DOI:
10.1039/C8RA04580A
(Paper)
RSC Adv., 2018,
8, 29321-29337
Photo-responsive polymeric micelles and prodrugs: synthesis and characterization
Received
29th May 2018
, Accepted 11th August 2018
First published on 17th August 2018
Abstract
Bio-recognizable and photocleavable amphiphilic glycopolymers and prodrugs containing photodegradable linkers (i.e. 5-hydroxy-2-nitrobenzyl alcohol) as junction points between bio-recognizable hydrophilic glucose (or maltose) and hydrophobic poly(α-azo-ε-caprolactone)-grafted alkyne or drug chains were synthesized by combining ring-opening polymerization, nucleophilic substitution, and “click” post-functionalization with alkynyl-pyrene and 2-nitrobenzyl-functionalized indomethacin (IMC). The block-grafted glycocopolymers could self-assemble into spherical photoresponsive micelles with hydrodynamic sizes of <200 nm. Fluorescence emission measurements indicated the release of Nile red, a hydrophobic dye, encapsulated by the Glyco-ONB-P(αN3CL-g-alkyne)n micelles, in response to irradiation caused by micelle disruption. Light-triggered bursts were observed for IMC-loaded or -conjugated micelles during the first 5 h. Following light irradiation, the drug release rate of IMC-conjugated micelles was faster than that of IMC-loaded micelles. Selective lectin binding experiments confirmed that glycosylated Glyco-ONB-P(αN3CL-g-alkyne)n could be used in bio-recognition applications. The nano-prodrug with and without UV irradiation was associated with negligible levels of toxicity at concentrations of less than 30 μg mL−1. The confocal microscopy and flow cytometry results indicated that the uptake of doxorubicin (DOX)-loaded micelles with UV irradiation by HeLa cells was faster than without UV irradiation. The DOX-loaded Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles effectively inhibited HeLa cells' proliferation with a half-maximal inhibitory concentration of 8.8 μg mL−1.
Introduction
In recent years, multifarious drug delivery systems (e.g., polymeric micelles, polymer drug conjugates, and polymeric nanoparticles) have been developed to address the problems associated with drug molecules, such as low aqueous solubility, short plasma circulation time, rapid in vivo degradation and systemic toxicity.1 Compared with free drugs, drugs incorporated into polymeric micelles exhibit favorable therapeutic advantages: enhanced drug solubility in water, prolonged circulation time by inhibiting phagocytic and renal clearance, and passive targeting to the tumor tissues by the enhanced permeability and retention effect.2–5 However, many drug delivery systems face strong limitations that potentially affect their further translation to clinical tests: the burst release in which a large fraction of adsorbed drug is rapidly released after administration and the inadequate drug loading which usually necessitates a high concentration of nanocarriers to obtain a noticeable therapeutic effect. These major obstacles may be overcome by applying the polymer prodrug approach, where the drug is covalently conjugated onto polymer backbones and side chains through labile linkers that can be cleaved in certain conditions. The drugs cannot be released before the degradable linking is cleaved. Therefore, polymer drug conjugates exhibit excellent storage stability, low systemic toxicity in circulation, and localized drug releases.6,7
Stimulus-responsive micelles formed from amphiphilic copolymers or polymer drug conjugates are promising materials for drug delivery and may lead to enhanced biological properties and efficiencies.8–15 Among the available stimuli, light is of particular interest because it can be highly localized in time and space.16 Photoactivable drug delivery systems are not only suitable for surface cancer treatment but also applicable to deep-seated cancer under endoscopic or optical fiber guidance, which is also used for photodynamic therapy.17–20 Recently, several research groups have demonstrated an irreversible response, involving the incorporation of the photocleavable units in the main chain of one of the blocks or in the hydrophobic cores conjugated with photocaged chemotherapeutic drugs, that induces the selective degradation of a specific micellar compartment. Among the many studied photolabile groups, o-nitrobenzyl (ONB) alcohol derivatives have received considerable attention in the area of synthetic polymer and polymer drug conjugates.21–23
In this study, we present a synthetic platform, which permits in situ construction of various light-triggered drug delivery systems from amphiphilic glycose block functionalized poly(α-azo-ε-caprolactone) (Glyco-ONB-PαN3CLn) copolymers with photocleavable ortho-nitrobenzyl (ONB) units in the middle moiety and side chain, in which the drug molecules to be delivered can be loaded either physically (i.e., the encapsulation approach) or chemically attached (i.e., the prodrug approach). For the encapsulation approach, the ONB linked the hydrophilic block glycose and the hydrophobic block P(αN3CL)n with the grafted alkyne, whereas the prodrugs were obtained by incorporating the linkers between the Glyco-ONB-PαN3CLn and conjugating a drug molecule in the hydrophobic block. A potent nonsteroidal anti-inflammatory drug, indomethacin (IMC), is suitable drug model for studying micelle formation.24 The block–graft copolymers were synthesized through ring-opening polymerization (ROP), nucleophilic substitution, and “click” reactions (Scheme 1). The physicochemical and photodegradable properties of these micelles in the aqueous phase were examined through fluorescence spectroscopy, dynamic light scattering (DLS), and transmission electron microscopy (TEM). The phototriggered controlled release from the drug-loaded micelles and micelle drug conjugates in the physiological condition was reported. Finally, to demonstrate their potential as fluorescent probes in optical bioimaging, these DOX-encapsulated prodrug micelles were internalized in the human cervical cancer cell line HeLa and analyzed through fluorescence imaging and cytotoxicity study.
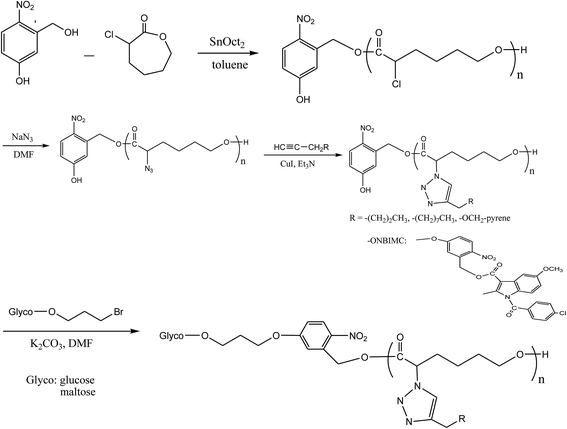 |
| | Scheme 1 Synthesis photo-triggered Glyco-ONB-P(αN3CL-g-alkyne)n and prodrug. | |
Experimental section
Materials
2-Chlorocyclohexanone (99%), 5-hydroxy-2-nitrobenzyl alcohol (97%), 1-hexyne, 1-decayne, sodium azide, pyrene (99%), dimethylamino-pyridine (DMAP, 99%), N,N′-dicyclohexyl-carbodiimide (DCC, >99%), IMC (99%), and Nile red (NR, 98%) were purchased from Aldrich Chemical Co. (Milwaukee, WI, USA). Moreover, m-chloroperoxybenzoic acid, α-D-pentaacetyl-glucopyranoside, D-(+)-maltose, and boron trifluoride diethyl etherate were purchased from Fluka Chemical Co. (Buchs, Switzerland). Stannous octoate (SnOct2, 95%) was purchased from Strem Chemical Inc. (Newburyport, MA, USA). 2-Chloro-ε-caprolactone (α-ClCL), 3-bromo-propyl-glucose, 3-bromo-propyl-maltose, 2-propargyoxymethyl were prepared according to previously described methods,22,25 but with modification. Doxorubicin hydrochloride (99%; Aldrich, Saint Louis, MO, USA) was deprotonated to obtain hydrophobic DOX as described previously.26 N,N-Dimethyl formamide (DMF) and toluene were distilled under calcium hydride. Other high-pressure liquid chromatography (HPLC) grade solvents, such as tetrahydrofuran (THF), dimethylsulfoxide (DMSO), methanol, chloroform (CHCl3), ethyl acetate (EA), and n-hexane were purchased from Merck KGaA (Darmstadt, Germany). A Milli-Q Plus system (Waters, Milford, MA, USA) was used to obtain ultrapure water. Dulbecco's modified Eagle's medium (DMEM), trypsin/EDTA, 100× antibiotic antimycotic, and Hoechst 33342 nuclei dye were purchased from Gibco (Invitrogen Corp. Carlsbad, CA, USA). Fetal bovine serum (FBS) was obtained from Biological Industry (Kibbutz Beit Haemek, Israel). A CellTiter 96® AQueuous One Solution kit was obtained from Promega (Fitchburg, WI, USA) Amiloride, chlorpromazine, methyl-β-cyclodextrin, and nystatin were purchased from Sigma Aldrich (Saint Louis, MO, USA).
Prepare of 2-propargyoxymethyl pyrene (Ppyrene). Pyrenemethanol (2 g, 8.61 mmol) was dissolved in toluene (40 mL) and added the KOH (9.64 g, 171.8 mmol). To the resulting suspension was slowly added propargyl bromide (25.6 g, 172.20 mmol) and stirred at 60 °C for 60 h. Then, the mixture was filtered and concentrated in vacuo. The residue was purified by flash column chromatography with hexane/EA (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 2-propargyoxymethyl pyrene (1.51 g, 65% yield). 1H NMR (CDCl3, δ): 8.01–8.49 (m, 9H), 5.35 (s, 2H), 4.29 (d, 2H), 2.48 (d, 1H).
:1) to give 2-propargyoxymethyl pyrene (1.51 g, 65% yield). 1H NMR (CDCl3, δ): 8.01–8.49 (m, 9H), 5.35 (s, 2H), 4.29 (d, 2H), 2.48 (d, 1H).
Prepare of 5-propargyoxy-2-nitrobenzyl indomethacinate (PONBIMC). 2-Nitro-5-propargyoxybenzyl alcohol27 (1.06 g, 5.10 mmol) and IMC (2.19 g, 6.12 mmol) was dissolved in THF (15 mL). Under N2 atmosphere, the DMAP (73 mg, 0.61 mmol) and DCC (3.2 g, 15.3 mmol) were added. The reaction mixture was stirred overnight at room temperature. Then, the mixture was filtered and concentrated in vacuo. The residue was purified by flash column chromatography with hexane/EA (5:1 to 2:1) to give 5-propargyoxy-2-nitrobenzyl indomethacinate (4.70 g, 86% yield). 1H NMR (CDCl3, δ): 8.18 (d, 1H), 7.79 (d, 2H). 7.49 (d, 2H), 7.0 (d, 1H), 6.98 (dd, 1H), 6.89 (d, 1H), 6.86 (d, 1H), 6.68 (d, 1H), 5.59 (s, 2H), 4.40 (s, 2H), 3.84 (s, 6H), 2.45 (s, 1H).
Synthesis of HONB-P(αN3CL)n polymers
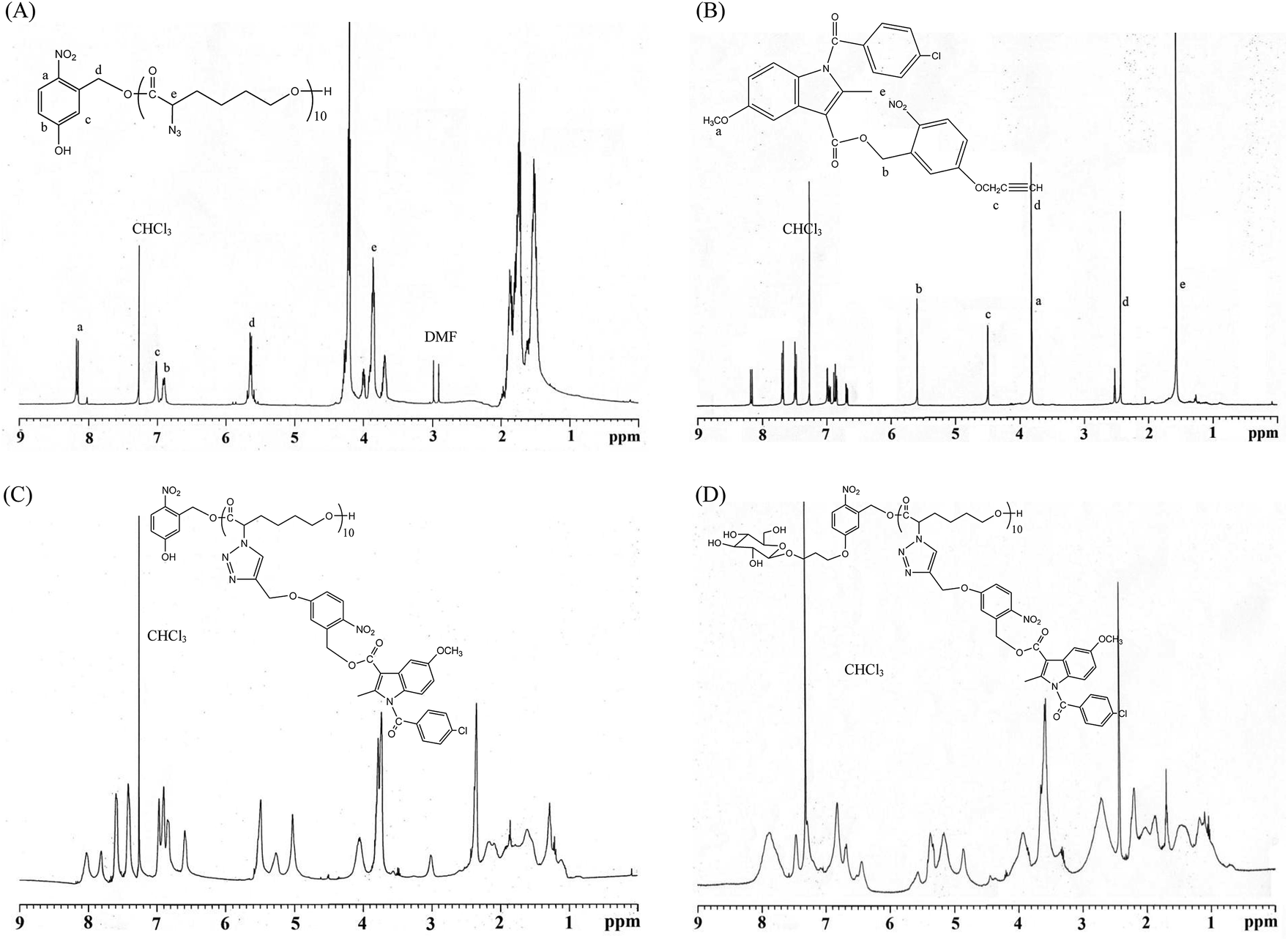
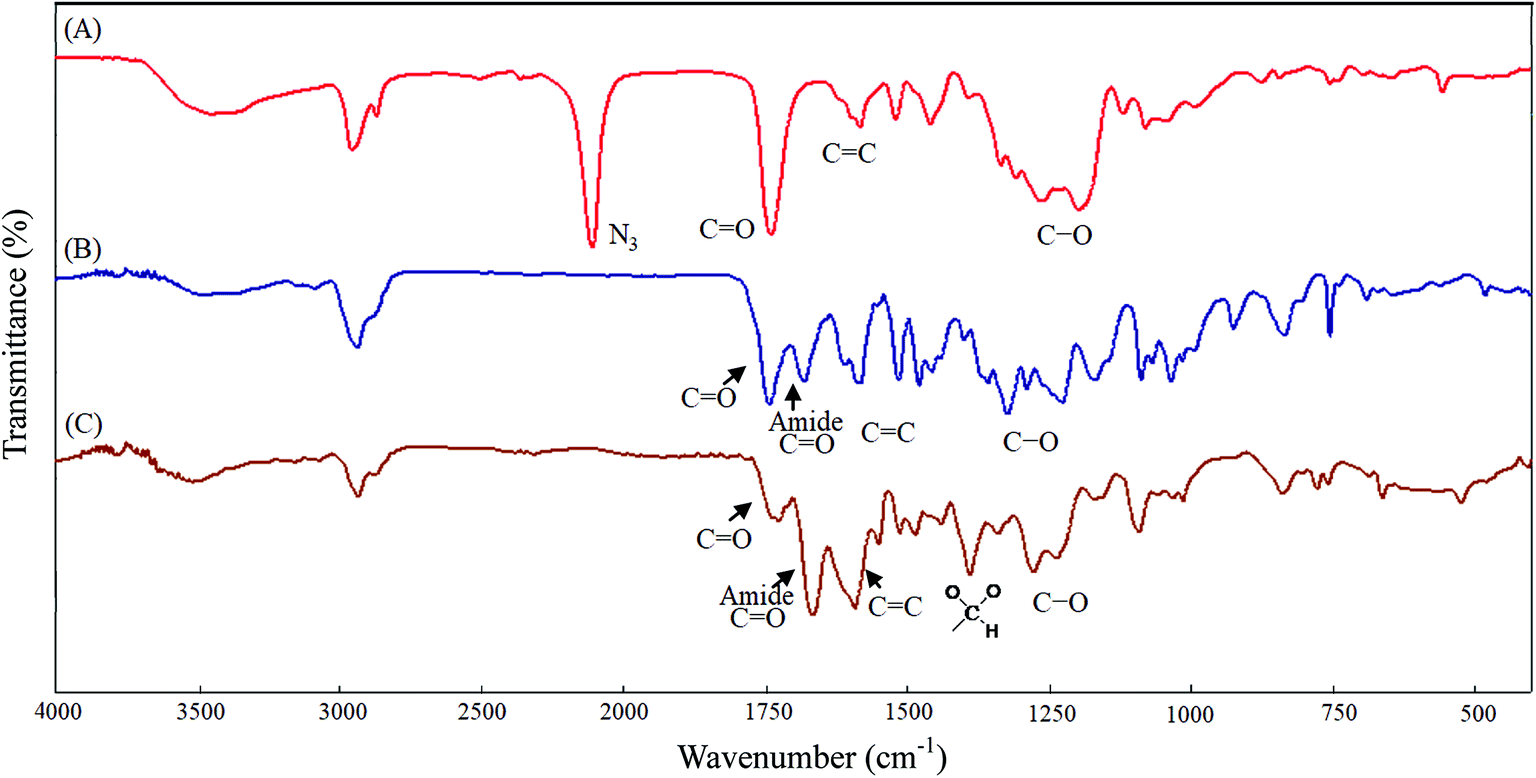
All glassware was dried in an oven and handled under a dry nitrogen stream. 5-Hydroxy-2-nitrobenzyl alcohol (HONB, 275.1 mg, 1.63 mmol), as an initiator, and αClCL (2.41 g, 16.27 mmol) were introduced into a flask, heated under a dry nitrogen stream, and dissolved in 50 mL of toluene. Subsequently, SnOct2 (36 mg, 1.5 wt%, based on the weight of HONB and αClCL) was added to the flask. The flask was purged with nitrogen and refluxed for 8 h, and the solution was vacuum-concentrated under reduced pressure. The resulting product (HONB-P(αClCL)10) was dissolved in CHCl3, and precipitated into excess n-hexane/diethyl ether (5:1 v/v) with stirring. The purified polymer was dried in vacuo at 50 °C for 24 h and analyzed subsequently. Yield: 85%. 1H NMR (400 MHz, CDCl3) δ ppm: 8.18 (d, ArH), 7.04 (d, ArH), 6.88 (dd, ArH), 5.60 (s, benzyl CH2–), 4.25 (t, –CαHCl–), 4.21 (t, –CH2O–), 1.91–2.15 (m, –CβH2–), 1.74 (m, –CδH2–), and 1.40–1.66 (m, –CγH2–). Subsequently, the chloro was substituted by the azide. HONB-P(αClCL)10 (2.68 g, 1.28 mmol, molar mass = 2090 g mol−1) was dissolved in 10 mL of dry DMF, followed by the addition of excess NaN3 (1.01 g, 15.53 mmol). The mixture was stirred at room temperature for 24 h, the solvent was completely removed through rotary evaporation under reduced pressure. The crude product was subsequently dissolved in CH2Cl2, and the insoluble salt was removed through filtration. After concentration, the modified polymer was precipitated in excess cold diethyl ether. The copolymer HONB-P(αN3CL)10 was obtained at a 91% yield. Fig. 1A and 2A depict the representative 1H NMR and infrared (IR) spectra of HONB-P(αN3CL)10, respectively.
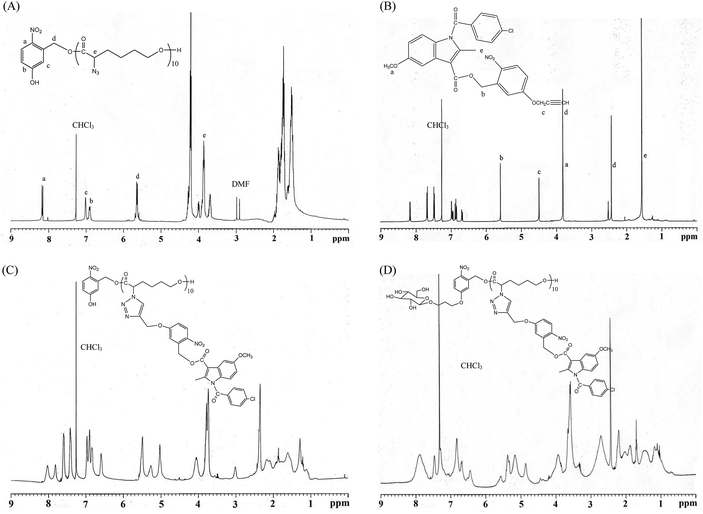 |
| | Fig. 1 Representative 1H NMR spectroscopy: (A) HONB-P(αN3CL)10, (B) (4-propargoxy-2-nitro)benzyl indomethacinoate (PONBIMC), (C) HONB-P(αN3CL-g-PONBIMC)10, and (D) Gluco-ONB-P(αN3CL-g-PONBIMC)10. | |
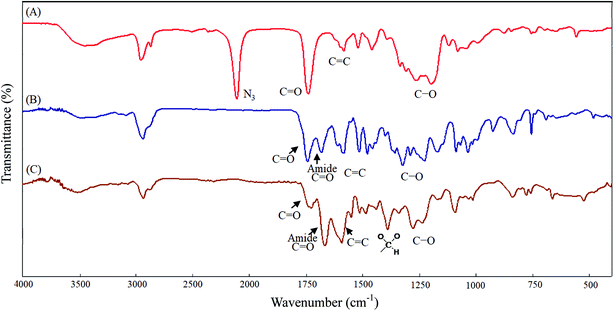 |
| | Fig. 2 IR spectra of (A) HONB-P(αN3CL)10, (B) HONB-P(αN3CL-g-PONBIMC)10, (C) Gluco-ONB-P(αN3CL-g-PONBIMC)10. | |
Synthesis of HONB-P(αN3CL-g-alkyne)n block-grafted polymer with pendant pyrene or IMC groups
HONB-P(αN3CL-g-alkyne)n was prepared through copper-catalyzed azide–alkyne cycloaddition. HONB-P(αN3CL)n (1.10 g) and various types of alkynes (such as 1-hexyne, 1-decayne, 2-propargyoxymethyl pyrene, and 5-propargyoxy-2-nitrobenzyl indomethacinate) were dissolved in THF (10 mL). Subsequently, CuI (0.01 eq.), and Et3N (0.1 eq.) were added under a nitrogen atmosphere. After one freeze pump thaw cycle, the click reaction was conducted at room temperature for 24 h and stopped by exposure to air. Copper ions were removed from the polymer solution by using a short alumina column. The polymer was purified by precipitating into an excess of cold diethyl ether and drying under reduced pressure. The yield of the copolymers was 83–92%. Fig. 1C and 2B depict the representative 1H NMR and IR spectra of HONB-P(αN3CL-g-PONBIMC)10, respectively.
Synthesis of Glycol-ONB-P(αN3CL-g-alkyne)n block-grafted polymers and prodrug
We used a typical procedure for coupling 3-bromopropyl-sugar with HONB-P(αN3CL-g-alkyne)n. First, a mixture of HONB-P(αN3CL-g-alkyne)n (0.17 mmol) and potassium carbonate (71.6 mg, 0.52 mmol) was stirred in DMF (5 mL) for 1 h at 60 °C. 3-Bromopropyl D-glucopyranoside (63.2 mg, 0.21 mmol) in DMF (3 mL) was subsequently added, and the mixture was stirred at 60 °C for 24 h. DMF was removed under reduced pressure. The residue was dissolved in CHCl3, and precipitated into an excess of cold diethyl ether while stirring. The obtained solids were further purified through dialysis [cellulose membrane, molecular weight cutoff (MWCO): 3500 Da] against CHCl3 for 24 h. The purified polymers Glyco-ONB-P(αN3CL-g-alkyne)n were dried in vacuo at 60 °C for 24 h to be obtained in 56–91% yield and analyzed. Fig. 1D and 2C depict the representative 1H NMR and IR spectra of Gluco-ONB-P(αN3CL-g-PONBIMC)10, respectively.
Ultraviolet irradiation
Glyco-ONB-P(αN3CL-g-alkyne)n (1.5 mg) in 1 mL of inhibitor-free THF or polymer micelles in phosphate buffer solution (PBS) (0.01 M, pH 7.4) were exposed to ultraviolet (UV) irradiation sourced from a UV light system (model PR-2000, Phnchum Co., Taiwan), equipped with a Hitachi FL8BL-B lamp (352 nm, 8 W × 8 W). To prevent UV absorption, the samples were placed in quartz cuvettes with a spot area of approximately 1 cm2 and irradiated at room temperature for an assigned duration. Radiation was applied vertically from the top of the cuvette.
Preparation of polymeric micelles
Polymeric micelles of Glyco-ONB-P(αN3CL-g-alkyne)n were prepared through dialysis. Briefly, a solution of Glyco-ONB-P(αN3CL-g-alkyne)n (30 mg) in 5 mL of DMF was transferred in a dialysis bag with a MWCO of 3500 Da. The solution was dialyzed against deionized water at an ambient temperature for 24 h. The water was replaced at 2 h intervals.
Determination of drug loading content and drug entrapment efficiency
Glyco-ONB-P(αN3CL-g-alkyne)n [50-fold critical micelle concentration (CMC) value] was dissolved in 6 mL of methylene chloride using the oil-in-water evaporation technique. IMC, the anti-inflammatory drug, served as a model drug and was added to the polymer at a 1:1 weight ratio. The solution was added dropwise to 150 mL of distilled water containing 1 wt% polyvinyl alcohol and stirred vigorously. Polyvinyl alcohol acted as a surfactant and reduced micellar aggregation. The mixture was sonicated for 1 h at an ambient temperature to reduce the droplet size. The resulting emulsion was stirred at an ambient temperature overnight to evaporate the methylene chloride. The unloaded IMC residue was removed through filtration using a Teflon filter (Whatman) with an average pore size of 0.45 μm. The micelles were obtained through vacuum drying. Later, a weighed amount of micelles was disrupted by adding a 10-fold excess volume of DMF. The drug content was assayed spectrophotometrically at 320 nm using a diode array UV-visible spectrophotometer. The following equations were used to calculate the drug loading content (DLC) and drug entrapment efficiency (DEE):| | |
DLC (%) = (weight of the drug in micelles/weight of micelles) × 100
| (1) |
| | |
DEE (%) = (weight of the drug in micelles/weight of the drug provided initially) × 100
| (2) |
Analysis of in vitro drug release
Appropriate amounts of IMC-incorporated or conjugated micelles (110.2 mg) were weighed and suspended in 10 mL of PBS (0.01 M, pH 7.4). The micellar solution was transferred to a dialysis membrane bag (MWCO = 3500 Da), and the bag was placed in 50 mL of a PBS release medium. The medium was exposed to UV irradiation at 37 °C. At predetermined intervals, 3 mL aliquots of the aqueous solution were withdrawn from the release medium, and an identical volume of the fresh buffer solution was added. The concentration of the released IMC was determined using a UV-visible spectrophotometer at a wavelength of 320 nm. The rate of controlled drug release was measured on the basis of the cumulatively released weight of the drug by using the calibration curve for the drug.
Carbohydrate–lectin binding recognition
The lectin recognition activity of the Glyco-ONB-P(αN3CL-g-alkyne)n solution was analyzed by assessing the change in turbidity at 600 nm at room temperature. A 2 mg mL−1sample of concanavalin A (Con A) lectin was prepared in PBS (0.01 M, pH 7.4). Subsequently, 600 μL of the lectin solution was transferred into a cuvette, and the baseline measurement was recorded. A solution of 60 μL of Glyco-ONB-P(αN3CL-g-alkyne)n at two concentrations (0.1 and 0.2 mg mL−1) in PBS was added to the cuvette containing the lectin solution. The solution in the cuvette was gently mixed using a pipette; thereafter, the absorbance was recorded immediately at 600 nm every 200 s. Control readings were obtained using lectin Con A and the PBS buffer solution without Glyco-ONB-P(αN3CL-g-alkyne)n under the same experimental conditions.
Determination of in vitro cellular viability
The CellTiter 96® AQueuous One Solution kit was used to determine cellular viability. The assay was conducted according to the manufacturer's instructions with minor modifications. HeLa cells were seeded in a 24 well plate (3 × 104 cells per well) overnight and subsequently treated with various concentrations of polymers (or DMSO vehicles) added to the DMEM/F12 1:1 medium with 1% FBS in a humidified incubator at 37 °C, supplied with 5% carbon dioxide. After 48 h, the medium in each well was removed and replaced with 350 μL of warm PBS and 35 μL of CellTiter 96® AQueuous One Solution. The mixture was incubated at 37 °C for 4 h. After incubation, 110 μL of supernatant was transferred from each well to a 96 well plate and absorbance was measured at 485 nm using an enzyme-linked immunosorbent assay reader (Hidex, Turku, Finland). Each experiment was conducted in triplicate.
Flow cytometric analysis of the uptake of doxorubicin-loaded micelles
HeLa cells were seeded in 35 mm dishes (1.5 × 105 cells per dish) and cultured overnight. Subsequently, after UV irradiation or without treated DOX-loaded Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles and free DOX (447 ng mL−1) dissolved in DMEM/F12 1:1 medium with 1% FBS, were added and the cells were incubated for 1, 5, and 60 min. Next, the cells were trypsinized and fixed with 4% paraformaldehyde for 15 min prior to analysis. A BD FACS-Calibur flow cytometer (equipped with a 488 nm argon laser) and CellQuest software were used for the analysis. An FL2 channel captured the fluorescence of the DOX. Each experiment was conducted in triplicate.
Results and discussion
Synthesis and characterization of the Glyco-ONB-P(αN3CL-g-alkyne)n block-grafted copolymer
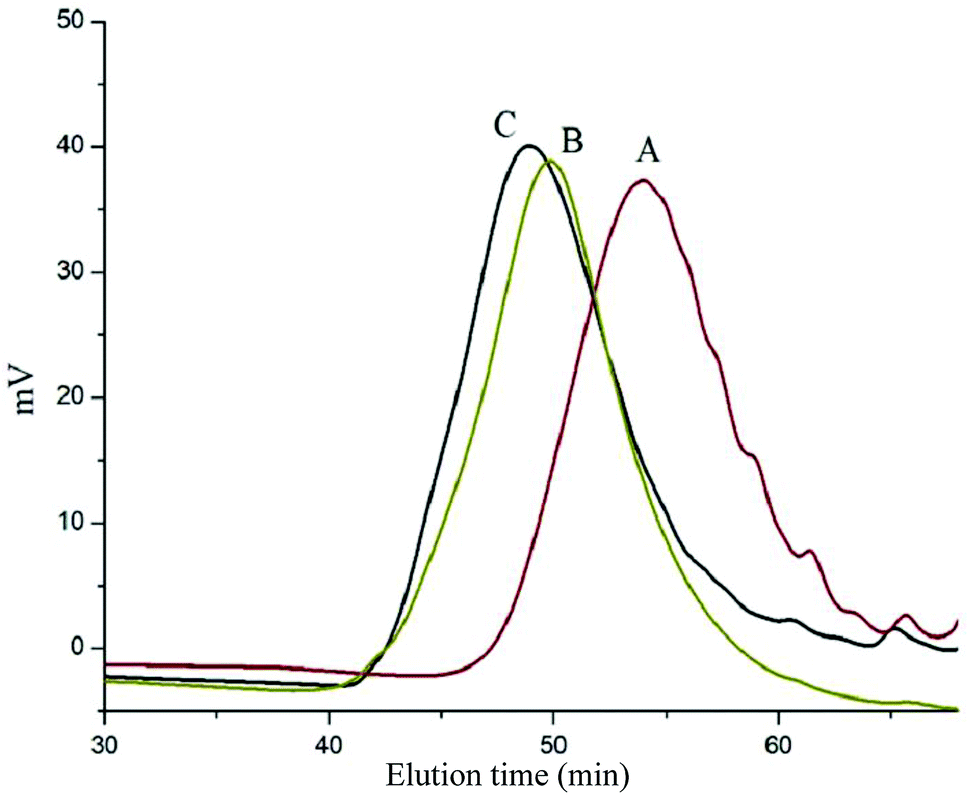
Scheme 1 depicts the strategy for synthesizing the biorecognizable and photocleavable Glyco-ONB-P(αN3CL-g-alkyne)n copolymer. 5-Hydroxy-2-nitrobenzyl alcohol, a difunctional initiator, which contains two hydroxyl groups, namely benzyl and phenolic hydroxyl groups, was used as a photoresponsive molecule because of its chemical stability and rapid cleavage in response to near-UV irradiation (wavelength > 320 nm).27,28 Being more nucleophilic than the phenolic hydroxyl group, the benzyl hydroxyl group initiated the ROP of αClCL catalyzed by SnOct2. The HONB-P(αClCL)n polymers with various P(αClCL)n lengths were obtained by varying the molar ratio of HONB to αClCL. The degrees of polymerization (DPn) of αClCL can be calculated on the basis of the area ratios of peaks at 4.25 ppm (CHCl of αClCL) to 5.80 (benzyl protons of HONB). Our calculation result indicates that the average DP of αClCL is approximately 10, and the number-average molecular weight (Mn) of the polymer is approximately 2 kDa. Subsequently, HONB-P(αN3CL)10 was prepared by a reaction of HONB-P(αClCL)10 and sodium azide. After the reaction, a strong absorption at 2106 cm−1, representing the stretching vibration of azide groups, was observed in the Fourier transform IR (FTIR) spectrum of the polymer (Fig. 2A). The 1H NMR result confirms the conversion of the pendent chlorides to azides. The signal at 4.25 ppm corresponding to the methyne protons next to the chloride groups disappears completely; furthermore, a peak at 3.85 ppm corresponding to the methyne protons next to the azide groups can be observed. Fig. 3A depicts the gel permeation chromatography (GPC) curves of HONB-P(αN3CL)10 (Mn = 2150 g mol−1, Mw/Mn = 1.19).
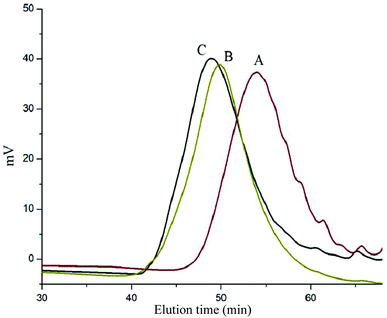 |
| | Fig. 3 GPC curve of (A) HONB-P(αN3CL)10, (B) HONB-P(αN3CL-g-PONBIMC)10, and (C) Gluco-ONB-P(αN3CL-g-PONBIMC)10. | |
The pendant alkyne groups were grafted to HONB-P(αN3CL)n backbone through copper-catalyzed azide–alkyne cycloaddition. The FTIR spectrum of HONB-P(αN3CL-g-PONBIMC)10 is shown in Fig. 2B. After the click reaction, the absorbance band at 2106 cm−1 disappeared completely, and new absorbance bands at 1680 cm−1, attributable to the amide carbonyl vibration of the IMC, were observed. The 1H NMR spectra of HONB-P(αN3CL-g-PONBIMC)10 is shown in Fig. 1C. New peaks at 5.30 ppm, corresponding to the methyne protons next to the triazole ring, and at 8.02 ppm, corresponding to the vinylic protons of triazole, were observed. Other peaks, including the signals at 7.81, 7.59, 5.61, 4.51, and 3.81 ppm of the IMC, were observed. The GPC trace of HONB-P(αN3CL-g-PONBIMC)10 is shown in Fig. 3B (Mn = 5490 g mol−1, Mw/Mn = 1.43), with unimodal distribution and a shift towards the higher molecular weight region compared with the original HONB-P(αN3CL)10.
Finally, the phenolic hydroxyl group of the resulting HONB-P(αN3CL-g-alkyne)n polymer was etherified using Glyco-(CH2)3Br (e.g., Gluco-(CH2)3Br and Malto-(CH2)3Br) through nucleophilic substitution in DMF at 60 °C to yield the Glyco-ONB-P(αN3CL-g-alkyne)n polymer. Table 1 presents the coupling results. The Mn,GPC of the block copolymers with different compositions ranged from 6100 to 10450 mg mol−1, and the polydispersity index (Mw/Mn) ranged from 1.08 to 1.79. The 1H NMR and FTIR results confirmed the effective coupling of Gluco-(CH2)3Br to yield Gluco-ONB-P(αN3CL-g-PONBIMC)10. The representative 1H NMR spectrum is depicted in Fig. 1D. The resonance peaks were assigned to the corresponding hydrogen atoms of the Gluco-blocks at δ 5.05–5.39, and 3.21–3.79 ppm. The presence of proton signals from a Gluco-unit suggested successful conjugation. The IR spectrum of Gluco-ONB-P(αN3CL-g-PONBIMC)10 indicated typical carbonyl absorption of an ester of ONB-P(αN3CL-g-PONBIMC)10 at 1720 cm−1, amide carbonyl absorption at 1680 cm−1, hemiacetal C–O absorption at 1400 cm−1, and alcohol C–O absorption at 1250 cm−1 for glucose (Fig. 2C). Fig. 3C depicts the representative GPC curves of Gluco-ONB-P(αN3CL-g-PONBIMC)10 (Mn = 6280 g mol−1, Mw/Mn = 1.40) and depicts a unimodal distribution with a shift in the peak towards the higher molecular weight region, compared with that of the HONB-P(αN3CL-g-PONBIMC)10.
Table 1 Results of the coupling of HONB-P(αN3CL-g-alkyne/or drug)n with Glyco(CH2)3Br
| Polymera |
Mn,thb |
Mn,NMRc |
Mn,GPCd |
Mw/Mn (PDI)d |
Isolated yield (%) |
| Abbreviations: Gluco = glucose; Malto = maltose; ONB = 5-hydroxy-2-nitrobenzyl alcohol; P(αN3CL) = poly(α-azo-ε-caprolactone); Hexy = 1-hexyne; Decy = 1-decayne; Ppyren = 2-propargyoxymethyl pyrene; PONBIMC = 5-propargoxy-2-nitro-benzyl indomethacinate. Mn,th = Mn,Glyco(CH2)3 + Mn,HONB-P(αN3CL-g-alkyne/or drug)n. Determined by 1H NMR. Determined by GPC. |
| Gluco-ONB-P(αN3CL-g-Hexy)18 |
4658 |
7735 |
8580 |
1.29 |
91 |
| Gluco-ONB-P(αN3CL-g-Decy)17 |
5370 |
8940 |
10450 |
1.79 |
69 |
| Gluco-ONB-P(αN3CL-g-Ppyren)12 |
5489 |
6236 |
6100 |
1.17 |
56 |
| Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) |
6968 |
7080 |
7910 |
1.35 |
76 |
| Malto-ONB-P(αN3CL-g-Ppyren2/-Hexy24) |
7131 |
6948 |
7510 |
1.08 |
86 |
| Gluco-ONB-P(αN3CL-g-PONBIMC)10 |
7400 |
6788 |
6280 |
1.40 |
83 |
Glyco-ONB-P(αN3CL-g-alkyne)n micelles
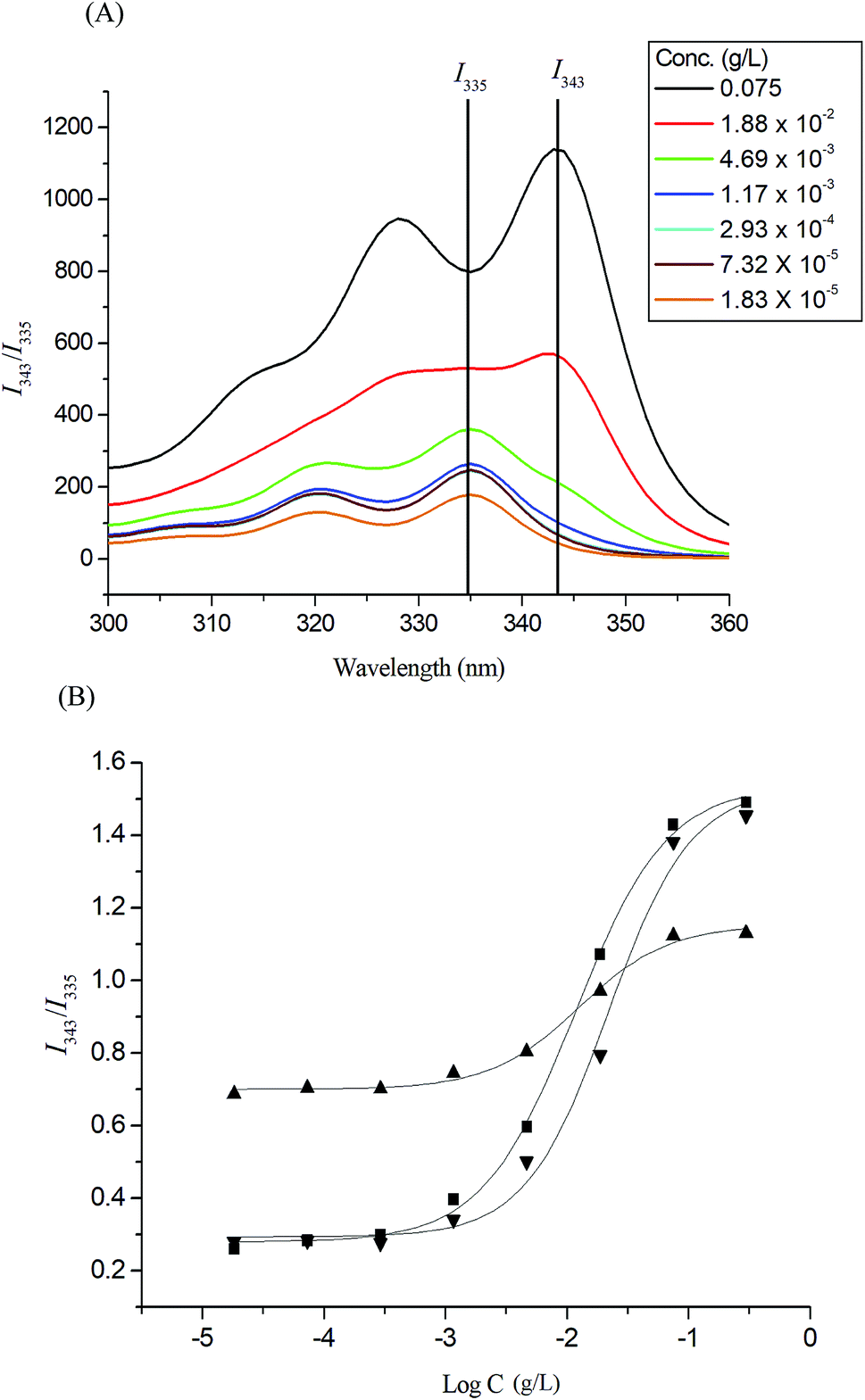
Pyrene molecules encapsulation into the Glyco-ONB-P(αN3CL-g-alkyne)n polymers was investigated through fluorescence spectroscopy. The ratio of the intensity of the third emission peak to that of the first emission peak (I343/I335) was used to characterize the microenvironment of pyrene, with a larger I343/I335 indicating a more hydrophobic environment for the pyrene molecules. The fluorescence intensity of the excitation spectrum of pyrene increased with the concentration of the Gluco-ONB-P(αN3CL-g-Ppyren)12 polymer (Fig. 4A). The characteristic feature of the pyrene excitation spectrum, a red-shift of the (0, 0) band from 335 nm to 343 nm during partitioning into a micellar hydrophobic core, was used to determine the CMC values of Gluco-ONB-P(αN3CL-g-Ppyren)12. Fig. 4B presents the intensity ratios I343/I335 of the pyrene excitation spectra and the logarithmic values of the Glyco-ONB-P(αN3CL-g-alkyne)n concentrations. The CMC value was determined according to the interaction between straight-line segments drawn through the points of the lowest polymer concentrations, which formed an almost horizontal line, and the points of the rapidly rising region of the plot. Table 2 lists the CMC values of various Glyco-ONB-P(αN3CL-g-alkyne)n polymers. The Glyco-ONB-P(αN3CL-g-alkyne)n polymers formed micelles in the aqueous phase, with the micellar CMC values ranging from 1.9 to 69.8 mg L−1. The Glyco-ONB-P(αN3CL-g-alkyne)n polymers exhibited lower CMC values than those of the surfactant (e.g., 2.3 g L−1 for sodium dodecyl sulfate in water), indicating thermodynamically favorable self-association in Glyco-ONB-P(αN3CL-g-alkyne)n polymers. The CMC values decreased as the hydrophobicity of the hydrophobic segment increased. When the hydrophobic pyrene or IMC molecules were grafted onto the P(αN3CL)n, the CMC values decreased significantly.
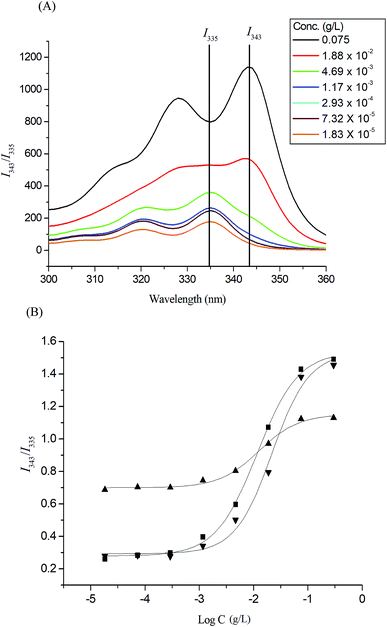 |
| | Fig. 4 (A) Excitation spectra of pyrene Gluco-ONB-P(αN3CL-g-Ppyren)12 micelles monitored at λem = 390 nm with different concentrations. (B) Plot of the I343/I335 intensity ratio (from pyrene excitation spectra: pyrene concentration = 6.1 × 10−7 M) versus the logarithm of the concentration (logC) of Glyco-ONB-P(αN3CL-g-alkyne)n: (■) Gluco-ONB-P(αN3CL-g-Ppyren)12, (▲) Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24), (▼) Malto-ONB-P(αN3CL-g-Ppyren2/-Hexy24). | |
Table 2 Properties of Glyco-ONB-P(αN3CL-g-alkyne)n polymeric micelles and prodrug
| Polymer |
CMC (mg L−1) |
Drug loading contenta (%) |
Drug entrapment efficiencya (%) |
Micelle sizeb (nm) |
| Blank |
PD |
Zeta potential (mv) |
With IMC |
PD |
Zeta potential (mv) |
| Feed weight ratio IMX/polymer = 1/1. Micelle size and particle size distribution (PD) determined by DLS. The IMC conjugation content (LC%) was calculated as LC% = WIMC/Wp × % where WIMC and Wp refer to the weight of conjugated IMC drug within micelles and the weight of prodrug micelles, respectively. |
| Gluco-ONB-P(αN3CL-g-Hexy)18 |
69.8 |
8.21 |
16.43 |
86.1 ± 50.1 |
0.15 |
−20.7 |
|
|
|
| Gluco-ONB-P(αN3CL-g-Decy)17 |
65.6 |
11.36 |
22.72 |
157.2 ± 45.1 |
0.05 |
−31.9 |
|
|
|
| Gluco-ONB-P(αN3CL-g-Ppyren)12 |
1.9 |
48.07 |
96.14 |
115.2 ± 33.7 |
0.25 |
−38.2 |
149.8 ± 40.2 |
0.12 |
−31.9 |
| Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) |
2.1 |
23.11 |
46.23 |
156.9 ± 38.3 |
0.04 |
−41.5 |
189.0 ± 82.6 |
0.17 |
−32.6 |
| Malto-ONB-P(αN3CL-g-Ppyren2/-Hexy24) |
4.4 |
17.88 |
35.76 |
105.0 ± 36.9 |
0.12 |
−28.4 |
106.0 ± 48.7 |
0.17 |
−27.3 |
| Gluco-ONB-P(αN3CL-g-PONBIMC)10 |
18.2 |
52.71c |
— |
74.5 ± 33.0 |
0.16 |
−31.6 |
|
|
|
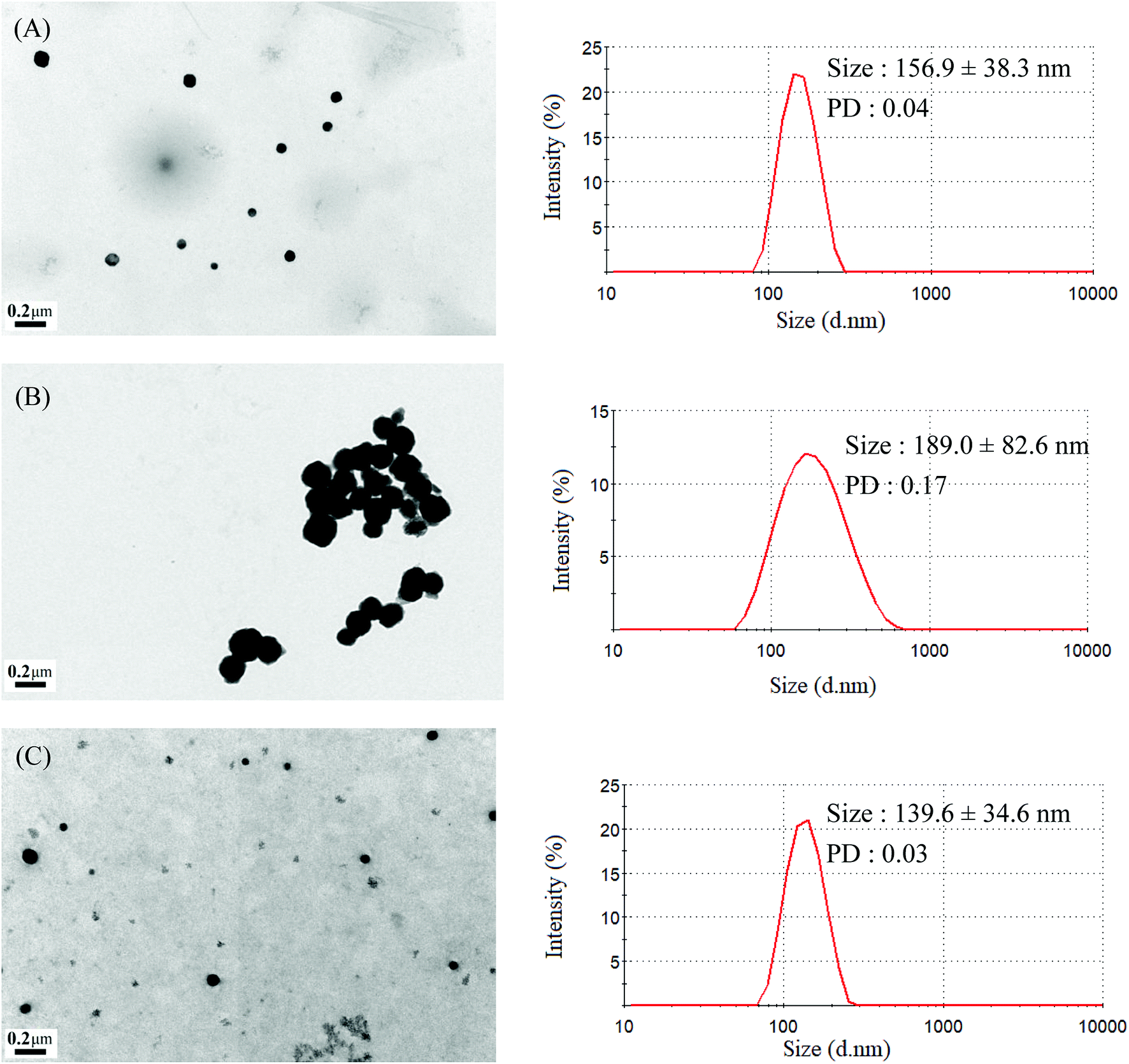
The mean hydrodynamic diameters of micelles ranged from 74.5 to 157.2 nm, with a narrow distribution, and demonstrated a particle size distribution (PD) of ≤0.25. The TEM and size distribution of Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) micelles are demonstrated in Fig. 5A. The spherical morphology was observed. When the drug was incorporated, the micellar size increased (Fig. 5B). The size of an IMC-incorporated Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) micelle was 189.0 nm larger than that of a blank micelle (156.9 nm). The increase in micelle size might be attributed to the increase of the hydrophobic core when the IMC is encapsulated into the copolymer, which is in agreement with the thermodynamic aggregation of the block copolymers (BCPs).29 However, the micelle size remained <200 nm for all formations. A suitable nanoparticle size (diameter: <200 nm) can reduce uptake in the reticulate endothelial system, minimize renal excretion, and increase micelle-encapsulated drug accumulation in tumors through increased permeability and retention.30 The average diameters measured through DLS were larger than those estimated using through TEM images. This is because DLS reveals the average dimensions of the micelles in aqueous solution, whereas TEM reveals the actual core dimensions of the micelles in a dry state. The TEM and PD of the prodrug Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles are presented in Fig. 6B and D, respectively.
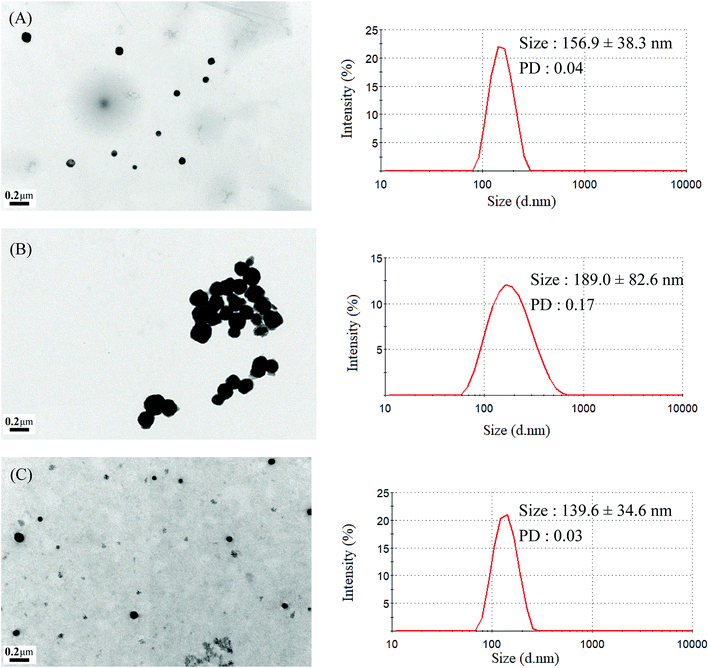 |
| | Fig. 5 TEM and size distribution of Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) micelles: (A) blank, (B) IMC-loaded, and (C) after UV irradiation 6 h. | |
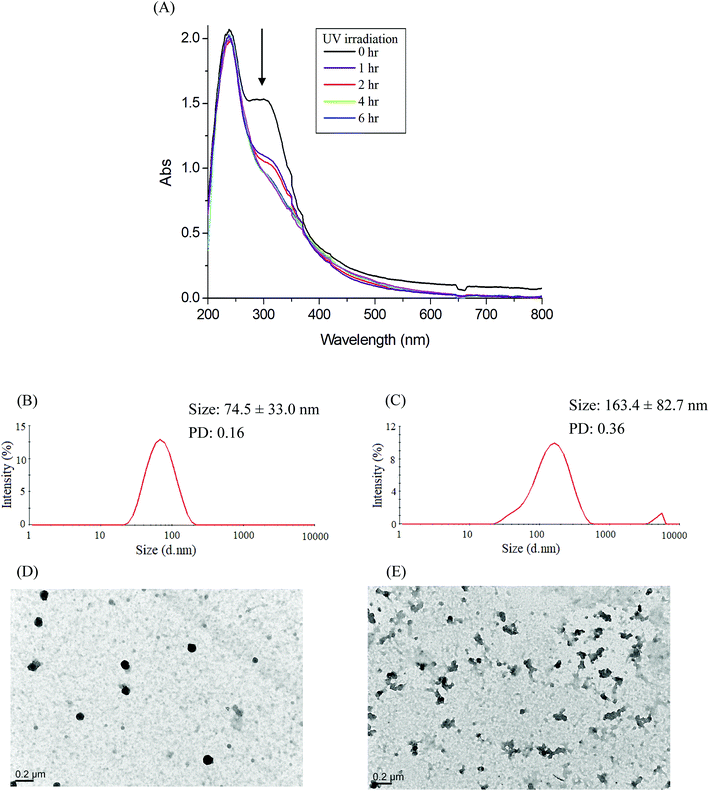 |
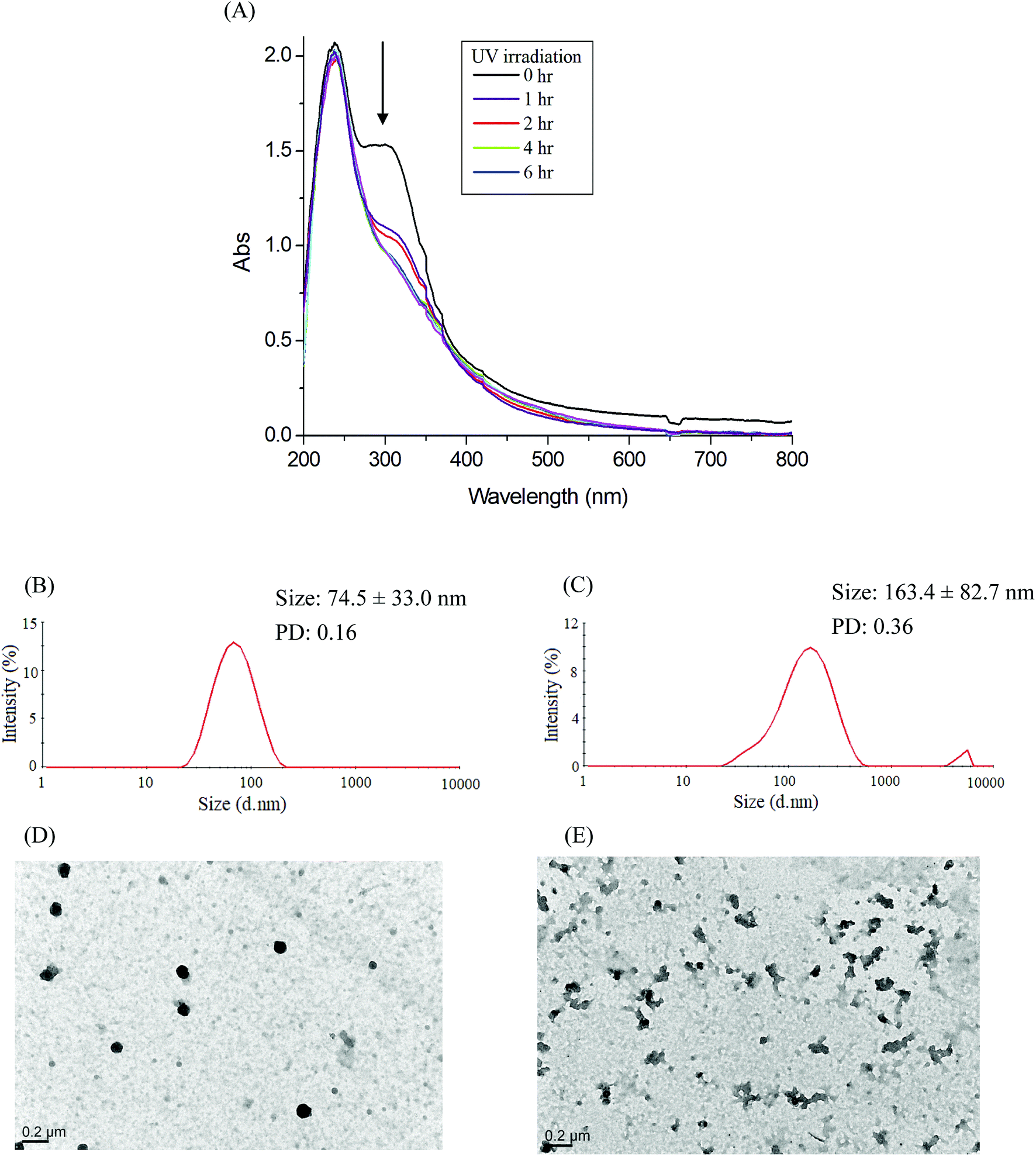
| | Fig. 6 (A) Time-dependent UV-vis spectra changes of the IMC-conjugate Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles treatment under UV irradiation (352 nm, 8 W × 8 W), and DLS and TEM changes of Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles: without irradiation (B and D), and with irradiation for 6 h (C and E). | |
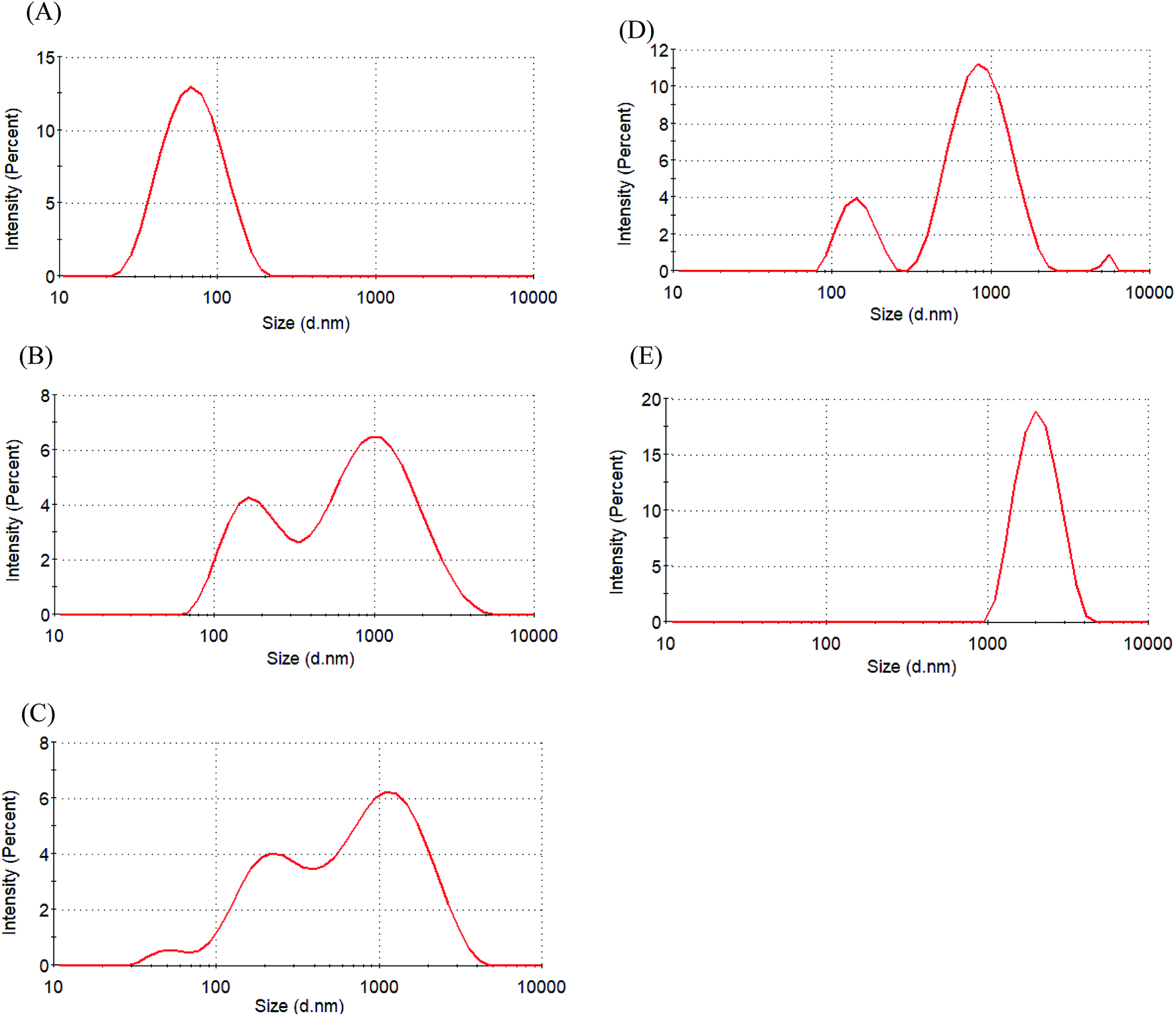
The zeta potential was used to assess micellar stability. When the absolute value of the zeta potential of a micelle was ≥30 mV, the micelle was stable.31 Table 2 indicates that the zeta potential of Glyco-ONB-P(αN3CL-g-alkyne)n micelles is in the range of −20.7 mV to −41.5 mV. The micellar surfaces were negatively charged with comparable zeta potentials. IMC loading lowered the degree of negative charges on the micellar surface because the hydroxyl groups of glycose molecules were protonated by the carboxylic acid group in the IMC molecules. For example, the zeta potential of the IMC-incorporated Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) micelles decreased from −41.5 mV to −32.6 mV. A less negatively charged surface is favorable for preventing uptake by the phagocytic cells, resulting in faster clearance from blood.32 However, to check the stability of the prodrug Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles in the culture medium, we dispersed the prodrug micelles with 10% bovine serum albumin (BSA) and incubated them at 37 °C for 14 day. The stability was studied by DLS measurement and presented in Fig. 7. BSA-treated prodrug micelles exhibited drastic increase in size to about 190 nm and 1130 nm with bimodal curve within 30 min. As the incubated time up to 14 day, the size of micelle increased to 2170 nm. These results imply that the prodrug micelle is easily absorption with protein.
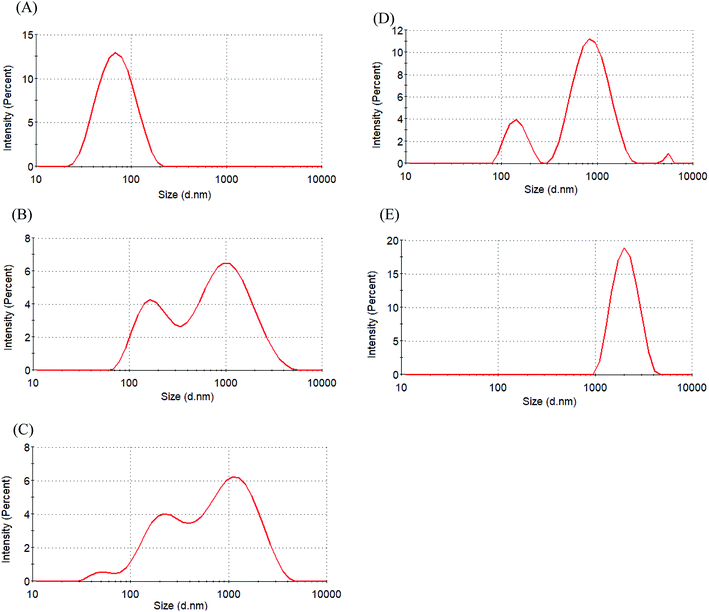 |
| | Fig. 7 Size distribution of Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelle in the presence of bovine serum albumin (10 wt%) for different time intervals: (A) blank, (B) 30 min, (C) 4 h, (D) 6 d, and (E) 14 d. | |
Photocleavable behaviors of micelles
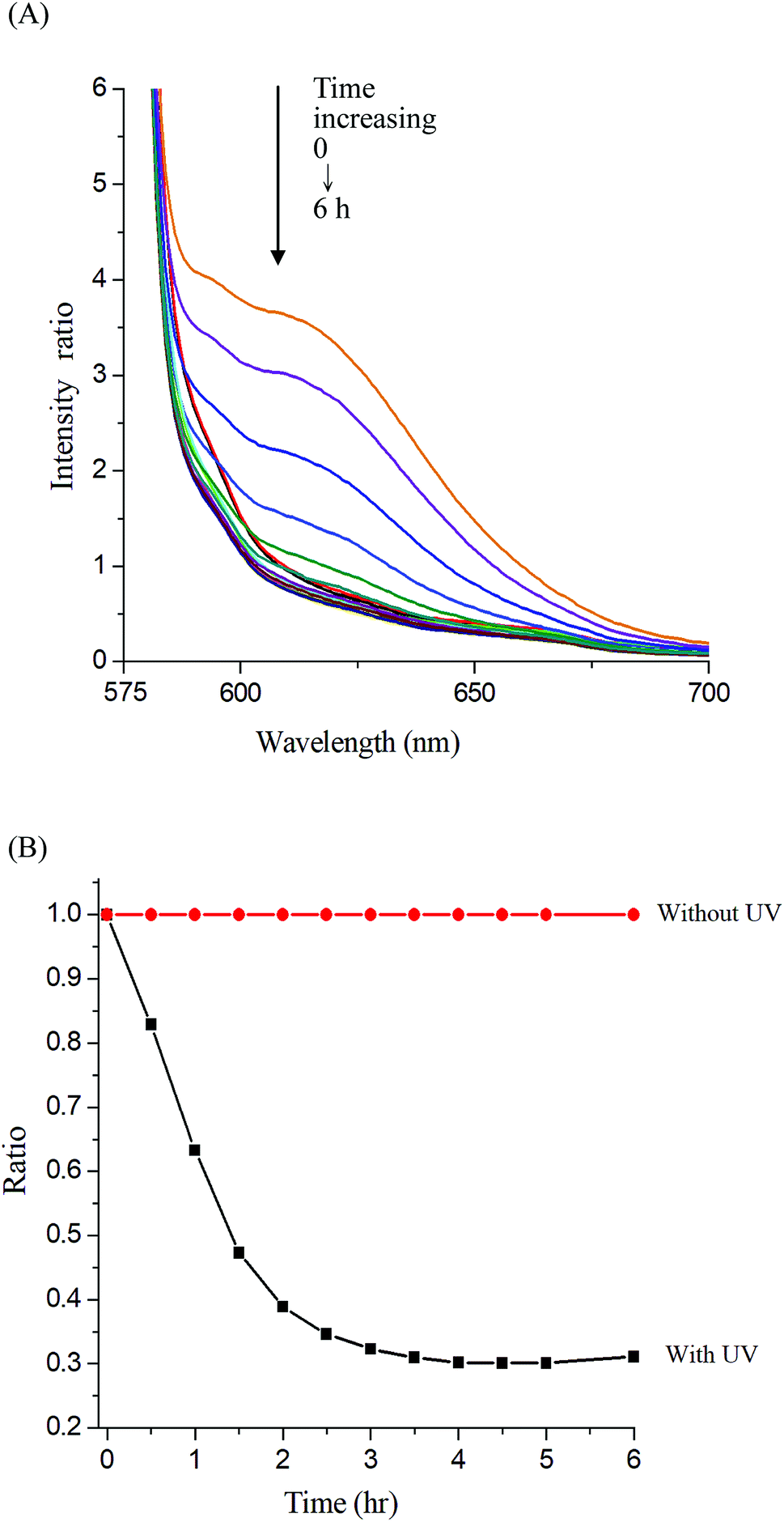
Photo-degradation of micelles was assessed at various irradiation times by monitoring changes in NR fluorescence.33 The reduction in the intensity of NR fluorescence at 610 nm was recorded. After dissolving the NR and Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) in THF (0.5 mg mL−1, NR: polymer ratio = 1:3), water was added to induce micelle formation and concomitant NR encapsulation by the micelle core. THF was subsequently removed through evaporation, and nonsolubilized NR was micro-filtered (0.2 μm pore filter). The final micellar concentration was adjusted to 0.2 mg mL−1. Fig. 8A depicts the fluorescence emission spectra of NR incorporated in the Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) micelles before and after UV irradiation at various time intervals. Fig. 8B plots normalized fluorescence against time; through UV irradiation of the solution, the emission intensity was reduced to 30% after 6 h of irradiation. The micelles exhibited photolabile properties in response to light activation. The TEM and PD of Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) micelles after 6 h UV irradiation are presented in Fig. 5C. The results indicated that the destruction of a micelle was not complete because of only one ONB moiety and the steric hindrance of polymer. The photo-degradability of the formed prodrug micelles was subsequently studied. The TEM and size distribution of Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles after 6 h UV irradiation are demonstrated in Fig. 6C and E, respectively. Before irradiation, micelles morphologies were uniform. However, after UV irradiation, disintegrated micelles with aggregation were observed, indicating that irradiation for a specific duration induced changes in the assembly state. When expose to UV light, the micelle size of Gluco-ONB-P(αN3CL-g-PONBIMC)10 increased from 74.5 (PD = 0.16) to 163.4 nm (PD = 0.36), with broader size distribution and larger aggregates with an average diameter > 750 nm. These aggregates are likely fragments of the hydrophobic backbone without Gluco, which arise from the backbone degradation and are insoluble in water. Compared with the photocleavage of Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) micelles, the Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles demonstrated easier photocleavage because they have greater numbers ONB moieties in the polymer.
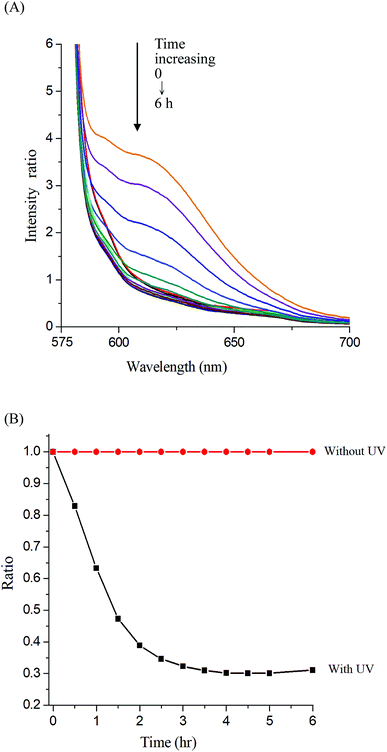 |
| | Fig. 8 (A) Fluorescence spectra change of Nile red-loaded Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) micelle in PBS (0.01 M, pH 7.4) in the presence of UV irradiation (352 nm) at 25 °C, (B) normalized fluorescence emission intensity vs. time of irradiation. | |
Evaluation of drug loading content, entrapment efficiency and in vitro release of IMC
The DLC and DEE of the polymeric micelles were determined through UV-visible absorption spectroscopy of IMC. IMC, a widely used hydrophobic, nonsteroidal anti-inflammatory drug, was used as a model drug to investigate drug loading in the hydrophobic core. The maximal absorption peak of IMC was proportional to its concentration at 320 nm. After releasing IMC and removing the polymer precipitate, the amount of loaded IMC was determined according to the absorbance at 320 nm. Table 2 lists the calculated drug loading content and entrapment efficiency. At a constant feed weight ratio (WIMC/Wcopolymer = 1:1), the DLC and DEE ranged from 8.21% to 48.07% and 16.43% to 96.14%, respectively, for the Glyco-ONB-P(αN3CL-g-alkyne)n series of polymers. The DLC and DEE increased with the hydrophobicity of the hydrophobic segment. When the longer hydrophobic alkyne or pyrene was grafted onto the hydrophobic block, which exhibits stronger interactions with guest molecules, higher DLC and DEE can therefore be achieved. For the Gluco-ONB-P(αN3CL-g-PONBIMC)10 prodrug, the DLC was calculated approximately 52.71%. Compare with the simple physical encapsulation system, the DLC of the prodrug is high.
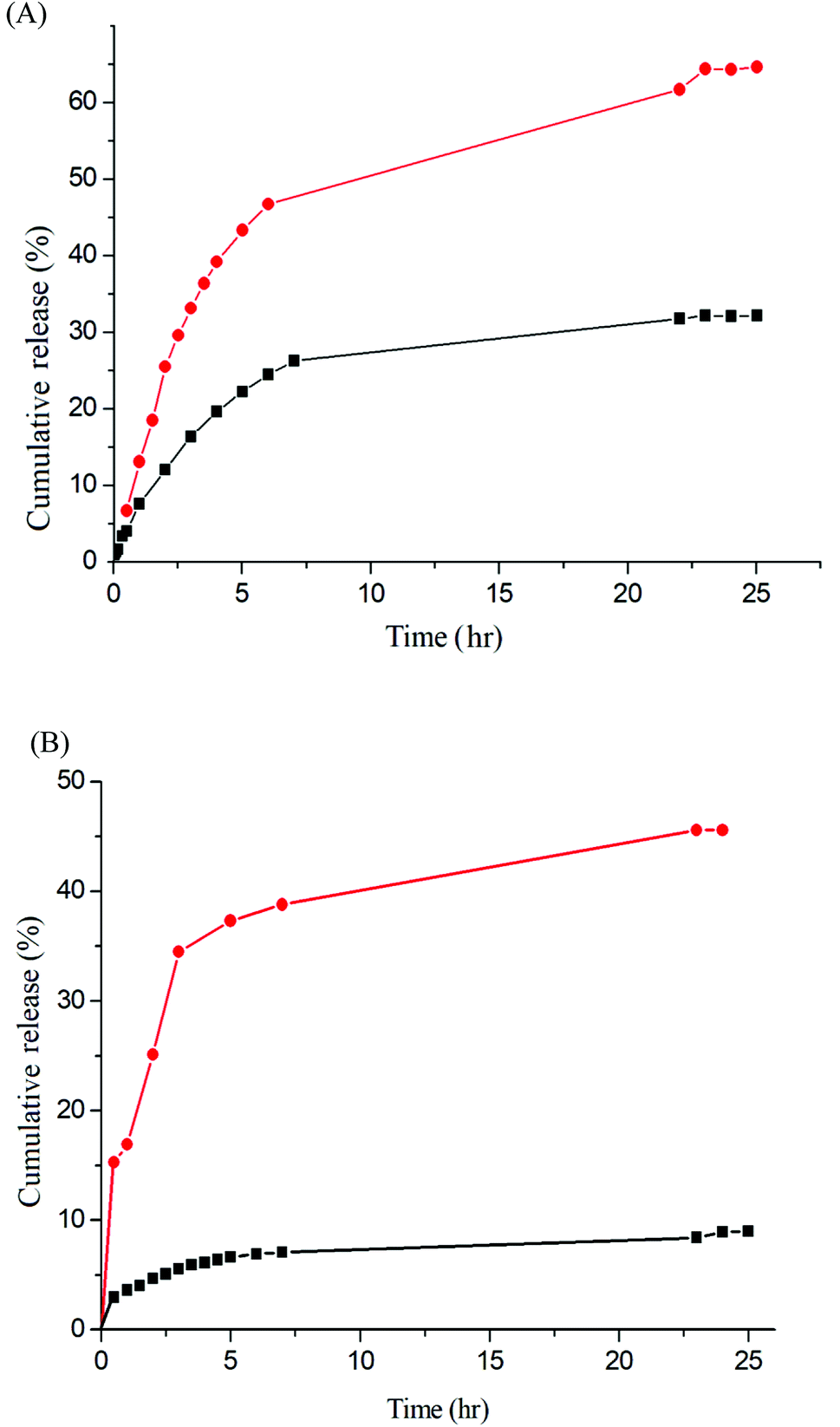
The release rate was monitored by determining the concentration of the total amount of released drug. Fig. 9 depicts the release profiles of IMC from the IMC-loaded micelles of Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) and IMC-conjugated micelles of Gluco-ONB-P(αN3CL-g-PONBIMC)10 prodrug. A biphasic release profile was observed, in which a rapid release stage was followed by a sustained release phase. Compared with the released rates in the absence of UV irradiation, the release rate of IMC was faster during UV irradiation at 37 °C, with approximately 45% of the encapsulated IMC released in a sustained manner during the first 5 h. After irradiation for 25 h, approximately 65% of the IMC was released, whereas only 26% release was exhibited without UV irradiation. For the IMC-conjugated Gluco-ONB-P(αN3CL-g-PONBIMC)10 prodrug system, the release rate of IMC was slower than the IMC-loading micelles of Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24). Approximately 45% of the IMC was released under UV irradiation for 25 h; only 8% release was observed without irradiation over the same period (Fig. 9B). This can be ascribed to the covalent linkage nature between IMC and polymer backbones within micellar cores. Therefore, those covalent linkages can be cleaved with UV irradiation.
 |
| | Fig. 9 IMC release (A) the IMC-loaded micelle of Gluco-ONB-P(αN3CL-g-Ppyren2/-Hexy24) in the presence of UV irradiation (●) and without irradiation (■), (B) the IMC-conjugate micelle of Gluco-ONB-P(αN3CL-g-PONBIMC)10 in the presence of UV irradiation (●) and without irradiation (■) in PBS (0.01 M, pH 7.4) at 37 °C. | |
Carbohydrate–lectin binding recognition
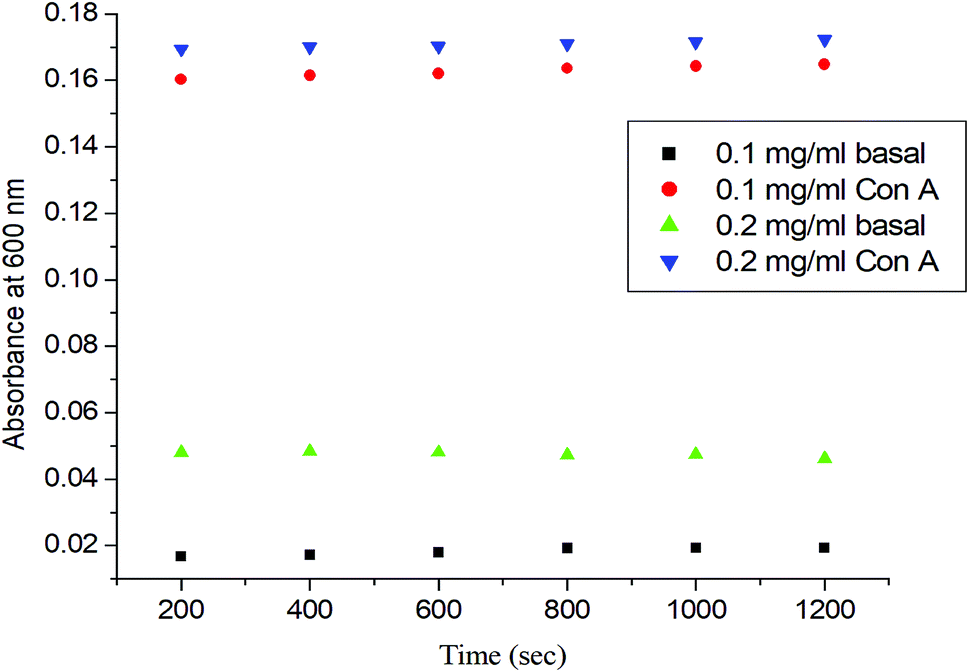
To determine the role of the synthetic glycopolymer, Glyco-ONB-P(αN3CL-g-alkyne)n, in drug targeting, the ability of the synthesized Gluco-ONB-P(αN3CL-g-PONBIMC)10 polymer to interact with the biological system was assessed. Carbohydrate is essential in several biological recognition events mediated by specific carbohydrate–lectin interactions. Although the exact mechanism of this interaction remains unknown, numerous studies have demonstrated that the mechanism is highly specific and noncovalent.34 Thus, an in vitro evaluation of this specific binding event provides an initial test of the ability of a synthetic Gluco-ONB-P(αN3CL-g-PONBIMC)10 polymer to interact with the biological systems, such as in drug delivery development, tissue engineering, and biomedical material synthesis. These tests are typically conducted by mixing Gluco-ONB-P(αN3CL-g-PONBIMC)10 with a lectin that is selective for the sugar conjugated to the polymer.35 A positive result is obtained when a precipitate appears because of lectin aggregation; this can be measured as the reduction in the transparency of the solution. Con A is a specific lectin used for selective binding to glucosyl residues. Therefore, we investigated the change in absorbance of solutions of Gluco-ONB-P(αN3CL-g-PONBIMC)10 with Con A at 600 nm. Fig. 10 illustrates that the absorbance (i.e., turbidity) increased with the concentration of Gluco-ONB-P(αN3CL-g-PONBIMC)10 because of the formation of larger aggregates. For a control reading, a PBS buffer solution without Con A was added to the glucose polymer. A marginal absorbance significantly lower than that for the sample containing Con A, was detected.36 These experiments confirmed that Gluco-ONB-P(αN3CL-g-PONBIMC)10 synthesized through the nucleophilic coupling of Gluco-(CH2)3Br to HONB-P(αN3CL-g-PONBIMC)10 entailed active biorecognition.
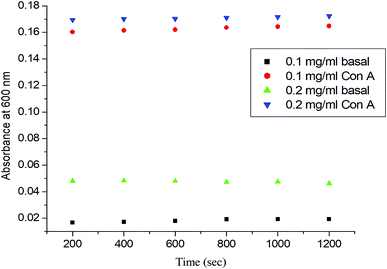 |
| | Fig. 10 The absorbance (450 nm) of the Gluco-ONB-P(αN3CL-g-PONBIMC)10 solution upon reaction with lectin Con A (2 mg mL−1) in PBS buffer: concentration 0.2 mg mL−1 with lectin Con A (▼) or without lectin Con A (▲), concentration 0.1 mg mL−1 with lectin Con A (●) or without lectin Con A (■). | |
In vitro cytotoxicities of the polymer
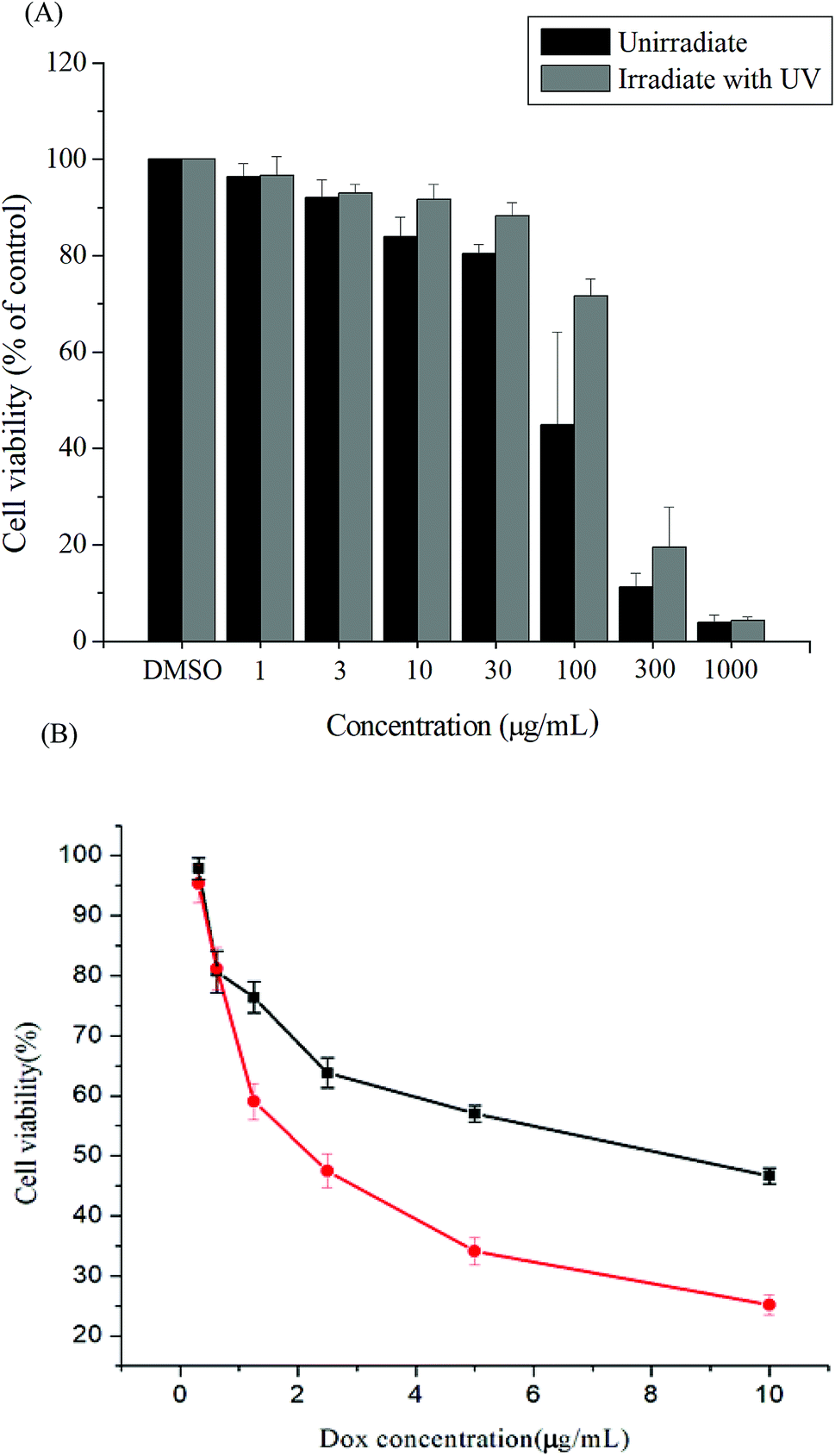
Cytotoxicity is a crucial consideration in drug carrier design. In vitro cytotoxicities of the Gluco-ONB-P(αN3CL-g-PONBIMC)10 polymer with and without UV irradiation were evaluated through a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay of HeLa cells treated with various polymer concentrations. To eliminate the undesirable cytotoxic effects of the polymer with and without UV irradiation, the viability of cells loaded with increasing amounts of Gluco-ONB-P(αN3CL-g-PONBIMC)10 was assessed using a Promega CellTiter 96® AQueuous One Solution kit. HeLa cells were incubated with various concentrations of Gluco-ONB-P(αN3CL-g-PONBIMC)10 for 48 h followed by a reaction with the MTS reagent, which was bioreduced to formazan because of the esterase in living cells, enabling spectrophotometric analysis absorbance at 485 nm. Fig. 11A depicts the relative viability percentages of cells treated with various concentrations of Gluco-ONB-P(αN3CL-g-PONBIMC)10 before UV exposure and after 1 h of UV irradiation for 48 h. Cell viability was 80% higher than the control at a polymer concentration ranging from 1 to 30 μg mL−1. The results demonstrate that the Gluco-ONB-P(αN3CL-g-PONBIMC)10 solution before UV exposure and after 1 h of UV irradiation were slightly cytotoxic. In addition, Fig. 11B presents the in vitro cytotoxicities of the DOX-loaded micelles and free DOX at various DOX dosages (0.125–10 μg mL−1). The DOX-loaded Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles effectively in-habited HeLa cell proliferation with a half-maximal inhibitory concentration (IC50) of 8.8 μg mL−1. The DOX-loaded micelles possess higher IC50 than the free DOX (2.4 μg mL−1), which is observed in many polymeric system.37 This is likely caused by the longer time required for DOX to be released from micelles into tumor cells.
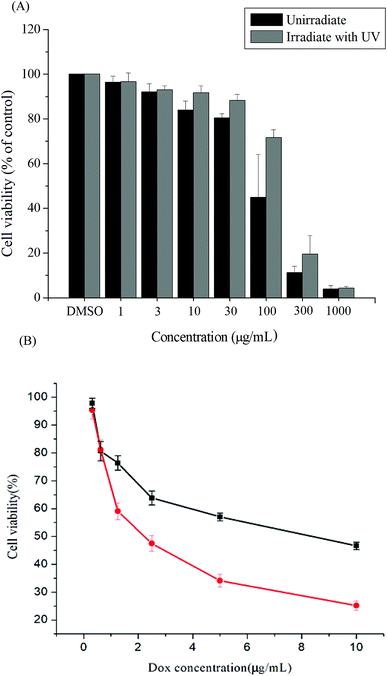 |
| | Fig. 11 The cell viabilities of HeLa cells treated: (A) with various concentrations of Gluco-ONB-P(αN3CL-g-PONBIMC)10 before UV exposure and after 1 h of UV irradiation, (B) with DOX-loaded Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles (■), and free DOX (●) for 48 h. Data are shown as mean ± S.E. (n = 3). | |
Cellular uptake profile of doxorubicin-incorporated micelles
The cellular uptake of prodrug micelles by HeLa cells was investigated through flow cytometry and confocal laser scanning microscopy (CLSM). Amphiphilic drug-conjugated polymer can transport drugs through micelle formation. We investigated the drug-carrying ability of the polymeric Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles using DOX, a potent antitumor drug. The self-fluorescence characteristic of this drug facilitated the identification and quantification of drug-incorporated micelles that entered the cells.38,39
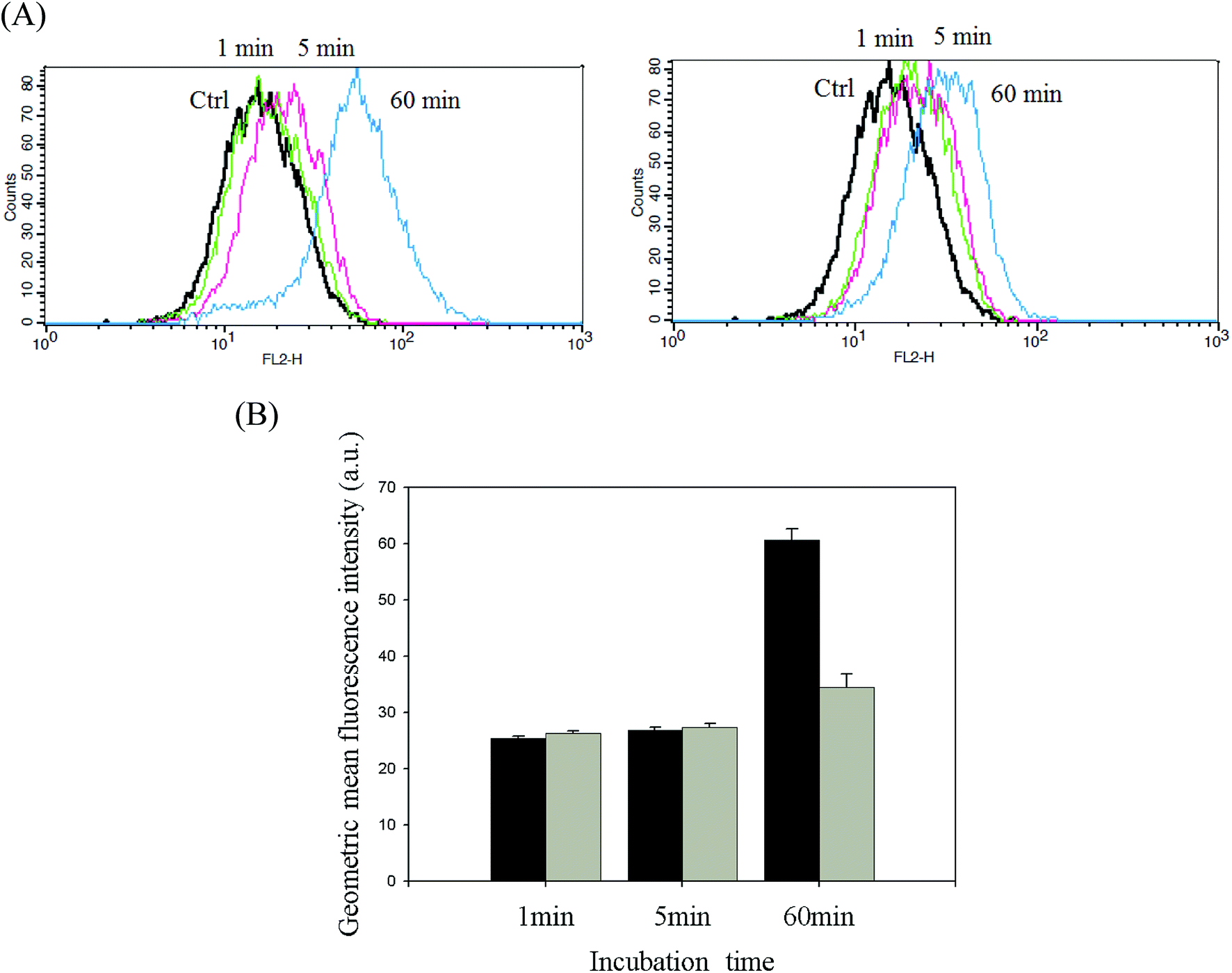
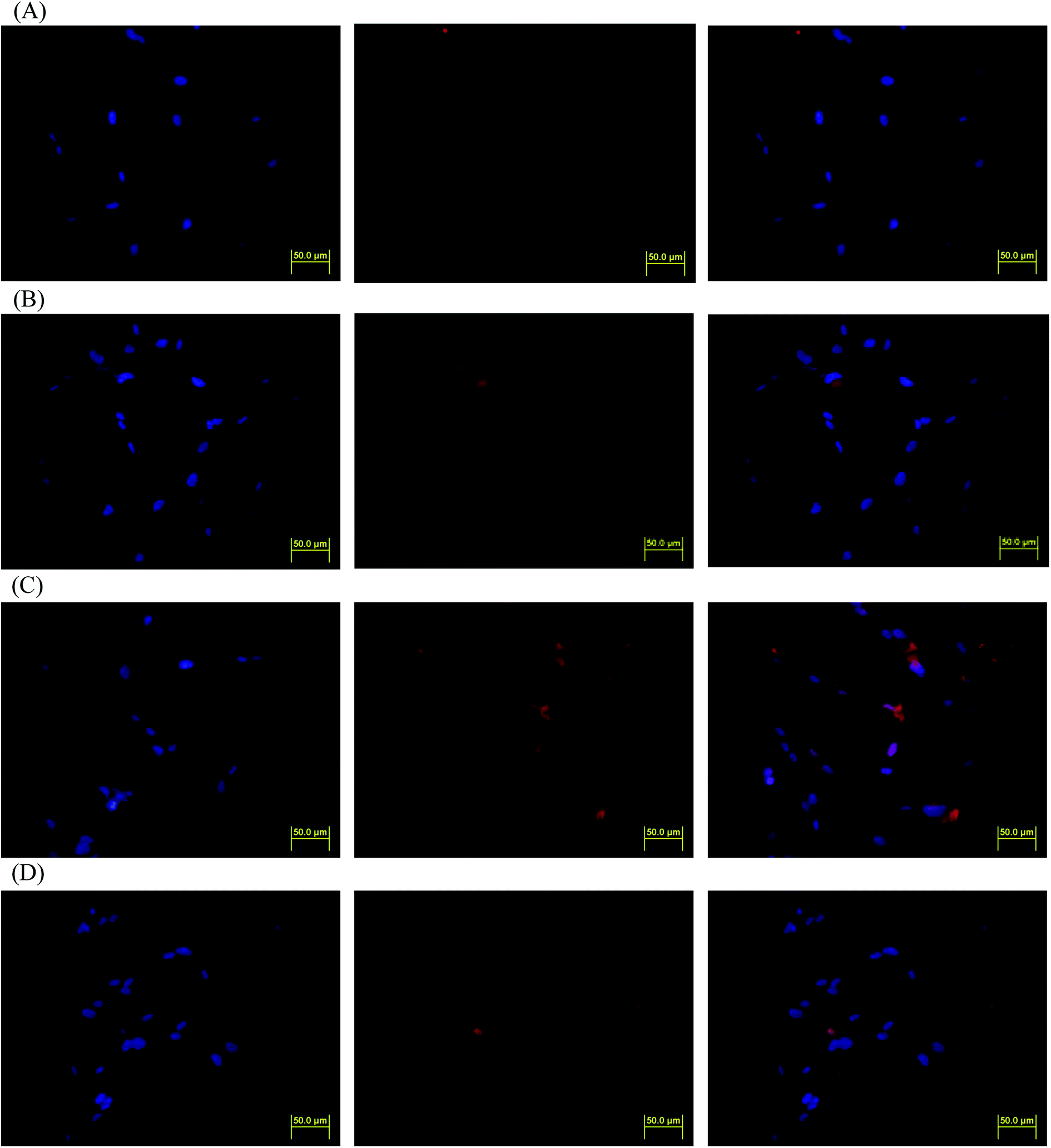
DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles were prepared through dialysis, and the uptake of micelles and free DOX in equal concentration (458.2 ng mL−1) at 1, 5, and 60 min, respectively, were recorded through flow cytometry. In the first 5 min, DOX uptake in the cells incubated with DOX-incorporated micelles was similar to that in those treated with free DOX (Fig. 12A). However, DOX in its free-form enters and accumulates into cells faster than the DOX encapsulated in micelles at incubation time of up to 60 min. The geometric mean fluorescence intensity in the HeLa cells treated with DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 was approximately 0.6-fold weaker than that in the HeLa cells treated with free DOX during the 60 min incubation time (Fig. 12B). Various fluorescence microscopic experiments were conducted to determine the intracellular distributions of DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles and free DOX following the cellular entry. Fig. 13 depicts the results of the nuclear stained Hoechst 33342 and DOX fluorescence as well as an overlay of the images (from left to right). These images indicate that free DOX and Gluco-ONB-P(αN3CL-g-PONBIMC)10-encapsulated DOX exhibited distinct temporal and spatial entry patterns. Free DOX accumulated in the cells at a substantially faster rate than did the Gluco-ONB-P(αN3CL-g-PONBIMC)10-encapsulated DOX, with a minimal fluorescence after 1 min of treatment, yielding visible fluorescence after 60 min of treatment (Fig. 13A and C). DOX fluorescence was predominantly concentrated in the cell nuclei, which could be an inherent tendency of the free DOX.40 By contrast, in the HeLa cells treated using DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 for 1 or 60 min, DOX fluorescence was concentrated in the cytoplasm with little to no DOX visible in the nucleus (Fig. 13B and D). These differences are attributable to the free DOX entering the cells through passive diffusion, whereas the Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelle-encapsulated DOX penetrated the plasma membrane through endocytosis.41 Although the uptake of the DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles was slow, the intensity of nuclear fluorescence in the HeLa cells treated with DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 was marginally higher after 60 min than after 1 min (Fig. 13B and D). Entry into the nuclei by DOX confirmed that Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelle-encapsulated DOX was released and successfully reached its pharmacological target. Low penetration of DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles into the extravascular tumor tissues may be attributed primarily to bulky micelle systems being substantial hindered from penetrating into tumor cells.42
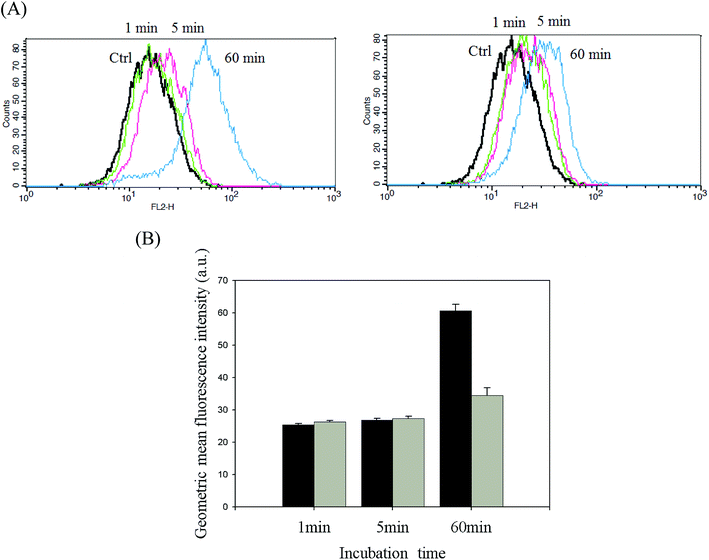 |
| | Fig. 12 (A) Flow cytometric histogram profiles of HeLa cells treated with free DOX (left), and DOX-loaded Gluco-ONB-P(αN3CL-g-PONBIMC)10 (right) for 1, 5, and 60 min. Control groups were cells that did not receive any treatment, representing basal fluorescent levels, and (B) geometric mean fluorescence intensities of free DOX (black) and DOX-loaded micelles (gray). Data shown mean ± S. E. (n = 3). | |
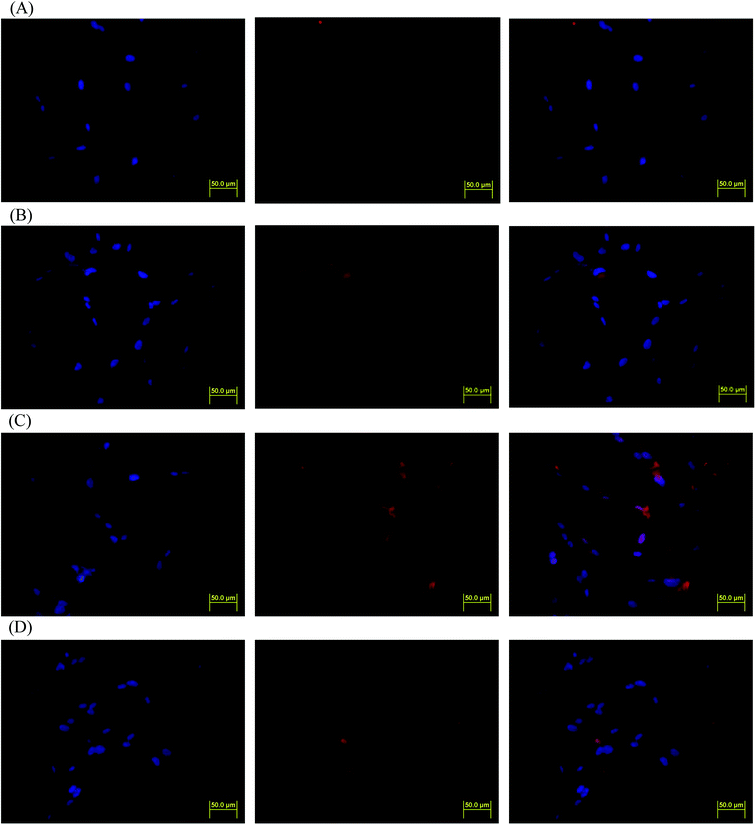 |
| | Fig. 13 Fluorescent microscopic images of HeLa cells incubated with free DOX (254.7 ng mL−1) or DOX-loaded Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles for different time intervals: (A) free DOX, and (B) DOX-loaded micelles for 1 min; (C) free DOX, and (D) DOX-loaded micelles for 60 min. For each row, images for left to right show the cells with Hoechst 33342 nuclear staining, DOX fluorescence, and the merged image (scale bar 50 μm; brightness not proportional to fluorescence intensity). | |
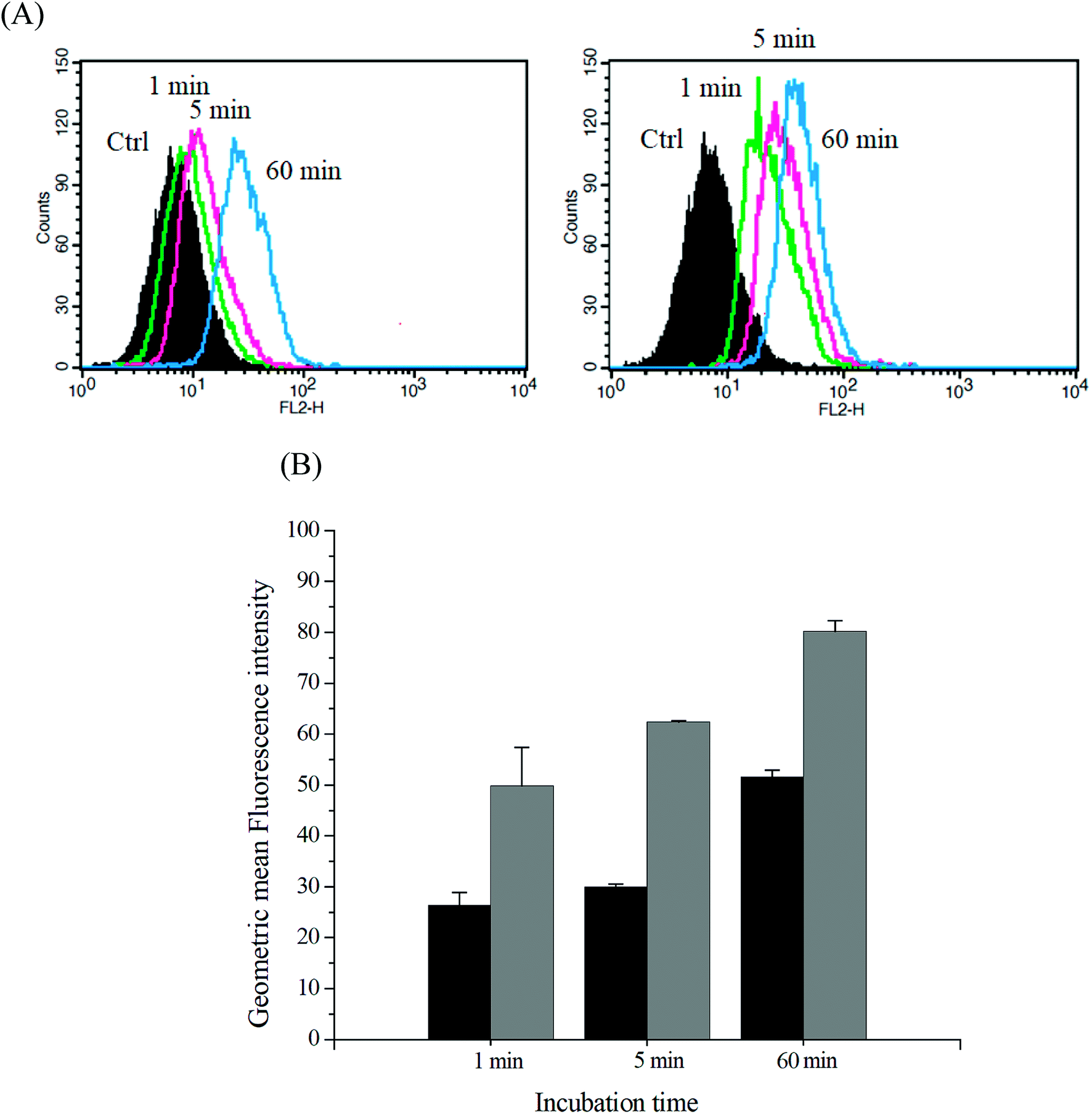
The intracellular DOX release from DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 under UV irradiation was also confirmed by flow cytometry and CLSM studies. As observed from Fig. 14 and 15, HeLa cells incubated with DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles after UV irradiation for 5 min displayed stronger intracellular DOX fluorescence intensity as compared to the free DOX. The DOX-incorporated micelles after UV irradiation for 5 min enters and accumulates into cells faster than the free-form DOX during the incubated time (Fig. 14A). The geometric mean fluorescence intensity in the HeLa cells treated with DOX-incorporated Gluco-ONB-P(αN3CL-g-PONBIMC)10 under UV irradiation was approximately 1.6-fold stronger than that in the HeLa cells treated with free DOX at incubation time of up to 60 min (Fig. 14B). Fig. 15 depicts the results of the nuclear stained Hoechst 33342 and DOX fluorescence as well as an overlay of the images (from left to right). These images indicate that the HeLa cells treated using DOX-incorporated micelles after UV irradiation for 60 min, DOX fluorescence was major concentrated in the cytoplasm. These results displayed that the cellular uptake of prodrug micelles under UV irradiation enters and accumulates into cells faster than without UV irradiation.43
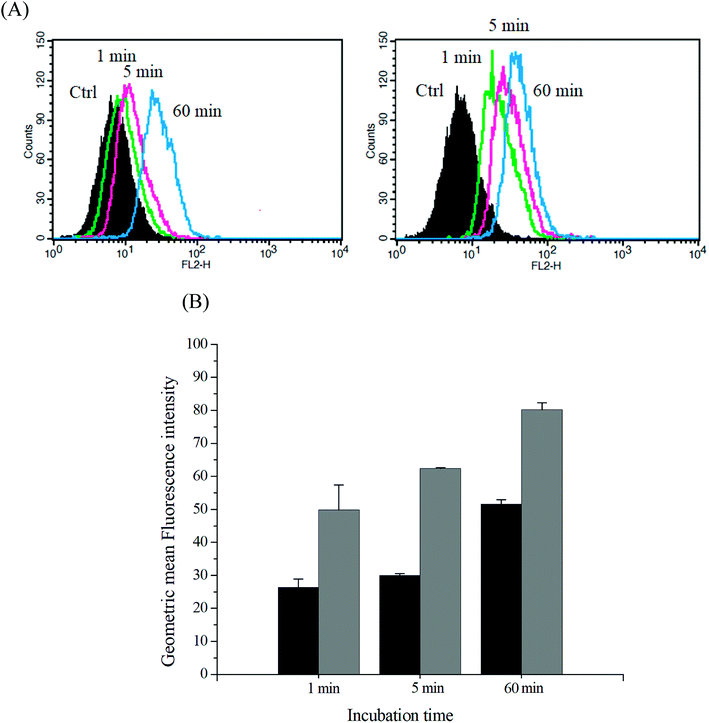 |
| | Fig. 14 (A) Flow cytometric histogram profiles of HeLa cells treated with free DOX (left), and after 5 min UV irradiation DOX-loaded Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles (right) for 1, 5, and 60 min. Control groups were cells that did not receive any treatment, representing basal fluorescent levels, and (B) geometric mean fluorescence intensities of free DOX (black) and after 5 min UV irradiation DOX-loaded micelles (gray). Data shown mean ± S. E. (n = 3). | |
 |
| | Fig. 15 Fluorescent microscopic images of HeLa cells incubated with free DOX (254.7 ng mL−1) or after 5 min UV irradiation DOX-loaded Gluco-ONB-P(αN3CL-g-PONBIMC)10 micelles for 60 min: (A) free DOX, and (B) after UV irradiation DOX-loaded micelles. For each row, images for left to right show the cells with Hoechst 33342 nuclear staining, DOX fluorescence, and the merged image (scale bar 50 μm; brightness not proportional to fluorescence intensity). | |
Conclusions
A family of phototriggered amphiphilic block-grafted copolymers and conjugated IMC in the hydrophobic segment prodrug were synthesized. The copolymers and prodrug with a photocleavable junction points between the hydrophilic glycose and the hydrophobic P(αN3CL-g-alkyne)n blocks were characterized through 1H NMR, FTIR, and GPC. The obtained copolymers and prodrug formed micelles with a spherical morphology in an aqueous solution. UV irradiation burst release of the encapsulated or conjugated drug. Cell viability was evaluated in response to these particles at concentrations of ≤1000 μg mL−1, with toxicity to HeLa cells when the concentration was ≧100 μg mL−1. After UV irradiation Gluco-ONB-P(αN3CL-g-PONBIMC)10 encapsulated DOX micelles entered and accumulated in the cells faster than its free-form counterpart and without UV irradiation. Thus, the results suggested that light-sensitive Gluco-ONB-P(αN3CL-g-PONBIMC)10 prodrug has potential targeted drug delivery applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research was supported by grants from Chang Gung Memorial Hospital (CMRPD5F0012) and the authors thank the Microscopy Center at Chang Gung University for technical assistance.
References
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef PubMed.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- A. Rösler, G. W. M. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef PubMed.
- A. Aoleimani, A. Borecki and E. R. Gillies, Polym. Chem., 2014, 5, 7062–7071 RSC.
- V. Delplace, P. Couvreur and J. Nicolas, Polym. Chem., 2014, 5, 1529–1544 RSC.
- Y. Bao, E. Guégain, J. Mougin and J. Nicolas, Polym. Chem., 2018, 9, 687–698 RSC.
- L. Zhou, M. Chen, Y. Guan and Y. Zhang, Polym. Chem., 2014, 5, 7081–7089 RSC.
- Z. Wang, Q. He, W. Zhao, J. Luo and W. Gao, J. Controlled Release, 2017, 264, 66–75 CrossRef PubMed.
- S. Chen, Q. Bian, P. Wang, X. Zheng, L. Lv, Z. Dang and G. Wang, Polym. Chem., 2017, 8, 6150–6157 RSC.
- Sauraj, S. U. Kumar, V. Kumar, R. Priyadarshi, P. Gopinath and Y. S. Negi, Carbohydr. Polym., 2018, 188, 252–259 CrossRef PubMed.
- C. Englert, I. Nischang, C. Bader, P. Borchers, J. Alex, M. Pröhl, M. Hentschel, M. Hartlieb, A. Traeger, G. Pohnert, S. Schubert, M. Gottschaldt and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 2479–2482 CrossRef PubMed.
- R. Zhang, X. Jia, M. Pei and P. Liu, React. Funct. Polym., 2017, 116, 24–30 CrossRef.
- T. Zhou, X. Jia, X. Zhao, J. Li and P. Liu, J. Mater. Chem. B, 2017, 5, 2840–2848 RSC.
- Y. Yu, C. K. Chen, W. C. Law, H. Sun, P. N. Prasad and C. Cheng, Polym. Chem., 2015, 6, 953–961 RSC.
- O. Bertrand and J. F. Gohy, Polym. Chem., 2017, 8, 52–73 RSC.
- Q. Jin, F. Mitschang and S. Agarwal, Biomacromolecules, 2011, 12, 3684–3691 CrossRef PubMed.
- Y. Shamay, L. Adar, G. Ashkenasy and A. David, Biomaterials, 2011, 32, 1377–1386 CrossRef PubMed.
- X. Tan, B. B. Li, X. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. Zhang, J. J. Zhao and K. Zhang, J. Am. Chem. Soc., 2015, 137, 6112–6115 CrossRef PubMed.
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842–1851 CrossRef PubMed.
- D. A. Ossipov, A. B. Romero and E. Ossipova, Carbohydr. Polym., 2018, 180, 145–155 CrossRef PubMed.
- S. Lenior, R. Riva, X. Lou, Ch. Detrembleur, R. Jérôme and P. Lecomte, Macromolecules, 2004, 37, 4055–4061 CrossRef.
- R. S. Lee, Y. C. Li and S. W. Wang, Carbohydr. Polym., 2015, 117, 201–210 CrossRef PubMed.
- C. Giacomelli, V. Schmidt and R. Borsali, Macromolecules, 2007, 40, 2148–2157 CrossRef.
- C. Lu, Q. Shi, X. Chen, T. Lu, Z. Xie, X. Hu, J. Ma and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3204–3217 CrossRef.
- H. Kuang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 CrossRef PubMed.
- X. Hu, J. Tian, T. Liu, G. Zhang and S. Liu, Macromolecules, 2013, 46, 6243–6256 CrossRef.
- D. R. Griffin, J. L. Schlosser, S. F. Lam, T. H. Nguyen, H. D. Maynard and A. M. Kasko, Biomacromolecules, 2013, 14, 1199–1207 CrossRef PubMed.
- Y. Dong and S. S. Feng, Biomaterials, 2004, 25, 2843–2849 CrossRef PubMed.
- A. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef PubMed.
- Y. Zhang, J. He, D. Cao, M. Zhang and P. Ni, Polym. Chem., 2014, 5, 5124–5138 RSC.
- C. Yang, A. B. Ebrahim attia, J. P. K. Tan, X. Ke, S. Gao and J. L. Hedrick, Biomaterials, 2012, 33, 2971–2979 CrossRef PubMed.
- C. Lv, Z. Wang, P. Wang and X. Tang, Langmuir, 2012, 28, 9387–9394 CrossRef PubMed.
- P. M. Antonik, A. M. Eissa, A. R. Round and N. R. Cameron, Biomacromolecules, 2016, 17, 2719–2725 CrossRef PubMed.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 RSC.
- J. Huang, G. Habraken, F. Audouin and A. Heise, Macromolecules, 2010, 43, 6050–6057 CrossRef.
- Y. Wu, D. Zhou, Y. Qi, Z. Xie, X. Chen, X. Jing and Y. Huang, RSC Adv., 2015, 5, 31972–31983 RSC.
- X. Dai, Z. Yue, M. E. Eccleston, J. Swartling, N. K. Slater and C. F. Kaminski, Nanomedicine, 2008, 4, 49–56 CrossRef PubMed.
- Y. Jin, L. Song, Y. Su, L. Zhu, Y. Peng and F. Qiu, Biomacromolecules, 2011, 12, 3460–3468 CrossRef PubMed.
- M. Sui, W. Liu and Y. Shen, J. Controlled Release, 2011, 155, 227–236 CrossRef PubMed.
- J. Yan, Z. Ye, M. Chen, Z. Liu, Y. Xiao and Y. Zhang, Biomacromolecules, 2011, 12, 2562–2572 CrossRef PubMed.
- S. M. Sagnella, H. Duong, A. MacMillan, C. Boyer, R. Whan and J. A. McCarroll, Biomacromolecules, 2013, 15, 262–275 CrossRef PubMed.
- Y. Zhang, C. Y. Ang, M. Li, S. Y. Tan, Q. Qu, Z. Luo and Y. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 18179–18187 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a