
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic structure and luminescence assets in white-light emitting Ca2V2O7, Sr2V2O7 and Ba2V2O7 pyro-vanadates: X-ray absorption spectroscopy investigations
Aditya Sharma *,
Mayora Varshney,
Keun-Hwa Chae and
Sung Ok Won*
*,
Mayora Varshney,
Keun-Hwa Chae and
Sung Ok Won*
Advance Analysis Centre, Korea Institute of Science and Technology (KIST), Seoul-02792, South Korea. E-mail: adityaiuac@gmail.com; sowon@kist.re.kr
First published on 24th July 2018
Abstract
Ca2V2O7, Sr2V2O7, and Ba2V2O7 pyro-vanadates were synthesized using a modified chemical precipitation method and annealing. Detailed crystal structure, morphology, electronic structure and optical properties were investigated by XRD, UV-visible absorption, FTIR, Raman, FE-SEM, XANES, and photoluminescence spectroscopy. Rietveld refinement on the XRD patterns of Ca2V2O7, Sr2V2O7, and Ba2V2O7 has confirmed the triclinic structure (space group; P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2)) of the pyro-vanadates. The band gap energy of Ca2V2O7, Sr2V2O7, and Ba2V2O7 is estimated to be ∼2.67 eV, ∼2.97 eV and ∼3.09 eV, respectively. XANES spectra at the Ca L-edge, Sr K-edge and Ba L-edge have confirmed the Ca2+, Sr2+ and Ba2+ ions in the Ca2V2O7, Sr2V2O7 and Ba2V2O7 compounds, respectively. V K-edge XANES spectra have strengthened the presence of sub-pentavalent V ions in all of the pyro-vanadates. O K-edge XANES spectra of Ca2V2O7, Sr2V2O7 and Ba2V2O7 have shown dominating tetrahedral symmetry of the V ions which is also corroborated with the V K-edge XANES. Broad-band emission spectra, ranging from 400 nm to 700 nm, have been observed from the charge-transfer transitions of VO4 tetrahedra. 3T1 → 1A1 and 3T2 → 1A1 transitions, from the VO4 tetrahedra, have provided two distinct emission peaks from the compounds which exhibit a red-shift with the decreasing ionic-radii of alkali-earth metal ions. The mixed compounds, with equal weight proportions, have shown remarkable emission characteristics towards the realization of rare-earth element free white-light-emitting devices.
(2)) of the pyro-vanadates. The band gap energy of Ca2V2O7, Sr2V2O7, and Ba2V2O7 is estimated to be ∼2.67 eV, ∼2.97 eV and ∼3.09 eV, respectively. XANES spectra at the Ca L-edge, Sr K-edge and Ba L-edge have confirmed the Ca2+, Sr2+ and Ba2+ ions in the Ca2V2O7, Sr2V2O7 and Ba2V2O7 compounds, respectively. V K-edge XANES spectra have strengthened the presence of sub-pentavalent V ions in all of the pyro-vanadates. O K-edge XANES spectra of Ca2V2O7, Sr2V2O7 and Ba2V2O7 have shown dominating tetrahedral symmetry of the V ions which is also corroborated with the V K-edge XANES. Broad-band emission spectra, ranging from 400 nm to 700 nm, have been observed from the charge-transfer transitions of VO4 tetrahedra. 3T1 → 1A1 and 3T2 → 1A1 transitions, from the VO4 tetrahedra, have provided two distinct emission peaks from the compounds which exhibit a red-shift with the decreasing ionic-radii of alkali-earth metal ions. The mixed compounds, with equal weight proportions, have shown remarkable emission characteristics towards the realization of rare-earth element free white-light-emitting devices.
Introduction
Analogous to the transition metal based pyro-vanadates, M2V2O7 (M = transition metal; Mn, Co, Ni and Cu etc.),1–5 the alkali earth metal based A2V2O7 (A = Ca, Sr and Ba etc.) oxides are of special interest because of their promising applications in rare earth element free white-light-emitting devices6–10 and microwave-dielectrics.11,12The electron correlation effects (because of the V–O4, V–O5 and V–O6 kinds of conceivable polyhedral configuration of V and O ions) make A2V2O7 oxides attractive candidates for probing their intriguing correlations among the crystal structure, electronic/atomic structure and diverse applications. The alkali earth metal based pyro-vanadates (i.e., Ca2V2O7, Sr2V2O7 and Ba2V2O7 etc.) obey the triclinic crystal structure. Their band structure, near the Fermi level, is characteristically formed by the hybridization of the frontier V d-orbitals and O 2p-orbitals. The bottom of the conduction band is shaped by the anti-bonding O 2p and V 3d orbitals and the top of the valence band mainly consists of non-bonding O 2p-orbitals.8,10 The orbitals of the alkali earth elements provide a large conduction band-width and do not contribute to the band structure near the Fermi level. The energy-band structure and, thus, the electronic/optical/magnetic properties of A2V2O7 oxides are not only sensitive to the amalgamation of foreign elements13–15 but are also expected to be modified by the distortion in the V–O polyhedra. A marginal distortion in the metal–oxygen polyhedra can affect the hybridization between the O 2p and metal d orbitals, which indeed regulate the band structure near the Fermi level and, consequently, the electronic/optical properties of the compounds.16,17
Optical properties and illumination performance of the compounds are categorically determined by the electronic structure around the stimulated ions and energy transfer processes among them.18–22 In case of vanadium-based optical materials, the (VO4)3− groups obey broad and strong charge-transfer (CT) absorption bands near the UV region and, thus, produce broad-emission spectra related to the electronic/atomic structure of the compound where the V ions are coordinated by the four O ligands.8–10,19,20 In previous reports, the Sr2V2O7 and Ba2V2O7 have shown broad emission characteristics ranging from 400 nm to 700 nm and have signified the concept of vanadium-based and rare-earth-free white-light emitting phosphor materials with high efficiency.8–10,19,20 However, synthesis of single phase A2V2O7 oxides is challenging because of unpredictable nature of V ions to form secondary phases of VO2, V2O5 etc. under the chemical and/or thermal treatment and, therefore, high-purity phase fabrication of A2V2O7 compounds is still a thirsty area of research for establishing the financially viable and straightforward synthesis approaches.
Besides the single-phase synthesis of A2V2O7 compounds, investigations on the crystal structure and electronic structure properties, which may have diverse V–O hybridization and complex electronic correlation, are also indispensable using the modern, non-destructive and element specific techniques. In this regard, X-ray photoelectron spectroscopy (XPS) had been applied to probe the electronic structure and chemical environment in pyro-vanadates.23–25 The XPS is sensitive to a few surface layers of the samples and an insulating/less-conducting sample undergoes severe charging effects during the measurements. Therefore, XPS is not ideally suited to correlate the structural features to the electronic/atomic structure properties of bulk of the material. In this study, in order to investigate the relationship among the crystal structure, orbital hybridization and optical properties of A2V2O7 pyro-vanadates, we have systematically applied the X-ray absorption near edge structure (XANES) spectroscopy technique at O K-edge, V L-edge, Ca L-edge, Sr K-edge, V K-edge and Ba L-edge. Besides providing the detailed experimental results on the absorption edges of constituent elements of the samples, the XANES investigations presented below address the several aspects. First, to specifically look into the dissimilarities in the hybridization of frontier orbitals of V and O ions in Ca2V2O7, Sr2V2O7 and Ba2V2O7, the low energy XANES spectra at O K-edge and V L-edge were studied. Secondly, to understand valence state of constituent elements in the samples, systematic, XANES spectra at Ca L-edge, Ba L-edge, Sr K-edge, V K-edge were collected and analyzed.
Experimental
The analytical grade calcium nitrate (Ca(NO3)2·4H2O), strontium nitrate (Sr(NO3)2), Barium carbonate (BaCO3), and ammonium meta-vanadate (NH4VO3) were chosen as starting reagents for the synthesis of Ca2V2O7, Sr2V2O7 and Ba2V2O7 ceramics. The stoichiometric proportions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were set for the alkali earth metal and vanadium precursors for preparing the A2V2O7 pyro-vanadates. The stoichiometric mixture of precursors was dispersed in 100 ml DI water under the sonication for 10 min. Then the solutions were stirred for 1 h with a bottom plate temperature of 100 °C. After fair dissolution of precursors, the pH of the solution was increased up to 9 by adding the NH4OH. After increasing pH, each sample was continuously stirred for one more hour at the same temperature. The precipitates were washed with 1 L water and then filtered. The filtered cake was dried in an oven at 80 °C for overnight. Such samples were crushed in the mortar-pestle and calcined at 800 °C for 4 hours. The X-ray diffraction measurements were performed using the Dmax-2500 (Rigaku) X-ray diffractometer. This machine uses the source of Cu Kα radiation (λ = 1.5418 Å), operated at the voltage of 40 kV and current of 200 mA. A wide scan range (5–140°) was scanned with the fixed time scan mode. For this 2 seconds was the fixed time for each 0.01° step. UV-visible absorption spectra were collected from the Cary500 (Agilent, US) UV-VIS-NIR spectrophotometer. This machine can produce the photon wavelength ranging from 3300 nm to 175 nm with dual beam method. Fourier-transformed infrared spectroscopy (FT-IR) measurements were performed using iS10FT-IR spectrophotometer (ThermoFisher Scientific). This instrument can provide a wide range of photon wavenumber ranging from 6500 cm−1 to 500 cm−1. The Raman spectra were collected by using the Renishaw (In-Via Raman) microscope with an Nd:YAG (neodymium-doped yttrium aluminum garnet) laser, which produces a photon beam of the wavelength of 532 nm. Scanning electron microscopy measurements were performed by applying the FEI (Inspect F50), scanning electron microscope with operational voltage < 30 kV. Photoluminescence was measured using a Varian-Cary eclipse fluorescence spectrophotometer equipped with a 150 W xenon lamp as the excitation source. The low energy XANES spectra at the O K-edge, V L-edge and Ca L-edge were collected in total electron yield (TEY) mode at the soft X-rays beamline, 10D-XAS-KIST, at Pohang Accelerator Laboratory (PAL), South Korea. The photon energy resolution of this beamline was better than 0.6 eV (at the O K-edge). The high energy XANES at V K-edge, Ba L-edge, Sr K-edge were collected from the 1D XRS KIST-PAL beamline. The procedure of data collection and uses of gas-mixtures were kept similar to our previous experiments.26–28
:1) were set for the alkali earth metal and vanadium precursors for preparing the A2V2O7 pyro-vanadates. The stoichiometric mixture of precursors was dispersed in 100 ml DI water under the sonication for 10 min. Then the solutions were stirred for 1 h with a bottom plate temperature of 100 °C. After fair dissolution of precursors, the pH of the solution was increased up to 9 by adding the NH4OH. After increasing pH, each sample was continuously stirred for one more hour at the same temperature. The precipitates were washed with 1 L water and then filtered. The filtered cake was dried in an oven at 80 °C for overnight. Such samples were crushed in the mortar-pestle and calcined at 800 °C for 4 hours. The X-ray diffraction measurements were performed using the Dmax-2500 (Rigaku) X-ray diffractometer. This machine uses the source of Cu Kα radiation (λ = 1.5418 Å), operated at the voltage of 40 kV and current of 200 mA. A wide scan range (5–140°) was scanned with the fixed time scan mode. For this 2 seconds was the fixed time for each 0.01° step. UV-visible absorption spectra were collected from the Cary500 (Agilent, US) UV-VIS-NIR spectrophotometer. This machine can produce the photon wavelength ranging from 3300 nm to 175 nm with dual beam method. Fourier-transformed infrared spectroscopy (FT-IR) measurements were performed using iS10FT-IR spectrophotometer (ThermoFisher Scientific). This instrument can provide a wide range of photon wavenumber ranging from 6500 cm−1 to 500 cm−1. The Raman spectra were collected by using the Renishaw (In-Via Raman) microscope with an Nd:YAG (neodymium-doped yttrium aluminum garnet) laser, which produces a photon beam of the wavelength of 532 nm. Scanning electron microscopy measurements were performed by applying the FEI (Inspect F50), scanning electron microscope with operational voltage < 30 kV. Photoluminescence was measured using a Varian-Cary eclipse fluorescence spectrophotometer equipped with a 150 W xenon lamp as the excitation source. The low energy XANES spectra at the O K-edge, V L-edge and Ca L-edge were collected in total electron yield (TEY) mode at the soft X-rays beamline, 10D-XAS-KIST, at Pohang Accelerator Laboratory (PAL), South Korea. The photon energy resolution of this beamline was better than 0.6 eV (at the O K-edge). The high energy XANES at V K-edge, Ba L-edge, Sr K-edge were collected from the 1D XRS KIST-PAL beamline. The procedure of data collection and uses of gas-mixtures were kept similar to our previous experiments.26–28
Results
(a) XRD, UV-visible absorption, FTIR, Raman and FE-SEM studies
Fig. 1 shows the Rietveld refined XRD patterns of pyro-vanadate ceramics sintered at 800 °C for 4 hours. All ceramics flawlessly matched with their relevant JCPDF files; #97-002-0609, #97-007-2200 and #97-003-4320 for Ca2V2O7, Sr2V2O7 and Ba2V2O7 respectively. The initial structure models in the Rietveld refinements of a pyro-vanadate compound were taken from the respective JCPDF files. The insets of Fig. 1 show the magnified view of the simulated patterns. The crystallographic data of Ca2V2O7, Sr2V2O7, and Ba2V2O7 is presented in Table 1. It is noticeable from the Table 1 that all three ceramics have a triclinic structure with P(2) space group symmetry without forming any secondary phase and, thus, are signifying the single phase formation of pyro-vanadate in this study. Moreover, the lattice parameters and cell volume are higher for the A2V2O7 ceramics with increasing the atomic number of the alkali earth element and are consistent with the previous reports.8–10
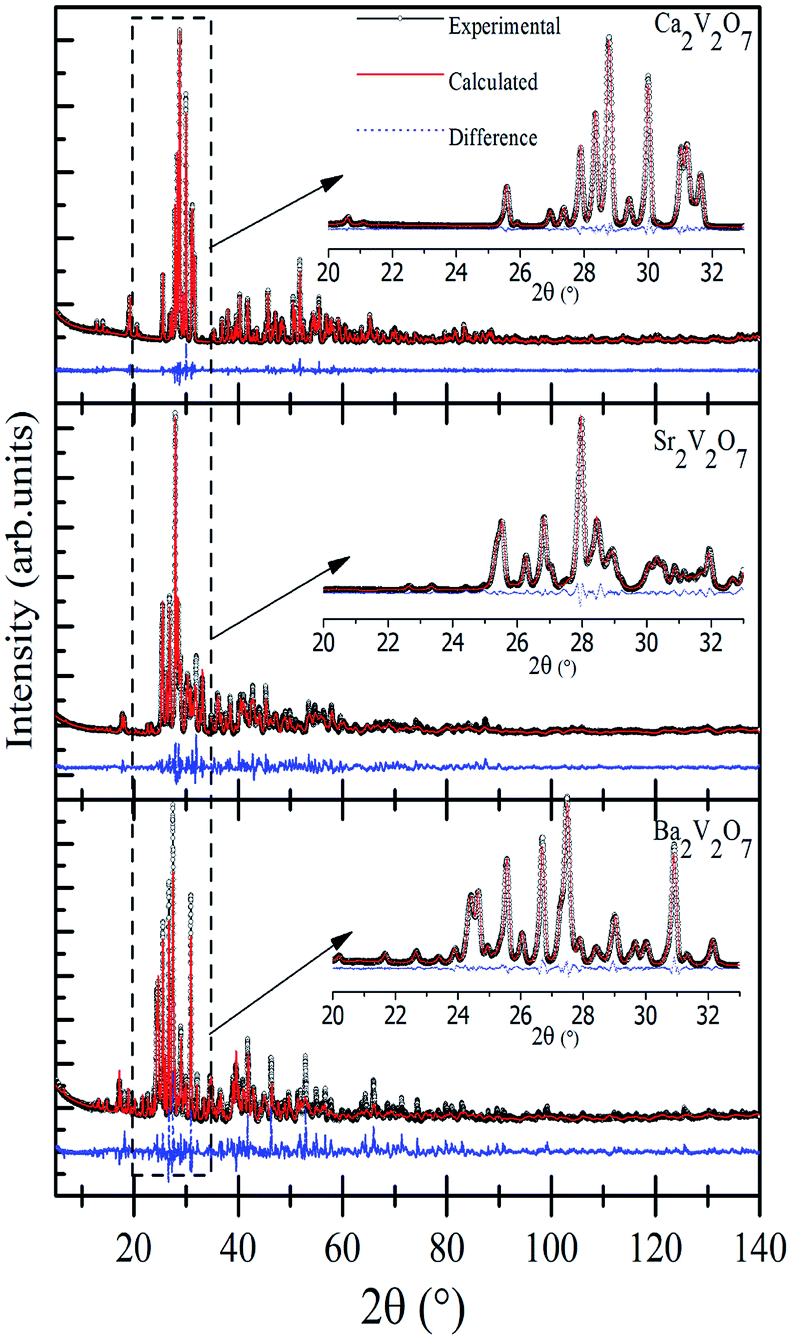 | ||
| Fig. 1 Experimental and Rietveld refined XRD patterns of Ca2V2O7, Sr2V2O7, and Ba2V2O7. The insets show the magnified view of simulated and experimental data. | ||
| Ca2V2O7 | Sr2V2O7 | Ba2V2O7 | |
|---|---|---|---|
| Symmetry | Triclinic | Triclinic | Triclinic |
| Space group | P(2) |
P(2) |
P(2) |
| a (Å) | 6.66766(8) | 7.0980(5) | 13.5675(1) |
| b (Å) | 6.92342(8) | 13.0029(9) | 7.3278(9) |
| c (Å) | 7.01801(8) | 7.0535(5) | 7.3297(9) |
| α (°) | 86.4432(5) | 93.725(3) | 90.132(5) |
| β (°) | 63.8597(4) | 90.750(3) | 99.391(3) |
| γ (°) | 83.6884(5) | 99.358(3) | 87.314(3) |
| Volume (Å)3 | 289.042(6) | 640.80(8) | 718.15(15) |
| R-Bragg (%) | 2.386 | 1.345 | 1.436 |
| Rwp (%) | 7.732 | 9.21 | 9.57 |
| GOF | 1.28 | 2.92 | 2.51 |
Fig. 2(a) shows the UV-visible absorption spectra of Ca2V2O7, Sr2V2O7, and Ba2V2O7 samples. It is noticeable from the Fig. 2 that the Ca2V2O7, Sr2V2O7, and Ba2V2O7 show a sharp absorption edge ∼465 nm, ∼418 nm and ∼402 nm, respectively. Fig. 2(b) shows the band-gap energy estimation from the UV-visible absorption spectra. The band-gap energy was estimated by plotting the hν versus (αhν)2 curves (Tauck's procedure, where α is the absorption coefficient and hν is the photon energy) and marking intersect at the energy scale. This gives band gap energy of 2.67 eV, 2.97 eV and 3.09 eV for the Ca2V2O7, Sr2V2O7, and Ba2V2O7, respectively. Thus the estimated band-gap energy of the compounds is comparable to the earlier reports.18,23,24
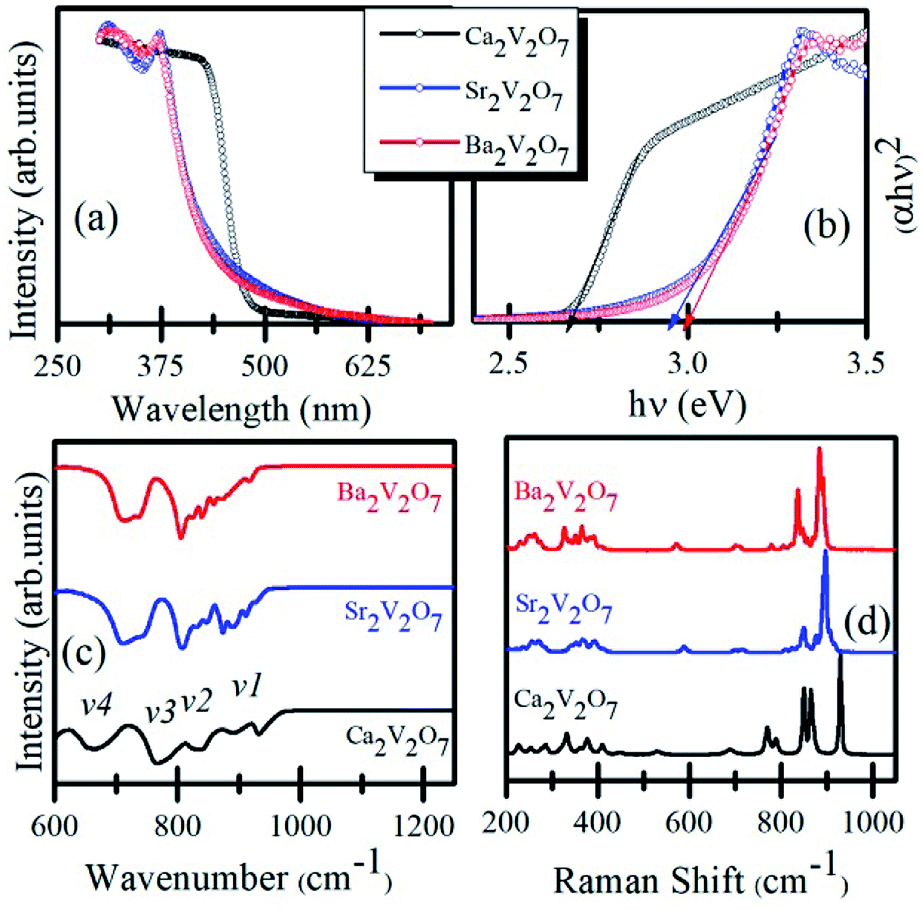 | ||
| Fig. 2 (a) UV-visible absorption spectra of Ca2V2O7, Sr2V2O7, and Ba2V2O7. (b) Shows the Tauck's plot for determining the band-gap energy. (c) FTIR spectra. (d) Raman spectra of the samples. | ||
Fig. 2(c) shows the FTIR spectra of the Ca2V2O7, Sr2V2O7, and Ba2V2O7 samples. All of the three samples show few dominating bands v1 (∼950–900 cm−1), v2 (830–810 cm−1), v3 (750–770 cm−1) and v4 (650–700 cm−1). These bands are mainly due to the stretching vibrations of metal-oxygen bonds and bending vibrations of VO43− groups in triclinic structured A2V2O7 pyro-vanadates.29,30 The v4 and v3 bands are the results of V–O–V bridge vibration and asymmetric stretching of longer V–O bond.31,32 The rest of the bands (v1 and v2) can be ascribed to symmetric stretching of the other shorter V–O bonds in the samples.31,32 Therefore, the feature-rich FTIR spectra convey the existence of VO43− groups in all of the pyro-vanadates.
Fig. 2(d) shows the Raman spectra of Ca2V2O7, Sr2V2O7, and Ba2V2O7 samples. Similar to the FTIR spectra, the Raman spectra have shown several vibration bands of V–O bonds. In all three samples, the Raman modes can be seen within the three distinct regions. The first region is between the 950–800 cm−1 and can be ascribed to the symmetric stretching vibrations of end VO3 groups.33–35 The second region is from 800 cm−1 to 400 cm−1 and attributed to stretching vibrations of bridging V–O–V band bending vibrations.33–35 The third region contains bands below the 400 cm−1 and may be due to the peripheral modes.33–35 Though the pyro-vanadates give their rich Raman bands due to the V–O or V–O–V bonds in the samples, however, their position is sensitive to the type of alkali metal element and defect chemistry.33–35
To investigate the surface morphology of the Ca2V2O7, Sr2V2O7 and Ba2V2O7 samples, systematic, FE-SEM images were collected and are presented in Fig. 3(a–c). It is noticeable from the micrographs that all pyro-vanadate have unusual and agglomerated morphology. This unusual morphology may arise due to the diffusion of particles during annealing of samples.36,37 The FE-SEM images ruled out the formation of fascinating morphologies (i.e., nano-rod, nano-wire or other structures) and, therefore, the observed luminescence properties are expected to be related only to the electronic/atomic structure properties but not due to the diverse surface/interface related phenomena in nanostructures.
 | ||
| Fig. 3 FE-SEM images of (a) Ca2V2O7, (b) Sr2V2O7 and (c) Ba2V2O7. | ||
(b) Electronic structure study by XANES
Formation of secondary phases in the host, lattice defects and variation in the coordination chemistry of the elements may facilitate ambiguous changes in the valence state of the metal ions in the pyro-vanadates. Therefore, to understand the valence state of Ca, Sr and Ba elements in the samples XANES spectra at Ca L3,2-edge, Sr K-edge and Ba L3,2-edge were collected and the normalized spectra, along with first derivative spectra, are presented in the Fig. 4 (a–c). The reference samples of CaO, SrO, and BaO, in which valence state of alkali earth cations is 2+ (i.e., Ca2+, Sr2+ and Ba2+), were also scanned under the similar experimental conditions and the spectra are plotted along with the A2V2O7 samples. It is noticeable from Fig. 4 that the edge-energy position and spectral features of reference CaO, SrO and BaO samples fairly resemble with the Ca2V2O7, Sr2V2O7, and Ba2V2O7, respectively, and are strengthening the 2+ valence state of the alkali earth cations in A2V2O7 pyro-vanadates of this study. Interestingly, Ba L2,3-edge XANES of Ba2V2O7 sample shows additional spectral features between the 5400 eV to 5600 eV. V K-edge spectral features originate within this energy range.16 In the present study, the existence of V ions in Ba2V2O7 sample is responsible for such spectral features. We have not seen any ambiguous spectral feature of secondary phases and, thus, the XANES spectra at Ca L-edge, Sr K-edge and Ba L-edge can convince the 2+ valence state of alkali earth elements and strengthened our XRD results of single phase nature of the samples.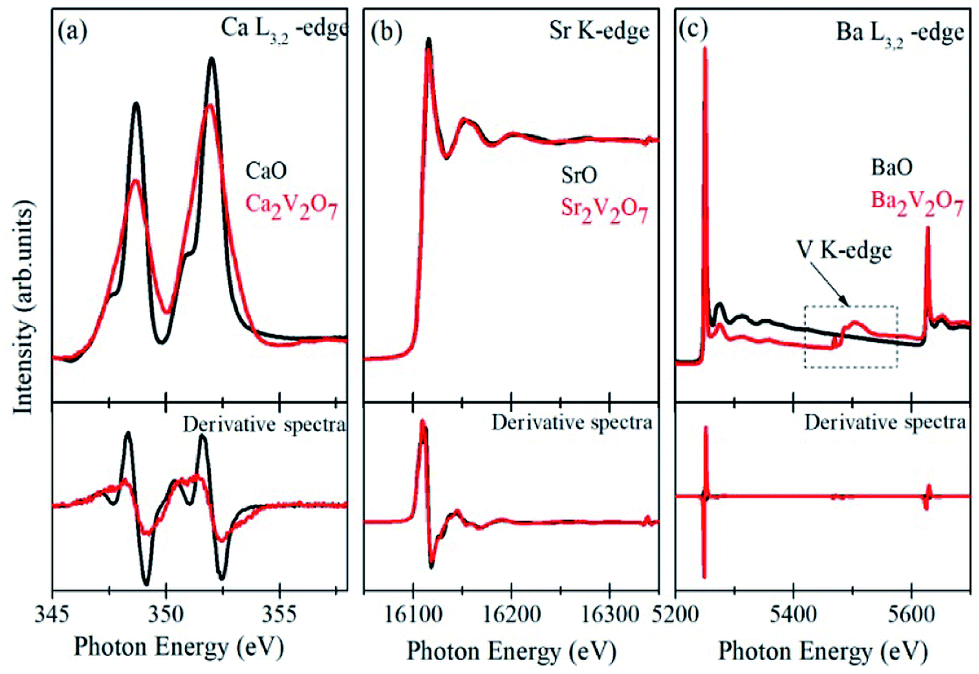 | ||
| Fig. 4 (a) Ca L-edge XANES of CaO and Ca2V2O7 samples, (b) Sr K-edge XANES of SrO and Sr2V2O7 samples and (c) Ba L-edge XANES of BaO and Ba2V2O7 samples. Lower panels show the first derivative spectra from the respective samples. | ||
Fig. 5(a) shows the pre/post-edge background subtracted and normalized V K-edge XANES spectra from the V metal foil, VO2, V2O5 and A2V2O7 samples. The first absorption feature between 5465–5475 eV, known as pre-edge peak, originates due to the transitions from 1s core levels to 3d state. Although, it is a dipole-forbidden transition but originates because of the combination of strong 3d–4p mixing and overlap of the metal 3d orbitals with the surrounding O 2p orbitals.16,38,39 Above the edge-energy, a strong peak is present in every spectrum which originates due to the dipole-allowed 1s–4p transitions followed by the other higher energy features from the transitions to higher np states and other multiple scattering contributions or, simply, the EXAFS signals.16,38,39 It is noticeable from the Fig. 5(a) that reference samples show a systematic edge-energy shift towards higher energy with increasing oxidation state of the V ions (i.e., energy position order is V2O5 > VO2 > V). The variation in the energy position is fairly visible from the second derivative spectra of the V K-edge XANES, presented in the Fig. 5(b) and inset therein. The edge-energy variation in the V K-edge spectra is corresponding to the oxidation state changes in the samples.
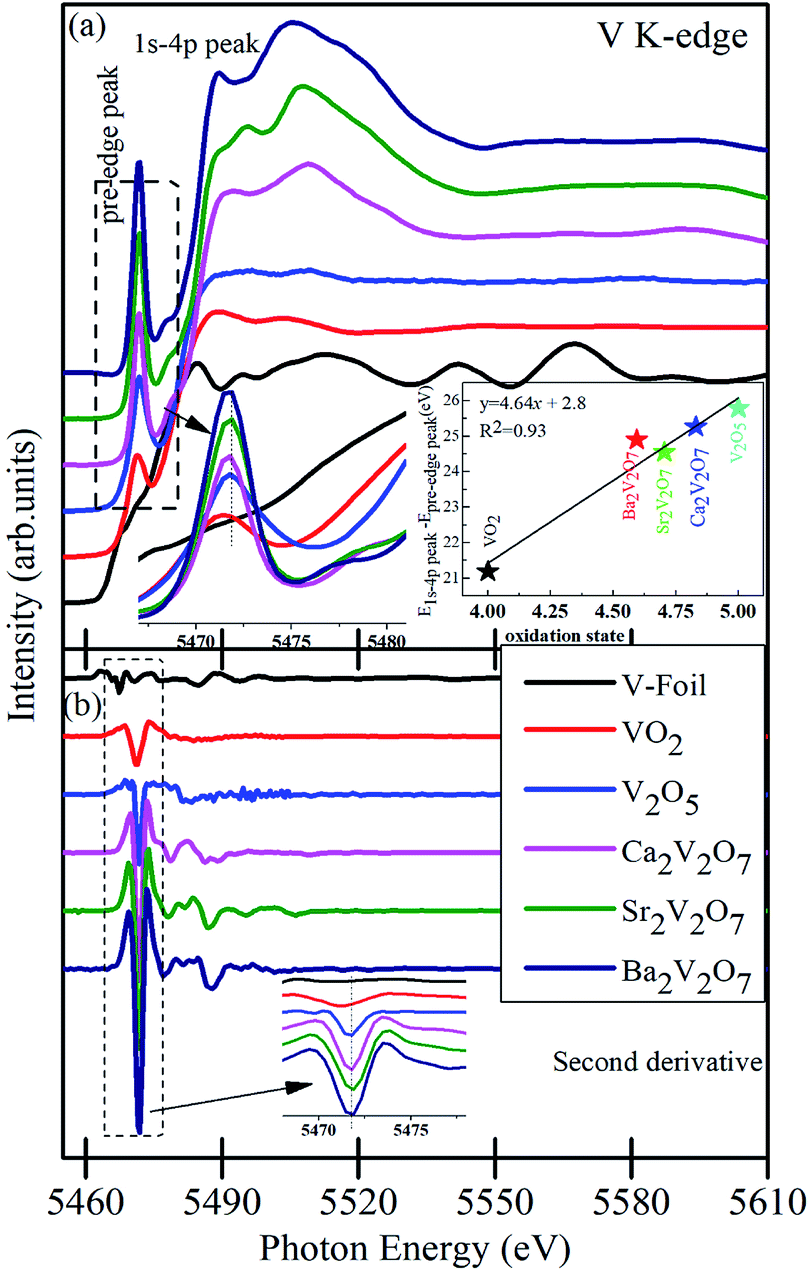 | ||
| Fig. 5 (a) V K-edge XANES of V, foil, VO2 and V2O5 reference samples along with A2V2O7 ceramics. (b) The second derivative of XANES spectra from the samples. The first inset of the (a) shows the magnified view of the pre-edge region and the second inset shows the oxidation state of the compounds. Inset of (b) shows the magnified view of the second derivative of XANES spectra. Spectra are vertically moved for the clarity of the presentation. | ||
The change in valence state has been estimated, quantitatively, by plotting the relative energy position of 1s-4p peak to the pre-edge peak as a function of formal oxidation state via deriving an approximate calibration curve16,38 and is presented in the inset of Fig. 5(a). It is noticeable that the A2V2O7 samples show the sub-pentavalent state of V ions. But the oxidation state of V ions in A2V2O7 samples is comparable (4.62–4.82), regardless of the variation in the alkali-metal element in the compounds. A little less oxidation of V ions, than the expected 5+ oxidation, could be due to the perturbation in the oxygen stoichiometry in the A2V2O7 samples under the given synthesis conditions.
Besides the oxidation state estimation of V ions, the pre-edge peak is prone to the electronic configuration (or empty density of states) and local symmetry of the V ions in various compound.38,39 The pre-edge intensity will be virtually zero in case of regular octahedral symmetry of the V ions but it enhances for the distorted symmetric compounds and reaches to maximum for the tetrahedral symmetric compounds.39 In the present study, the pre-edge intensity of A2V2O7 compounds is superior to the VO2 or V2O5 samples and is strengthening the more distorted octahedral symmetry or existence of tetrahedral symmetry of V ions in the pyro-vanadates. The Ca2V2O7 compound is exhibiting a lower intensity of pre-edge peak, while compared with Sr2V2O7 and Ba2V2O7, which may indicate lesser transition probabilities from the V 1s core levels to 3d states in this compound compared with other two pyro-vanadates. Moreover, the pre-edge intensity of V K-edge XANES spectra of A2V2O7 may be subjected to the diverse 3d-4p mixing and overlapping of O 2p with the V 3d orbitals under the influence of alkali metal orbitals. Therefore, to understand the local symmetry of V ions, surrounded by the O ligands in A2V2O7 compounds, it is advantageous to investigate the O K-edge XANES because it gives precise measure of bottom of the conduction band of a compound via probing the unoccupied density of states in t2g and eg orbitals along with the measure of crystal field splitting.
Fig. 6(a) and (b) present the V L-edge and O K-edge XANES spectra of Ca2V2O7, Sr2V2O7, and Ba2V2O7 compounds. As stated above, the d0 electronic configuration of V d orbitals provides 5+ oxidation state of V ions in A2V2O7 compounds which is similar to the oxidation state of V ions in V2O5 compound (i.e., V5+). Therefore, spectral features and energy position of A2V2O7 compounds were compared with the V L-edge and O K-edge XANES spectrum of the reference V2O5 compound.
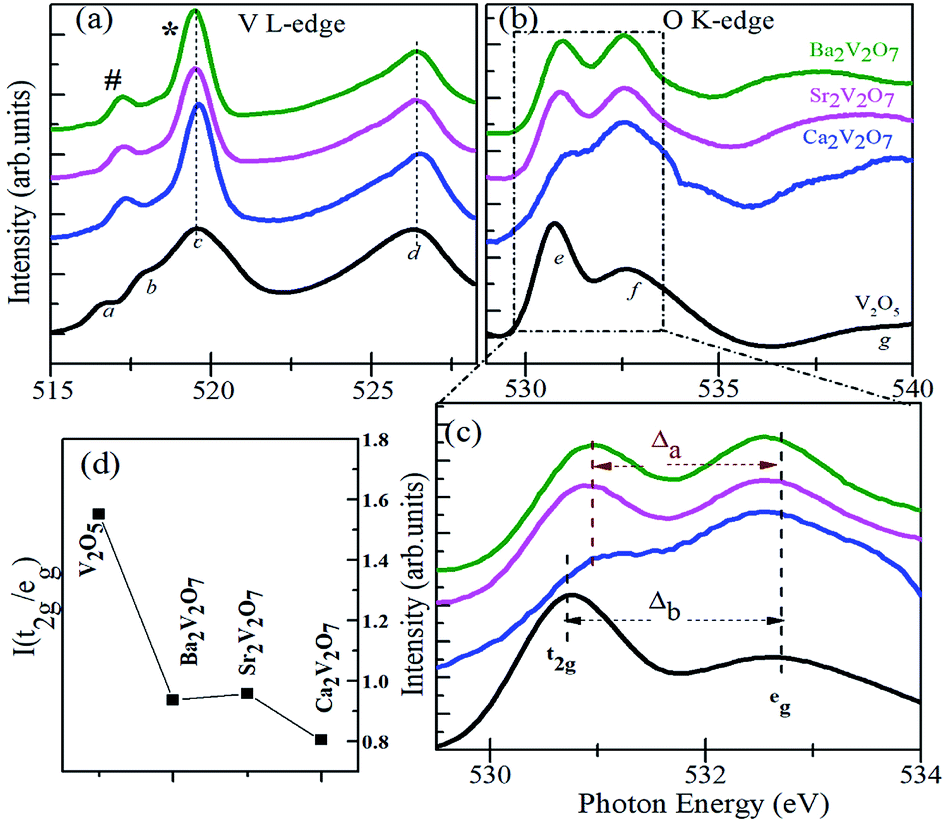 | ||
| Fig. 6 V L-edge XANES spectra (a) and O K-edge XANES spectra (b) of Ca2V2O7, Sr2V2O7 and Ba2V2O7 along with the reference VO2 and V2O5 samples. (c) Shows the crystal field splitting (Δa) for the pyro-vanadates and (Δb) for the reference vanadium oxide samples. (d) Shows intensity ratio of t2g and eg peaks. | ||
According to the dipole selection rules (Δl = ±1), XANES spectra at V L-edge and O K-edge are originated due to the d-projected unoccupied density of states and p-projected density of states of the valence levels, respectively.39–42 There are two distinct peaks in the V L-edge spectra at ∼519.5 eV (peak c; known as L3-edge) and 526.5 eV (peak d; known as L2-edge), which are corresponding to the electronic transitions from the V 2p3/2 and V 2p1/2 core levels, respectively, to the un-occupied 3d levels.16,38–40 The V L3 and L2-edges of vanadium compounds obey several spectral features which originate from the crystal field and/or atomic-multiplet effects.39–42 However, the V L2-edge is less important for the analysis because of its broadened nature via the Coster–Kronig Auger decay process into a 2p3/2 hole.39 Therefore well-resolved V L3-edge features are important to understand the electronic structure assets of the compounds. According to the band-structure calculations of vanadium oxides by Maganas et al., the c feature exhibits more of the 3dz2 character of the V ions whereas the b and a features arise from the 3dxz + 3dyz and 3dxy components, respectively.43 However, better-resolved spectra of vanadium oxides have also shown another spectral feature between the b and c peaks and it was assigned to the V 2p to V 3dx2−y2 transitions.39 In the present study, we observed two distinct peaks in the V L3-edge spectra of A2V2O7 compounds (i.e., # and * peaks in the Fig. 6(a)), instead of three features in the V2O5 sample. This indicates that V L3-edge of A2V2O7 compounds is composed of two main transitions between the V 2p3/2 and 3d orbitals. The # and * peak can be assigned to the transitions between V 2p3/2 → 3d (xy, yz, xz) orbitals and V 2p3/2 → 3d (x2, y2 and z2) orbitals. This presumably due to the different crystal field or multiplet effects in the sub-pentavalent A2V2O7 with triclinic crystal structure compared with pentavalent V2O5 with an orthorhombic crystal structure. However, there are no measurable changes in the V L3,2-edge spectral features of Ca2V2O7, Sr2V2O7 and Ba2V2O7 samples and are indicating the similar local symmetry (tetrahedral) and oxidation state of V ions as suggested by the V K-edge XANES.
The feature-rich O K-edge spectra are seen in the Fig. 6(b), which are originated from the electronic transitions from O 1s to O 2p hybridized V 3d orbitals and are known to provide valuable information on the crystal structure, local symmetry of metal ions and ligand-field effects.38–42 In an octahedral environment, the metal eg (dz2, dx2−y2) orbitals are pointed directly towards the O ligands to form strongly bonded σ bonds with the O pz orbital. On the other hand, the metal t2g (dxy, dxz, dyz) orbitals points between the O ligands and make weak π bonds with the O px and py orbitals.42 Consequently, the energy hierarchy of the anti-bonding orbitals is t2g < eg and their intensity ratio is 3/2 for an octahedrally coordinated compound.42 In contrast to the octahedral ligand field, both the symmetries of molecular orbitals and their energy hierarchy are reversed in case of tetrahedrally coordinated compounds.42 As a result of the tetrahedral interaction of ligands, the hybridized molecular orbitals are characterized by lower lying eg (π-type) and higher energy-lying t2g (σ-type) orbitals with their intensity ratio of 2/3.42
As stated above, the V ions in V2O5 obey the distorted octahedral symmetry, therefore, the e and f peaks in the O K-edge XANES can be assigned to the t2g and eg, respectively. However, the V ions form (VO4)3− groups in A2V2O7 compounds with tetrahedral symmetry. Therefore, the energy hierarchy of t2g and eg peaks and their intensity ratios are expected to be reversed for the Ca2V2O7, Sr2V2O7 and Ba2V2O7 samples. It can be seen from the Fig. 6(d) that the I(t2g/eg) ratio is nearly 3/2 for the V2O5 and is significantly lowered for the A2V2O7 samples. However, the I(t2g/eg) ratio for the A2V2O7 samples is not reached up to 2/3 (for the ideal tetrahedral symmetry). This could be due to the availability of octahedral locations to a few of the V ions in A2V2O7 samples along with their tetrahedral occupancy. It is noticeable that, the intensity of the second peak in Fig. 6(c) is higher for the Ca2V2O7 sample (which gives minimum I(t2g/eg) ratio) compared with remaining two samples of pyro-vanadates. This is indicating the much-unoccupied density of states were available for the second peak of Ca2V2O7 sample compared with Sr2V2O7 or Ba2V2O7. In the present study, the Δa (crystal field splitting parameter for the pyro-vanadates) is marginally lesser than the reference V2O5 sample (Δb). In general, the octahedral ligand field (with six O ions) gives much crystal field splitting (Δ) values while the tetrahedral ligand field (with four O ions) leads to lesser Δ values to a given compound.42 Therefore, the observed decrease in the Δa reinforces the tetrahedral symmetry in the pyro-vanadates as suggested by the V K-edge XANES.
Overall, the V K-edge and O K-edge spectra of pyro-vanadates samples strengthened tetrahedral occupancy of the V ions. However, the Ca2V2O7 sample point a distinct difference in the electronic hybridization of the frontier orbitals of Ca2V2O7 sample compared with Sr/Ba based pyro-vanadates. This could be due to the dissimilar distribution of V ions at tetrahedral/octahedral sites and their hybridization with O 2p orbitals in this compound (compared with other two pyro-vanadate).
(c) Photoluminescence study
Fig. 7(a–c) shows the PL spectra of Ca2V2O7, Sr2V2O7 and Ba2V2O7 samples along with their CIE diagrams (g). Fig. 7(d–f) show the emission spectra with the Gaussian fittings of emission bands (at energy axis). It is noticeable from the Fig. 8(a) that the Ca2V2O7 shows the intense emission bands near to the yellow-orange color (see spot S1 in CIE diagram) and in accordance with the previous reports.7–10 The broad emission band centered ∼ 530 nm and ∼507 nm is observed for the Sr2V2O7 (Fig. 7(b)) and Ba2V2O7 (Fig. 7(c)) samples, respectively. This kinds of PL spectra have resulted in the blue-green and blue-indigo emission characteristic in the CIE diagram (Fig. 7g) of Sr2V2O7 (S2) and Ba2V2O7 (S3) samples, respectively.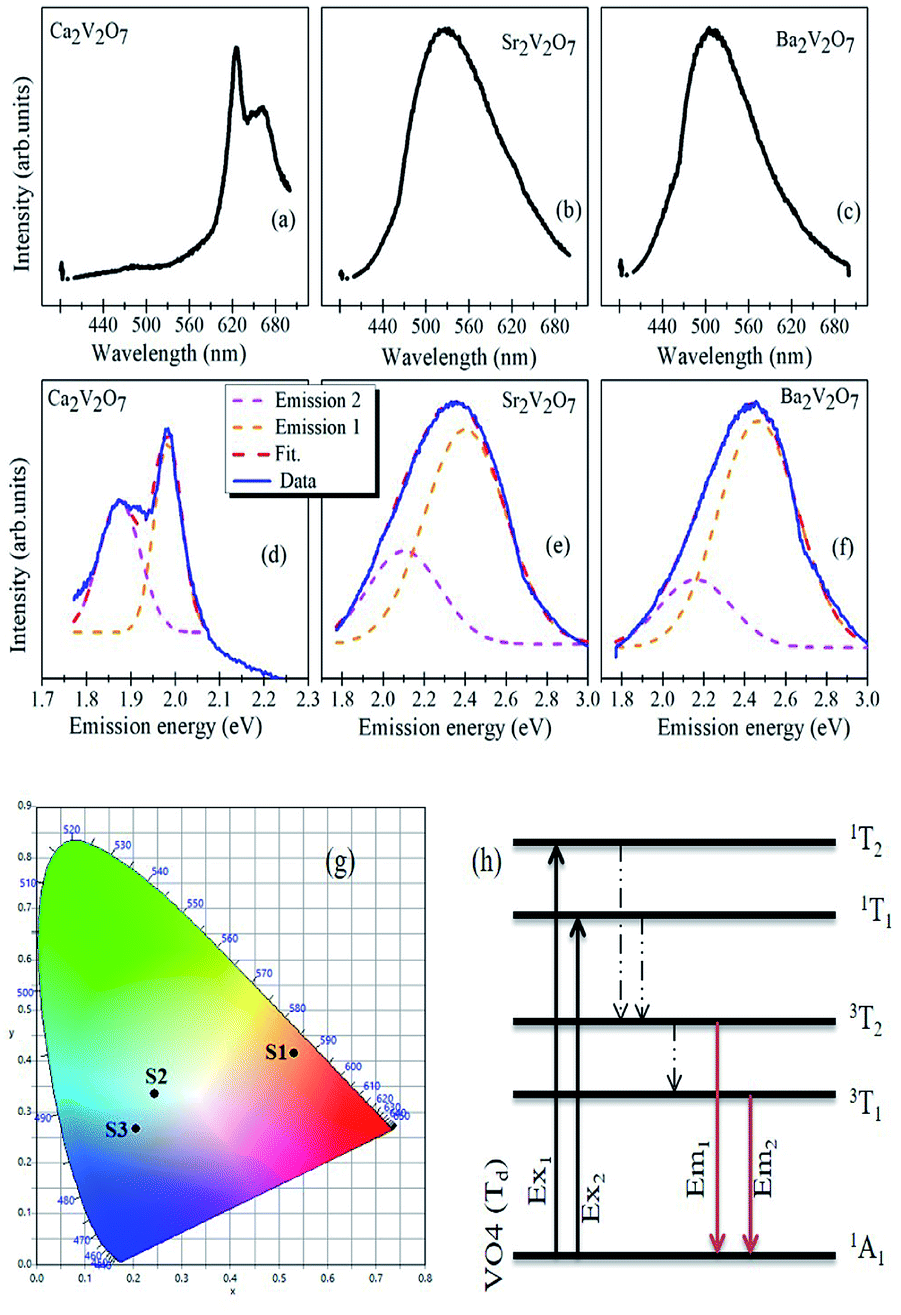 | ||
| Fig. 7 (a–c) Emission spectra (λex = 375 nm) of A2V2O7 samples. (d–f) shows the deconvolution of the emission-energy spectra with two Gaussian peaks related to the two distinct emission bands from the samples. (g) Show the CIE diagram for S1 (Ca2V2O7), S2 (Sr2V2O7) and S3 (Ba2V2O7) samples. (h) Schematic for the absorption and emission characteristics of VO4 tetrahedron with Td symmetry in pyro-vanadates. | ||
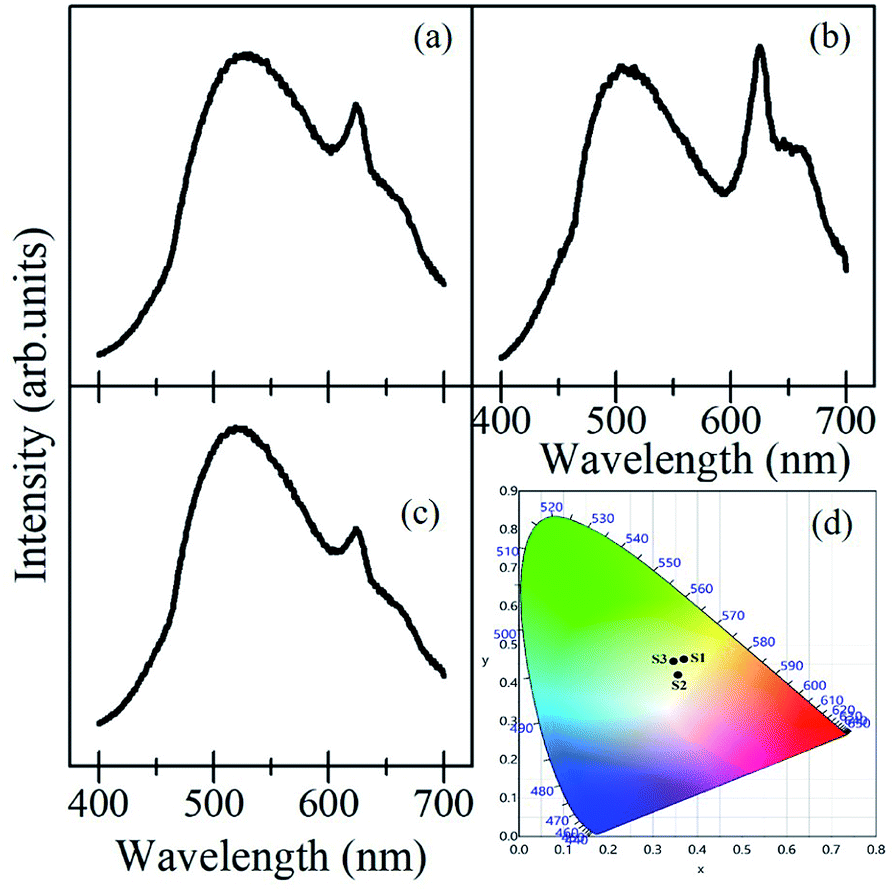 | ||
| Fig. 8 Emission spectra (λex = 375 nm) of mixed A2V2O7 samples with their equal weight proportions; (a) Ca2V2O7 + Sr2V2O7, (b) Ca2V2O7 + Ba2V2O7, (c) Ca2V2O7 + Sr2V2O7 + Ba2V2O7. (d) Shows the CIE diagram for S1 (Ca2V2O7 + Sr2V2O7), S2 (Ca2V2O7 + Ba2V2O7) and S3 (Ca2V2O7 + Sr2V2O7 + Ba2V2O7) samples. | ||
One of the possibilities of broad-emission spectra could be the CT of an electron from O 2p orbitals to the vacant 3d orbitals of V5+ ions under the tetragonal symmetry of V–O4 groups.7–10 In previous reports, it was also assumed that the forbidden luminescence process with the perfect tetragonal symmetry of V–O4 groups would be partially allowed due to the distorted structure as well as the spin-orbit interaction.8,10 In addition to that, theoretical calculations were also performed to explain the broad emission spectra of Sr2V2O7:Eu crystals.44 In such calculations, a 10 × 10 energy matrix was successfully established, based on an effective operator Hamiltonian, including free ion and crystal field interactions. According to the crystal field theory, the pentavalent vanadium (i.e. V5+ ions with 3d0 electronic configuration) in the alkali earth metal based pyro-vanadates may not result in any d–d transitions. The d–d electronic transition based emission-characteristics can be achieved from the V5+ ion if it gets one electron from the surrounding oxygen atoms and attains 3d1 electronic configuration via formation of V4+ ions (i.e., V5+O42− → V4+O42− (ref. 44)). Based on the theoretical calculations, it was conveyed that the broad PL properties of Sr2V2O7:Eu compound originate from four different VO4 groups with C3v symmetry and not from the f–d transitions of the Eu2+ ions. In contrary to this, f–d transition induced broad luminescence has previously reported for the Eu3+ and Eu2+ dual emission centers.45 Thus, the mechanistic concept of the broad luminescence spectra from alkali earth metal based pyro-vanadates is still under development.
In the present study, all of the samples have exhibited broad PL properties which can be assigned to the charge transfer (CT) transitions from the O 2p orbitals to the V 3d orbitals within the VO4 tetrahedra.8,10 The molecular orbitals of the V5+ ions, with the tetrahedral symmetry, obey 1A1 ground state and four (3T1, 3T2, 1T1 and 1T2) excited states as shown in the Fig. 8(h).8,10 Though the all three pyro-vanadates of this study obeyed the triclinic crystal structure, with a space group of P(2), there are local structural dissimilarities. In the Ca2V2O7, VO4 dimers approach to each other within the shorten unit cell (compared with the Sr2V2O7 and Ba2V2O7) and the VO5 pyramids are formed by the manifestation of edge-sharing at adjacent VO4 tetrahedra.8,10 Therefore miscellaneous CT transitions are expected for the Ca2V2O7 compound which, indeed, facilitate yellow-orange emission properties. However, this kind of emission from the A2V2O7 needs electronic structure correlations.
In case of A2V2O7 compounds, the valence band is derived from the fully occupied O 2p orbitals and the conduction band is mainly consist of empty V 3d orbitals. The O K-edge XANES of Sr2V2O7 and Ba2V2O7 samples has shown similar crystal field splitting and nearly the same t2g/eg ratio which indicates iso-structured electronic structure properties in these two compound. On the other hand, the t2g/eg ratio is significantly decreased for the Ca2V2O7 sample. This indicates that more unoccupied 3d density of states were made available for this compound and could be due to the mixed occupancy of V ions (tetrahedral and/or octahedral). The adverse charge-transfer of an electron from O 2p orbitals to the vacant 3d orbitals of V ions under the distorted tetragonal symmetry may obey the distinct emission properties from the Ca2V2O7. The diverse splitting of V 3d orbitals had previously studied for the Ca2V2O7 (but not seen for the Sr2V2O7 and Ba2V2O7) by the partial density of state (PDOS) calculations18 and supports our XANES investigation.
The broad emission from the A2V2O7 compounds consist of two distinct emission peaks (higher energy peak (emission 1) and lower energy peak (emission 2)) which are corresponding to the transitions from 3T2 and 3T1 to the ground state 1A1 (see Fig. 7(h)). It is evident from the Fig. 7(d–f) that the emission 1 is stronger than the emission 2 and consistent of the previous reports.8,10,18 Moreover, the peak position of emission 1 and emission 2 was red-shifted with decreasing the ionic radii of alkali earth ions. The luminescent colour coordinates of A2V2O7, which were obtained from the CIE chromaticity diagram, were changed from yellow-orange (x = 0.5308, y = 0.4151; for Ca2V2O7), green-blue (x = 0.2445, y = 0.3357; for Sr2V2O7) to blue-indigo (x = 0.2053, y = 0.2666; for Ba2V2O7). The variation in the color coordination in A2V2O7 compounds could be due to the dissimilarities in the electronic/atomic structure as discussed above. In the present study, the CIE coordinates of Sr2V2O7 are quite close to the CEI coordinate of white light (i.e., x = 0.333, y = 0.333). Moreover, advanced emission characteristics may be anticipated by mixing A2V2O7 pyro-vanadates in certain weight/molar ratio and has been attempted in the present study by mixing the individual powders in their equal weight percentage.
Fig. 8(a–c) show the PL emission spectra from the mixture of the samples ((a); Ca2V2O7 + Sr2V2O7, (b); Ca2V2O7 + Ba2V2O7, and (c); Ca2V2O7 + Sr2V2O7 + Ba2V2O7) along with their CIE coordinate diagram in Fig. 8(d). It is evidenced from the Fig. 8 that the mixed samples have exhibited much fascinating spectral features and contain the emission bands from constituent compounds. It is noticeable that the intensity of 600 nm–700 nm peaks, which is characteristic yellow-orange emission of Ca2V2O7, is significant in case of the mixture of Ca2V2O7 + Ba2V2O7 sample but has been less dominating in case of other two combinations. This could be due to the superior emission of Ca2V2O7 over the Ba2V2O7 sample. In the Fig. 8(d), samples are named as S1 (for Ca2V2O7 + Sr2V2O7 mixture) S2 (for Ca2V2O7 + Ba2V2O7 mixture) and S3 (for Ca2V2O7 + Sr2V2O7 + Ba2V2O7 mixture). Thus the obtained CIE colour coordinates for S1 (x = 0.3696, y = 0.4617), S2 (0.3561, y = 0.4207) and S3 (0.3462, y = 0.4562) are quite diverse to that of single component pyro-vanadates. The obtained colour coordinates from the mixed compounds are very close to the white-light colour coordinates. Therefore, we may conclude that the vanadium-based compounds of the present study are proficient for the white-light-emitting devices without adding the rare-earth or other dopants.
Conclusions
Single phase pyro-vanadate (Ca2V2O7, Sr2V2O7, and Ba2V2O7) ceramics were successfully synthesized using a modified chemical precipitation method followed by air annealing of the samples. Ca2+, Sr2+, and Ba2+ ions were confirmed in the Ca2V2O7, Sr2V2O7 and Ba2V2O7 samples, respectively, using the XANES study at the Ca L-edge, Sr K-edge and Ba L-edge. Sub-pentavalent vanadium ions have been confirmed in all three pyro-vanadates of this study by analyzing the V K-edge XANES spectra. O K-edge and V K-edge XANES have confirmed the tetrahedral occupancy in the pyro-vanadate samples. However, the Ca2V2O7 exhibits a smaller t2g/eg peak intensity ratio in the O K-edge which indicates a much-unoccupied density of states in O 2p and V 3d hybridized orbitals under the mixed tetrahedral/octahedral V locations in this sample. Charge-transfer transitions in VO4 tetrahedra have established broad photoluminescence characteristics in pyro-vanadates. Yellow-orange (CEI colour coordinates; x = 0.5308, y = 0.4151), green-blue (CEI colour coordinates; x = 0.2445, y = 0.3357) and blue-indigo (CEI colour coordinates; x = 0.2053, y = 0.2666) emissions have been observed from the Ca2V2O7, Sr2V2O7 and Ba2V2O7 samples, respectively. The emitted colour coordinates could be tailored by mixing of the compounds and has resulted in advanced photoluminescence properties which are comprehensively suitable for the rare-earth element free white-light-emitting devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Korea Institute of Science and Technology, Seoul, Korea (KIST Project nos. 2V04081 and 2V04611). Authors are also thankful to Dr Jitendra Pal Singh (KIST-PAL beamline at Pohang Accelerator Laboratory, South Korea) and Dr Ankush Vij (Department of Physics, Amity University, Gurgaon, India) for fruitful discussion on EXAFS and PL data analysis.Notes and references
- Z. He and Y. Ueda, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 052402 CrossRef.
- Z. He, J. Yamaura, Y. Ueda and W. Cheng, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 092404 CrossRef.
- S. Zhou, J. Wang, Y. Weng, Z. Wu, Z. Ni, Q. Xu, J. Du and S. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 26213 CrossRef PubMed.
- M. V. Rotermel, A. Yu. Suntsova, T. I. Krasnenko, R. F. Samigullin and E. D. Pletnev, Bull. Russ. Acad. Sci.: Phys., 2016, 80, 668 Search PubMed.
- J. Wang, J. Pei, K. Hua, D. Chen, Y. Jiao, Y. Hu and G. Chen, ChemElectroChem, 2018, 5, 737 CrossRef.
- S. K. Gupta, K. Sudarshan and R. M. Kadam, Mater. Des., 2017, 130, 208 CrossRef.
- S.-P. Kuang, Y. Meng, J. Liu, Z.-C. Wu and L.-S. Zhao, Optik, 2013, 124, 5517–5519 CrossRef.
- T. Nakajima, M. Isobe, T. Tsuchiya, Y. Ueda and T. Kumagai, Nat. Mater., 2008, 7, 735 CrossRef PubMed.
- Y. Matsushima, T. Koide, M. H. Oka, M. Shida, A. Sato, S. Sugiyama and M. Ito, J. Am. Ceram. Soc., 2015, 98, 1236 CrossRef.
- T. Nakajima, M. Isobe, T. Tsuchiya, Y. Ueda and T. Manabe, J. Phys. Chem. C, 2010, 114, 5160 CrossRef.
- M.-R. Joung, J.-S. Kim, M.-E. Song, S. Nahmw and J.-H. Paik, J. Am. Ceram. Soc., 2009, 92, 3092 CrossRef.
- M.-R. Joung, J.-S. Kim, M.-E. Song, S. Nahmw, J.-H. Paik and B.-H. Choi, J. Am. Ceram. Soc., 2009, 92, 1621 CrossRef.
- F. Li, H. Fang and Y. Chen, J. Rare Earths, 2017, 35, 135 CrossRef.
- V. Kumar, A. K. Bedyal, J. Sharma, V. Kumar, O. M. Ntwaeaborwa and H. C. Swart, Appl. Phys. A, 2014, 116, 1785 CrossRef.
- J. Gu and B. Yan, J. Alloys Compd., 2009, 476, 619 CrossRef.
- A. Sharma, M. Varshney, W. C. Lim, H. J. Shin, J. P. Singh, S. O. Won and K. H. Chae, Phys. Chem. Chem. Phys., 2017, 19, 6397 RSC.
- A. Sharma, M. Varshney, H. J. Shin, K. H. Chae and S. O. Won, RSC Adv., 2017, 7, 52543 RSC.
- R. Yu, M. Yuan, T. Li, Q. Tu and J. Wang, RSC Adv., 2016, 6, 90711 RSC.
- M. Takahashi, M. Hagiwara and S. Fujihara, Inorg. Chem., 2016, 55, 7879 CrossRef PubMed.
- T. Nakajima, M. Isobe, T. Tsuchiya, Y. Ueda and T. Manabe, Opt. Mater., 2010, 32, 1618 CrossRef.
- H. Fang, X. Wei, S. Zhou, Y. Chen, C. Duan and M. Yin, Inorg. Chem., 2016, 55, 9284 CrossRef PubMed.
- W. Guo, W. D. Chemelewski, O. Mabayoje, P. Xiao, Y. Zhang and C. B. Mullins, J. Phys. Chem. C, 2015, 119, 27220 CrossRef.
- R. Karthik, J. V. Kumar, S.-M. Chen, P. S. Kumar, V. Selvam and V. Muthuraj, Sci. Rep., 2017, 7, 7254 CrossRef PubMed.
- R. Yu, N. Xue, S. Huo, J. Li and J. Wang, RSC Adv., 2015, 5, 63502 RSC.
- Y. Sun, J. Wang, M. Chen, C. Li, Z. Hu and S.-L. Chou, J. Mater. Chem. C, 2017, 5, 6336 RSC.
- A. Sharma, M. Varshney, H. J. Shin, K. H. Chae and S. O. Won, Spectrochim. Acta, Part A, 2017, 173, 549 CrossRef PubMed.
- A. Sharma, M. Varshney, H. J. Shin, K. H. Chae and S. O. Won, Curr. Appl. Phys., 2016, 16, 1342 CrossRef.
- A. Sharma, M. Varshney, J. Park, T. K. Ha, K. H. Chae and H. J. Shin, Phys. Chem. Chem. Phys., 2015, 17, 30065 RSC.
- A. C. Dhaussy, F. Abraham, O. Mentre and H. Steinfink, J. Solid State Chem., 1996, 126, 328 CrossRef.
- I. O. Mazali and O. L. Alves, J. Braz. Chem. Soc., 2004, 4, 464 CrossRef.
- V. B. Taxak, S. Dayawati and S. P. Khatkar, Curr. Appl. Phys., 2013, 13, 594 CrossRef.
- H. Pan, L. Zhang, L. Jin, B. Zhang and W. Yang, J. Electron. Mater., 2015, 44, 3465 CrossRef.
- A. N. Unnimaya, E. K. Suresh, J. Dhanya and R. Ratheesh, J. Mater. Sci.: Mater. Electron., 2014, 25, 1127 CrossRef.
- M. L. Occelli, R. S. Maxwell and H. Eckert, Microporous Mater., 1994, 3, 305 CrossRef.
- M. Aureliano, C. A. Ohlin, M. O. Vieira, M. Paula, M. Marques, W. H. Caseye and L. A. E. Batista de Carvalho, Dalton Trans., 2016, 45, 7391 RSC.
- F. Ravaux, N. S. Rajput, J. Abed, L. George, M. Tiner and M. Jouiad, RSC Adv., 2017, 7, 32087 RSC.
- N. T. H. An, M. T. Islam, J. M. Park, Y.-S. Gal, K. P. Johnston and K. T. Lim, Mater. Lett., 2012, 75, 111 CrossRef.
- Y. Wu, L. Fan, W. Huang, S. Chen, S. Chen, F. Chen, C. Zou and Z. Wu, Phys. Chem. Chem. Phys., 2014, 16, 17705 RSC.
- Y. R. Lu, H. H. Hsu, J. L. Chen, H. W. Chang, C. L. Chen, W. C. Chou and C. L. Dong, Phys. Chem. Chem. Phys., 2016, 18, 5203 RSC.
- S. Zhang, B. Shang, J. Yang, W. Yan, S. Wei and Y. Xie, Phys. Chem. Chem. Phys., 2011, 13, 15873 RSC.
- C. Zou, L. Fan, R. Chen, X. Yan, W. Yan, G. Pan, Z. Wu and W. Gao, CrystEngComm, 2012, 14, 631 Search PubMed.
- J. G. Chen, Surf. Sci. Rep., 1997, 30, 1–152 CrossRef.
- D. Maganas, M. Roemelt, M. Havecker, A. Trunschke, A. K. Gericke, R. Schlogl and F. Neese, Phys. Chem. Chem. Phys., 2013, 15, 7260 RSC.
- W.-Q. Yang, H.-G. Liu, M. Gao, Y. Bai, J.-T. Zhao, X.-D. Xu, B. Wua, W.-C. Zheng, G.-K. Liu and Y. Lin, Acta Mater., 2013, 61, 5096 CrossRef.
- D. K. Patel, A. Sengupta, B. Vishwanadh, V. Sudarsan, R. K. Vatsa, R. Kadam and S. K. Kulshreshtha, Eur. J. Inorg. Chem., 2012, 1609 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |