
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent outcomes of gut microbiota alteration upon use of spectrum antibiotics in high sugar diet-induced diabetes in rats†
Bhumika Prajapatia,
Prasant Kumar Jenaab,
Sweta Patela and
Sriram Seshadri *a
*a
aInstitute of Science, Nirma University, Sarkhej-Gandhinagar Highway, Chharodi, Ahmedabad-382481, Gujarat, India. E-mail: sriram.seshadri@nirmauni.ac.in; sriramsjpr@gmail.com; Fax: +91 2717 241916; Tel: +91 2717 241901-04 ext. 752
bDepartment of Pathology and Laboratory Medicine, University of California, Davis, Sacramento, USA
First published on 20th July 2018
Abstract
Background: A sugar rich diet induces inflammation and insulin resistance (IR) mainly through gut microbiota alteration. Gut dysbiosis increases lipopolysaccharide (LPS) and reduces propionate and butyrate levels to impair the insulin signalling cascades by different molecular pathways, which progresses into IR. The present study was designed to investigate the effect of spectrum specific antibiotics on the modulation of gut microbiota and its signalling pathways to prevent diet-induced diabetes. Methods: Healthy male Wistar rats were divided into a non-diabetic group with a control diet (CD), a diabetic group with a high sucrose diet (HSD) and two antibiotic fed groups (linezolid and cefdinir; administered by oral gavage) along HSD. Physiological, biochemical, inflammatory and microbiome parameters were examined. Results: Cefdinir administration in HSD rats reduced fasting glucose, serum triglyceride, and cholesterol levels compared to HSD alone. In addition, cefdinir reduced serum LPS by decreasing the population of Gram-negative phyla, that is, Bacteroidetes and Proteobacteria in the fecal content. Furthermore, cefdinir treatment decreased hepatic/ileal/colonic Tlr4, Nlr1, and Nf-κB at the mRNA level. Moreover, cefdinir-treated rats had shown increased fecal butyrate and propionate and reduced acetate levels compared to HSD alone. Cefdinir treatment also induced ileal/colonic Gpr43 and Glut4 at the mRNA level after 12 weeks of administration. Conclusions: Taken together, these data suggest that administration of a Gram-negative spectrum antibiotic, that is, cefdinir, has modulated the gut microbiota, and reduced serum LPS and triglycerides, which prevented the progression of IR and inflammation in HSD rats.
1. Introduction
A combination of environmental factors, including high sugar diets and sedentary lifestyles, has led to the dramatic increases in the incidence of metabolic disorders worldwide.1 The chronic low-grade inflammation in obesity and type 2 diabetes (T2D) is characterized by increased levels of pro-inflammatory cytokines, acute-phase reactants and activation of inflammatory signalling pathways.2 Recent evidence suggests that the gut microbiota is important for body weight control and energy homeostasis.1 Several mechanisms have been identified for this interaction, but they are still not well understood. A transplantation of the gut microbiota from lean mice to germ free animals resulted in rapid weight gain, increased epididymal fat content, reduction in lean body mass, lower food intake and development of insulin resistance (IR).3 This suggests the important role of the gut microbiota in energy harvest. Focusing on gut microbiota profiles, Turnbaugh and colleagues4 showed different bacterial prevalence in lean vs. obese mice as well as in humans. Metagenomic analysis had shown increased energy harvest in the gut microbiota of obese individuals. The gut microbiota plays an important role in body weight gain and glucose homeostasis through energy harvest, but the mechanism of induction is still not clear. Researchers have been focusing on gut microbiota profiling and have compared gut metagenomic composition between T2D and healthy animal groups and observed an increase in Lactobacillus species with a decrease in Clostridium species in the T2D group.5 Real time quantitative PCR (qPCR) investigation has shown a significantly reduced proportion of phylum Firmicutes and Clostridia class in the T2D group as compared to controls. Moreover, the β proteobacteria class was highly enriched in the diabetic group as compared to the non-diabetic control group.3 Such findings provide significant knowledge for developing strategies to control the pathogenesis of diabetes through altering the gut microbiota.The gut microbiota regulates insulin sensitivity through various factors, but most importantly it works through the gut derived lipopolysaccharide (LPS) and short chain fatty acids (SCFAs). These molecules can act through the different signalling pathways of innate immune receptors and G-protein-coupled receptors (GPCRs) to develop IR.6 A high sucrose diet (HSD) can induce T2D through alterations in various gut dominant microbial species in mice.7 Antibiotics, probiotics and prebiotics can modulate the gut microbiota considerably by reducing the number of bacterial pathogens to prevent the development of metabolic diseases.8 It has been established that a HSD can modulate gut microbiota in mice, altering the percentages of particular species of bacteria. In addition, antibiotics can downregulate the gut microbiota dramatically, reducing the number of colonies and also modulating some bacterial species. Linezolid and cefdinir were selected due to their Gram-positive and Gram-negative antimicrobial spectrum, respectively. Moreover, these drugs have complete oral bioavailability. Long term antibiotic usage can result in antimicrobial resistance which is the cause of severe infections, treatment complications and mortality. Therefore, the aim of the present study was not to prove antibiotic usage as a treatment strategy, but to use this approach to modulate the gut microbiota for investigating the correlations between changes in the microbiota and insulin sensitivity and signalling, and, at the same time, the possible mechanism that accounts for these effects.
2. Materials and methods
2.1 Drugs and consumables
The active pharmaceutical ingredients of linezolid and cefdinir were obtained as a gift from Macleod Pharmaceuticals ltd (Mumbai, India). Synthesized oligonucleotides (IDT, USA), emerald green PCR master mix (Takara, South Korea), cDNA synthesis kit (Thermo Scientific, USA), QIAamp Stool DNA Kit (Qiagen, Germany), biochemical reagent kits (Accucare Ltd), SYBR green master mix (Takara, South Korea) were purchased.2.2 Diets and experimental animals
Experimental diet including rodent control diet (CD) (Amrut agro foods, Mumbai) and HSD which contains 65% sucrose was prepared in our laboratory for diabetes induction.9 The composition of the experimental diets is presented in Table S1.† Around 8- to 10-week-old male Wistar rats, weighing about 150–200 g, were purchased from the animal research facility (Cadila Pharmaceutical Limited, Ahmedabad, India) with approval of the Institutional animal ethics committee with protocol no. (IS/BT/PhD11-12/1004) following the committee for the purpose of control and supervision of experiments on animal's guidelines. They were maintained in the animal house of the Institute of Pharmacy (Nirma University, India). All experimental animals having a similar weight were divided into four experimental groups with 4 animals each, as follows: a non-diabetic group with CD, a diabetic group with HSD, a diabetic group with HSD and linezolid (10 mg kg−1) and a diabetic group with HSD along with cefdinir (10 mg kg−1). The antibiotics dosage was determined based on previously reported studies which suggested the optimum MIC50 dose based on animal experimentation using agar dilution methods10,11 as well as following the US Food and Drug Administration (USFDA) manuals. Animals were fed their respective diets and antibiotics were administered daily for 80 days, while body weight gain was observed on a weekly basis.2.3 Oral glucose tolerance test (OGTT) assay
Animals were examined using the OGTT after 12 h of fasting. Briefly, after 12 h fasting, animals were given a glucose load (2 g kg−1) orally. Blood samples were collected from the animal's tail vein at 0 min (before glucose administration), 15, 30, 60, 90 and 120 min after glucose administration. Glucose concentration was determined using the CareSens N blood glucose monitor and glucose strips. The area under the curve for glucose (AUCglucose) was calculated using the trapezoidal rule.122.4 Blood, fecal and tissue sample collection
The experimental animals were euthanized at the end of the study and blood was collected through heart puncture followed by serum and plasma separation. Fecal samples were collected the day before euthanasia from all animals and stored at −80 °C for further processing. The liver, adipose tissue, small intestine (SI), and colon from each animal were excised, weighed and stored at −80 °C for analysis.2.5 Standard bacterial strains
Standard strains of Lactobacillus casei (MTCC 1423), Escherichia coli (MTCC 443), and Clostridium perfringens (MTCC 450) were obtained from the microbial type culture collection (MTCC, Chandigarh, India), whereas Bifidobacterium bifidum (NCDC229) was obtained from the national collection of dairy culture (NDRI, Karnal, India). The strain of Bacteroides vulgatus (ATCC 25285) was obtained from the American Type Culture Collection (ATCC, USA). The standard strains were used for construction of standard curves for qPCR. Anaerobic culture methods were maintained by using an O2-free anaerobic glass chamber with air absorbing capsules (Himedia, USA) for cultivation and maintenance of Clostridium perfringens.2.6 Microbial quantification by qPCR
The specific 16s rDNA primers targeting different bacterial phyla and genera were used to quantify the fecal microbiota by qPCR (Table S2†)13. The experiments were performed in the Applied biosystems 7500 Fast Real-Time PCR system using SYBR® Premix Ex Taq ™ II (Tli RNaseH Plus) from TAKARA bio Inc, Japan. Each qPCR reaction was carried out in duplicate with a final volume of 20 μL containing 1 μL of each fecal isolated DNA and 10 μM of each primer as shown in Table S2.† The thermal cycling conditions used were as follows: an initial DNA denaturation step at 95 °C for 30 s, 40 cycles of denaturation at 95 °C for 3 s, primer annealing and extension at 60 °C for 30 s. Standard curves for all bacteria were constructed for each experiment using serial tenfold dilutions of isolated bacterial genomic DNA (of known concentration) from pure cultures, corresponding to 101 to 1010 16S rRNA gene copies per g of faeces. The specific bacterial concentration of each fecal sample was calculated by comparing the cycle threshold (Ct) values obtained from the standard curves with applied biosystems 7500 Fast Real-Time PCR System software v2.0.6. The copy numbers of bacterial genome were calculated using the Avogadro constant and assuming the mean molecular weight of a base pair to be 660 g mol−1. The data presented are the mean values of duplicate qPCR analysis.2.7 SCFA estimation
Short chain fatty acids such as acetate, butyrate, and propionate from fecal samples were determined by HPLC analysis (ZORBAX, eclipse plus Phenyl-Hexyl, Agilent, USA).14 0.1 M phosphate buffer solution having a pH of 2.5 was used as a mobile phase. Fresh fecal samples were homogenized with mobile phase, centrifuged at 8000 × g for 15 min at 4 °C and filtered through a 4 micrometer HPLC filter with a 0.45 micrometer nylon membrane (Millipore) directly into vials, which were immediately preserved and analysed. The analysis was performed at a flow rate of 0.5 mL min−1 at 25 °C, with 10 mM H2SO4 as eluent and an injection volume of 50 μL. The mean metabolite concentrations were expressed in millimolar (mM). Short-chain fatty acids such as sodium acetate, sodium propionate, and sodium butyrate (Sigma-Aldrich, USA) were used as internal standards for the estimation.2.8 Tissue biochemistry
Liver tissue was washed with physiological buffer or saline solution, blotted dry and weighed before being used for tissue biochemical analysis for glycogen, TC and TG. Liver glycogen was determined as previously described.15 In brief, liver tissue (200 mg) was finely ground with 20 mL of 5% trichloroacetic acid using a homogenizer. The clear filtrate was obtained by filtering out the protein precipitates and analysed with anthrone reagent. The colour was developed and the OD was read within 2 h at 650 nm. Liver cholesterol content was determined after homogenization with Folch solution (chloroform/methanol ratio = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The lipids in the frozen liver were extracted according to the protocol mentioned in the previous study.16 Triglyceride was first hydrolysed in basic solution (0.5 N KOH in ethanol) and then measured using a commercial enzymatic triglyceride analysis kit.17 The histopathological analysis of the adipose tissue of all experimental groups was performed using hematoxylin and eosin staining as per description.9
:1). The lipids in the frozen liver were extracted according to the protocol mentioned in the previous study.16 Triglyceride was first hydrolysed in basic solution (0.5 N KOH in ethanol) and then measured using a commercial enzymatic triglyceride analysis kit.17 The histopathological analysis of the adipose tissue of all experimental groups was performed using hematoxylin and eosin staining as per description.9
2.9 Gene expression using real time PCR (RT-PCR)
Total RNA was isolated from fresh tissue samples (SI, colon, liver, and adipose tissue) using TRI reagent (Sigma-Aldrich, USA) according to the manufacturer's protocols. An extracted RNA was quantified by determining optical density at 260 nm, while it's purity was determined by calculating the ratio at 260 nm and 280 nm using Nanodrop spectrophotometer. The mRNA expression of various inflammatory genes, such as Tlr2, Tlr4, Nlr1, Nlr2, Nf-κB, Tnf-α, and Il6 was assessed. Gene expression analyses have also been performed for Glut4, Glp1, Gpr43 and all the primer sequences are listed in Table S3.†18 β-Actin served as an internal control. Reverse transcription was performed using a first strand cDNA synthesis kit (Thermo Scientific, USA) according to the protocol mentioned by manufacturer. The RT-PCR reaction was performed on the Applied Biosystems 7500 Fast Real-Time PCR system using SYBR Premix Ex Taq™ II (Tli RNaseH Plus) (Takara Bio Inc, Japan). Relative quantification of gene expression was performed using the comparative threshold cycle method (ΔΔCt), in which the expression level of each target gene was normalized to the levels of β-actin.19 Data output was expressed as fold changes of mRNA expression levels.3. Results
3.1 Effects of gut microflora modulation on OGTT and physiological parameters
Incremental changes in the plasma glucose concentration of experimental rats following oral glucose intake were mentioned (Fig. S1a†). The blood glucose levels of HSD rats were significantly higher than those for the CD group at 15, 30 and 60 min. There were no significant differences in incremental glucose levels at 120 min between the HSD and the linezolid-treated HSD groups, while cefdinir administration to HSD rats had significantly reduced the glucose levels at 120 min. As shown (Fig. S1b†), the glucose AUCs during the OGTT of the HSD group were elevated significantly (p < 0.001) compared with the CD group, while the cefdinir-treated HSD group showed significantly (p < 0.001) decreased glucose AUC levels compared with HSD alone. HSD-fed rats had significantly increased body weight (p < 0.05), liver (p < 0.05) and adipose tissue weight (p < 0.05) as compared to the CD group after 12 weeks of diet intervention and these changes were reversed by cefdinir administration. HSD-fed rats treated with linezolid had not shown any significant changes with respect to all these parameters (Fig. S1†).3.2 Effects of gut microflora modulation on biochemical and inflammatory parameters
HSD administration resulted in significantly increased fasting glucose (p < 0.001), serum triglyceride (p < 0.001) and serum total cholesterol (p < 0.05) levels as compared with the CD group (Table 1). Cefdinir-treated HSD rats had reduced fasting glucose (p < 0.05), serum triglyceride (p < 0.001) and serum TC (p < 0.05) levels compared with HSD controls, while linezolid administration did not significantly affect these parameters, as shown in Table 1. Rats that were HSD-fed for 12 weeks had significantly (p < 0.05) elevated serum ALT and serum AST levels as compared with the CD group and cefdinir-treated HSD rats showed a significant (p < 0.05) decrease in serum AST levels as compared with HSD controls, as shown in Table 1. HSD-fed animals had increased (p < 0.01) CRP and LPS levels compared to the CD group and cefdinir administration reversed those changes, while linezolid did not significantly change these parameters. HSD-fed rats had significantly increased hepatic cholesterol (p < 0.05), triglyceride (p < 0.001), and glycogen (p < 0.05) levels compared with the CD group and cefdinir administration had reduced liver triglyceride and glycogen levels. In contrast, cefdinir treatment did not affect liver cholesterol. Linezolid administration had slightly increased the liver cholesterol and glycogen levels, but had no effect on liver triglycerides (Table 1).| Parameters | Experimental groups | |||
|---|---|---|---|---|
| CD | HSD | HSD + Lin | HSD + Cef | |
| a CD: control diet, HSD: high sucrose diet, Lin: linezolid, Cef: cefdinir, SGOT: serum glutamic oxaloacetic transaminase, SGPT: serum glutamate-pyruvate transaminase , CRP: C-reactive protein, LPS: lipopolysccharide *Compared with CD, #compared with HSD. Data is presented as means ± SD (n = 4). One-way ANOVA was carried out using GraphPad Prism 6 where *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.05 and ##p < 0.01. | ||||
| Blood biochemical parameters | ||||
| Fasting glucose (mg dL−1) | 85.4 ± 15.67 | 142.4 ± 11.77*** | 153.45 ± 23.5 | 120.7 ± 15.67# |
| Triglycerides (mg dL−1) | 50.7 ± 8.67 | 174.3 ± 23.17*** | 145.1 ± 25.3 | 125.6 ± 23.1## |
| Total cholesterol (mg dL−1) | 100.4 ± 30.6 | 181.1 ± 35.12* | 161.2 ± 35.11 | 150.9 ± 11.6# |
|
||||
| Inflammatory parameters | ||||
| SGOT (IU mL−1) | 28.46 ± 5.02 | 53.64 ± 4.22* | 54.23 ± 4.98 | 39.76 ± 2.67 |
| SGPT (IU mL−1) | 17.79 ± 6.82 | 40.34 ± 10.11* | 36.78 ± 5.56 | 28.56 ± 9.02# |
| CRP (mg dL−1) | 2.34 ± 1.81 | 8.18 ± 2.16** | 6.56 ± 2.78 | 3.99 ± 0.97## |
| LPS (IU mL−1) | 0.37 ± 0.02 | 0.65 ± 0.08** | 0.80 ± 0.11# | 0.482 ± 0.32# |
|
||||
| Liver biochemical parameters | ||||
| Glycogen (mg g−1) | 4.23 ± 0.96 | 8.54 ± 2.15* | 9.23 ± 2.54 | 5.76 ± 0.23# |
| Triglyceride (mg g−1) | 11.23 ± 2.18 | 33.48 ± 6.18*** | 25.89 ± 3.18 | 19.98 ± 3.67# |
| Cholesterol (mg g−1) | 2.27 ± 1.62 | 7.18 ± 2.15* | 9.24 ± 1.67 | 5.98 ± 0.98 |
3.3 Effects of gut microflora modulation on gut dominant microflora
The effects of gut microflora modulation using antibiotics was studied on three major gut dominant phyla, Firmicutes, Bacteroidetes, and Proteobacteria. HSD and antibiotic administration had shown divergent effects on the microbial phyla. HSD administration had decreased Firmicutes (18.37%), while increasing Bacteroidetes (17.06%), and Proteobacteria (1.3%) copy numbers as compared with the CD group. Linezolid administration had further decreased the Firmicutes (9.15%) copy number with a concurrent increase in the Bacteroidetes (7.34%) and Proteobacteria (1.82%) populations compared to the HSD control. Cefdinir administration to HSD rats had increased Firmicutes (11.73%) with a decrease in Bacteroidetes (11.08%) and Proteobacteria (0.83%) compared with HSD controls (Fig. 1A and B). The effects of gut microflora alteration were also studied on four gut dominant genera, Lactobacilli, Bifidobacteria, Escherichia, and Clostridia (Fig. 2). HSD-fed rats had significantly altered gut dominant microbiota and reduction in the Lactobacilli (7.15%), Bifidobacteria (12.48%), and Clostridia (4.07%) with an increased Escherichia (23.91%) copy number as compared to CD rats. Linezolid-treated HSD rats shown decrease in the Bifidobacteria (3.09%) and Clostridia (2.0%) with an increased Escherichia (5.0%) copy number, but it did not show any effect on Lactobacilli copy numbers. Cefdinir reduced the copy number of Escherichia (11.54%) with an increase in Bifidobacteria (7.39%), Lactobacilli (2.12%) and Clostridia (1.83%) copy numbers compared to HSD controls (Fig. 2A and B).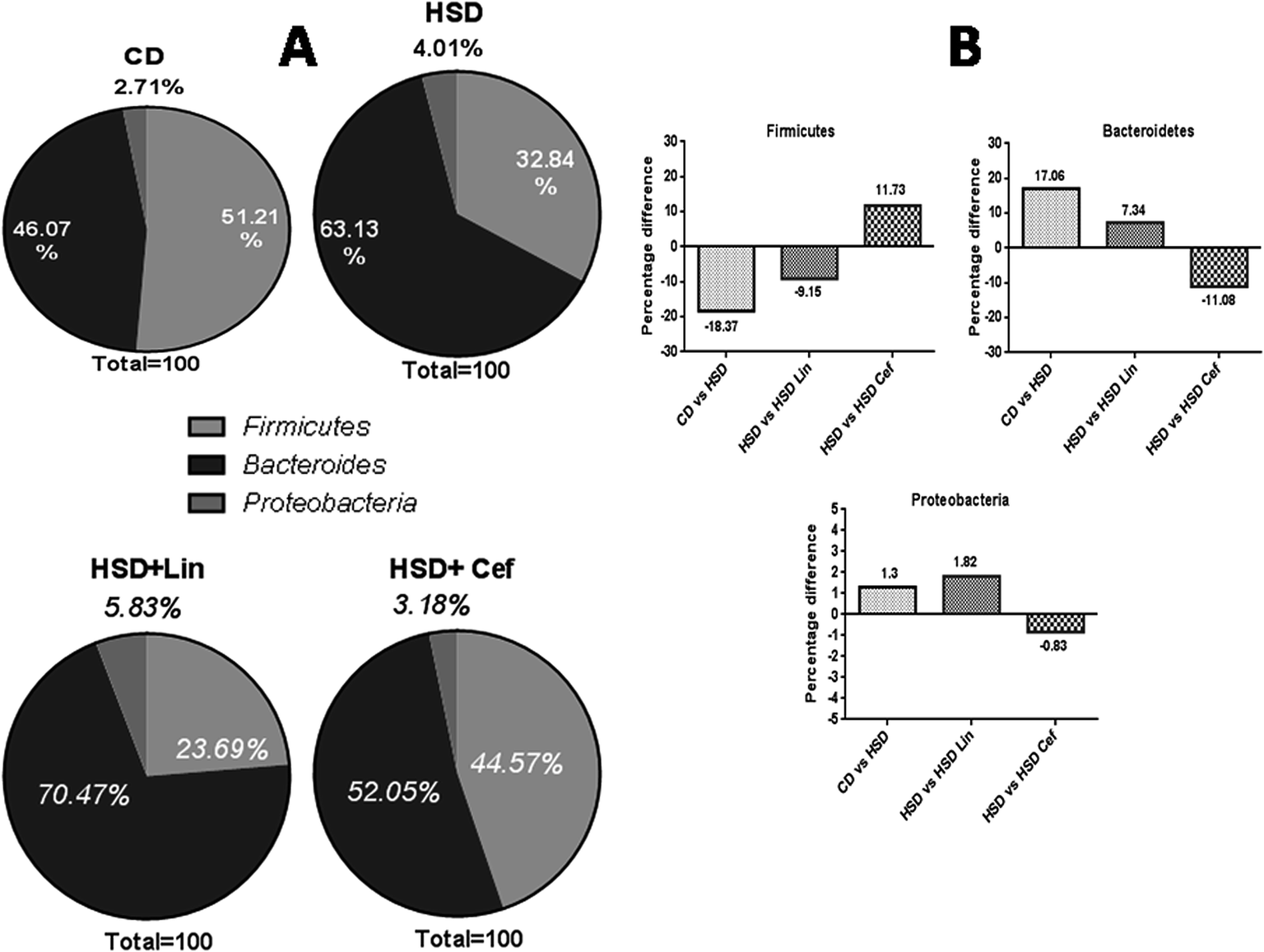 | ||
| Fig. 1 Effects of gut microflora alteration on gut dominant phyla: effects of altering the gut microflora using spectrum specific antibiotics (linezolid and cefdinir) were studied on three major gut dominant phyla, Firmicutes, Bacteroidetes and Proteobacteria, where CD: control diet, HSD: high sucrose diet, Lin: linezolid and Cef: cefdinir. (A) The percentage microflora is presented by calculating the difference between initial and final microbial copy numbers. (B) The respective changes in bacterial copy numbers amongst the experimental groups. | ||
 | ||
| Fig. 2 Effects of gut microflora alteration on gut dominant genera: effects of altering the gut microflora using spectrum specific antibiotics were studied on four major gut dominant genera, where CD: control diet, HSD: high sucrose diet, Lin: linezolid and Cef: cefdinir. (A) The percentage microflora is presented by calculating the difference between initial and final microbial copy numbers. (B) The respective changes in bacterial copy numbers amongst the experimental groups. | ||
3.4 The effects of gut microbiota alteration on GPCR and insulinotropic modulation
The effects of gut microbiota alteration by spectrum specific antibiotic administration on major microbial metabolites, the SCFAs, including acetate, propionate, and butyrate, was studied (Fig. 3A). HSD-fed rats had significantly increased fecal acetate levels (p < 0.05), while decreased propionate and butyrate levels (p < 0.05) as compared to the CD group. Linezolid had increased the acetate levels (p < 0.05) but did not change propionate and butyrate levels as compared to HSD controls. Cefdinir supplementation to HSD rats resulted in significant reduction in the acetate level (p < 0.05) while increased propionate (p < 0.05) and butyrate (p < 0.05) levels as compared with HSD controls, as shown in Fig. 3A. The histopathological analysis of adipose tissue showed that the mean area of adipocytes was enlarged in HSD-fed rats and those administered linezolid compared with the CD group. The cefdinir-treated HSD group showed decreased adipocyte size compared to HSD controls as shown in Fig. S2.† The mRNA expression of Gpr43 and glucose transporter 4 (Glut4) was studied from adipose tissue, while Glp1 was studied from colonic tissue, according to their highest expression. HSD-fed rats had significantly (p < 0.01, p < 0.01 and p < 0.001, respectively) decreased mRNA expression of Gpr43, Glut4, and Glp1 compared with the CD group. Cefdinir administration had increased the mRNA expression of Gpr43 (p < 0.01) and Glut4 (p < 0.05) as compared with HSD controls. Linezolid administration did not have any significant effects on the mRNA expression of these genes (Fig. 3B).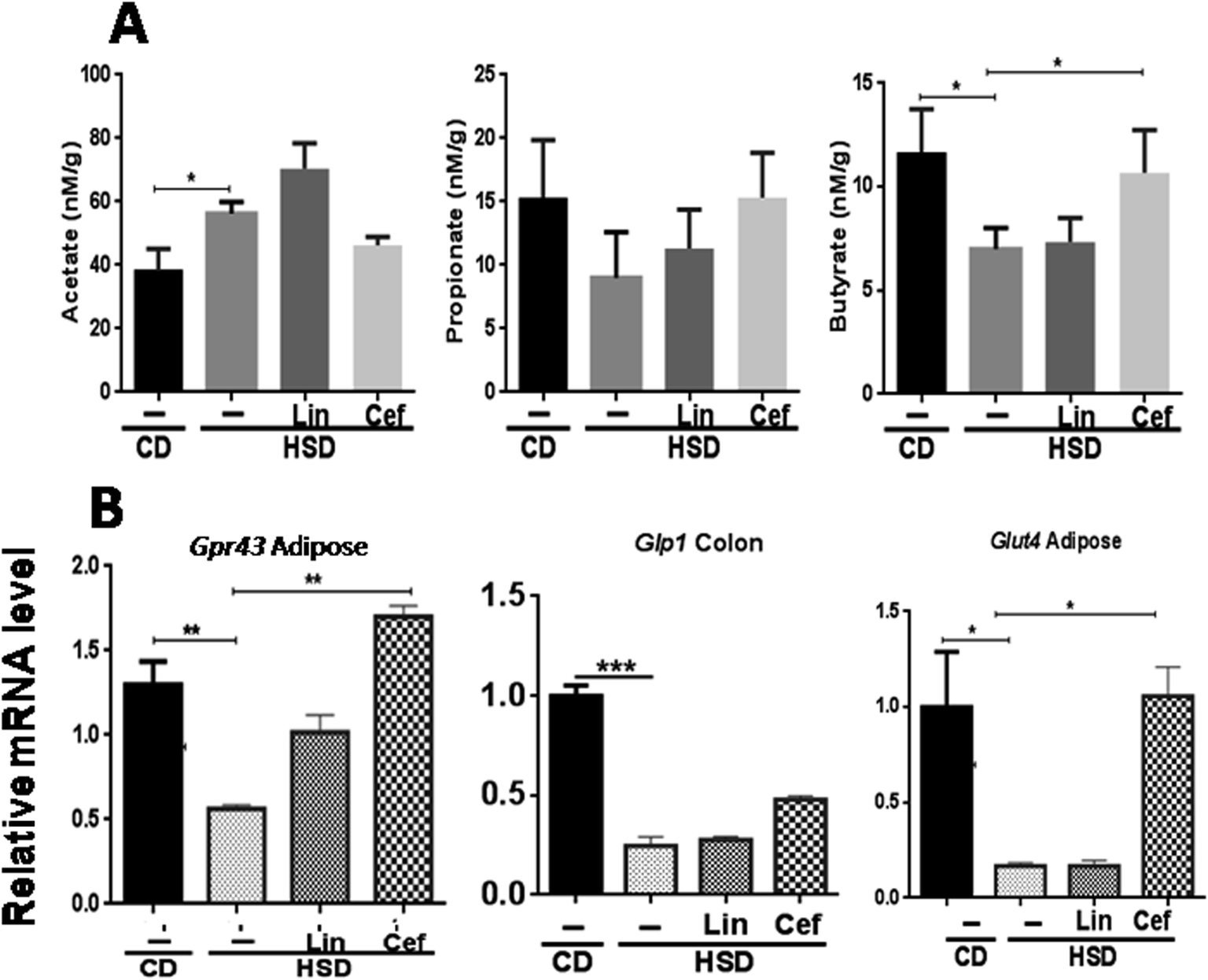 | ||
| Fig. 3 Effects of gut microflora alteration on short chain fatty acids (SCFAs) and insulinotropic parameters: Effects of altering the gut microflora using spectrum specific antibiotics were studied on (A) major microbial metabolites, that is, SCFAs such as acetate, propionate and butyrate and (B) mRNA expression of SCFA receptors, that is, Gpr43 and Glp1 as well as Glut4. Data are presented as means with SD (n = 4). One-way ANOVA was performed using GraphPad Prism 6 where *p < 0.05, **p < 0.01 and ***p < 0.001 ; CD: control diet, HSD: high sucrose diet, Lin: linezolid and Cef: cefdinir. | ||
3.5 The effects of gut microbiota alteration on innate immune receptor expression
The effects of gut microbiota alteration were studied on the expression of major pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs) in the SI and colon. In the SI, Tlr2 (p < 0.05) and Nlr1 (p < 0.01) mRNA expression was found to be increased, while Nlr2 mRNA expression was found to be significantly decreased (p < 0.05) in the HSD group compared to the CD group. Cefdinir administration to HSD-fed rats had significantly decreased Tlr2 (p < 0.01) and Nlr1 (p < 0.05) mRNA expression compared with HSD alone. Linezolid administration significantly increased Nlr1 mRNA expression compared with HSD controls (Fig. 4). In the colon, Tlr4 and Nlr1 (p < 0.05) mRNA expression was found to be significantly increased in the HSD group as compared to the CD group. Cefdinir administration had significantly (p < 0.05) decreased Tlr4 mRNA expression compared with HSD controls. Linezolid application had significantly (p < 0.05) increased Nlr1 mRNA expression in HSD controls (Fig. 4).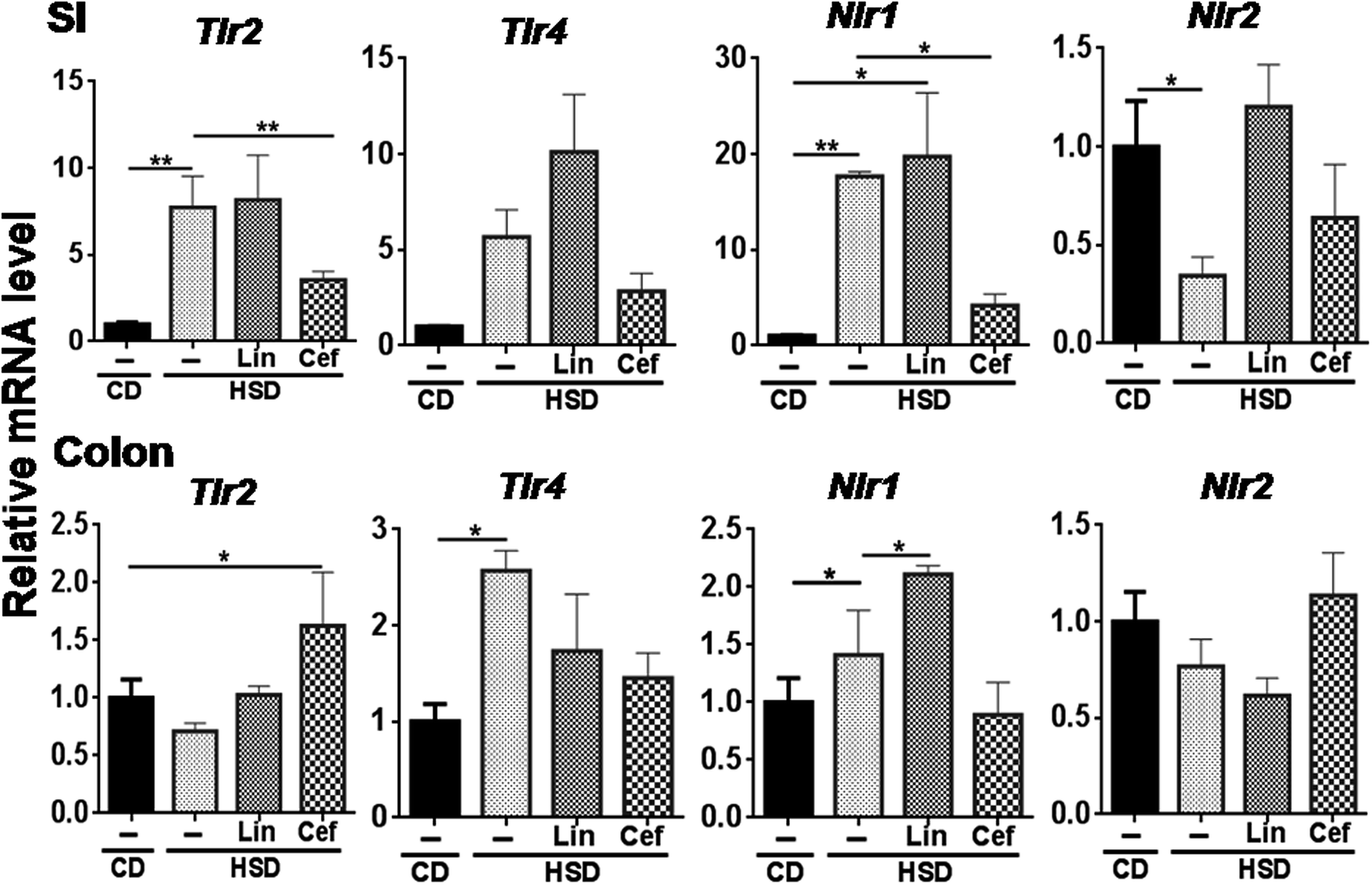 | ||
| Fig. 4 Effects of gut microflora alteration on TLR and NLR expression: Effects of altering the gut microflora using antibiotics were studied on mRNA expression of TLRs such as Tlr2 and Tlr4 as well as NLRs such as Nlr1 and Nlr2. CD: control diet, HSD: high sucrose diet , SI: small intestine, Lin: linezolid and Cef: cefdinir. One-way ANOVA was carried out (n = 4) where *p < 0.05 and **p < 0.01. | ||
3.6 The effects of gut microbiota alteration on inflammation receptors
The effects of gut microbiota modulation were studied on inflammatory mediators, that is, Nf-κB and inflammatory cytokines such as Il6 and Tnf-α from the SI, colon, and liver. HSD administration had significantly increased mRNA expression of Nf-κB, Il6 and Tnf-α in the liver (p < 0.01, p < 0.01 and p < 0.001, respectively); the SI (p < 0.001, p < 0.05 and p < 0.001, respectively) and the colon (p < 0.01, p < 0.001 and p < 0.05, respectively) and cefdinir administration reversed those changes. Linezolid administration did not significantly alter expression of these inflammatory genes compared with HSD controls (Fig. 5).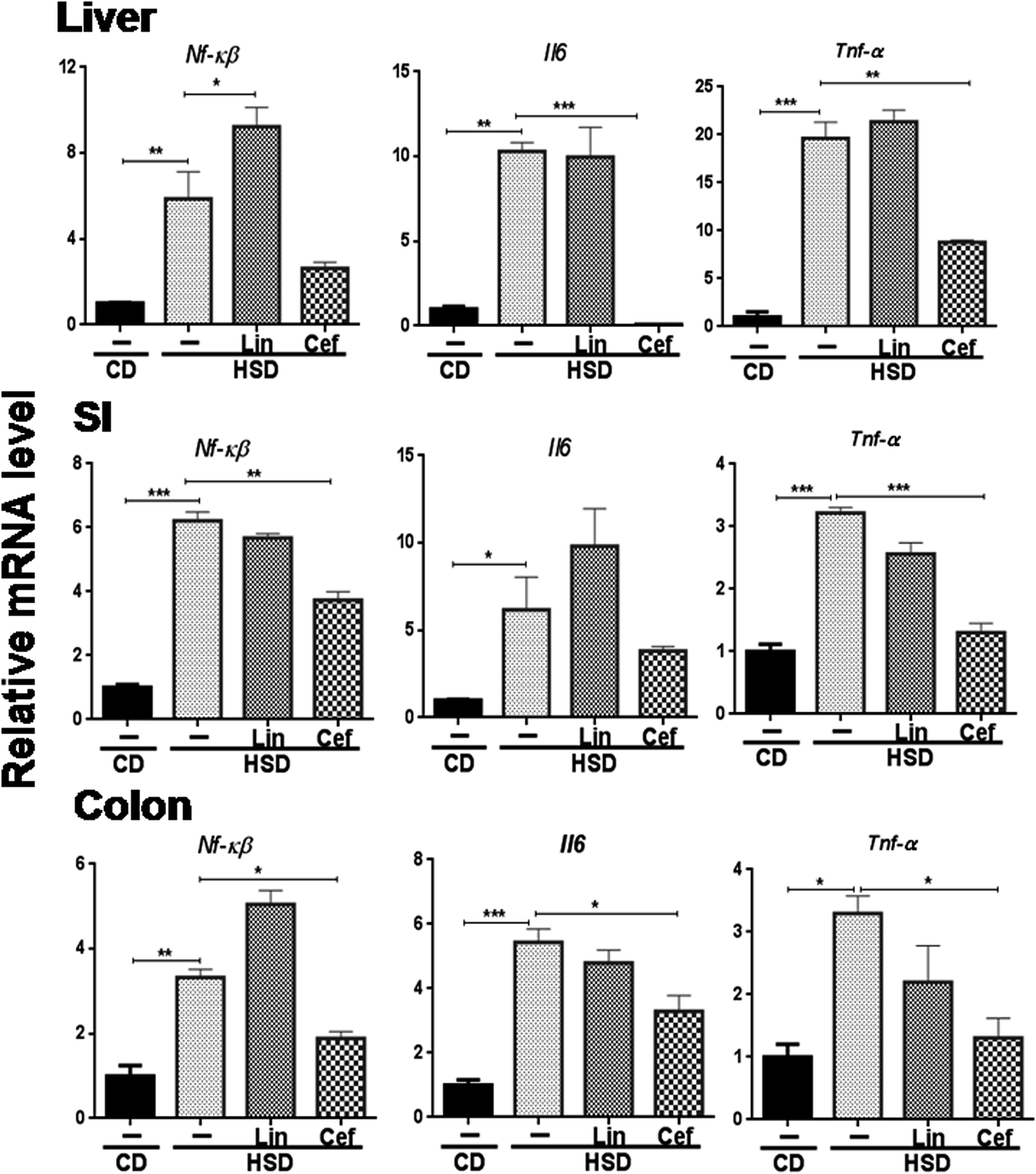 | ||
| Fig. 5 Effects of gut microflora alteration on inflammatory gene expression: effects of altering the gut microflora using antibiotics were studied on mRNA expression of inflammation genes, such as Nf-κB, Il6 and Tnf-α. CD: control diet, HSD: high sucrose diet; SI: small intestine, Lin: linezolid and Cef: cefdinir. One-way ANOVA was carried out (n = 4) where *p < 0.05, **p < 0.01, ***p < 0.001. | ||
4. Discussion
The increased incidence of metabolic complications cannot only be ascribed to alterations in genetic profile, nutritional habits and physical activity in day-to-day life, but lately identified gut microbiota being the most important environmental factor involved in this. Gut microbiota modulation using antibiotics could be a novel therapeutic approach due to its noticeable involvement in human health and disease progression.20 The present investigation was focused on the effect of gut microbiota modulation using different spectrum antibiotics on the progression of HSD-induced hyperglycaemia. Daily co-administration of antibiotics to HSD rats for 12 weeks resulted in major alterations at physiological, biochemical, histopathological and microbiome levels. Gut microbiota composition contributes to the regulation of body weight and glycemic control through an association with energy extraction and storage, intestinal integrity, fat metabolism and inflammation. The dietary pattern is considered to be one of the most important environmental factors that influences the gut microbiota composition within a host, affecting the functional interrelationships.21 Furthermore, antibiotics have recently being used to alter the gut microbiome composition in rodent models for the prevention of obesity and diabetes.22 The non-specificity of the antibiotics with regard to their antimicrobial spectrum was the major limitation of previous gut microbiome studies involving administration of various antibiotics for gut microbiota modulation. The use of narrow antimicrobial spectrum antibiotics for selectively “knock-down” of gut microbial species in rat model systems might help to understand the microbiome-disease causality. The major aim of the current investigation was to examine the relationship between specific gut microbial communities and the HSD-induced obesity/diabetes phenotype. To fulfil this aim, we tried gut microbiome modulation in the HSD-induced diabetic rat using two different spectrum antibiotics, linezolid and cefdinir. Major changes in the rat metabolic and inflammatory phenotypes, physiology, biochemistry and gut microbiome 16S rDNA gene profiles were observed following antibiotic administration. Linezolid belongs to the oxazolidinone class which specifically inhibits cell wall synthesis in Gram-positive bacteria with very little effect on Gram-negative bacteria due to differences in their outer membrane structures.23 In contrast, cefdinir is a third generation cephalosporin which inhibits cell wall synthesis mainly in Gram-negative bacteria.24 Feeding with a HSD provides a dietary model of T2D associated with IR, hyperinsulinemia and hypertriglyceridemia.25,26 A HSD can be a source of inflammation, which causes weight gain. An overload of fructose in the liver perturbs the glucose metabolism and glucose uptake pathways, leading to an enhanced rate of de novo lipogenesis and triacylglycerol synthesis through the high flux of glycerol and acyl molecules from fructose catabolism, ultimately inducing the IR commonly observed in humans and animals.27We have observed the reduced number of Firmicutes with an increased Bacteroidetes and Proteobacteria population upon HSD administration (Fig. 1). This can be correlated with previous reports, which suggested that western style diets, that is, high in sugar and low in fibre reduce the amount of beneficial Firmicutes that ferment the dietary plant-derived polysaccharides to SCFAs, while increase the mucosa-associated Proteobacteria and enteric pathogens.28 Previous microbiome studies in mice showed that a high-fat or sugar diet increases the ratio of Gram-negative to Gram-positive microbes leading to an elevated bacterial LPS and proinflammatory responses.19 Our results showed a significant increase in body weight gain, adipose tissue and liver weight in the HSD group, while the antibiotics group have shown a divergence with respect to these parameters (Fig. S1†). Cefdinir administration had decreased the Gram-negative bacteria load in the HSD group due to its antimicrobial spectrum against Gram-negative microorganisms, which further decreased LPS-induced inflammation and prevented HSD-mediated weight gain. Linezolid did not have any significant effect on body weight due to its Gram-positive antimicrobial spectrum. The intestinal bacterial diversity and abundance would be lowered by application of these both antibiotics but the positive effects of cefdinir in the prevention of diabetes cannot be explained by a general “cleansing” of the gastrointestinal microbiome. Instead, the positive effects of cefdinir on weight loss and hyperglycaemic control indicate changes in the specific gut microbiome composition. Cefdinir-treated rats showed a higher proportion of the Gram-positive bacteria, Firmicutes and a lower abundance of the Gram-negative bacteria, Bacteroidetes and Proteobacteria. We have observed a significant reduction in Lactobacilli, Bifidobacteria and Clostridia genera in HSD rats compared to CD rats, which can be correlated with a decrease in hepatic cholesterol and triglyceride levels. One study has reported on the role of probiotic bacterial species, such as Lactobacillus and Bifidobacteria, in the production of SCFAs through fermentation of food-derived non-digestible carbohydrates in the colon. These metabolites play a role in the clearance of blood lipids by inhibiting cholesterol synthesis in the liver or redistributing it from plasma to the liver. Animal studies have demonstrated the efficacy of Lactobacillus in lowering serum cholesterol levels, presumably by breaking down bile in the gut, thus inhibiting its reabsorption.29 We have observed reduced levels of major SCFAs, such as butyrate, in the fecal samples of HSD animals compared to CD animals, while it was found to be increased upon cefdinir administration (Fig. 3). Butyrate plays a major role in decreasing mucosal inflammation and oxidative stress, altering intestinal motility and visceral sensitivity, regulating transepithelial fluid transport and maintaining the epithelial defence barrier. In addition, its role in the prevention and inhibition of inflammation has been stressed by an increasing number of studies.30 Alterations in the SCFA levels can be directly correlated with the microbial species dominating in the human gut. A decrease in the Clostridia number resulting from a HSD has been directly correlated with a decrease in butyrate from fecal samples.31 This can be explained by the fact that Clostridium species play a crucial role in the metabolic welfare of colonocytes by releasing butyrate as an end-product of fermentation and this also inhibits the activation of the transcription factor Nf-κB in gut cells. Increased production of Il-10 by T cells and decreased expression of Nf-κB might lead to a consequent intestinal and systemic anti-inflammatory effect.32
An increase in the amount of butyrate in fecal samples from cefdinir-treated animals can be correlated with an increase in Firmicutes levels in this group. One study has stated that colon-residing Gram-positive anaerobic bacteria have demonstrated maximum ability to produce butyrate in the human. Faecalibacterium prausnitzii, which belongs to the Clostridium leptum (or clostridial cluster IV) cluster, and Eubacterium rectale/Roseburia spp., which belong to the Clostridium coccoides (or clostridial cluster XIVa) cluster of Firmicutes bacteria appear to be two of the most important groups of butyrate producers.33 We have observed a decrease in propionate levels in HSD rats, which were increased following cefdinir administration. Propionate has potential health-promoting effects including anti-lipogenic, cholesterol-lowering, anti-inflammatory and anti-carcinogenic properties.34 The molecular mechanism through which SCFAs mediate an effect on the progression of metabolic complications is still not understood. However, several studies have stated that SCFAs might play a role in disease progression through SCFA receptor pathways, that is, via GPCRs.35 We found a decrease in the expression of Gpr43 in the HSD group, while cefdinir treatment had increased its mRNA expression (Fig. 3). Gpr43 is reported to be involved in energy regulation in response to SCFAs produced from gut microbiota. The body weight and fat pad weight of Gpr43 knockout mice are significantly increased compared to wild-type mice, and this difference is abolished in germ-free conditions.36 We have observed a decrease in incretin, that is, Glp1, and Glut4 expression in HSD rats compared with the CD group. An increase in Glp1 levels through Glp1 receptor agonists contributes to the reduction in storage and secretion of triglycerides in the hepatic system. Cefdinir administration increased the expression of Glut4 in HSD-receiving rats. Glut4 is a facilitative diffusion glucose transporter and is the major insulin-regulated glucose transporter in skeletal muscle, heart, and adipocytes. It is downregulated in adipocytes of both humans and rodents with obesity or T2D, and this downregulation is one of the earliest events in the pathogenesis of T2D.37 Recent findings indicated that knockdown of Glut4 in adipocytes resulted in IR and its overexpression reduced fasting glycaemia and improved glucose tolerance.38 An increased percentage of Bacteroidetes and Proteobacteria was found to be associated with an increase in circulating LPS which decreased the whole body insulin tolerance. When these animals were treated with cefdinir as a strategy for intestinal microflora modulation, they showed a marked reduction in Bacteroidetes and Proteobacteria along with circulating LPS, which attenuated Tlr4 and Nlr1 activation and resulted in improved insulin sensitivity. The available data indicate that translocation of Tlr4 and Nlr1 agonists such as LPS and iE-DAP, respectively, from the gut is an important feature in metabolic diseases promoting inflammation and IR.39 An intracellular muramyl dipeptide (MDP), a ligand for Nlr2, is predominantly present in Gram-positive strains.40 In the CD group, due to symbiosis in the Gram-positive and Gram-negative bacteria, there is an increased Nlr2 expression response. Following HSD induction, Gram-negative flora increases which results in decreased Gram-positive flora, hence a subsequent drop in MDP decreases expression of Nlr2. On the contrary, as linezolid is Gram-positive specific, it caused degradation of strains and elevated levels of MDP and hence increased expression of Nlr2 was observed in the SI (Fig. 4).
Previous studies have shown the importance of LPS in inflammation and the induction of IR using LPS injection and sepsis models.41 The mechanism involving LPS is initiated when it binds to Tlr4, its receptor, which, in turn, associates with MyD88 and triggers its signalling pathway; this, in turn, inhibits Nf-κB kinase subunit β (IKKß) through IL-1 receptor-associated kinase (IRAK), leading to serine phosphorylation of IRS-1, nuclear Nf-κB activation, transcription of proinflammatory cytokines and IR.42 Nlr1 senses iE-DAP, which is found in the peptidoglycan of all Gram-negative bacteria.43 Ligand-bound Nlr1 and Nlr2 oligomerize and signal via the serine/threonine RIP2 (RICK, CARDIAK) kinase through CARDCARD homophilic interactions.44 Once activated, RIP2 mediates ubiquitination of NEMO/IKKγ leading to the activation of Nf-κB and production of inflammatory cytokines. Therefore, our results have shown an increase in the expression of the proinflammatory cytokines Tnf-α and Il6 in HSD rats.
Gut microbiota modulation through cefdinir lowered the expression of HSD-induced proinflammatory cytokines. The reduction in the Gram-negative microbial load through cefdinir application reduced circulatory LPS levels in the gut, which decreased the expression of innate immune receptors such as the TLRs and NLRs. Downregulation of these pathways led to the reduction in Nf-κB activity and thus reduced the expression of proinflammatory cytokines which are directly associated with progression of inflammation-induced IR. The expression of various pro-inflammatory cytokines such as Tnf-α, Il-1 and Il6 has been found to be linked to systemic inflammation and associated with IR.45 In a previous study, treatment with a combination of three antibiotics (ampicillin, neomycin and metronidazole) greatly modified the gut microbiota with a significant reduction in overall bacterial count and circulating plasma LPS levels. This manipulation also reduced Tnf-α and Il6 along with reduced activity of Tlr4, JNK, and IKK-β which improved glucose tolerance and insulin action in metabolically active tissues.8 It has been reported that proinflammatory cytokines can interfere to downregulate the IRS pathway. Proinflammatory cytokines phosphorylate the threonine moiety of the insulin receptor instead of the serine, which changes the structural conformation of the receptor. Thus, insulin cannot bind to the receptor leading to the downregulation of the IRS pathway and progression into IR.46 Downstream signalling pathways of the GPCRs induced the release of incretin, that is, Glp1, which had increased the glucose uptake through Glut4 and increased insulin sensitivity. Gut microbiota modulation by reducing Gram-negative microflora has been shown to have a preventative effect in diet-induced diabetes. Such modulation can be done by using spectrum specific antibiotics, probiotics, and prebiotics. Our previous data showed that the polymer chitosan can reduce the pathogenesis of diet-induced diabetes through increasing the beneficial gut microbiota (for example, Lactobacilli and Bifidobacteria) population and decreasing the Gram-negative microflora.47 Our results are in agreement with earlier work showing that Gram-negative bacteria-targeted antibiotics caused both weight loss as well as a favourable glycemic profile for glucose and insulin.8 A combination of polymyxin B and neomycin application had dramatically reduced glucose levels by decreasing Gram-negative bacteria.48 Taken together, it has been shown that targeted inhibition of Gram-negative bacteria reduced the progression of diabetes and metabolic disorders in rats and mice.
5. Conclusion
In conclusion, cefdinir (Gram-negative specific) exhibited interesting diabetic preventative properties in HSD-induced diabetic rats. In addition, cefdinir intervention had prevented the progression of IR and normalized HSD-mediated gut dysbiosis by decreasing the relative copy number of Proteobacteria and Bacteroidetes. Furthermore, cefdinir treatment had decreased metabolic endotoxemia-mediated inflammation and might have activated insulin receptor signalling (IRS) by decreasing the expression of the pro-inflammatory cytokines. It had further increased the amount of the major beneficial bacteria in the gut, that is, Bifidobacteria and Clostridia, which had been found to increase the amount of butyrate in the colon. Cefdinir had induced expression of the SCFA receptor Gpr43 at the mRNA level. Thus, selective manipulation of the gut microbiota using spectrum specific antibiotics can be an emerging therapeutic strategy for the prevention of IR.Contributors
S. S. designed the study; B. P., P. K. J., and S. P. performed experiments; B. P., P. K. J., S. P., and S. S. analysed and interpreted the data; B. P., P. K. J., and S. S. wrote the manuscript; all authors commented and approved the final manuscript.Conflicts of interest
The authors who have taken part in this paper declared that they do not have any disclosures regarding funding or conflicts of interest.Acknowledgements
The authors are thankful to Nirma Education and Research Foundation (NERF), Ahmedabad for financial support.References
- S. E. Kahn, R. L. Hull and K. M. Utzschneider, Nature, 2006, 444(7121), 840–846 CrossRef PubMed.
- K. E. Wellen and G. S. Hotamisligil, J. Clin. Invest., 2005, 115(5), 1111–1119 CrossRef PubMed.
- F. Backhed, H. Ding, T. Wang, L. V. Hooper, G. Y. Koh, A. Nagy, C. F. Semenkovich and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(44), 15718–15723 CrossRef PubMed.
- P. J. Turnbaugh, R. E. Ley, M. A. Mahowald, V. Magrini, E. R. Mardis and J. I. Gordon, Nature, 2006, 444(7122), 1027–1031 CrossRef PubMed.
- N. Larsen, F. K. Vogensen, F. W. J. van den Berg, D. S. Nielsen, A. S. Andreasen, B. K. Pedersen, W. A. Al-Soud, S. J. Sorensen, L. H. Hansen and M. Jakobsen, PLoS One, 2010, 5(2), e9085 CrossRef PubMed.
- P. J. Turnbaugh, F. Backhed, L. Fulton and J. I. Gordon, Cell Host Microbe, 2008, 3(4), 213–223 CrossRef PubMed.
- E. F. Murphy, P. D. Cotter, A. Hogan, O. O'Sullivan, A. Joyce, F. Fouhy, S. F. Clarke, T. M. Marques, P. W. O'Toole, C. Stanton, E. M. Quigley, C. Daly, P. R. Ross, R. M. O'Doherty and F. Shanahan, Gut, 2013, 62(2), 220–226 CrossRef PubMed.
- B. M. Carvalho, D. Guadagnini, D. M. Tsukumo, A. A. Schenka, P. Latuf-Filho, J. Vassallo, J. C. Dias, L. T. Kubota, J. B. Carvalheira and M. J. Saad, Diabetologia, 2012, 55(10), 2823–2834 CrossRef PubMed.
- P. K. Jena, S. Singh, B. Prajapati, G. Nareshkumar, T. Mehta and S. Seshadri, Appl. Biochem. Biotechnol., 2014, 172(8), 3810–3826 CrossRef PubMed.
- A. Louie, W. Liu, R. Kulawy and G. L. Drusano, Antimicrob. Agents Chemother., 2011, 55(7), 3453–3460 CrossRef PubMed.
- S. Iwata, K. Kawahara, M. Ikeda, E. Isohata, Y. Kin, Y. Kusumoto, Y. Sato, H. Akita, S. Nanri and T. Oikawa, et al., Jpn. J. Antibiot., 1992, 45(1), 28–47 Search PubMed.
- F. E. Harlass, G. B. McClure, J. A. Read and K. Brady, J. Reprod. Med., 1991, 36(2), 147–150 Search PubMed.
- L. E. Ritchie, K. F. Burke, J. F. Garcia-Mazcorro, J. M. Steiner and J. S. Suchodolski, Vet. Microbiol., 2010, 144(1–2), 140–146 CrossRef PubMed.
- V. Cleusix, C. Lacroix, S. Vollenweider and G. Le Blay, FEMS Microbiol. Ecol., 2008, 63(1), 56–64 CrossRef PubMed.
- J. Van Der Vies, Biochem. J., 1954, 57(3), 410–416 CrossRef PubMed.
- P. Robinet, Z. Wang, S. L. Hazen and J. D. Smith, J. Lipid Res., 2010, 51(11), 3364–3369 CrossRef PubMed.
- C. L. Mendenhall, J. Lipid Res., 1972, 13(2), 177–183 Search PubMed.
- A. R. Kurland, H. Schreiner and G. Diamond, J. Periodontal Res., 2006, 41(6), 567–572 CrossRef PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef PubMed.
- P. D. Cani, R. Bibiloni, C. Knauf, A. Waget, A. M. Neyrinck, N. M. Delzenne and R. Burcelin, Diabetes, 2008, 57(6), 1470–1481 CrossRef PubMed.
- D. K. Rajpal, J. L. Klein, D. Mayhew, J. Boucheron, A. T. Spivak, V. Kumar, K. Ingraham, M. Paulik, L. Chen, S. V. Horn, E. Thomas, G. Sathe, G. P. Livi, D. J. Holmes and J. R. Brown, PLoS One, 2015, 10(12), e0145499 CrossRef PubMed.
- M. Membrez, F. Blancher, M. Jaquet, R. Bibiloni, P. D. Cani, R. G. Burcelin, I. Corthesy, K. Mace and C. J. Chou, FASEB J., 2008, 22(7), 2416–2426 CrossRef PubMed.
- D. M. Livermore, J. Antimicrob. Chemother., 2003, 51(2), ii9–16 Search PubMed.
- J. C. Rodriguez, M. Ruiz, M. Lopez and G. Royo, Int. J. Antimicrob. Agents, 2002, 20(6), 464–467 CrossRef PubMed.
- P. I. Moreira, Curr. Opin. Clin. Nutr. Metab. Care, 2013, 16(4), 440–445 CrossRef PubMed.
- L. P. Musselman, J. L. Fink, K. Narzinski, P. V. Ramachandran, S. S. Hathiramani, R. L.Cagan and T. J. Baranski, Dis. Models & Mech., 2011, 4(6), 842–849 Search PubMed.
- H. Basciano, L. Federico and K. Adeli, Nutr. Metab., 2005, 2(1), 5 CrossRef PubMed.
- H. L. Simpson and B. J. Campbell, Review article: dietary fibre-microbiota interactions, Aliment. Pharmacol. Ther., 2015, 42(2), 158–179 CrossRef PubMed.
- D. I. Pereira and G. R. Gibson, Crit. Rev. Biochem. Mol. Biol., 2002, 37(4), 259–281 CrossRef PubMed.
- R. B. Canani, M. D. Costanzo, L. Leone, M. Pedata, R. Meli and A. Calignano, World J. Gastroenterol., 2011, 17(12), 1519–1528 CrossRef PubMed.
- V. D. Abbeele, C. Belzer, M. Goossens, M. Kleerebezem, W. M. DeVos, O. Thas, R. D. Weirdt, F. M. Kerckhof and T. V. Wiele, ISME J., 2013, 7(5), 949–961 CrossRef PubMed.
- L. R. Lopetuso, S. Chowdhry and T. T. Pizarro, Front. Immunol., 2013, 4, 181 Search PubMed.
- P. Louis, P. Young, G. Holtrop and H. J. Flint, Environ. Microbiol., 2010, 12(2), 304–314 CrossRef PubMed.
- M. A. Vinolo, H. G. Rodrigues, R. T. Nachbar and R. Curi, Nutrients, 2011, 3(10), 858–876 CrossRef PubMed.
- A. Ichimura, S. Hasegawa, M. Kasubuchi and I. Kimura, Front. Pharmacol., 2014, 5, 236 Search PubMed.
- B. S. Samuel, A. Shaito, T. Motoike, F. E. Rey, F. Backhed, J. K. Manchester, R. E. Hammer, S. C. Williams, J. Crowley, M. Yanagisawa and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(43), 16767–16772 CrossRef PubMed.
- B. M. Carvalho, D. Guadagnini, D. M. Tsukumo, A. A. Schenka, P. Latuf-Filho, J. Vassallo, J. C. Dias, L. T. Kubota, J. B. Carvalheira and M. J. Saad, Diabetologia, 2012, 55(10), 2823–2834 CrossRef PubMed.
- E. D. Abel, O. Peroni, J. K. Kim, Y. B. Kim, O. Boss, E. Hadro, T. Minnemann, G. I. Shulman and B. B. Kahn, Nature, 2001, 409(6821), 729–733 CrossRef PubMed.
- J. Sun and E. B. Chang, Genes Dis., 2014, 1(2), 132–139 CrossRef PubMed.
- Z. A. Nabhani, G. Dietrich, J. Hugot and F. Barreau, PLoS Pathog., 2017, 13(3), e1006177 CrossRef PubMed.
- A. G. Oliveira, B. M. Carvalho, N. Tobar, E. R. Ropelle, J. R. Pauli, R. A. Bagarolli, D. Guadagnini, J. B. Carvalheira and M. J. Saad, Diabetes, 2011, 60(3), 784–796 CrossRef PubMed.
- M. T. Nguyen, S. Favelyukis, A. K. Nguyen, D. Reichart, P. A. Scott, A. Jenn, R. Liu-Bryan, C. K. Glass, J. G. Neels and J. M. Olefsky, J. Biol. Chem., 2007, 282(48), 35279–35292 CrossRef PubMed.
- M. Chamaillard, S. E. Girardin, J. Viala and D. J. Philpott, Cell. Microbiol., 2003, 5(9), 581–592 CrossRef PubMed.
- K. Kobayashi, N. Inohara, L. D. Hernandez, J. E. Galan, G. Nunez, C. A. Janeway, R. Medzhitov and R. A. Flavell, Nature, 2002, 416(6877), 194–199 CrossRef PubMed.
- V. Wieser, A. R. Moschen and H. Tilg, Arch. Immunol. Ther. Exp., 2013, 61(2), 119–125 CrossRef PubMed.
- J. C. Winer, T. L. Zern, S. E. Taksali, J. Dziura, A. M. Cali, M. Wollschlager, A. A. Seyal, R. Weiss, T. S. Burgert and S. Caprio, J. Clin. Endocrinol. Metab., 2006, 91(11), 4415–4423 CrossRef PubMed.
- B. Prajapati, P. Rajput, P. K. Jena and S. Seshadri, Curr. Pharm. Biotechnol., 2016, 17(2), 173–184 Search PubMed.
- C. J. Chou, M. Membrez and F. Blancher, Nestle Nutr. Inst. Workshop Ser., Pediatr. Program, 2008, 62, 127–137 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03774d |
| This journal is © The Royal Society of Chemistry 2018 |