
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast interface charge transfer dynamics on P3HT/MWCNT nanocomposites probed by resonant Auger spectroscopy†
Yunier Garcia-Basabe *ae,
Denis Ceolinb,
Aldo J. G. Zarbinc,
Lucimara S. Romand and
Maria Luiza M. Roccoa
*ae,
Denis Ceolinb,
Aldo J. G. Zarbinc,
Lucimara S. Romand and
Maria Luiza M. Roccoa
aInstituto de Química, Universidade Federal do Rio de Janeiro, 21941-909, Rio de Janeiro, RJ, Brazil
bSynchrotron Soleil, L'Orme des Merisiers Saint-Aubin, BP 48 91192 Gif-sur-Yvette, France
cDepartamento de Química, Universidade Federal do Paraná, 81531-990, Curitiba, PR, Brazil
dDepartamento de Física, Universidade Federal do Paraná, 81531-990, Curitiba, PR, Brazil
eInstituto Latino Americano de Ciências da Vida e da Natureza, Universidade Federal da Integração Latino-Americana, UNILA, 85866-900, Foz do Iguaçu, PR, Brazil. E-mail: yunier.basabe@unila.edu.br; Fax: +55-21-3938-7265; Tel: +55-45-3576-2113
First published on 24th July 2018
Abstract
The interfacial electronic structure and charge transfer dynamics of poly-3-hexylthiophene (P3HT) and multi-walled carbon nanotube (Fe-MWCNT) nanocomposites were investigated by near-edge X-ray absorption fine structure (NEXAFS) and resonant Auger (RAS) spectroscopies around the sulfur K-edge. Nanocomposites with 5 wt% (P3HT/Fe-MWCNT-5%) and 10 wt% (P3HT/Fe-MWCNT-10%) of Fe-MWCNT species were prepared and compared with pristine P3HT film. The quantitative NEXAFS analysis shows a strong π–π interchain interaction of the pristine P3HT polymer film, which is reduced by the presence of the Fe-MWCNT. S–KL2,3L2,3 RAS spectra were measured at photon energies corresponding to the main electronic transitions appearing in the S–K edge NEXAFS spectrum. Ultrafast charge transfer times were estimated from the RAS spectra using the core-hole clock approach with the S 1s core-hole lifetime as an internal clock. The π–π interchain charge transfer time increases from 4.7 fs on pristine P3HT polymer to 6.5 fs on the P3HT/Fe-MWCNT-5% nanocomposite. The electronic coupling between P3HT and Fe-MWCNT species occurs mainly through the P3HT π* molecular orbital. The increase of Fe-MWCNT concentration from 5 to 10 wt% reduces the charge transfer rate at the resonance maximum due probably to Fe-MWCNT aggregation, reducing the P3HT and Fe-MWCNT electronic coupling.
1. Introduction
Organic-based electronic devices have emerged in the last decades as a viable alternative to silicon inorganic counterparts due to their low-cost processing, simple manufacturing process and flexibility.1–5 The organic active layers, π-conjugated semiconductor-based polymers, have been extensively explored in organic light emitting diodes, field effect transistors, sensors and organic solar cells applications.6–9 Among semiconductor polymers, the regioregular poly[3-hexylthiophene-2,5-diyl] (P3HT) has been the prototypical polymer used to investigate organic optoelectronic device applications.10,11 The P3HT polymer combined with some carbon-based nanomaterials like fullerene (C60 and its derivative PCBM), graphene and multiwalled carbon nanotubes (MWCNT) has been extensively used in heterojunction blend configurations.12–21 Specifically, carbon nanotubes (CNTs) have attracted much attention thanks to their high carrier mobility and excellent chemical and mechanical stability.22,23 Many reports about the morphological, optical and electronic properties of P3HT–CNT nanocomposites and their relationship with preparation routes can be found in the literature.19,20,24–28 In this sense, it is known that the interfacial charge transfer process between P3HT and CNTs species has an important role in the device performance, considering photodiodes, sensors or OFETs. On the other hand, this process can be notably affected by many factors such as interface hybridization and film ordering and morphology. Therefore, understanding how charge transfer between the P3HT and CNT interface is affected by the concentration of each component and by film ordering is necessary to control these factors and improve applicability. Some studies about ultrafast charge and energy transfer in P3HT–CNT interfaces employing laser excited pump-probe spectroscopy were previously reported.29–34 However, this method is limited to processes occurring at tens of femtosecond (10−15 s) timescale due to the laser bandwidth and delay time between pump and probe lasers.35,36 An alternative capable to reach charge transfer dynamics in the attosecond (10−18 s) or subfemtosecond timescale is the core-hole clock (CHC) method.37–39 CHC is an elemental sensitive synchrotron-based spectroscopy that uses the core-hole lifetime as an internal clock.Our motivation to study the P3HT/Fe-MWCNT nanocomposites with concentrations of 5 and 10 wt% of Fe-MWCNT species is based on previous results reported by Canestraro et al.12 They reported an improvement in the photovoltaic device efficiency for P3HT/Fe-MWCNT film as active layer when the concentration of Fe-MWCNT species increases from 1 to 5 wt%. However, for films prepared with concentration of 10 wt% of Fe-MWCNT species the photoconversion efficiency is reduced significantly. Similarly, more recently, Rathore et al.20 investigated the influence of MWCNT doping on performance of the bulk heterojunction based devices. In this study they found that the devices performance increases with addition of MWCNT for MWCNT weight concentration up to 4 wt% while as weight concentration increases beyond 4 wt% the devices performance is significantly deteriorated. Therefore, in this paper we present near-edge X-ray absorption fine structure (NEXAFS) and resonant Auger (RAS) studies on the poly-3-hexylthiophene (now on: P3HT) and nanocomposites thin films made of poly-3-hexylthiophene and multi-walled carbon nanotubes (MWCNT) filled with iron/iron-oxide (now on: P3HT/Fe-MWCNT), obtained at the sulfur K-edge. The dynamic of charge transfer was determined using the CHC approach with S 1s core-hole lifetime as the internal clock, showing that this parameter is notably affected by the presence of Fe-MWCNT species. A strong charge transfer rate through π–π interchain interaction was measured in pristine P3HT, which is reduced by the presence of MWCNT. The increase of the concentration from 5 to 10 wt% decreases the rate of the charge transfer process across P3HT and MWCNT interface due to nanotube aggregation, reducing the interfacial electronic coupling between these two species.
2. Experimental
2.1. Sample preparation
The multi-walled carbon nanotubes filled with iron/iron oxide (Fe-MWCNT), used in this work, were prepared through the pyrolysis of ferrocene in a poor oxygen-containing atmosphere (for details about synthesis, structure and properties see ref. 40). They have external diameter varying from 8 to 140 nm (50% of them between 23 and 48 nm) and length of 2–5 mm. Also, in their majority the tubes are filled with Fe or Fe3O4. In order to study the spectroscopic properties of P3HT/Fe-MWCNT composites films we follow similar procedure in the fabrication of photovoltaic devices as in ref. 41 The polymer/Fe-MWCNT mixture was produced by dispersing a known quantity of Fe-MWCNT (5 and 10 wt% in relation to the polymer amount) in chloroform. The Fe-CNT dispersion was sonicated for approximately 2 h. Then, a fixed quantity of poly-3-hexylthiophene (P3HT, from Merck) was added in each bottle, resulting in a 5 mg mL−1 polymer solution. The polymer/Fe-MWCNT nanocomposites were deposited by spin coating onto ITO/glass substrate from the chloroform solution described above.2.2. Characterization
The experiments were performed at the French synchrotron facility SOLEIL, GALAXIES beamline,42 on the hard X-ray photoelectron spectroscopy (HAXPES) end station.43 A U20 undulator covering the 2.3–12 keV photon energy range provides linearly polarized light, monochromatized by a Si(111) double-crystal. Electrons are analyzed by a large acceptance angle EW4000 Scienta hemispherical analyzer, whose lens axis is set collinear to the polarization. The XPS and RAS spectra were recorded at 45° incidence/emission angle, and the NEXAFS spectra were recorded by measuring the current on the sample while scanning the photon energy.3. Results and discussion
Fig. 1a and b shows S 1s NEXAFS spectra for poly-3-hexylthiophene (P3HT) deposited on ITO/glass substrate and nanocomposite thin film of P3HT/multi-walled carbon nanotube (P3HT/Fe-MWCNT-5%). The spectra are similar to previously reported X-ray absorption data on thiophene-based polymers: polythiophene (PT),44–46 poly-3-methylthiophene (P3MT),47 and also P3HT and P3HT/Fe-MWCNT nanocomposite (with 10% of Fe-MWCNT).48 It presents an intense peak, which was assigned to the overlapping of the S 1s → π* and S 1s → σ*(S–C) transitions, followed by weak structures. This is a well establish assignment corroborated by resonant Auger spectroscopy (RAS) tuned around S 1s edge for polythiophene45,46 and also by recent theoretical calculations.49 Due to the overwhelming experimental resolution after close inspection of the NEXAFS spectrum of Fig. 1a it is possible to identify a shoulder at the low photon energy side, corresponding to the π* symmetry state due to strong π–π interchain interaction.50,51 This peak can be also observed in the angular dependent study at the S–K edge NEXAFS spectra of P3HT film obtained by doctor blade casting52 and also for the P3HT:PCBM blend.53 The other feature with maximum at around 2480 eV above sulfur 1s ionization potential corresponds to higher energy excitations. The small peak appearing on the pre-absorption edge region (around 2469 eV) may be attributed to atomic sulfur impurity accordingly to previous report.54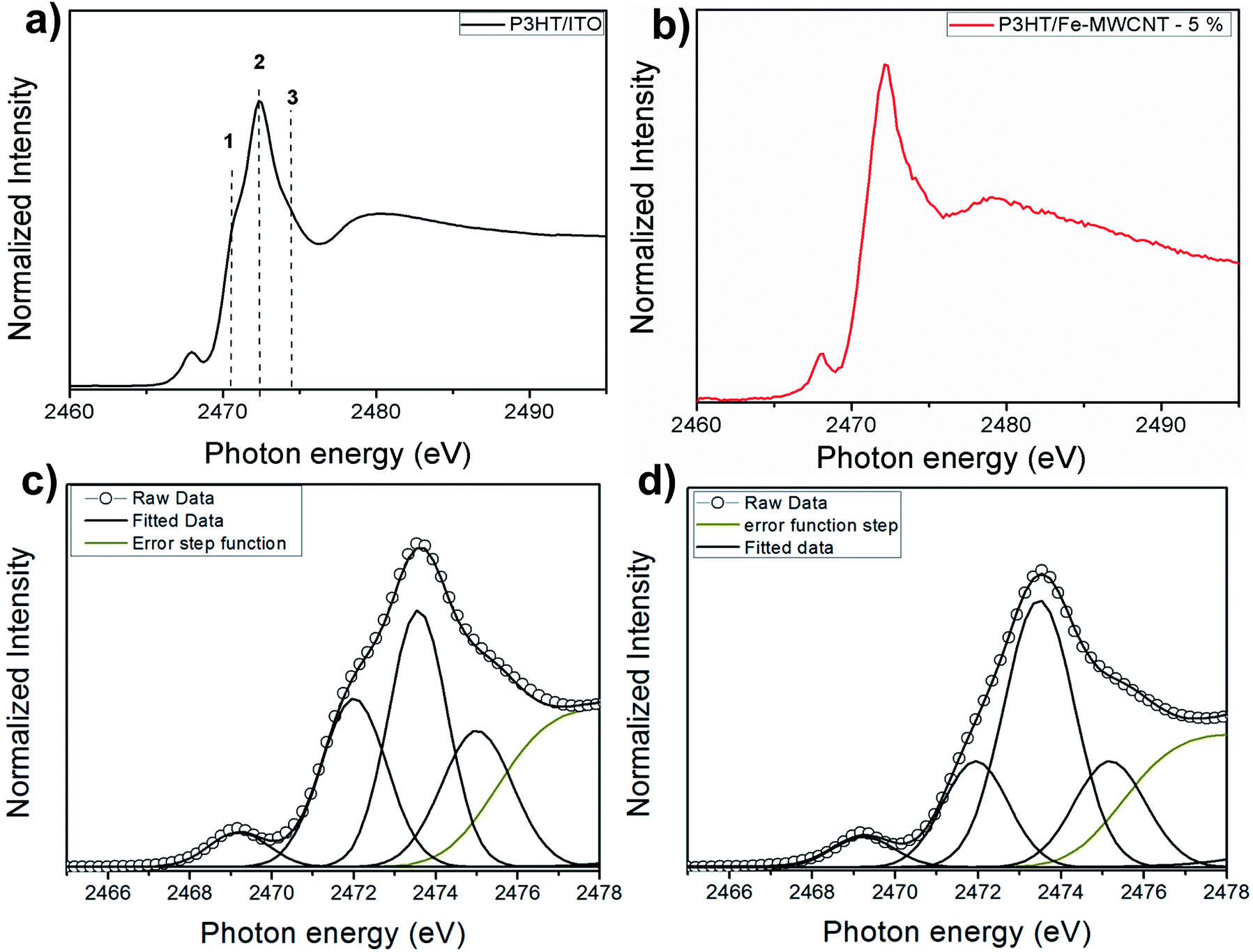 | ||
| Fig. 1 Sulfur 1s NEXAFS spectra for: (a) P3HT/ITO; (b) P3HT/Fe-MWCNT-5%. (c) and (d) Fitted NEXAFS spectra of P3HT/ITO and P3HT/Fe-MWCNT-5%, respectively. The labels 1–3 represent the photon energies used to obtain the RAS spectra. | ||
A quantitative analysis was performed through NEXAFS spectra fitting as shown in Fig. 1c and d. The spectra were deconvoluted using Gaussian profile function to three features in the photon energy region of the S–K edge resonance. The quantitative results are presented in Table 1. The most notable effect in S–K edge NEXAFS spectra when passing to the nanocomposite is the decrease of peak 1 intensity due to the presence of the carbon nanotubes. Comparing S 1s NEXAFS spectra of regioregular P3HT film and polycrystalline powder (RR-P3HT) it was shown a reduction in the LUMO energy level which the authors suggested as being directly associated to the π–π intermolecular interaction present for the regioregular polymeric film.55 This would explain the remarkable signal related to the π* transition well visualized in the spectrum of Fig. 1c for pristine P3HT, which also demonstrate the quality of the film deposition. Moreover, according to several morphological studies on P3HT/Fe-MWCNT nanocomposites the polymer wraps around the nanotube.24–26,41,56 Thus, this configuration between P3HT and Fe-MWCNT may reduce the π–π interchain stacking of P3HT polymer and consequently affect the intensity of peak 1 as observed by the S 1s NEXAFS spectrum.
| Peak | P3HT | P3HT/Fe-MWCNT-5% | ||||
|---|---|---|---|---|---|---|
| Photon energy (eV) | FWHM (eV) | Area (%) | Photon energy (eV) | FWHM (eV) | Area (%) | |
| 1 | 2471.9 | 1.89 | 26 | 2471.9 | 1.93 | 12 |
| 2 | 2473.2 | 1.63 | 30 | 2473.6 | 1.86 | 46 |
| 3 | 2474.4 | 2.01 | 20 | 2474.5 | 1.94 | 21 |
Ultrafast charge delocalization dynamics was evaluated using the core-hole clock (CHC) method through sulfur–KL2,3L2,3 Auger decay spectra. Fig. 2 shows the results of the deconvolution process for three energies labeled as 1–3 in Fig. 1. The decay Auger spectra are composed of 1S and 1D Auger multiplets.37 Similarly to previous data reported by Rocco et al.46 on polythiophene films, it is possible to separate the contributions of two spectator Auger channels: S 1s → π* (SP1) and S 1s → σ*(S–C) (SP2) peaks in the Auger decay spectra. Rydberg signal appearing at higher kinetic energy is also observed in the RAS spectra for these polymeric films with enhancement of their intensities at higher photon energies close to the ionization potential. The contribution appearing at ∼2112.5 eV kinetic energy labelled as NA (due to final state with two holes in the L shell similar to the Normal Auger spectrum after a core-ionized state) in Fig. 2 is attributed to electron transfer Auger decay channel.46
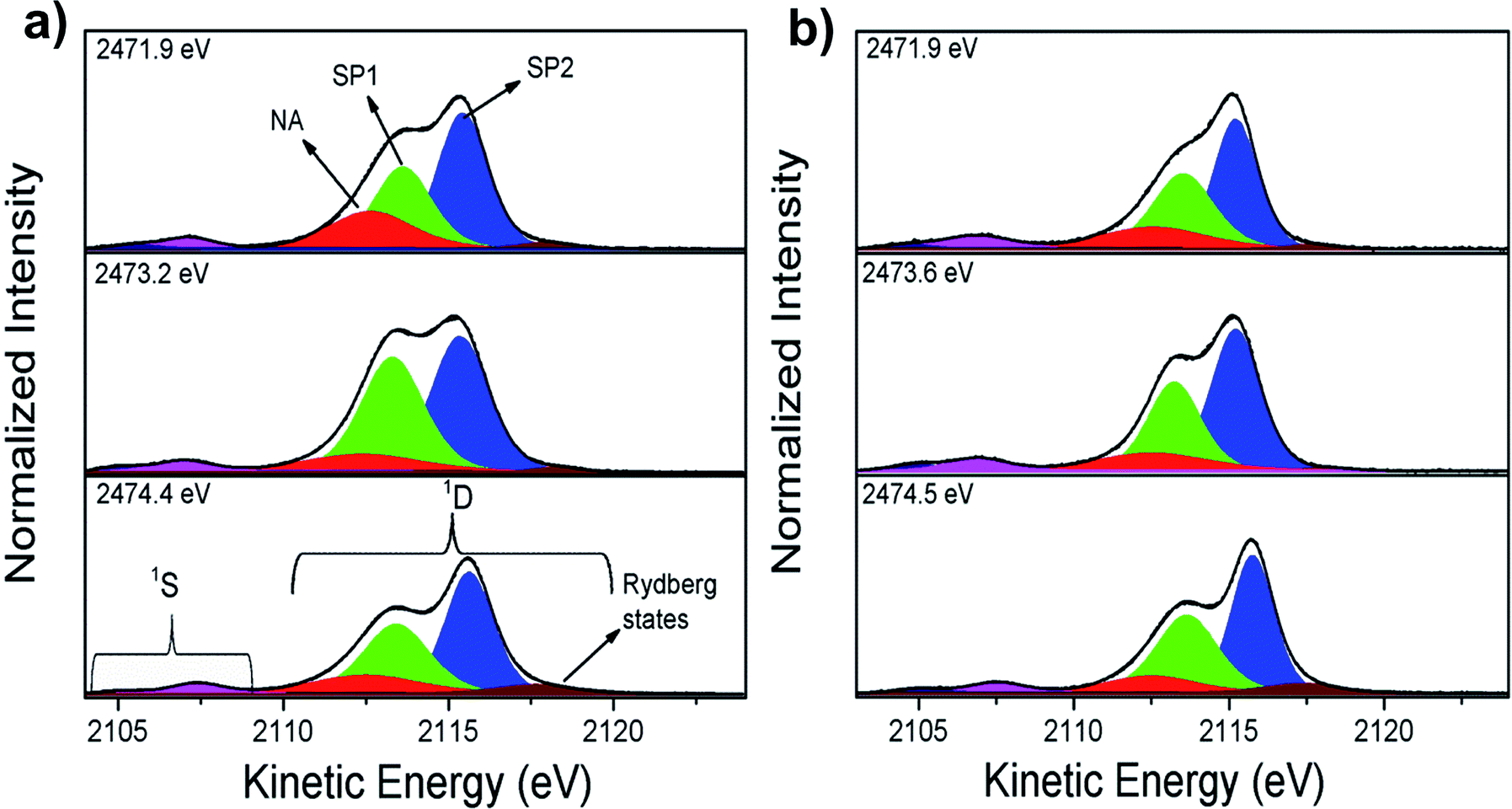 | ||
| Fig. 2 S–KL2,3L2,3 resonant Auger spectra and deconvolution results for: (a) P3HT and (b) P3HT/Fe-MWCNT-5% films. The spectator and normal Auger decay channels are leveled as SP1 (green), SP2 (blue) and NA (red), respectively. The Rydberg state is represented by wine color. The regions corresponding to 1S and 1D Auger multiplets are also delimited. | ||
As an evidence of the electron delocalization degree in P3HT/ITO and P3HT/Fe-MWCNT-5% films, the spectator shift parameter was estimated from RAS spectra measured at photon energies (hν) 2471.9 eV, 2473.2 eV and 2474.4 eV. The spectator shift effect is due to the screening of the core-hole by the excited electron acting as spectator in the decay process; therefore a small value for this parameter is indicative of delocalization of the excited electron away from the core-hole in timescales comparable to those of core-hole lifetimes. This parameter is calculated as the difference between kinetic energies of the SP features and non-resonant Auger decay (also called as normal Auger, NA). At hν = 2471.9 eV the spectator shift of the SP1 decay channel is 1.03 eV for P3HT and 1.42 eV for P3HT/Fe-MWCNT-5% films. However, at hν = 2473.2 eV the SP1 spectator shifts are 1.30 eV and 0.84 eV for P3HT/ITO and P3HT/Fe-MWCNT-5%, respectively, while at hν = 2474.4 eV the spectator shift parameter increases from 1.14 eV for P3HT/ITO to 1.45 eV for P3HT/Fe-MWCNT-5%. The analysis of the SP1 spectator shift parameters of the RAS spectra measured at these three photon energies shows that the electron delocalization degree of electron excited of π* symmetry (2471.9 eV) and σ* (2474.4 eV) states is higher for P3HT/ITO than P3HT/Fe-MWCNT-5%. This σ* transition presents a transition moment perpendicular to the polymer backbone, as previously discussed by H. Ikeura-Sekiguchi et al.55 and R. Onoki et al.57 However, at photon energy corresponding to the resonance maximum (2473.2 eV) the electron delocalization degree is higher for the P3HT/Fe-MWCNT-5% nanocomposite compared to P3HT/ITO. These results show that the electronic coupling between P3HT and Fe-MWCNT depends on the character of the electronic excited states.
The overall evolution of spectator and electron transfer decay channels when the incident photon energy is tuned across the S 1s excitation resonance for P3HT/ITO and P3HT/Fe-MWCNT-5% films is shown in Fig. 3. The sum of the two spectator contributions (Fig. 3a and c) for both films reproduces qualitatively the profile of the NEXAFS spectrum. A maximum is observed for the electron transfer decay channel (NA) for the P3HT film at lower photon energy (∼2471.9 eV) corresponding to quick electron transfer due to strong π–π interchain stacking of the P3HT polymer. The increased maximum at higher photon energy means that the electron delocalization effect is energetically favorable for excitation energies close to the ionization potential. On the other hand, the relative intensity of the NA channel in P3HT/Fe-MWCNT-5% film (Fig. 3d) shows a maximum around 2473.6 eV photon energy corresponding to the overlapping of the S 1s → π* and S 1s → σ*(S–C) transitions.
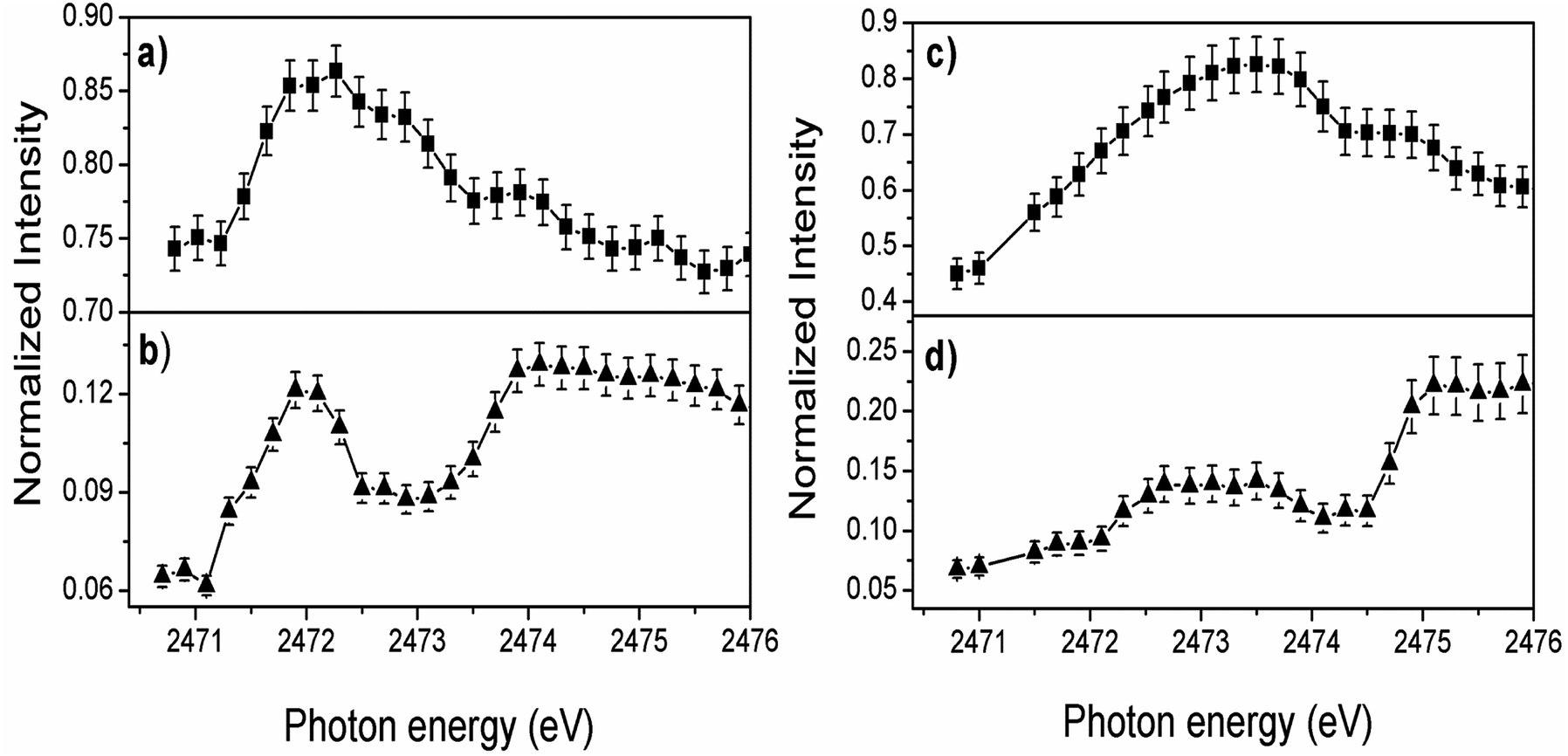 | ||
| Fig. 3 Normalized intensities plotted against incident photon energy of (a) sum of spectators (SP1 + SP2) and (b) normal Auger components for P3HT/ITO film. (c) and (d) Show the sum of spectators (SP1 + SP2) and normal Auger components for P3HT/Fe-MWCNT-5% film. The error bars represent uncertainties of 5% from the fit of the spectator and normal Auger peak areas. | ||
For a quantitative evaluation of the data, charge transfer times (τCT) was calculated for the two samples from the ratio between spectators and normal Auger (with constant kinetic energy of ∼2112.5 eV) signals by τCT = (Ispectator/Inormal) × τCH equation38 and using S 1s core-hole lifetime τCH = 1.27 fs as internal clock.58 The results are presented in Table 2 for the photon energies labelled as 1–3 in Fig. 1. The description and application of the CHC approach to thiophene-based polymers can be found elsewhere45,46,53,59 and is summarized in the ESI.†
| Peak | τCT (fs) | ||
|---|---|---|---|
| P3HT | P3HT/Fe-MWCNT-5% | P3HT/Fe-MWCNT-10% | |
| a The τCT standard deviation values are shown in parentheses. | |||
| 1 | 4.7(2) | 6.5(3) | 6.1(3) |
| 2 | 8.9(3) | 5.3(2) | 8.1(3) |
| 3 | 5.5(3) | 7.6(3) | 7.4(3) |
From the comparative analysis, we can observe that charge transfer times are quite different for P3HT/Fe-MWCNT-5% and for pristine P3HT/ITO films. Firstly, we can see a faster charge transfer pathway through π–π interchain stacking (4.7 fs) in pristine P3HT polymer. As discussed previously from the NEXAFS data the presence of Fe-MWCNT species reduces notably the π–π interchain stacking of the P3HT polymer causing an increase of the charge transfer time to 6.5 fs for the P3HT/Fe-MWCNT-5% sample. This increase may be due to the fact that in this composite the MWCNT are wrapped around by P3HT polymer.41 This morphological configuration could hinder interlayer π–π interchain charge transfer pathways of P3HT film. From S–K L2,3L2,3 RAS deconvolution showed in Fig. 2 we can observe that the main factor causing faster charge transfer process at the resonance maximum (2473.2 eV photon energy) in P3HT/Fe-MWCNT-5% is the decrease of the SP1 decay channel, which reflects the NEXAFS data. Quantitatively, the Raman fraction (f = ISP1/(ISP1 + INA))38,39 of this spectator contribution determined at the resonance maximum decreases from 0.69 in P3HT to 0.49 in the nanocomposite. The SP1 contribution is related to the S 1s → π* transition, as previously discussed. Therefore, we can conclude that the Fe-MWCNT are interacting with the P3HT through thiophene π* electronic states. Similar result was reported by Jin et al.60 in the study of carboxylic MWCNT and regioregular P3HT nanocomposites. They found a photoluminescence quenching in this system which was associated with the possibility of electron transfer between P3HT and MWCNT due to the π–π interaction of these two species. The charge transfer between P3HT and MWCNT interface is also observed by time-resolved pump-probe absorption transient spectroscopy.31 They found a photoinduced absorption signal assigned to charge states in P3HT that increases with the presence of MWCNT.
The effect of the carbon nanotube concentration in the ultrafast electron transfer delocalization was also investigated here for P3HT/Fe-MWCNT-10%. S–K-edge NEXAFS spectrum and deconvolution results are presented in Fig. S1 and Table S1 in the ESI.† The S–KL2,3L2,3 resonant Auger spectra measured at three features labeled as 1, 2 and 3 photon energies are shown in Fig. S2.† Charge transfer times reported in Table 2 increase from 5.3 to 8.1 fs for higher amount of Fe-MWCNT measured at resonance maximum (peak 2). However, for the other two excitation energies (1 and 3) the τCT is unchanged with the increase from 5 to 10 wt% of Fe-MWCNT species. These results reinforce that the electronic coupling between P3HT and Fe-MWCNT species occurs mainly through P3HT π* molecular orbital. The increase in the τCT of P3HT/Fe-MWCNT-10% compared with P3HT/Fe-MWCNT-5% nanocomposite may be also associated with MWCNT species aggregation, resulting in poor polymer/MWCNT interface.19,20 Similar result was reported by Lioudakis et al.61 for exciton dissociation studies in P3HT/PCBM system by pump-probe spectroscopy. They found that exciton dissociation decreases with the increase of PCBM concentration and it was associated to the loss of P3HT crystallinity. PCBM like MWCNT tends to form easily undesired aggregates,62,63 therefore similar effects here should be expected. The aggregation of Fe-MWCNT species (due to van der Waals interactions) in the nanocomposites reduces the electronic coupling with P3HT polymer. The effect of the increase of Fe-MWCNT concentration from 5 to 10 wt% was also reported by Canestraro et al.12 showing a decrease of the photoconversion efficiency in an organic solar cell device. They explained this behavior due to a decrease of the internal field in the polymer, necessary to dissociate the exciton.
4. Conclusions
In this study we explored the interfacial electronic structure and the ultrafast charge transfer dynamics in P3HT/Fe-MWCNT nanocomposites. The charge transfer times were evaluated by the core-hole clock (CHC) approach. The electronic interaction between P3HT and Fe-MWCNT and the polymer ordering in the nanocomposites were investigated by S–K-edge NEXAFS spectroscopy. Quantitative NEXAFS analysis shows that the presence of Fe-MWCNT reduces π–π interchain stacking in P3HT polymer. This result was corroborated by the CHC approach where the charge transfer time between π–π interchain increase from 4.7 fs on pristine P3HT polymer to around 6–7 fs on P3HT/Fe-MWCNT-5% and 10% nanocomposites. The more efficient electron transfer channel at P3HT/Fe-MWCNT interface occurs through π electrons. This result was corroborated by NEXAFS and CHC techniques. The increase of Fe-MWCNT concentration from 5 to 10 wt% reduces the charge transfer rate due to Fe-MWCNT aggregation, reducing the P3HT and Fe-MWCNT electronic coupling.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Experiments were performed at the GALAXIES beamline, SOLEIL Synchrotron, France (Proposal No. 20120010). The authors are grateful to the SOLEIL staff for assistance during the beamtime. M. L. M. R., L. S. R. and A. J. G. Zarbin would like to thank CNPq for financial support. A. J. G. Zarbin would like INCT Nanocarbon for finantla support. We would also like to thank CNPq for a PDJ scholarship (Y. Garcia-Basabe).References
- W. Cao and J. Xue, Energy Environ. Sci., 2014, 7, 2123–2144 RSC.
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef PubMed.
- J. D. Myers and J. Xue, Polym. Rev., 2012, 52, 1–37 CrossRef.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef PubMed.
- P. Hudhomme, EPJ Photovoltaics, 2013, 4, 1–11 CrossRef.
- M. Eising, C. E. Cava, R. V. Salvatierra, A. J. G. Zarbin and L. S. Roman, Sens. Actuators, A, 2017, 245, 25–33 CrossRef.
- A. R. Aiyar, J. I. Hong, R. Nambiar, D. M. Collard and E. Reichmanis, Adv. Funct. Mater., 2011, 21, 2652–2659 CrossRef.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, M. Alamoudi, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- C. D. Canestraro, M. S. Schnitzler, A. J. G. Zarbin, M. G. M. da Luz and L. S. Roman, Appl. Surf. Sci., 2006, 252, 5575–5578 CrossRef.
- M. D. Irwin, D. B. Buchholz, A. W. Hains, R. P. Chang and T. J. Marks, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2783–2787 CrossRef.
- C. Yang, J. K. Lee, A. J. Heeger and F. Wudl, J. Mater. Chem., 2009, 19, 5416–5423 RSC.
- S. Ren, M. Bernardi, R. R. Lunt, V. Bulovic, J. C. Grossman and S. Gradecak, Nano Lett., 2011, 11, 5316–5321 CrossRef PubMed.
- Q. Liu, Z. Liu, X. Zhang, L. Yang, N. Zhang, G. Pan, S. Yin, Y. Chen and J. Wei, Adv. Funct. Mater., 2009, 19, 894–904 CrossRef.
- D. Yu, K. Park, M. Durstock and L. Dai, J. Phys. Chem. Lett., 2011, 2, 1113–1118 CrossRef PubMed.
- J. Huang, D. R. Hines, B. J. Jung, M. S. Bronsgeest, A. unnell, V. Ballarotto, H. E. Katz, M. S. Fuhrer, E. D. Williams and J. Cumings, Org. Electron., 2011, 12, 1471–1476 CrossRef.
- P. Rathore, C. M. S. Negi, A. S. Verma, A. Singh, G. Chauhan, A. R. Inigo and S. K. Gupta, Mater. Res. Express, 2017, 4, 085905 CrossRef.
- P. Rathore, C. M. S. Negi, A. Yadav, A. S. Verma and S. K. Gupta, Optik, 2018, 160, 131–137 CrossRef.
- N. Boulanger, V. Yu, M. Hilke, M. F. Toney and D. R. Barbero, Phys. Chem. Chem. Phys., 2018, 20, 4422–4428 RSC.
- P. C. Mahakul, K. Sa, B. Das and P. Mahanandia, Mater. Chem. Phys., 2017, 199, 477–484 CrossRef.
- C. J. An, Y. C. Lee, Y. H. Kang and S. Y. Cho, Carbon, 2017, 124, 662–668 CrossRef.
- M. Giulianini, E. R. Waclawik, J. M. Bell, M. De Crescenzi, P. Castrucci, M. Scarselli and N. Motta, Appl. Phys. Lett., 2009, 95, 1–3 Search PubMed.
- M. Giulianini, E. R. Waclawik, J. M. Bell, M. Scarselli, P. Castrucci, M. De Crescenzi and N. Motta, Polymers, 2011, 3, 1433–1446 CrossRef.
- C. Caddeo, C. Melis, L. Colombo and A. Mattoni, J. Phys. Chem. C, 2010, 114, 21109–21113 CrossRef.
- A. P. P. Alves, J. P. C. Trigueiro, H. D. R. Calado and G. G. Silva, Electrochim. Acta, 2016, 209, 111–120 CrossRef.
- S. Agbolaghi and S. Zenoozi, Org. Electron., 2017, 51, 362–403 CrossRef.
- A. Nakamura, T. Koyama, Y. Miyata and H. Shinohara, J. Phys. Chem. C, 2016, 120, 4647–4652 CrossRef.
- N. Banerji, S. Cowan, E. Vauthey and A. J. Heeger, J. Phys. Chem. C, 2010, 115, 9726–9739 CrossRef.
- L. Lüer, S. Hoseinkhani, M. Meneghetti and G. Lanzani, Phys. Rev. B, 2010, 81, 1–6 CrossRef.
- D. J. Bindl, N. S. Safron and M. S. Arnold, ACS Nano, 2010, 4, 5657–5664 CrossRef PubMed.
- S. D. Stranks, C. Weisspfennig, P. Parkinson, M. B. Johnston, L. M. Herz and R. J. Nicholas, Nano Lett., 2011, 11, 66–72 CrossRef PubMed.
- J. M. Holt, A. J. Ferguson, N. Kopidakis, B. A. Larsen, J. Bult, G. Rumbles and J. L. Blackburn, Nano Lett., 2010, 10, 4627–4633 CrossRef PubMed.
- M. Cho, Chem. Rev., 2008, 108, 1331–1418 CrossRef PubMed.
- D. L. Andrews and A. A. Demidov, An introduction to laser spectroscopy, Springer Science & Business Media, 2012 Search PubMed.
- A. Föhlish, S. Vijayalakshmi, F. Hennies, W. Wurth, V. R. R. Medicherla and W. Drube, Chem. Phys. Lett., 2007, 434, 214–217 CrossRef.
- D. Menzel, Chem. Soc. Rev., 2008, 37, 2212–2223 RSC.
- R. Friedlein, S. Braun, M. P. de Jong, W. Osikowicz, M. Fahlman and W. R. Salaneck, J. Phys. Chem. C, 2011, 183, 101–106 Search PubMed.
- M. C. Schnitzler, M. M. Oliveira, D. Ugarte and A. J. G. Zarbin, Chem. Phys. Lett., 2003, 381, 541–548 CrossRef.
- M. Koehler, C. D. Canestraro, M. S. Schnitzler, M. M. Oliveira, A. J. G. Zarbin, M. G. M. da Luz and L. S. Roman, Europhys. Lett., 2007, 79, 1–6 CrossRef.
- J.-P. Rueff, J. M. Ablett, D. Céolin, D. Prieur, T. Moreno, V. Balédent, B. Lassalle-Kaiser, J. E. Rault, M. Simon and A. Shukla, J. Synchrotron Radiat., 2015, 22, 175–179 CrossRef PubMed.
- D. Céolin, J. M. Ablett, D. Prieur, T. Moreno, J.-P. Rueff, T. Marchenko, L. Journel, R. Guillemin, B. Pilette, T. Marin and M. Simon, J. Electron Spectrosc. Relat. Phenom., 2013, 190, 188–192 CrossRef.
- H. Ikeura-Sekiguchi and T. Sekiguchi, Surf. Interface Anal., 2008, 40, 673–675 CrossRef.
- Y. Garcia-Basabe, B. G. A. L. Borges, D. C. Silva, L. Micaroni, L. S. Roman and M. L. M. Rocco, Org. Electron., 2013, 14, 2980–2986 CrossRef.
- C. Arantes, B. Beck, B. G. A. L. Borges, G. Araújo, L. S. Roman and M. L. M. Rocco, J. Phys. Chem. C, 2013, 117, 8208–8213 CrossRef.
- J. R. Santa Rita, C. Arantes, G. Araújo, L. S. Roman, L. Micaroni and M. L. M. Rocco, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 265–269 CrossRef.
- G. Araújo, C. Arantes, L. S. Roman, A. J. G. Zarbin and M. L. M. Rocco, Surf. Sci., 2009, 603, 647–652 CrossRef.
- G. N. George, M. J. Hackett, M. Sansone, M. L. Gorbaty, S. R. Kelemen, R. C. Prince, H. H. Harris and I. J. Pickering, J. Phys. Chem. A, 2014, 118, 7796–7802 CrossRef PubMed.
- P. K.-H. Ho, L.-L. Chua, M. Dipankar, X. Gao, D. Qi, A. T.-S. Wee, J.-F. Chang and R. H. Friend, Adv. Mater., 2007, 19, 215–221 CrossRef.
- X.-T. Hao, T. Hosokai, N. Mitsuo, S. Kera, K. K. Okudaira, K. Mase and N. Ueno, J. Phys. Chem. B, 2007, 111, 10365–10372 CrossRef PubMed.
- U. Aygül, D. Batchelor, U. Dettinger, S. Yilmaz, S. Allard, U. Scherf, H. Peisert and T. Chassé, J. Phys. Chem. C, 2012, 116, 4870–4874 CrossRef.
- B. G. A. L. Borges, A. G. Veiga, L. Tzounis, A. Laskarakis, S. Logothetidis and M. L. M. Rocco, J. Phys. Chem. C, 2016, 120, 25078–25082 CrossRef.
- Y. Takata, Y. Kitajima, H. Aga, S. Yagi, T. Asahi, T. Yokoyama, K. Tanaka and T. Ohta, Jpn. J. Appl. Phys., 1993, 32, 350–352 CrossRef.
- H. Ikeura-Sekiguchi and T. Sekiguchi, Jpn. J. Appl. Phys., 2014, 53, 1–4 CrossRef.
- M. Bernardi, M. Giulianini and J. C. Grossman, ACS Nano, 2010, 4, 6599–6606 CrossRef PubMed.
- R. Onoki, G. Yoshikawa, Y. Tsuruma, S. Ikeda, K. Saiki and K. Ueno, Langmuir, 2008, 24, 1605–11610 CrossRef PubMed.
- J. L. Campbell and T. Papp, At. Data Nucl. Data Tables, 2001, 77, 1–56 CrossRef.
- Y. Garcia-Basabe, C. F. N. Marchiori, B. G. A. L. Borges, N. A. D. Yamamoto, A. G. Macedo, M. Koehler, L. S. Roman and M. L. M. Rocco, J. Appl. Phys., 2014, 115, 1–7 CrossRef.
- H.-D. Jin, F. Zheng, W.-L. Xu, W.-H. Yuan, M.-Q. Zhu and X.-T. Hao, J. Phys. D: Appl. Phys., 2014, 47, 1–6 Search PubMed.
- E. Lioudakis, I. Alexandrou and A. Othonos, Nanoscale Res. Lett., 2009, 4, 1475–1480 CrossRef PubMed.
- P. C. Mahakul and P. Mahanandia, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 178, 1–8 Search PubMed.
- N. J. Alley, K.-S. Liao, E. Andreoli, S. Dias, E. P. Dillon, A. W. Orbaek, A. R. Barron, H. J. Byrne and S. A. Curran, Synth. Met., 2012, 162, 95–101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04629h |
| This journal is © The Royal Society of Chemistry 2018 |