
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferences in gut microbiota between silkworms (Bombyx mori) reared on fresh mulberry (Morus alba var. multicaulis) leaves or an artificial diet†
Hui-Ling Dong‡
ab,
Sheng-Xiang Zhang‡c,
Zhuo-Hua Chenab,
Hui Taoab,
Xue Liab,
Jian-Feng Qiuab,
Wen-Zhao Cuia,
Yang-Hu Simaabd,
Wei-Zheng Cui*c and
Shi-Qing Xu *abd
*abd
aSchool of Biology and Basic Medical Sciences, Medical College, Soochow University, Suzhou 215123, China. E-mail: szsqxu@suda.edu.cn
bInstitute of Agricultural Biotechnology & Ecology (IABE), Soochow University, Suzhou 215123, China
cCollege of Forestry, Shandong Agricultural University, Taian, Shandong 271018, China. E-mail: wzcui88@163.com
dNational Engineering Laboratory for Modern Silk (NEAER), Soochow University, Suzhou 215123, China
First published on 20th July 2018
Abstract
Artificial diets for silkworms have many potential applications and they are important in sericulture. However, the challenges of weak larvae and low silk protein synthesis efficiency in silkworms reared on artificial diets have not been resolved. Here, we used high-throughput sequencing to analyse the differences between the gut microbiota of 5th-instar larvae reared on mulberry leaves and larvae reared on an artificial diet. The results showed that at the phylum level, Cyanobacteria, Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria are the dominant bacteria in the intestines of silkworm larvae of all the strains. But the abundance of dominant bacteria in the gut microbiota differed between the silkworm strains that were reared on mulberry leaves, as well as between the silkworm strains that were reared on the artificial diet, while the gut microbiota diversity was lower in the silkworm strains that were reared on the artificial diet. Prediction of the functions of the gut microbiota in the hosts indicated that there was no significant difference between the silkworm strains that were reared on mulberry leaves, while there were significant differences between silkworm strains reared on the artificial diet. When the silkworm diet changed from mulberry leaves to the artificial diet, changes in gut microbiota in the silkworms affected host nutrient metabolism and immune resistance. These changes may be related to the adaptation of silkworms to their long evolutionary history of eating mulberry leaves.
Introduction
Currently, China and other countries use fresh mulberry (Morus alba L.) leaves to rear silkworms (Bombyx mori). The production of silkworm cocoons using all-weather factory farming is very challenging because M. alba is deciduous and loses its leaves in winter. Many studies suggest that rearing silkworms on mulberry leaves have become a bottle-neck to sericulture development, and it continues to weaken the viability of sericulture.1–3Establishing an artificial diet to rear silkworms in all-weather factory farming, as found in the livestock and poultry industries, is of great importance.1,4,5 However, even though sericulturists have long been concerned about breeding silkworm varieties with good ingestive abilities for artificial diets6,7 and artificial diet formulae are constantly being improved,8–10 issues such as physical weakness and low silk yield in B. mori reared on artificial diets have not been resolved, to date, and the metabolic utilization of artificial diets in silkworms is still less than that of mulberry leaves.1,11
The effect of the composition and activity of the gut microbiota on host animals cannot be ignored.12–14 Many reports suggest that a host's gut microbiota is highly susceptible to changes in diet and the environment.15–17 Sterile feeding environments, which are related closely to the development of silkworms reared on artificial diets, and the addition of antibiotics to artificial diets are also considered to have significant adverse effects on the intestinal microbial diversity of B. mori larvae, and even resistance to B. mori cytoplasmic polyhedrosis virus infection.18,19 Studies of B. mori infection by B. mori cytoplasmic polyhedrosis virus,20 Pseudomonas aeruginosa and Bacillus bombysepticus,21 and Staphylococcus aureus and Escherichia coli,22 as well as fluoride poisoning,23 have shown that the gut microbiota affects the infection of hosts by viruses, as well as resistance to xenobiotic toxins, by altering the immunity of B. mori. Also, some reports have indicated that the gut microbiota is involved directly in nutrient uptake and metabolism in B. mori.24,25 It is notable that the gut microbiota is involved in regulating the growth and development of Asian corn borer larvae (Ostrinia furnicalis).26
Bombyx mori is an economically important lepidopteran insect that has been domesticated and reared on a large scale.2,27 Therefore, research of its gut microbiota is of great importance. To date, it is not clear what effects artificial diets have on the gut microbiota of B. mori. Two important questions are: (1) what, if any, differences are there in the gut microbiota of silkworms reared on mulberry leaves compared with that of silkworms reared on an artificial diet, and (2) how can we explain the physical weakness and low efficiency of silk protein synthesis in silkworms reared on an artificial diet, as well as the significant changes in metabolic levels arising from changes in the gut microbiota? To answer these questions, we used high-throughput sequencing to analyze the gut microbiota of silkworms with different artificial diet ingestive habits, which were reared on mulberry leaves or an artificial diet.28,29
Experimental
Preparation of animals
Four silkworm strains were used in this study. They were the Guangshi (Gs) and Lu7 (L7) strains, a Jingsong strain with high ingestive habit (Hi) for artificial diets, and a Jingsong strain with a low ingestive habit (Lo) for artificial diets. Almost 100% of the larvae from the Hi and Gs strains ingested the artificial diet and developed well, while almost 100% of the larvae from the Lo or L7 strains did not ingest the artificial diet and starved to death. The Gs and L7 strains are two natural mutants that differ in terms of their ingestion of artificial diets, while the Hi and Lo strains were obtained after seven years of continuous selection of silkworm larvae with different artificial diet ingestive habits. However, it should be noted that the development, viability, and efficiency of silk protein synthesis of the Hi and Lo strains did not differ when larvae were reared on mulberry leaves.1In this study, we used four silkworm strains that were reared on fresh mulberry (Morus alba var. multicaulis) leaves, named HiM, GsM, LoM, and L7M, and two strains that were reared on an artificial diet, HiA and GsA. The larvae of all strains were fed ad libitum and maintained at 26 °C ± 1 °C, a relative humidity of 70% ± 5%, and a 12 h light/12 h dark photoperiod. The artificial diet composition (w/w) was described in our previous report.1 Briefly, the artificial diet comprised equal amounts of 35% mulberry leaf powder of the Husang strain and soybean powder. The contents of mulberry green twig powder, starch, vitamin C, vitamin B complex, citric acid, crotonic acid, and choline chloride were 15%, 9.4%, 1.5%, 1.5%, 2%, 0.4%, and 0.2%, respectively. The powdered ingredients were mixed with 1.9 times (w/w) as much ultrapure water and boiled for 60 min, after which they were placed into preservation bags and stored at 4 °C. Fresh mulberry leaves were picked from mulberry trees of the Husang strain in the Dushuhu campus of Soochow University (Jiangsu, China).
Similarly-sized 5 day-old, 5th-instar larvae were selected to assess their gut microbiota. The head and tails of the larvae were ligated with cotton thread to prevent the excretion of digestive juice and feces, and then the larvae were soaked in 70% ethanol for 5 min for surface disinfection. Their complete digestive tracts were removed under sterile conditions. After Malpighian tubules and fat bodies were removed quickly, the tracts were placed in sterilized centrifuge tubes and stored at −80 °C. Complete digestive tracts, including food particles in the gut, from 10 randomly selected male and female larvae were used as a sample for each gut microbiota test, which was repeated five times for each sample.
DNA extraction and polymerase chain reaction (PCR) amplification
Microbial DNA was extracted from the silkworm larval digestive tract samples using the E.Z.N.A.® Stool DNA Kit (Omega Bio-tek, Norcross, GA, USA) following the manufacturer's protocol. The primers 343F (5′-barcode-TACGGRAGGCAGCAG-3′) and 798R (5′-AGGGTATCTAATCCT-3′) were used to amplify the V3–V4 region of bacterial 16S rDNA.PCRs were conducted as follows: a 95 °C initial denaturation for 2 min, followed by 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, followed by a final extension at 72 °C for 5 min. The PCRs were performed in triplicate in a 20 μL reaction containing 4 μL of 5× FastPfu buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu polymerase, and 10 ng of template DNA.
Illumina MiSeq sequencing
Amplicons were extracted from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer's instructions and quantified using QuantiFluor™-ST (Promega, Madison, WI, USA). Then, the purified amplicons were pooled in equimolar amounts and paired-end sequenced (2 × 300) on an Illumina (San Diego, CA, USA) MiSeq platform according to standard protocols. The raw reads were deposited into the Sequence Read Archive database (Accession number: SRP128452).Processing of sequencing data
Raw sequencing data were in the FASTQ format. Paired-end reads were preprocessed using Trimmomatic software to detect and cut off ambiguous bases (N).30 The processing also cut off low-quality sequences with average quality scores below 20 using the sliding window trimming approach. After trimming, the paired-end reads were assembled using FLASH software.31 The parameters of assembly were: 10 bp of minimal overlap, 200 bp of maximum overlap, and 20% of the maximum mismatch rate. Sequences were subjected to further denoising as follows: reads with ambiguous, homologous sequences or those less than 200 bp were abandoned; reads with 75% of bases above a quality score of 20 were retained. Then, chimeric reads were detected and removed. These two steps were achieved using QIIME software (version 1.8.0).32Clean reads were subjected to primer sequences removal and clustering to generate operational taxonomic units (OTUs) using the Cluster Database at High Identity with Tolerance method with a 97% similarity cutoff. The representative read of each OTU was selected using the QIIME package. All representative reads were annotated and used in Basic Local Alignment Search Tool searches of the Greengenes database (16S rDNA) using the Ribosomal Database Project classifier (the confidence threshold was 70%).33 A heat map was generated using the heat map function of the R software package (http://www.r-project.org/). A principal coordinate analysis (PCoA) was conducted using QIIME. Prediction of the microbial function was performed using a phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt, version 1.0.0).34 PICRUSt is a tool designed to infer metagenomics information from 16S rRNA sequencing data. The obtained OTU table was normalized by 16S rRNA copy number, and functional genes were predicted from the Kyoto Encyclopedia of Genes and Genomes (KEGG) catalogue.
Results
Genomes assembled from the silkworm gut microbiome
Over 1.396 million sequencing reads obtained by 16S rDNA sequencing were analyzed from 30 samples, resulting in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 245800 optimized reads after cleaning and trimming. The number of clean reads per library ranged from 32995 to 98382. The length of the reads distribution was 300–499 bp, and 96.29% of the reads were in the 400–449 bp range. Good's coverage percentage of all samples ranged from 99.7% to 100% (mean value = 99.9%). A total of 2229 OTUs were obtained at the 97% sequence similarity cut-off level, with a mean number of OTUs per sample of 174 ± 120 (range: 20 to 402) (Fig. S1†).
245800 optimized reads after cleaning and trimming. The number of clean reads per library ranged from 32995 to 98382. The length of the reads distribution was 300–499 bp, and 96.29% of the reads were in the 400–449 bp range. Good's coverage percentage of all samples ranged from 99.7% to 100% (mean value = 99.9%). A total of 2229 OTUs were obtained at the 97% sequence similarity cut-off level, with a mean number of OTUs per sample of 174 ± 120 (range: 20 to 402) (Fig. S1†).
We used several different metrics to calculate alpha diversity, including the OTU species count (Fig. 1a); the Chao1 index for microbial richness (Fig. 1b); the Shannon index for biodiversity (Fig. 1c), and phylogenetic diversity (Fig. 1d). Rarefaction curves for phylogenetic diversity plateaued after 8000 reads per sample, approximating a saturation phase. The Shannon index indicated that there was a much higher gut microbiota diversity within the samples obtained from the silkworms that were reared on mulberry leaves compared with those obtained from the silkworms that were reared on the artificial diet.
 | ||
| Fig. 1 Rarefaction curves of different alpha diversity metrics. Similarity thresholds of 97% (genus level) were considered. The observed metrics were (a) OTUs, (b) the Chao1 measure of microbial richness, (c) the Shannon index of biodiversity, and (d) Faith's phylogenetic diversity index (whole-tree phylogenetic diversity). The HiM, GsM, LoM, and L7M silkworm strains were reared on fresh mulberry leaves, and the HiA and GsA strains were reared on the artificial diet. Each measurement was repeated five times. | ||
Core gut microbiome composition in the silkworms that were reared on fresh mulberry leaves
Based on the phylogenetic classification, OTUs could be assigned to 36 phyla in the four silkworm strains that were reared on mulberry leaves. Here, the shared taxa by all samples in each sampling group (strain) were deemed to be the core gut microbiome. We determined the core bacteria for these four sampling groups, and the number of OTUs shared by all samples within each sampling group was 228, 291, 356, and 172 for the GsM, HiM, LoM, and L7M strains, respectively. The gut microbiota compositions of the HiM and LoM strains, which have the same genetic background, were very similar (Fig. 2a). According to weighted and unweighted UniFrac distances, the gut microbiotas in the HiM, LoM, and L7M strains were quite different from those in the GsM strain (Fig. 2a–c).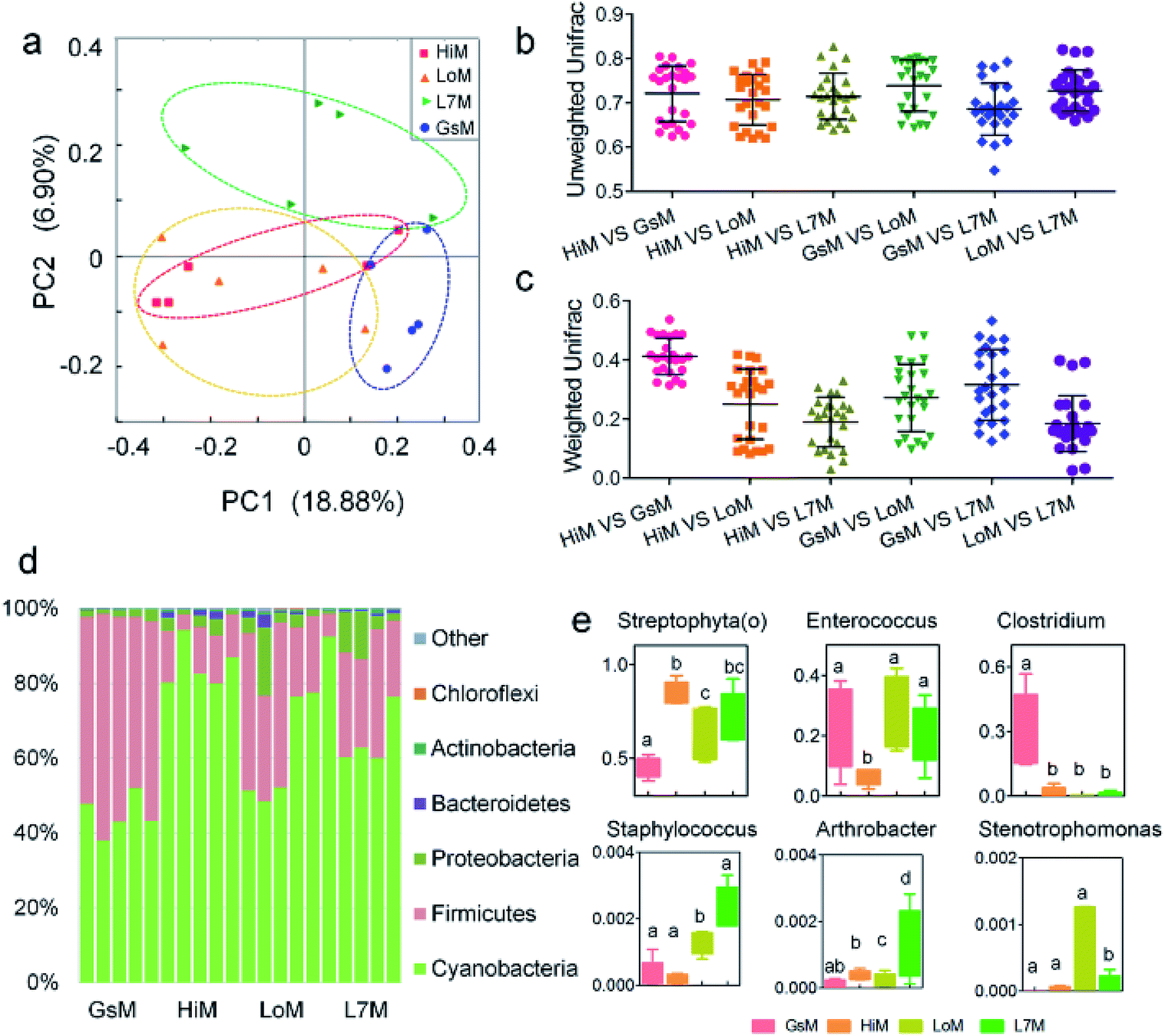 | ||
| Fig. 2 Relative abundance of bacteria comprising the gut microbiota in the four silkworm strains that were reared on mulberry leaves. (a) A PCoA of the gut microbiota structure based on the unweighted UniFrac metric. Each symbol represents the gut microbiota of one sample; each sample group is represented by its respective color. (b) Average unweighted and (c) weighted Unifrac distances among the strains. (d) Relative abundance of phylum-classified gut microbiota. Histograms are based on the proportion of OTUs per sample. Colors were assigned for all phyla with a relative abundance >0.1% in at least 10% of the samples. (e) Relative abundance of genus-classified gut microbiota. The same letter indicates there was no significant difference (P > 0.5). | ||
A phylum-level analysis of the gut microbiota compositions of all four strains revealed that Cyanobacteria was the most abundant phylum, followed by Firmicutes, Proteobacteria, and Bacteroidetes, and the sum of the abundance of Cyanobacteria and Firmicutes accounted for 94.4% ± 5.4% of the total microbiotas. In the GsM strains, the abundance of Cyanobacteria and Firmicutes were similar, accounting for 44.8% ± 5.3% and 52.9% ± 5.6% of the total microbiotas, respectively. In the LoM and L7M strains, the abundance ratio of Cyanobacteria to Firmicutes increased to 2:1 and 3.1:1, respectively, while in the HiM strain, the abundance of Cyanobacteria increased to 84.7% ± 6.1%, and the abundance ratio of Cyanobacteria to Firmicutes reached 7.8:1 (Fig. 2d).
An analysis at the genus level showed that the dominant bacteria ranked in terms of abundance, were clade Streptophyta(o) and genera Enterococcus and Clostridium. Streptophyta(o) had the highest abundance in the HiM strain and the lowest abundance in the GsM strain. The abundance of Enterococcus in HiM intestines was lower than that in the other silkworm strains, while Clostridium was dominant with high abundance only in the GsM strain (Fig. 2e). Thus, the genetic differences of silkworms with different ingestive habits affected the composition of the gut microbiota in larvae reared on mulberry leaves, which was evidenced mainly as a difference in the abundance of dominant bacteria, while differences in the diversity of gut microbiota were not obvious (Fig. 2d).
PICRUSt was further used to predict the impact of the gut microbiota on the host. Level 1 results showed that in the four silkworm strains reared on mulberry leaves, the effects of the gut microbiota on the host were fundamentally similar. There was no significant difference (P > 0.5) in the effects on metabolism, genetic information processing, and environmental information processing (Fig. 3a), and only the effect of the gut microbiota on cellular processes in the GsM strain was significant (P < 0.5) (Fig. 3b). Level 2 results showed that the gut microbiota provided dozens of functions, including amino acid metabolism, carbohydrate metabolism, membrane transport, energy metabolism, metabolism of cofactors and vitamins, xenobiotics biodegradation and metabolism, lipid metabolism, nucleotide metabolism, transcription, and translation for B. mori, but there was no significant difference in the effect of the gut microbiota on host functions in the different silkworm strains that were reared on mulberry leaves (Fig. 3c).
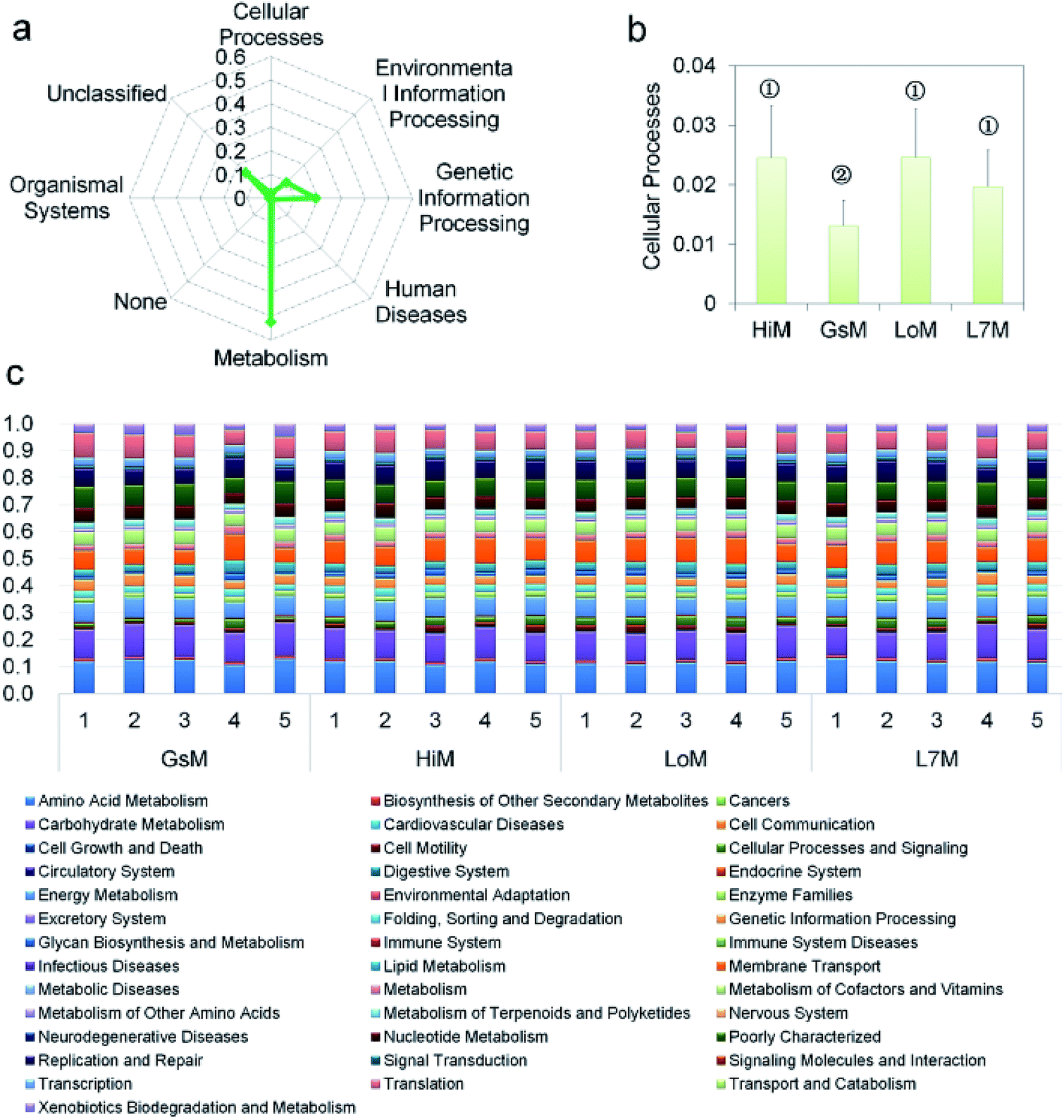 | ||
| Fig. 3 PICRUSt analysis results of the gut microbiota in the silkworm larvae that were reared on fresh mulberry leaves. (a) Metabolic function classification at Level 1. (b) Differences of cellular processes in the four silkworm strains. Samples marked with the same number did not differ significantly from each other, P > 0.05 (n = five replicate samples). (c) Relative abundance of predicted functions at Level 2. Level 1 or Level 2 of the Kyoto Encyclopedia of Genes and Genomes (KEGG) ortholog hierarchy based on the PICRUSt dataset. Each stacked bar represents the relative abundance of the predicted functions of a sample. | ||
Core gut microbiome composition in the silkworms that were reared on the artificial diet
Of the two silkworm strains that were reared on the artificial diet, only the larvae of the Hi and Gs strains ingested the artificial diet. However, there was a significant difference between the gut microbiotas of the HiA and GsA strains, as determined by a PCoA (Fig. 4a) and the UniFrac distance (Fig. 4b). Four hundred forty-seven and 92 types of microorganisms were detected in the intestines of the HiA and GsA strains, respectively, among which 392 and 37 types were unique to each silkworm strain, respectively, and 55 types were common to both strains (Fig. 4c).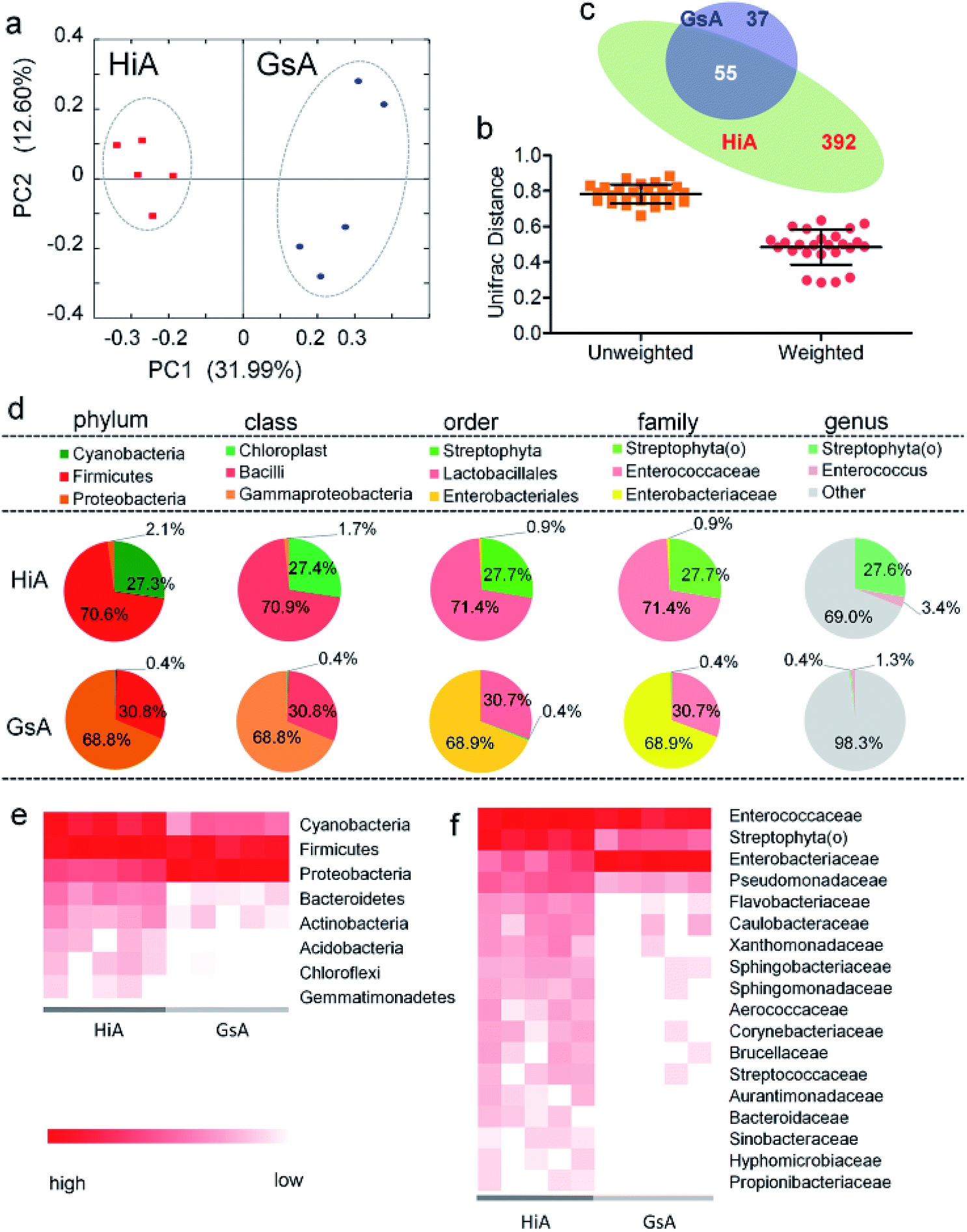 | ||
| Fig. 4 Gut microbiota relative abundance in the two silkworm strains that were reared on the artificial diet. (a) A PCoA based on unweighted UniFrac distances. (b) Average UniFrac distances of the GsA and HiA strains. (c) Venn diagram showing the unique and shared genera. (d) 16S rDNA gene survey of the gut microbiotas. Relative abundance of the members of the gut microbiotas reported at the phylum, class, order, family, and genus levels. The pie charts are based on the proportion of OTUs per sample. Colors were assigned for all the samples detected, and for all microorganisms with a relative abundance ≥1% in at least 10% of the samples. (e) Phylum-classified and (f) family-classified microorganisms of the gut microbiota whose relative abundance differed between the HiA and GsA strains (P < 0.05, n = 5). | ||
A comparison at the phylum level showed that Firmicutes were abundant in both the HiA and GsA strains, while Cyanobacteria and Proteobacteria were the most abundant members of the gut microbiota in the HiA and GsA strains, respectively (Fig. 4d). Because most of the detected microorganisms comprising the gut microbiotas were not named at the genus level, and their proportions in the GsA and HiA strains were as high as 98.3% and 69%, respectively, we added comparisons at the class, order, and family levels. The results in Fig. 4d show that, at the family level, Enterococcaceae and Streptophyta(o) were the two most abundant microorganisms in HiA intestines, while Enterobacteriaceae and Enterococcaceae had the highest abundance in GsA intestines. The sums of abundance of these two dominant microorganisms all reached 99% of the gut microbiota in both silkworm strains.
The results in Fig. 4e show that the abundance of the dominant microorganisms in the GsA and HiA strains differed greatly. At the phylum level, eight types of microorganisms had the most significant differences in abundance, namely Cyanobacteria, Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, Acidobacteria, Chloroflexi, and Gemmatimonadetes. The specific abundance values are presented in Table S1.† At the family level, there were 18 types of dominant microorganisms whose abundance was greater than 1% and whose differences were significant, including Enterococcaceae, Enterobacteriaceae, Streptophyta(o), and Pseudomonadaceae (Fig. 4f and Table S2†). Therefore, not only did the abundance of the dominant microorganisms in the intestines of the different silkworm strains that were reared on the artificial diet change significantly, the gut microbiota diversity changed as well, and it differed from those of the silkworms that were reared on mulberry leaves.Gut microbiota functions in the silkworms that were reared on the artificial diet were predicted by PICRUSt based on microbial 16S rDNA sequences and the KEGG database. A cluster analysis showed that the effect of the gut microbiota differed significantly in the different silkworm strains (Fig. 5a). Level 1 horizontal function prediction results showed that the main functions of the gut microbiota in the silkworms that were reared on the artificial diet had three fundamental functions, namely metabolism, genetic information processing, and environmental information processing, and there was no significant difference between the different silkworm strains (P > 0.05). However, there were statistically significant differences (P < 0.05) in cellular processes and organismal systems (Fig. 5b).
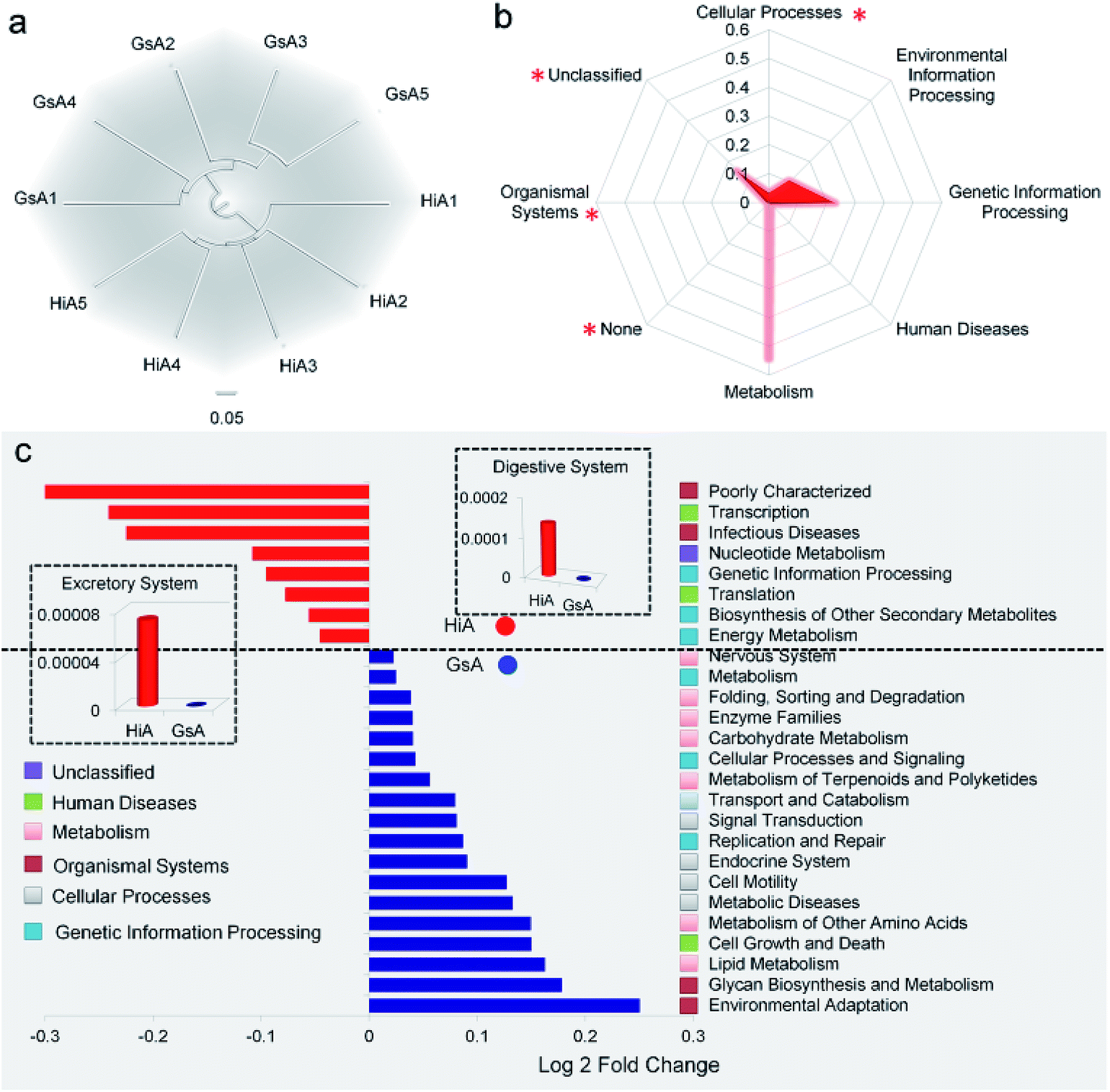 | ||
| Fig. 5 Predicted functional analysis of the gut microbiota using the PICRUSt software tool. (a) Cluster analysis. The numbers 1, 2, 3, 4, and 5 after GsA and HiA are the numbers of repeats in the cluster analysis tree that was based on European clustering, the longest distance method. (b) Metabolic function classification. (c) Predictive metabolic function with significant differences between the HiA and GsA strains. The bar graph shows a significant difference in the metabolic pathway, which was verified by one-way analysis of variance (P < 0.05, n = 5). The fold-change represents the value of the GsA strain divided by that of the HiA strain. | ||
Level 2 function prediction results (Fig. 5c) showed that compared with the gut microbiota in the GsA strain, the gut microbiota in the HiA strain had a much greater impact on poorly characterized transcription and infectious diseases in the host, and, also, it had a greater impact on nucleotide metabolism, genetic information processing, translation, biosynthesis of secondary metabolites, and energy metabolism in the host. The gut microbiota had the greatest influence on environmental adaptation in the GsA strain, and it also had an impact on multiple physiological and metabolic functions, including glycan biosynthesis and metabolism, lipid metabolism, cell growth and death, metabolism of amino acid, metabolic diseases, and cell motility. These results indicated that the effect of the gut microbiota on functions in the different silkworm strains that were reared on the artificial diet differed significantly.
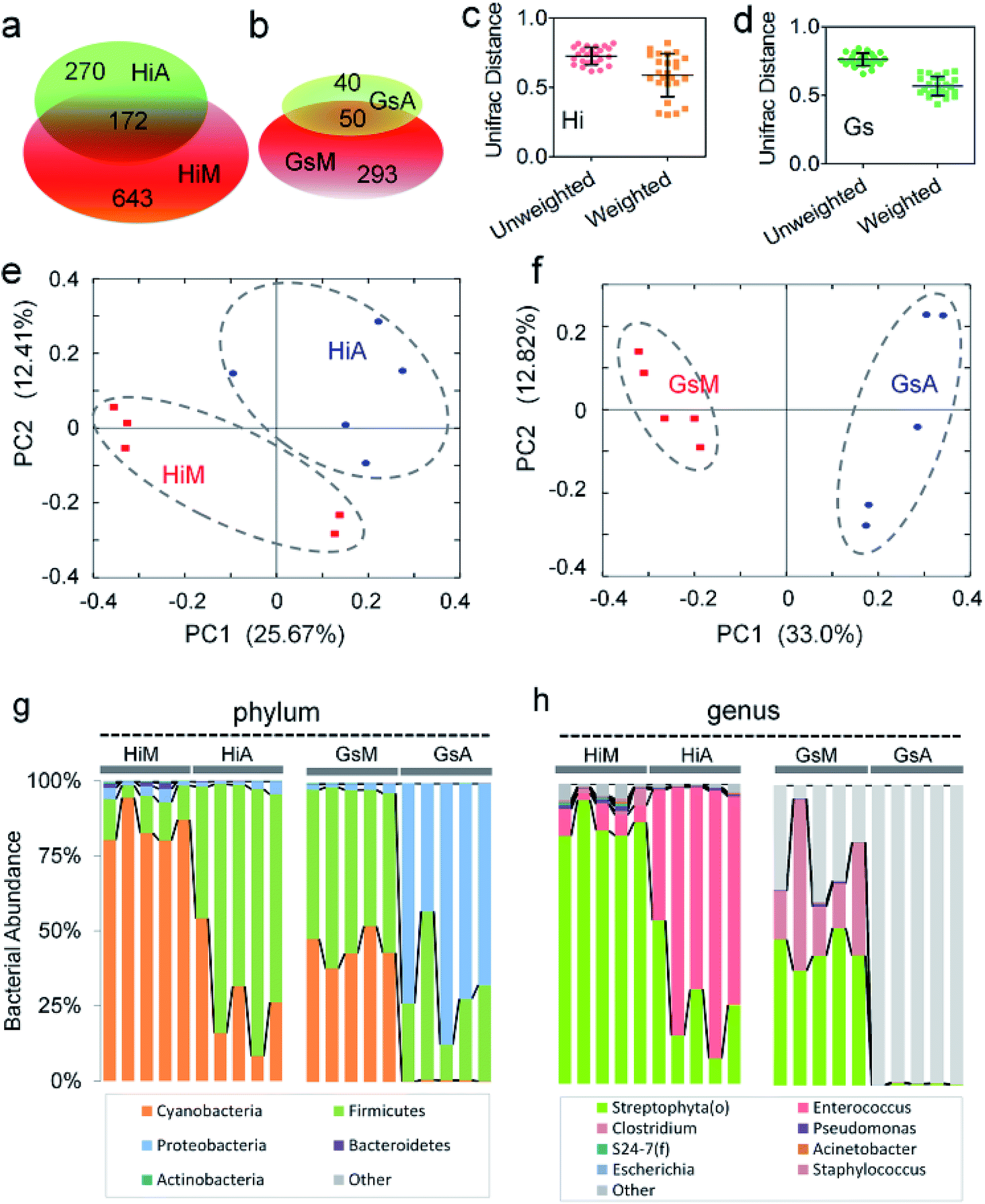 | ||
| Fig. 6 Effect of mulberry leaf and artificial diets on the composition of intestinal microbes in the same silkworm strains. (a and b) Venn analysis of different microbes in different silkworm strains. Average UniFrac distance differences between the (c) HiM and HiA and (d) GsM and GsA strains. PCoA analysis of the (e) HiM and HiA and (f) GsM and GsA strains. The compositions of microbes at the (g) phylum and (h) genus levels. | ||
An analysis at the phylum level showed that the abundance of Cyanobacteria and Actinobacteria in the HiA silkworms were 67.9% ± 20.7% and 83.6% ± 10.9% lower than those in HiM silkworms, respectively, while the abundance of Firmicutes increased by 5.5 ± 1.6-fold. The members of the gut microbiota whose abundance were much lower in GsA silkworms, compared with those in GsM silkworms, were Cyanobacteria (99.1% ± 0.6%), Firmicutes (41.7% ± 30.7%), Actinobacteria (97.0% ± 2.8%), AD3 (100.0% ± 0.0%), Acidobacteria (97.4% ± 5.8%) and Bacteroidetes (94.2% ± 4.4%), while the abundance of Proteobacteria increased by 33.9 ± 8.1-fold (Fig. 6g). The relative content changes of three types of intestinal bacteria which had the highest abundance were analyzed. The ratio of Firmicutes:Cyanobacteria:Proteobacteria was 26:22:1 in HiA intestines, while it was 79:1:175 in GsA intestines, and the abundance of the dominant Cyanobacteria and Actinobacteria in the silkworms that were reared on mulberry leaves were significantly lower than those in the silkworms that were reared on the artificial diet. These results indicated that gut microbiota compositions and bacterial types differed greatly between the silkworms that were reared on the artificial diet and those that were reared on mulberry leaves.
An analysis at the genus level further showed that the top three dominant bacteria in the HiM silkworms were Streptophyta(o), Enterococcus, and Pseudomonas, whose abundance were 84.5% ± 6.1%, 6.3% ± 2.6%, and 0.9% ± 0.5%, respectively, and their relative ratio was 93:7:1. Meanwhile, the top three dominant bacteria in the HiA silkworms were Enterococcus, Streptophyta(o), and Pseudomonas, whose abundance were 70.1% ± 17.6%, 27.2% ± 17.5%, and 0.5% ± 0.2%, respectively, and their relative ratio was 54:139:1 (Fig. 6h). An analysis of the Gs silkworm strains showed that the dominant bacteria in the GsM silkworms were Streptophyta(o) (44.68% ± 5.31%) and Clostridium (28.14% ± 18.52%). In the GsM intestines, 25.60% ± 13.89% of the gut microbiota comprised unnamed, unknown microorganisms, while in the GsA intestines, this type of microorganism accounted for as much as 99.31% ± 0.48% of the gut microbiota (Fig. 6h), which further confirmed that the compositions of the gut microbiota and the abundance of dominant microorganisms differed significantly between the silkworms that were reared on the artificial diet and those that were reared on mulberry leaves.
In this study, the results of a microbiological investigation (Fig. 7) showed that the abundance of Enterococcus in the intestines of the Hi strains that were reared on the artificial diet was 11.1-fold higher than that in the silkworms which were reared on mulberry leaves (Fig. 7h), and the abundance of Enterococcus, which is a member of the Enterobacteriaceae, was 64.97-fold higher in the Gs strains. Clostridium (Fig. 7a), Pseudomonas (Fig. 7d) and Escherichia (Fig. 7g), which were abundant in the silkworms that were reared on mulberry leaves, were present at significantly lower levels in the silkworms that were reared on the artificial diet; only traces of Lactococcus existed in the GsA intestines (Fig. 7e). These results indicate that the difference in the gut microbiota between the silkworms that were reared on the artificial diet is related to immune resistance in silkworms.
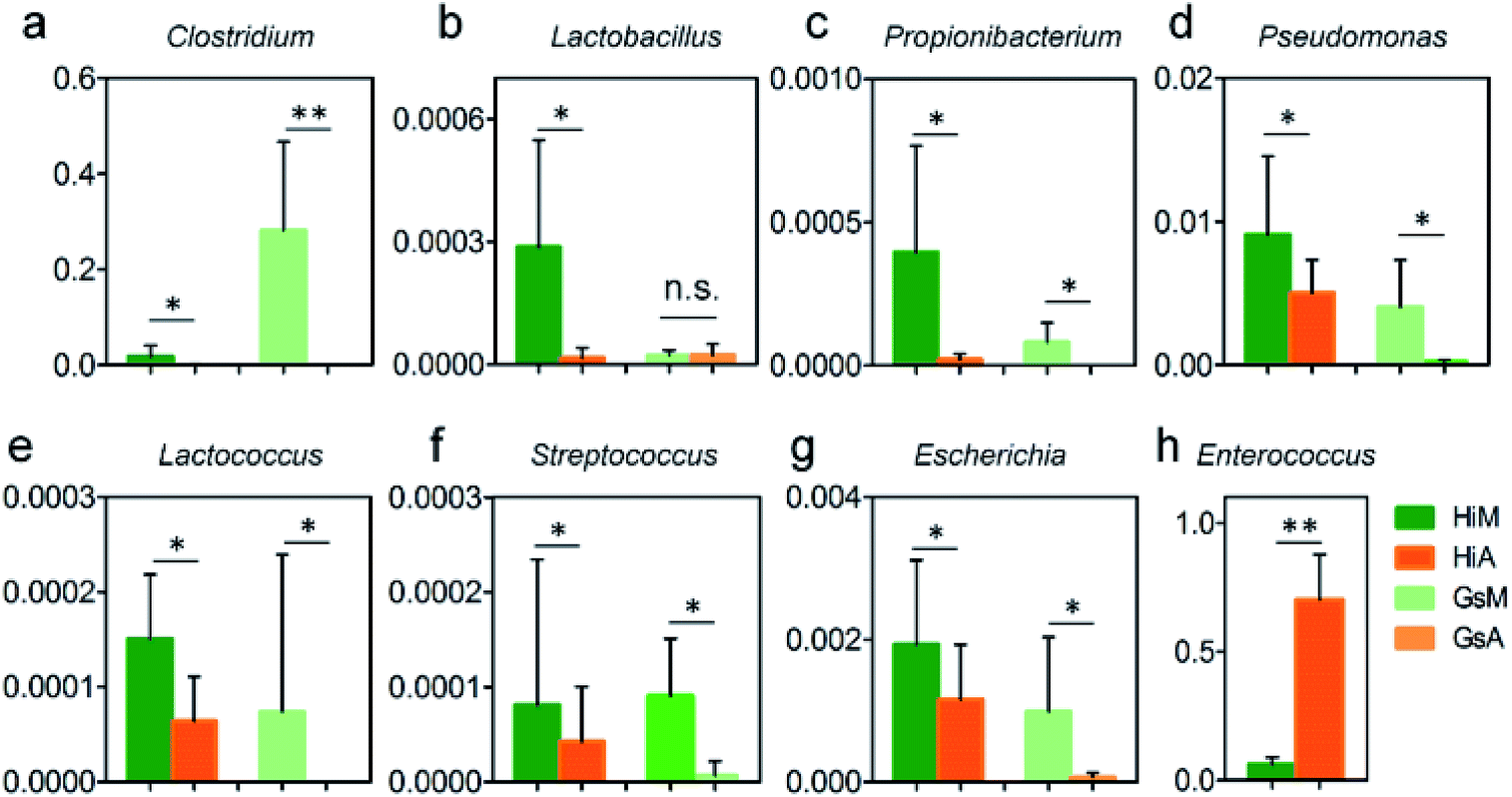 | ||
| Fig. 7 Relative abundance differences of members of the gut microbiota relating to immunization in the silkworms that were reared on the artificial diet. Units of the y-axis are relative abundance. * and ** indicate that the difference between the two groups reached the significance levels of P < 0.5 and P < 0.1 (n = 5), respectively. | ||
Constituents of the gut microbiotas whose abundance differed significantly between the silkworms that were reared on mulberry leaves and those that were reared on the artificial diet are presented in Fig. 8. Whether at the phylum (Fig. 8a and b) or genus levels (Fig. 8c and d), microorganisms with abundance differences in the Gs strains whose ingestion habit was naturally acquired, were greater than those in the Hi strains whose ingestion habits were selected systematically. At the phylum level, the abundance of Cyanobacteria and Actinobacteria in the Gs and Hi silkworms which were reared on the artificial diet were significantly lower than those in the Gs and Hi silkworms which were reared on mulberry leaves (P < 0.05) (Fig. 8a and b). At the family level, the abundance of six types of microorganisms, namely Veillonellaceae, Streptophyta(o), Sphingomonadaceae, Ruminococcaceae, Pseudomonadaceae, and Lachnospiraceae, were significantly lower in the Gs and Hi silkworms that were reared on the artificial diet compared with those of the silkworms that were reared on mulberry leaves (P < 0.05) (Fig. 8c and d).
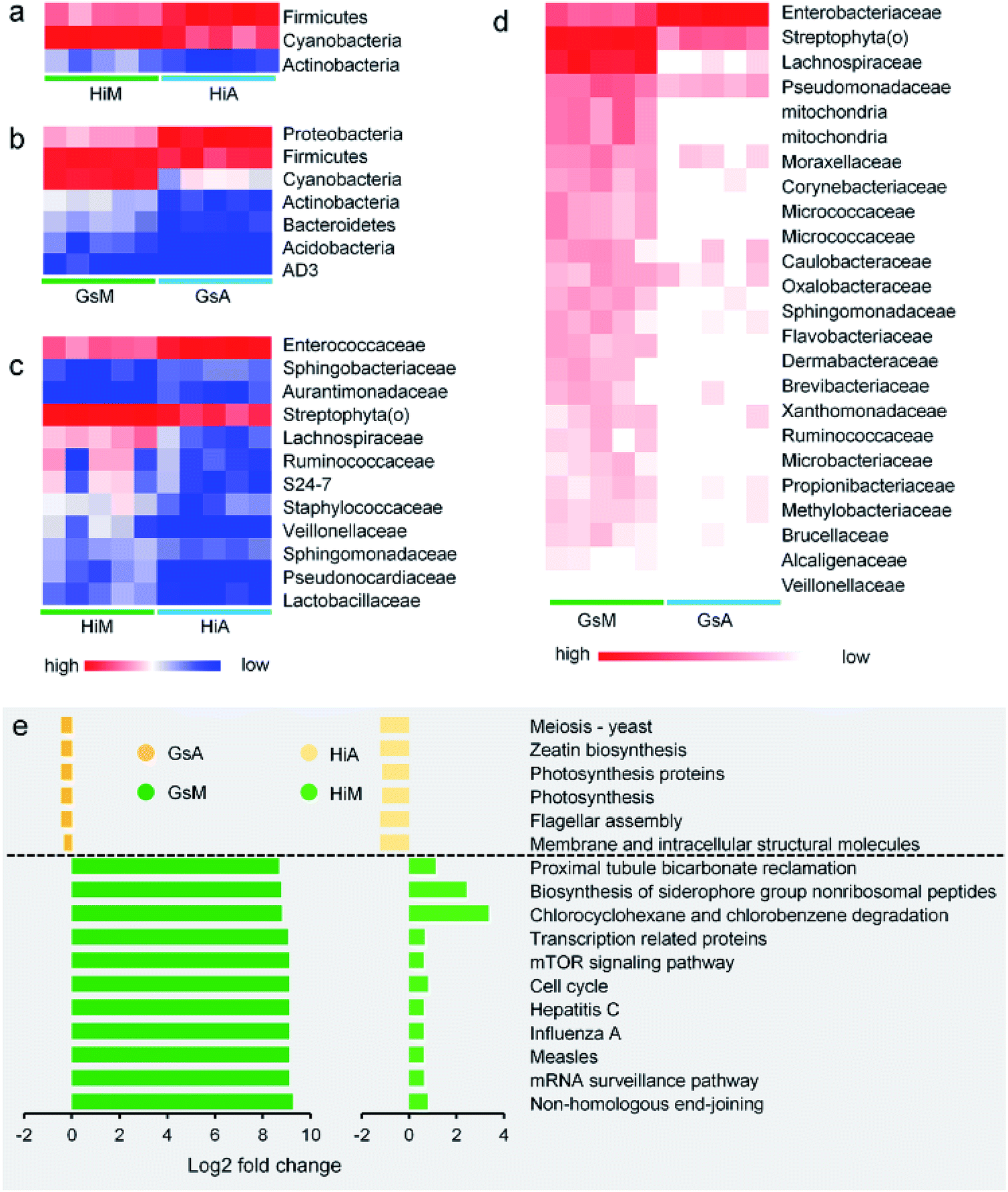 | ||
| Fig. 8 Comparison of the impact of differences between the gut microbiota of the silkworms that were reared fresh mulberry leaves or the artificial diet. (a and b) Phylum-classified and (c and d) family-classified gut microbiota whose relative abundance differed between the silkworms that were reared on fresh mulberry leaves or the artificial diet (the relative abundance of the HiM vs. HiA strains and the GsM vs. GsA are shown, P < 0.05, n = 5). (e) Predicted functional analysis of the silkworm gut microbiomes using the PICRUSt software tool. The bar plot shows the pathways that were enriched significantly by the artificial diet (P < 0.05), as determined by a one-way analysis of variance. The fold-change represents the values of the HiM strain divided by those of the HiA strain, or the values of the GsM strain divided by those of the GsA strain. | ||
The functions of the gut microbiota were predicted using PICRUSt. There were 44 types of function that differed significantly and consistently between the silkworm strains, of which 17 functional levels were reduced, while 27 were increased (Fig. S2†). Seventeen types of function with the most significant differences are presented in Fig. 8e (P < 0.05, the fold-change was greater than 2). The silkworms that were reared on mulberry leaves were used as a control, and the differences of the gut microbiota of the silkworms that were reared on the artificial diet affected 11 types of function, including non-homologous end-joining, whose functional levels were downregulated, and six types of function, including membrane and intracellular structural molecules, whose functional levels were upregulated. In the silkworms that were reared on the artificial diet, the degrees of decreased functional levels in the Gs strains were greater than those in the Hi strains, while the degrees of increased functional levels in the Gs strains were lower than those in the Hi strains. These results indicated that the Gs strains were more strongly affected than the Hi strains when reared on the artificial diet, compared with the same strains that were reared on mulberry leaves.
In a previous study, we analyzed the metabolome of silkworms1 and the results showed that the contents of multiple vitamins, including thiamine and riboflavin, in the hemolymph of silkworms reared on an artificial diet were significantly lower than those in silkworms reared on mulberry leaves. The contents of six essential and non-essential amino acids, including methionine and lysine, were also significantly lower, and multiple metabolic pathways, including serine metabolic pathways associated with silk protein synthesis, were inhibited. Meanwhile, although the contents of key substances in the metabolic pathways of uric acid and urea formed from amino acids were significantly higher, uric acid and urea were heavily enriched in the hemolymph. In the present study, more dominant microorganisms that were positively correlated with the metabolism of vitamins, including thiamine and riboflavin, appeared in the gut microbiota of the silkworms that were reared on the artificial diet, compared with those in the silkworms that were reared on mulberry leaves (Fig. 9a), while the abundance of microorganisms that were negatively correlated with key substances in the metabolic pathways of uric acid and urea were much higher (Fig. 9b). While microorganisms that were associated with the metabolic pathways of amino acids in the host appeared to occur randomly, dominant microorganisms that were positively associated with some metabolites were closely related to those that were negatively correlated with other metabolites (Fig. 9c). This can be explained by the fact that the abundance of microorganisms involved in the synthesis of vitamins, such as thiamine and riboflavin, in the host were significantly higher, and the functions that help the host eliminate toxins and xenobiotics were significantly lower, while the differences in microorganisms involved in amino acid metabolism in the host were not consistent. These results indicate that the difference in the gut microbiota of silkworms reared on the artificial diet was highly correlated with metabolic changes in the host. Therefore, we conclude that the differences in the diversity of gut microbiota and dominant bacteria in the silkworms that were reared on the artificial diet could be related to the adaptation of silkworms to the artificial diet.
 | ||
| Fig. 9 Co-occurrence network analysis among gut microbial populations and metabolites involved in the enriched pathways in the silkworm hemolymph. Correlation network of gut bacteria and (a) vitamins and (b) metabolites related to the urea cycle, and (c) amino acids. Colored circle nodes represent metabolites and microbial populations. Each co-occurring pair among the microbial populations and metabolites has an absolute Spearman rank correlation > 0.90 (red line, positive correlation, R > 0.90; blue line, negative correlation, R < −0.90) with a false discovery rate-corrected significance level less than 0.5. | ||
Discussion
Intestinal microorganisms in the silkworms that were reared on the artificial diet differ significantly from those of the silkworms that were reared on mulberry leaves
Changes in diet can affect gut microbiota diversity and relative gut microbial contents in insects, as demonstrated in Riptortus clavatus and Schistocerca gregaria.35,36 A similar phenomenon has also been found in Phlebotomus argentipes37 and Coptotermes formosanus20,38 which efficiently digest cellulose. Also, it has been demonstrated repeatedly in humans39–41 and mice.42,43Mulberry leaves are almost the only food that silkworms have adapted to over a long period. It has been reported that Cyanobacteria, Firmicutes, and Proteobacteria are the dominant bacteria in the intestines of silkworm larvae of the Dazao variety that were reared on mulberry leaves.20 It is worth mentioning that the similar bacterial phyla were observed in the various beetles (Coleoptera).44,45 In this study, we investigated four silkworm strains that were reared on mulberry leaves, and the dominant bacteria in their intestines were also Cyanobacteria, Firmicutes, and Proteobacteria, indicating that the gut microbiota of silkworms reared on mulberry leaves is quite stable. The functions of the intestinal bacteria in the gut microbiota, as predicted by PICRUSt, showed that genetic differences between the silkworm strains with different artificial diet ingestive habits, which were selected systematically or naturally occurring, did not have an impact on the functions of silkworms that were reared on mulberry leaves. We further found that the gut microbiota of the silkworms that were reared on the artificial diet differed greatly from that of the silkworms that were raised on mulberry leaves, as the most dominant bacteria in their intestines were the Firmicutes or Proteobacteria. This difference in silkworms is similar to that in chickens,46 pigs,47 and horses48 when they are reared on an artificial diet such as soybean meal. In this study, the prediction results of the functions of the intestinal bacteria showed that the gut microbiota functions in the different silkworm strains that were reared on the artificial diet differed significantly, indicating that the impact of the gut microbiota on the silkworms that were reared on the artificial diet may be greater and more varied than that on the silkworms that were reared on mulberry leaves. These results suggest that the microorganisms harbored in the silkworm intestines may co-evolve with the selection of an artificial diet ingestive habit in the silkworm, and they may be involved in a variety of metabolic functions in the host.
Differences of intestinal microorganisms affect immune resistance in the silkworms that were reared on the artificial diet
A number of reports suggest that gut microbiota can regulate the expression of host genes and affect the proliferation of host cells49 and the development of tissues and organs,50 and that it even plays a key role in the maturation and homeostasis of the host immune system.51–53 Furthermore, the gut microbiota can protect the host from excessive growth of pathogens,54 and it can react to or modify specific drugs and eliminate xenobiotic toxins.55,56 Studies in silkworms reported that the abundance of Enterococcus in the intestines of silkworms reared on mulberry leaves is associated with increased immunity.20 Lactobacillus 11/19-B1 enhanced the survival rate of larvae infected with P. aeruginosa by activating innate immunity in B. mori.57 The colonization of ice-nucleation active Enterobacteriaceae in the intestines of mulberry pyralid larvae significantly reduced the cold hardiness of the larvae.58 Endogenous P. aeruginosa, which colonizes B. mori intestines, promotes the binding of ExoS to FXYD3 by expressing the pvdE gene, which mediates penetration of bacteria through the intestinal epithelial cell barrier from infected B. mori hemolymph to achieve bacterial translocation.59 Escherichia coli and Beauveria bassiana affect the immune response in silkworm intestines by increasing the expression of the signal-transduction-mediating transmembrane protein BmToll9 during the innate immune response.22 These results indicate that different bacteria induce different immune responses in B. mori.Differences of the gut microbiota affect nutrient metabolism in the silkworms that were reared on the artificial diet
Studies have found that the gut microbiota can regulate the synthesis of neurotransmitters in the host,60 as well as intestinal endocrine hormones,61 vitamins62 and the digestion of complex carbohydrates.63,64 It also affects specific physiological functions and provides energy to the host.65–67The insect gut microbiota is thought to be involved in the decomposition and digestion of food.68 Research on B. mori has revealed that the abundance of Cyanobacteria in the intestines of silkworms reared on mulberry leaves may be related to the digestion of chlorophyll and other ingredients that are abundant in fresh mulberry leaves.20 Bacillus circulans, Proteus vulgaris, E. coli, and Citrobacter freundii can produce digestive enzymes to help degrade carbohydrates, and Alkaliphilic bacteria found and isolated from B. mori intestines can degrade polysaccharides, while the number of cellulose-decomposing bacteria in the intestines increases with increasing larval age.24 The proteases secreted by Stenotrophomonas maltophilia can improve the digestion and absorption of mulberry leaves in B. mori intestines.69 Also, it has been reported that the abundance of lipase-producing bacteria in the gut microbiota of silkworms reared on an artificial diet changed significantly.25
In summation, gut microbiota diversity and dominant bacteria in the silkworms that were reared on the artificial diet differed greatly compared with those of the silkworms that were reared on mulberry leaves, which may be related to multiple issues, including development, metabolism, and disease resistance in the silkworms that were reared on the artificial diet.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (no. 31472149 and 31672492), the China Agriculture Research System (CARS) (no. CARS-22), the Provincial Key R & D Program of Jiangsu (no. BE2015317), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The manuscript was revised by a native English speaker from International Science Editing Scientific Services.References
- H. L. Dong, S. X. Zhang, H. Tao, Z. H. Chen, X. Li, J. F. Qiu, W. Z. Cui, Y. H. Sima, W. Z. Cui and S. Q. Xu, Sci. Rep., 2017, 7, 10972 CrossRef PubMed
.
- Q. Xia, S. Li and Q. Feng, Annu. Rev. Entomol., 2014, 59, 513–536 CrossRef PubMed
- H. A. Yanagawa, K. Watanabe and M. Nakamura, J. Insect Biotechnol. Sericology, 2010, 58, 401–406 Search PubMed
- R. Chinnaswamy, H. Lakshmi, S. S. Kumari, C. M. Anuradha and C. S. Kumar, J. Insect Sci., 2012, 12, 3 CrossRef PubMed
- Y. Horie and S. Nakasone, J. Insect Physiol., 1971, 17, 1441–1450 CrossRef PubMed
- T. Kanda, T. Tamura and H. Inoue, J. Insect Biotechnol. Sericol., 1988, 57, 489–494 Search PubMed
- K. Asaoka and Y. Mano, J. Seric. Sci. Jpn., 1992, 1, 1–5 Search PubMed
- L. Cappellozza, S. Cappellozza, A. Saviane and G. Sbrenna, Appl. Entomol. Zool., 2005, 40, 405–412 CrossRef
- H. Chikara, S. Masahiro and S. Hiroshi, J. Insect Physiol., 1999, 45, 15–20 CrossRef
- A. Saviane, L. Toso, C. Righi, C. Pavanello, V. Crivellaro and S. Cappellozza, Bulletin of Insectology, 2014, 67, 167–174 Search PubMed
- Z. H. Zhou, H. J. Yang, M. Chen, C. F. Lou, Y. Z. Zhang, K. P. Chen, Y. Wang, M. L Yu, F. Yu, J. Y. Li and B. X. Zhong, J. Proteome Res., 2008, 7, 5103–5111 CrossRef PubMed
- F. Fransen, A. A. van Beek, T. Borghuis, B. Meijer, F. Hugenholtz, C. van der Gaast-de Jongh, H. F. Savelkoul, M. I. de Jonge, M. M. Faas, M. V. Boekschoten, H. Smidt, S. El Aidy and P. de Vos, Front. Immunol., 2017, 8, 754 CrossRef PubMed
- S. M. Jandhyala, R. Talukdar, C. Subramanyam, H. Vuyyuru, M. Sasikala and D. Nageshwar Reddy, World J. Gastroenterol., 2015, 21, 8787–8803 CrossRef PubMed
- J. K. Nicholson, E. Holmes, J. Kinross, R. Burcelin, G. Gibson, W. Jia and S. Pettersson, Science, 2012, 336, 1262–1267 CrossRef PubMed
- C. De Filippo, D. Cavalieri, M. Di Paola, M. Ramazzotti, J. B. Poullet, S. Massart, S. Collini, G. Pieraccini and P. Lionetti, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14691–14696 CrossRef PubMed
- W. W. Wang, C. He, J. Cui, H. D. Wang and M. L. Li, J. Insect Sci., 2014, 14, 111 Search PubMed
- G. D. Wu, J. Chen, C. Hoffmann, K. Bittinger, Y. Y. Chen, S. A. Keilbaugh, M. Bewtra, D. Knights, W. A. Walters, R. Knight, R. Sinha, E. Gilroy, K. Gupta, R. Baldassano, L. Nessel, H. Li, F. D. Bushman and J. D. Lewis, Science, 2011, 334, 105–108 CrossRef PubMed
- S. Cappellozza, A. Saviane, G. Tettamanti, M. Squadrin, E. Vendramin, P. Paolucci, E. Franzetti and A. Squartini, J. Invertebr. Pathol., 2011, 106, 386–393 CrossRef PubMed
- X. Liang, Y. Fu, L. Tong and H. Liu, Appl. Microbiol. Biotechnol., 2014, 98, 3769–3776 CrossRef PubMed
- Z. Sun, Y. Lu, H. Zhang, D. Kumar, B. Liu, Y. Gong, M. Zhu, L. Zhu, Z. Liang, S. Kuang, F. Chen, X. Hu, G. Cao, R. Xue and C. Gong, PLoS One, 2016, 11, e0146313 CrossRef PubMed
- L. Zhang, Y. W. Wang and Z. Q. Lu, J. Zhejiang Univ., Sci., B, 2015, 16, 875–882 CrossRef PubMed
- S. Wu, X. Zhang, Y. He, J. Shuai, X. Chen and E. Ling, Dev. Comp. Immunol., 2010, 34, 1191–1198 CrossRef PubMed
- G. N. Li, X. J. Xia, W. C.Tang and Y. Zhu, Appl. Microbiol. Biotechnol., 2016, 100, 6715–6724 CrossRef PubMed
- A. A. Anand, S. J. Vennison, S. G. Sankar, D. I. Prabhu, P. T. Vasan, T. Raghuraman, C. J. Geoffrey and S. E. Vendan, J. Insect Sci., 2010, 10, 107 CrossRef PubMed
- W. Feng, X. Q. Wang, W. Zhou, G. Y. Liu and Y. J. Wan, J. Insect Sci., 2011, 11, 135 CrossRef PubMed
- Y. Y. Li, J. Tang, K. H. Fu, S. G. Gao, Q. Wu and J. Chen, J. Environ. Sci. Health, Part B, 2012, 47, 622–630 CrossRef PubMed
- H. Wang, L. Wang, Y. Wang, H. Tao, W. Yin, Y. SiMa, Y. Wang and S. Xu, Sci. Rep., 2015, 5, 13839 CrossRef PubMed
- S. V. Lynch and O. Pedersen, N. Engl. J. Med., 2016, 375, 2369–2379 CrossRef PubMed
- S. T. Williams, P. G. Foster and D. T. Littlewood, Gene, 2014, 533, 38–47 CrossRef PubMed
- A. M. Bolger, M. Lohse and B. Usadel, Bioinformatics, 2014, 30, 2114–2120 CrossRef PubMed
- D. Reyon, S. Q. Tsai, C. Khayter, J. A. Foden, J. D. Sander and J. K. Joung, Nat. Biotechnol., 2012, 30, 460–465 CrossRef PubMed
- J. G. Caporaso, J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman, E. K. Costello, N. Fierer, A. G. Peña, J. K. Goodrich, J. I. Gordon, G. A. Huttley, S. T. Kelley, D. Knights, J. E. Koenig, R. E. Ley, C. A. Lozupone, D. McDonald, B. D. Muegge, M. Pirrung, J. Reeder, J. R. Sevinsky, P. J. Turnbaugh, W. A. Walters, J. Widmann, T. Yatsunenko, J. Zaneveld and R. Knight, Nat. Methods, 2010, 7, 335–336 CrossRef PubMed
- Q. Wang, G. M. Garrity, J. M. Tiedje and J. R. Cole, Appl. Environ. Microbiol., 2007, 73, 5261–5267 CrossRef PubMed
- M. G. Langille, J. Zaneveld, J. G. Caporaso, D. McDonald, D. Knights, J. A. Reyes, J. C. Clemente, D. E. Burkepile, R. L. Vega Thurber, R. Knight, R. G. Beiko and C. Huttenhower, Nat. Biotechnol., 2013, 31, 814–821 CrossRef PubMed
- Y. Kikuchi, T. Hosokawa and T. Fukatsu, Appl. Environ. Microbiol., 2007, 73, 4308–4316 CrossRef PubMed
- R. Dillon and K. Charnley, Res. Microbiol., 2002, 153, 503–509 CrossRef PubMed
- H. Hillesland, A. Read, B. Subhadra, I. Hurwitz, R. McKelvey, K. Ghosh, P. Das and R. Durvasula, Am. J. Trop. Med. Hyg., 2008, 79, 881–886 Search PubMed
- A. Hayashi, H. Aoyagi, T. Yoshimura and H. Tanaka, J. Biosci. Bioeng., 2007, 103, 358–367 CrossRef PubMed
- F. Bäckhed, J. Roswall, Y. Peng, Q. Feng, H. Jia, P. Kovatcheva-Datchary, Y. Li, Y. Xia, H. Xie, H. Zhong, M. T. Khan, J. Zhang, J. Li, L. Xiao, J. Al-Aama, D. Zhang, Y. S. Lee, D. Kotowska, C. Colding, V. Tremaroli, Y. Yin, S. Bergman, X. Xu, L. Madsen, K. Kristiansen, J. Dahlgren and J. Wang, Cell Host Microbe, 2015, 10, 852 CrossRef PubMed
- S. L. Schnorr, M. Candela, S. Rampelli, M. Centanni, C. Consolandi, G. Basaglia, S. Turroni, E. Biagi, C. Peano, M. Severgnini, J. Fiori, R. Gotti, G. De Bellis, D. Luiselli, P. Brigidi, A. Mabulla, F. Marlowe, A. G. Henry and A. N. Crittenden, Nat. Commun., 2014, 5, 3654 CrossRef PubMed
- G. D. Wu, C. Compher, E. Z. Chen, S. A. Smith, R. D. Shah, K. Bittinger, C. Chehoud, L. G. Albenberg, L. Nessel, E. Gilroy, J. Star, A. M. Weljie, H. J. Flint, D. C. Metz, M. J. Bennett, H. Li, F. D. Bushman and J. D. Lewis, Gut, 2016, 65, 63–72 CrossRef PubMed
- N. Marungruang, F. Fåk and E. Tareke, Nutr. Metab., 2016, 13, 22 CrossRef PubMed
- G. H. Norris, C. Jiang, J. Ryan, C. M. Porter and C. N. Blesso, J. Nutr. Biochem., 2016, 30, 93–101 CrossRef PubMed
- E. E. Ziganshina, W. S. Mohammed, E. I. Shagimardanova, P. Y. Vankov, N. E. Gogoleva and A. M. Ziganshin, BioMed Res. Int., 2018, 6765438 Search PubMed
- M. Kaltenpoth and S. Steiger, Mol. Ecol., 2014, 23, 1251–1267 CrossRef PubMed
- J. Ding, L. Zhao, L. Wang, W. Zhao, Z. Zhai, L. Leng, Y. Wang, C. He, Y. Zhang, H. Zhang, H. Li and H. Meng, Genet., Sel., Evol., 2016, 48, 93 CrossRef PubMed
- Q. Niu, P. Li, S. Hao, Y. Zhang, S. W. Kim, H. Li, X. Ma, S. Gao, L. He, W. Wu, X. Huang, J. Hua, B. Zhou and R. Huang, Sci. Rep., 2015, 5, 9938 CrossRef PubMed
- Y. Zhao, B. Li, D. Bai, J. Huang, W. Shiraigo, L. Yang, Q. Zhao, X. Ren, J. Wu, W. Bao and M. Dugarjaviin, Asian-Australas. J. Anim. Sci., 2016, 29, 1345–1352 CrossRef PubMed
- N. Ijssennagger, C. Belzer, G. J. Hooiveld, J. Dekker, S. W. van Mil, M. Müller, M. Kleerebezem and R. van der Meer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10038–10043 CrossRef PubMed
- C. Reinhardt, M. Bergentall, T. U. Greiner, F. Schaffner, G. Ostergren-Lundén, L. C. Petersen, W. Ruf and F. Bäckhed, Nature, 2012, 483, 627–631 CrossRef PubMed
- M. Fulde and M. W. Hornef, Immunol. Rev., 2014, 260, 21–34 CrossRef PubMed
- Human Microbiome Project Consortium, Nature, 2012, 486, 207–214 CrossRef PubMed
- J. F. Rawls, B. S. Samuel and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4596–4601 CrossRef PubMed
- N. Kamada, G. Y. Chen, N. Inohara and G. Núñez, Nat. Immunol., 2013, 14, 685–690 CrossRef PubMed
- A. S. Devlin and M. A. Fischbach, Nat. Chem. Biol., 2015, 11, 685–690 CrossRef PubMed
- H. J. Haiser, D. B. Gootenberg, K. Chatman, G. Sirasani, E. P. Balskus and P. J. Turnbaugh, Science, 2013, 341, 295–298 CrossRef PubMed
- S. Nishida, Y. Ono and K. Sekimizu, Drug Discoveries Ther., 2016, 10, 49–56 CrossRef PubMed
- K. Watanabe and M. Sato, Cryobiology, 1999, 38, 281–289 CrossRef PubMed
- J. Okuda, M. Okamoto, N. Hayashi, S. Sawada, S. Minagawa and N. Gotoh, J. Infect. Chemother., 2012, 18, 332–340 CrossRef PubMed
- J. M. Yano, K. Yu, G. P. Donaldson, G. G. Shastri, P. Ann, L. Ma, C. R. Nagler, R. F. Ismagilov, S. K. Mazmanian and E. Y. Hsiao, Cell, 2015, 161, 264–276 CrossRef PubMed
- H. Neuman, J. W. Debelius, R. Knight and O. Koren, FEMS Microbiol. Rev., 2015, 39, 509–521 CrossRef PubMed
- T. Yatsunenko, F. E. Rey, M. J. Manary, I. Trehan, M. G. Dominguez-Bello, M. Contreras, M. Magris, G. Hidalgo, R. N. Baldassano, A. P. Anokhin, A. C. Heath, B. Warner, J. Reeder, J. Kuczynski, J. G. Caporaso, C. A. Lozupone, C. Lauber, J. C. Clemente, D. Knights, R. Knight and J. I. Gordon, Nature, 2012, 486, 222–227 CrossRef PubMed
- M. S. Gilmore and J. J. Ferretti, Science, 2003, 299, 1999–2002 CrossRef PubMed
- H. M. Wexler, Clin. Microbiol. Rev., 2007, 20, 593–621 CrossRef PubMed
- A. M. O'Hara and F. Shanahan, EMBO Rep., 2006, 7, 688–693 CrossRef PubMed
- C. E. Stevens and I. D. Hume, Physiol. Rev., 1998, 78, 393–427 CrossRef PubMed
- E. E. Canfora, J. W. Jocken and E. E. Blaak, Nat. Rev. Endocrinol., 2015, 11, 577–591 CrossRef PubMed
- A. C. Wong, A. J. Dobson and A. E. Douglas, J. Exp. Biol., 2014, 217, 1894–1901 CrossRef PubMed
- Z. Wang, L. Sun, J. Cheng, C. Liu, X. Tang, H. Zhang and Y. Liu, Biotechnol. Appl. Biochem., 2016, 63, 292–299 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04627a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |