respectively. By using three extraction stages, the extraction percentages could be increased up to 100% for cerium and 98.8% for lanthanum. The higher extraction percentages obtained from the diluted MSA leachate can be explained with the fact that D2EHPA is an acidic extractant, therefore an increase in the pH, by dilution of the acid in the leachate, leads to an increase of metal extraction (the equilibrium is shifted to the right). The extractant D2EHPA was used in its undiluted form because, as shown in Table 1, this gave the highest extraction efficiencies for lanthanum and cerium. Furthermore, the higher extraction obtained for the heavy REEs compared to those for the light REEs can be explained with the decreasing ionic radii of these elements and the consequent increased charge density.30 It is well known that D2EHPA is a strong extractant for heavy REEs.31 In fact, extraction of the elements at the end of the lanthanides series (ytterbium and lutetium) is so strong that it is very difficult to strip these REEs from the loaded organic phase. For a detailed discussion of the extraction mechanism of REEs by D2EHPA, the reader is referred to the recent literature.32
Stripping
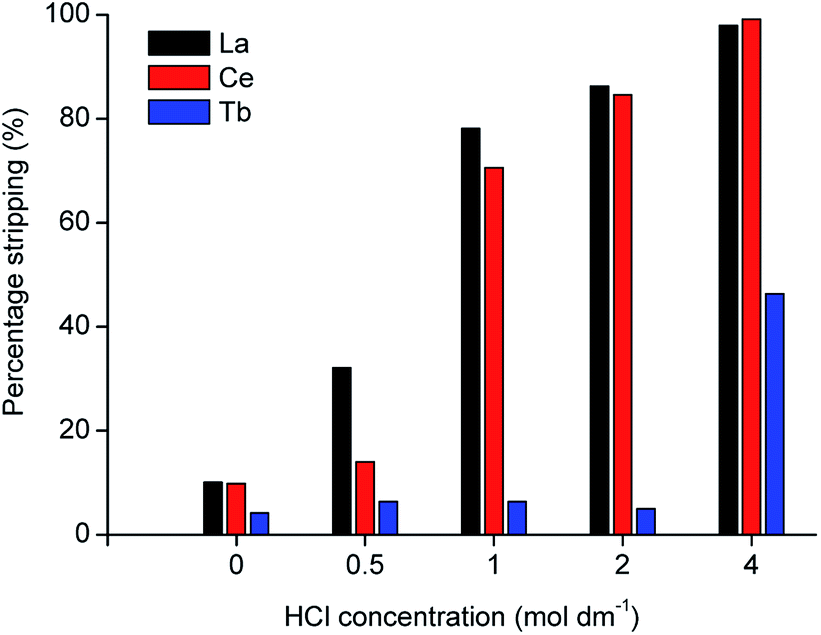
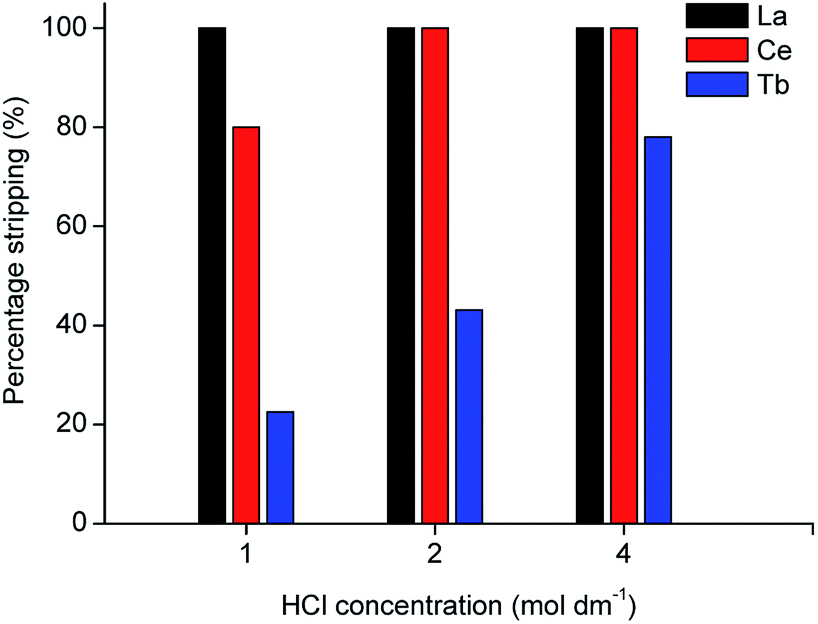
In the proposed process, two organic phases were obtained after extraction of the REEs from the MSA leachate: a 70 vol% D2EHPA solution containing terbium and an undiluted D2EHPA solution containing cerium and lanthanum. Metal recovery from the loaded organic phase was first tested by stripping with HCl solution. The D2EHPA was contacted with several solutions of different HCl concentrations for two stages, as shown in Fig. 4 and 5.
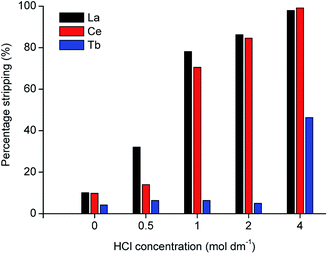 |
| | Fig. 4 Stripping efficiency (%) of La, Ce and Tb from 40 times diluted MSA leachate as a function of HCl concentration (phase ratio = 1, T = 298 K, t = 1 h, first stripping stage). | |
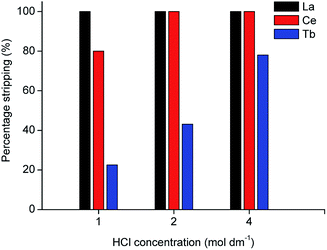 |
| | Fig. 5 Stripping efficiency (%) of La, Ce and Tb from 40 times diluted MSA leachate as a function of HCl concentration (phase ratio = 1, T = 298 K, t = 1 h, second stripping stage). | |
It was observed that even with 4 mol dm−3 HCl, terbium is not completely stripped after 2 stages. Precipitation tests with a stoichiometric amount of pure oxalic acid were performed, but even with high dilutions of the leachate, the precipitation efficiency was below 50%.
Both phases were thus contacted with an aqueous solution containing a large excess of oxalic acid compared to the rare-earth concentration, namely 1.5 mol dm−3. It was found that by using an organic-to-aqueous (O/A) phase ratio of 20, quantitative precipitation of lanthanum and cerium could be achieved, while terbium precipitation was about 90% (T = 298 K, t = 40 min), as shown in Table 3. The undiluted D2EHPA (containing the remaining 10% Tb), the 70 vol% D2EHPA and the remaining oxalic acid solutions can be recycled back into the process. The REEs could be recovered as their oxalates, which can be transformed to the corresponding oxides (Tb4O7 and CeO2 + La2O3) by calcination (t = 4 h, T = 973 K).
Table 3 Precipitation tests with 1.5 mol dm−3 oxalic acid (T = 298 K, t = 40 min)
| O/A phase ratio |
Tb precipitation efficiency, % |
La and Ce precipitation efficiency, % |
| 1 |
95 |
100 |
| 10 |
91 |
100 |
| 20 |
90 |
>99.5 |
Recovery process applied on the real residue
The process was then tested on a residue obtained from the HydroWEEE process, by leaching of the lamp phosphor waste by a sulphuric acid solution.9 The concentration of the main elements present in this residue was determined by borate fusion, followed by ICP-OES analysis (Table 4). This residue contains also silicon, zinc and sulphur, but the behaviour of these elements was not investigated in this work. The results show that the residue contains, besides the expected terbium, gadolinium, lanthanum and cerium, also significant concentrations of yttrium and europium. This is caused by reaction of the yttrium and europium brought in solution upon dissolution of the YOX phosphor with the phosphate ions of the dissolved HALO phosphor, forming poorly soluble REE phosphates.
Table 4 Composition of the residue left after leaching of lamp phosphor waste with H2SO4
| Element |
Concentration (wt%) |
| Y |
2.2 |
| La |
1.2 |
| Ce |
1.1 |
| Tb |
0.5 |
| Eu |
0.3 |
| Gd |
0.1 |
Table 5 shows the results of the leaching tests performed with concentrated MSA at 473 K for 1 h. About 74% terbium, 78% cerium and 95% lanthanum could be leached from the residue. By increasing the leaching time to more than 1 h, the increase in the leaching efficiency of terbium, cerium and lanthanum was negligible, whilst the leaching of europium and gadolinium reached 100% after 24 h. Therefore, a leaching time of 1 h was selected as the optimal time. In the work performed by Tunsu et al., several leaching agents were tested for rare earths recovery from spent fluorescent lamps (HNO3, HCl, H2SO4 and MSA) at different concentrations (0.5–4 mol dm−3).14 It was found that very long leaching times were required to achieve significant dissolution of Ce, La and Tb and the leaching efficiencies increase by increasing acid concentration. In particular, when using 4 mol dm−3 MSA, 37.3% Ce, 6.6% La and 45.1% Tb dissolved after 168 h. These long leaching times are due to the fact that the leaching was carried out with diluted acids and at low temperature (293 ± 1 K). The fact that not all the lanthanum, cerium and terbium could be recovered from the residue of the HydroWEEE process can be explained by the fact that not all the green phosphors is LaPO4:Ce3+,Tb3+ (LAP); some of the terbium is in the phosphors CeMgAl11O19:Tb3+ (CAT) and (Ce, Gd)MgB5O10:Tb3+ (CBT), which cannot be dissolved by concentrated MSA. This hypothesis is supported by the fact that the recovery of lanthanum is much higher than that of cerium and terbium. Indeed, all the lanthanum is present in the LAP phosphor, whereas cerium and terbium are present in the LAP, CAT and CBT phosphors.
Table 5 Leaching efficiencies of the lamp phosphor residue with concentrated MSA (LS = 15 cm3 g−1, T = 473 K, t = 1 h)
| Element |
Leaching efficiency, % |
| La |
94.7 |
| Y |
82.7 |
| Ce |
78.3 |
| Tb |
74.3 |
| Eu |
58.2 |
| Gd |
51.8 |
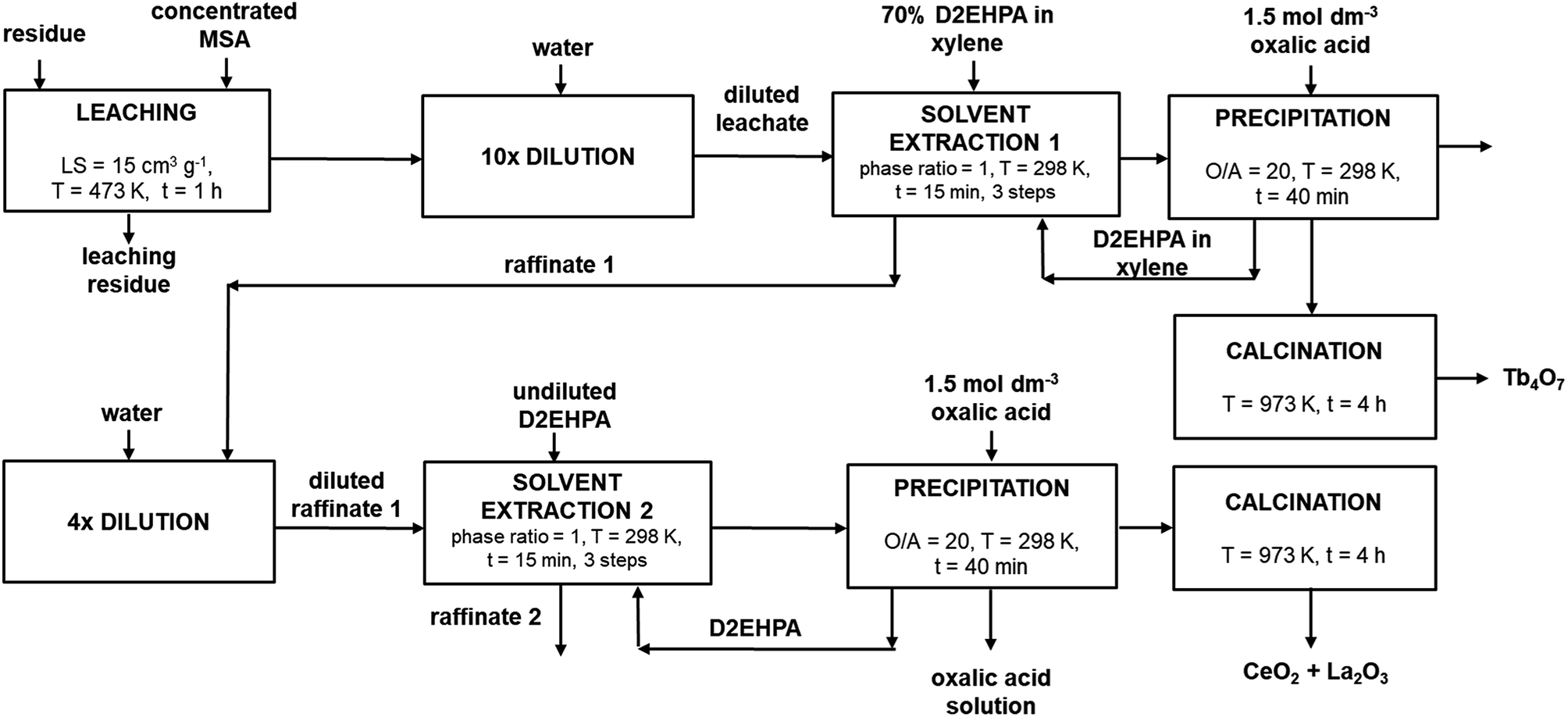
Solvent extraction, oxalate precipitation and calcination conditions described for the synthetic LAP phosphor leachate were subsequently applied to the leachate of the residue and were found to be well suited. Moreover, it was found that terbium could be efficiently extracted by using a lower phase ratio (0.5 instead of 1), without decreasing the extraction efficiency. Table 6 shows the composition of the mixed REE oxides obtained after calcination of the oxalate precipitates, calculated by measuring the concentration in the organic phase before and after precipitation. Mixed oxide 1 refers to the oxide which contains terbium, while mixed oxide 2 refers to the oxide rich in lanthanum and cerium. The low purity of the terbium oxide is mainly due to the co-extraction of the leached europium, gadolinium and yttrium, as shown in Table 5. A detailed flow-sheet of the new process is shown in Fig. 6.
Table 6 Composition (wt%) of the mixed REE oxides recovered from the lamp phosphor residue
| Oxide |
Mixed oxide 1 |
Mixed oxide 2 |
| Y2O3 |
76.2 |
0.0 |
| Tb4O7 |
13.1 |
0.4 |
| Eu2O3 |
8.6 |
0.0 |
| Gd2O3 |
1.8 |
0.0 |
| La2O3 |
0.4 |
55.8 |
| CeO2 |
0.0 |
43.8 |
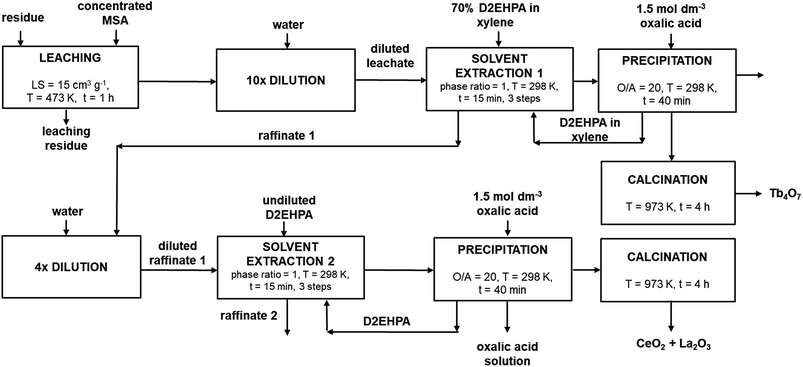 |
| | Fig. 6 Detailed process flow-sheet for REEs recovery from the LAP phosphor. | |
Conclusions
A process with a solvometallurgical leaching step was developed to recover the rare earths present in the residue of the HydroWEEE process for valorising lamp phosphor waste.9 The process makes use of the green organic acid methanesulphonic acid as lixiviant and allows obtaining high leaching efficiencies (74% Tb, 78% Ce and 95% La) in a relatively short time (1 h). The results show that most of the REEs present in the green phosphor LaPO4:Ce3+,Tb3+ (LAP) could be recovered, whereas methanesulphonic acid could not dissolve the green phosphors CeMgAl11O19:Tb3+ (CAT) and (Ce,Gd)MgB5O10:Tb3+ (CBT). By carefully selecting the subsequent solvent extraction conditions, it was possible to achieve high selectivity for terbium over lanthanum and cerium for experiments on LAP phosphor. Yttrium, europium and gadolinium, which were also present in the residue of the HydroWEEE process as impurities, were co-extracted as well so that mixed REE oxides were obtained instead of a pure product. These mixed REE oxides can be further purified by solvent extraction.33–35 The advantage of the process is that the leaching can be carried out at lower temperatures than digestion in concentrated sulphuric acid or fused alkali. The process takes advantage of the much higher solubility of the rare-earth methanesulphonates compared to the corresponding sulphates, so that solvent extraction can be performed directly on the leachate after dilution, without the need of several additional steps to convert the rare-earth sulphates into chlorides or nitrates.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has received funding from the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement No. 680629 (REMAGHIC: New Recovery Processes to produce Rare Earth-Magnesium Alloys of High Performance and Low Cost) (project website: http://www.remaghic-project.eu). The authors acknowledge Relight Srl (Rho, Italy) for providing the residue.
Notes and references
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef.
- K. Binnemans and P. T. Jones, J. Rare Earths, 2014, 32, 195–200 CrossRef.
- C. Tunsu, M. Petranikova, M. Gergoric, C. Ekberg and T. Retegan, Hydrometallurgy, 2015, 156, 239–258 CrossRef.
- Q. Tan, J. Li and X. Zeng, Crit. Rev. Environ. Sci. Technol., 2015, 45, 749–776 CrossRef.
- D. Dupont and K. Binnemans, Green Chem., 2016, 18, 176–185 RSC.
- C. Tunsu, C. Ekberg and T. Retegan, Hydrometallurgy, 2014, 144, 91–98 CrossRef.
- C. Tunsu, J. B. Lapp, C. Ekberg and T. Retegan, Hydrometallurgy, 2016, 166, 98–106 CrossRef.
- C. Tunsu, M. Petranikova, C. Ekberg and T. Retegan, Sep. Purif. Technol., 2016, 161, 172–186 CrossRef.
- V. Innocenzi, I. De Michelis, F. Ferella and F. Veglio, Int. J. Miner. Process., 2017, 168, 87–94 CrossRef.
- M. A. Rabah, Waste Manag., 2008, 28, 318–325 CrossRef PubMed.
- I. De Michelis, F. Ferella, E. F. Varelli and F. Veglio, Waste Manag., 2011, 31, 2559–2568 CrossRef PubMed.
- C. H. Lee, C. H. Liao, S. R. Popuri and C. E. Hung, J. Mater. Cycles Waste Manage., 2017, 19, 1235–1243 CrossRef.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- C. Tunsu, C. Ekberg, M. Foreman and T. Retegan, Trans. Inst. Min. Metall., Sect. C, 2016, 125, 199–203 Search PubMed.
- N. M. Ippolito, V. Innocenzi, I. De Michelis, F. Medici and F. Veglio, J. Cleaner Prod., 2017, 153, 287–298 CrossRef.
- V. Innocenzi, N. M. Ippolito, I. De Michelis, F. Medici and F. Veglio, J. Environ. Manage., 2016, 184, 552–559 CrossRef PubMed.
- L. Chunfa, L. Zhenyuan, Z. Yanliang, C. Jingyuan, Z. Liqin and W. Li, J. Rare Earths, 2017, 35, 1008–1013 CrossRef.
- S. G. Zhang, M. Yang, H. Liu, D. A. Pan and J. J. Tian, Rare Met., 2013, 32, 609–615 CrossRef.
- S. Van Loy, K. Binnemans and T. Van Gerven, J. Cleaner Prod., 2017, 156, 226–234 CrossRef.
- Q. Tan, C. Deng and J. Li, J. Cleaner Prod., 2017, 142, 2187–2191 CrossRef.
- Q. Tan, C. Deng and J. Li, Sci. Rep., 2016, 6, 19961 CrossRef PubMed.
- G. Song, W. Yuan, X. Zhu, X. Wang, C. Zhang, J. Li, J. Bai and J. Wang, J. Cleaner Prod., 2017, 151, 361–370 CrossRef.
- S. Cagno, L. Gijsemans, V. Tyrpekl, T. Cardinaels, M. Verwerft and K. Binnemans, J. Sustain. Metall., 2017, 3, 659–667 CrossRef.
- M. Gernon, M. Wu, T. Buszta and P. Janney, Green Chem., 1999, 1, 127–140 RSC.
- K. Binnemans and P. T. Jones, J. Sustain. Metall., 2017, 3, 570–600 CrossRef.
- S. Riaño, M. Regadío, K. Binnemans and T. Vander Hoogerstraete, Spectrochim. Acta, Part B, 2016, 124, 109–115 CrossRef.
- M. Regadío, S. Riaño, K. Binnemans and T. Vander Hoogerstraete, Anal. Chem., 2017, 89, 4595–4603 CrossRef PubMed.
- BASF SE, Lutropur® – the friendly acid. The purest form of MSA methanesulfonic acid made, 2011.
- N. K. Batchu and K. Binnemans, Hydrometallurgy, 2018, 177, 146–151 CrossRef.
- M. Gergoric, C. Ekberg, B. M. Steenari and T. Retegan, J. Sustain. Metall., 2017, 3, 601–610 CrossRef.
- T. Sato, Hydrometallurgy, 1989, 22, 121–140 CrossRef.
- J. Zhang, B. D. Zhao, B. Schreiner, Separation Hydrometallurgy of Rare Earth Elements, Springer International Publishing, 2016 Search PubMed.
- D. Fontana and L. Pietrelli, J. Rare Earths, 2009, 27, 830–833 CrossRef.
- V. Innocenzi, N. M. Ippolito, L. Pietrelli, M. Centofanti, L. Piga and F. Vegliò, J. Cleaner Prod., 2018, 172, 2840–2852 CrossRef.
- C. Tunsu, C. Ekberg, M. Foreman and T. Retegan, Solvent Extr. Ion Exch., 2014, 32, 650–668 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Federica Forte
,
Federica Forte