
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of an Ag@AgBr nanoparticle-decorated K4Nb6O17 photocatalyst with improved photocatalytic properties
Lingyu Tian,
Kelei Sun,
Yulan Rui,
Wenquan Cui and
Weijia An*
and
Weijia An*
College of Chemical Engineering, North China University of Science and Technology, Tangshan 063210, P. R. China. E-mail: anweijia@ncst.edu.cn; Tel: +86-315-8805466
First published on 17th August 2018
Abstract
An Ag@AgBr nanoparticle-decorated K4Nb6O17 (Ag@AgBr/K4Nb6O17) photocatalyst was prepared via the oil-in-water self-assembly method. The Ag@AgBr nanoparticles, with average diameters of 20 nm, were uniformly deposited on the K4Nb6O17 surface. The as-prepared Ag@AgBr/K4Nb6O17 composites exhibited high visible light absorption, high photocurrent intensity, and high charge transfer efficiency, thus enhancing the photocatalytic performance for methyl-blue (MB) dye degradation. The Ag@AgBr (20 wt%)/K4Nb6O17 composite displayed the highest photocatalytic activity, degrading 96% of the MB solution under visible light irradiation for 60 min, which was 2.3-times and 8.5-times that of the bulk Ag@AgBr and K4Nb6O17, respectively. The excellent photocatalytic activity of the Ag@AgBr/K4Nb6O17 composites is due to the synergistic effect between Ag@AgBr and K4Nb6O17, where the Ag@AgBr nanoparticles not only enhanced visible light absorption efficiency due to the Ag nanoparticles' SPR, but also greatly accelerated the separation of the photogenerated electron–hole pairs. From the UV-vis spectra, the Ag@AgBr nanoparticles greatly extend the composites' visible light absorption. The data collected from photoluminescence (PL), photocurrent and electrochemical impedance spectra (EIS) were consistent and confirmed the rapid separation of charge carriers. Moreover, the composite exhibited a larger specific surface area, which was also beneficial for the photocatalytic activity. In addition, the roles of the radical species were investigated, and the holes and ·O2− radicals were hypothesized to dominate the photocatalytic process. Based on the characterization analysis and experimental results, a possible photocatalytic mechanism for the enhancement of photocatalytic activity is proposed.
1. Introduction
Photocatalytic technology is considered as an effective means of dealing with organic pollutants with the help of photogenerated electrons and holes.1–3 Among the various photocatalysts studied, those containing Ag nanoparticles have attracted considerable attention due to their incorporated surface plasmon resonance (SPR), which can greatly increase the utilization of visible light.4–7 Moreover, it is widely accepted that noble metal nanoparticles can act as electronic capture centers, which is favorable for separating the photogenerated carriers effectively and accelerating the redox reaction.8 Till date, many Ag-based photocatalysts, such as Ag3PO4,9 Ag2Mo2O7,10 Ag2CO3,11 Ag2O,12 and Ag/AgX,13,14 have been increasingly studied due to the high photocatalytic activity for the photodegradation of organic pollutants. Among them, Ag/AgX (X = Cl, Br, I), as a new type of plasmon resonance photocatalyst, combines the surface plasmon resonance effect of noble metals and the properties of metal semiconductor contacts and semiconductor photocatalytic materials. Consequently, Ag/AgX significantly enhances the absorption of visible light, which effectively solves the problem of low utilization rate of traditional solar photocatalysts and exhibits excellent catalytic performance in the degradation of organic pollutants.15 However, Ag@AgX suffers from the issues of easy particle agglomeration, poor stability and high photo-electron–hole recombination rates. To further improve its photocatalytic efficiency, two key issues need to be solved: (1) efficient separation of photogenerated charges in visible light and (2) improve the dispersibility of the catalyst, increase the active adsorption sites and promote the transfer of photo-generated charges.In fact, semiconductor-modified Ag@AgX composites with appropriate band positions could greatly accelerate the separation of charges due to the coupled energy levels that provide good internal potential driving forces to separate the photogenerated charges.16,17 For example, the plasmonic photocatalyst Z-scheme Ag2CO3/Ag/AgBr, which was fabricated by Hu et al., displayed much higher photocatalytic activity compared to that of pure Ag2CO3 and Ag/AgBr under visible light irradiation.18 Moreover, combining Ag@AgX composites with the material having a π–π conjugated structure, such as C3N4 (ref. 19 and 20) and graphene,21,22 could effectively inhibit the combination of photo-generated carriers due to the special conductivity of the conjugate material.
Recently, our group prepared various types of Ag@AgX-based heterostructured composites, all of which promoted rapid charge separation and demonstrated improved photocatalytic activity.23–27 In this study, the Ag@AgBr nanoparticle-decorated K4Nb6O17 was prepared via the oil-in-water self-assembly method at room temperature. The Ag@AgBr nanoparticles, with average diameters of 20 nm, were successfully deposited on the K4Nb6O17 surface. Benefiting from the unique structural features and the SPR property, the Ag@AgBr/K4Nb6O17 composites provided more active sites, extended the range of visible light absorption, and showed higher charge transfer efficiency and enhanced photocatalytic performance. The dependence of the photocatalytic activity on AgBr content in the K4Nb6O17 and the reactive species involved in the photodegradation process were studied.
2. Experimental
2.1 Materials
All the reagents used in our experiment were of analytical grade and used without further purification. All of the materials, including niobium pentoxide (Nb2O5), potassium carbonate (K2CO3), silver nitrate (AgNO3), potassium bromide (KBr), cetyl trimethyl ammonium bromide (CTAB), and carbon tetrachloride (CCl4), were purchased from Sinopharm Chemical Reagent Co. Ltd.2.2 Synthesis of K4Nb6O17
K4Nb6O17 powder was synthesized using a procedure reported by our group previously.25 Typically, a mixture of K2CO3 (3.81 g) and Nb2O5 (10 g) with the molar ratio of 2.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 was ground sufficiently in an agate mortar with a known amount of absolute ethyl alcohol. The mixture was then calcined at 1000 °C for 2 h, and the product was further ground to a fine powder for subsequent use.
:3 was ground sufficiently in an agate mortar with a known amount of absolute ethyl alcohol. The mixture was then calcined at 1000 °C for 2 h, and the product was further ground to a fine powder for subsequent use.
2.3 Fabrication of Ag@AgBr/K4Nb6O17 composites
The Ag@AgBr nanoparticle-decorated K4Nb6O17 samples were prepared via an oil-in-water self-assembly method. In a typical procedure, 0.5 g K4Nb6O17 powder and 0.12 g AgNO3 powder was dissolved in 30 mL deionized water under magnetic stirring at room temperature for 30 min and 0.38 g CTAB (nAgNO3:nCTAB = 1:1.5) was dissolved in 20 mL carbon tetrachloride solution. Subsequently, the CTAB solution was added dropwise to the solution containing K4Nb6O17 and AgNO3 under vigorous magnetic stirring. Following CTAB addition, the reaction mixture was magnetically stirred for another 30 min and then, the reaction mixture was irradiated with a 250 W metal halide lamp (Philips) for an hour. The resulting sample Ag@AgBr (20 wt%)/K4Nb6O17 was washed with distilled water and anhydrous ethanol to remove surfactants, and the final products were dried at 80 °C for 8 h. The Ag@AgBr/K4Nb6O17 composites with different ratios of Ag@AgBr to K4Nb6O17 were prepared according to the typical procedure described above. Pure Ag@AgBr was synthesized similarly using an oil-in-water self-assembly method with AgNO3 and CTAB.
For comparison, a Ag@AgBr/K4Nb6O17 sample was prepared via a precipitation method with K4Nb6O17 powder, AgNO3 and CTAB. The dosage and preparation were the same, except CTAB was dissolved in 20 mL deionized water. The as-prepared catalyst was denoted as P–Ag@AgBr/K4Nb6O17.
2.4 Characterization
The crystal phases of the catalysts were evaluated by powder X-ray diffraction (XRD, D/MAX2500PC, Cu Kα, 40 kV, 100 mA) scanning over the two-theta range of 5–80°. The morphologies of the catalysts were examined by scanning electron microscopy (SEM, Hitachi, s-4800) and transmission electron microscopy (TEM, JEM-2010, 200 kV). UV-vis diffuse reflectance spectra were obtained using a Puxi, UV1901 spectrometer with BaSO4 as a reference, and the chemical compositions of the catalysts were studied by energy dispersive X-ray spectroscopy (EDX, Thermo Noran 7). Surface areas of the samples were determined using a Brunauer–Emmett–Teller (BET) estimation based on the N2 sorption isotherms collected using a Quantachrome Nova 4200e automatic analyzer (USA). X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos Axis Ultra XPS systems using monochromated Al Kα radiation to examine the chemical state of the samples. The Ag+ concentration in the solution was determined by Inductively Coupled Plasma (ICP) (HORIBA, ULTIMA2). To study the recombination of photo-induced charge carriers, photoluminescence (PL, Hitachi F-7000, 250 nm) spectra were collected. The photocurrent was measured on an electrochemical system (CHI-660E, China). A standard three-electrode cell with a working electrode (as-prepared photocatalyst), a platinum wire as the counter electrode, and a standard calomel electrode (SCE) as the reference electrode were used in the photoelectric studies. The electrolyte solution was 0.1 M Na2SO4. The visible light irradiation was obtained from a 500 W Xe lamp with a 420 nm cut off filter.2.5 Photocatalytic activity
The photocatalytic activities of the Ag@AgBr/K4Nb6O17 catalysts were evaluated by the decomposition of MB and phenol solution under visible light irradiation. Visible light was provided by a 250 W halide lamp (Philips) equipped with a 420 nm cut off filter. In the MB and phenol degradation test, 0.1 g catalyst was added into a 100 mL (10 mg L−1) solution at room temperature. Prior to irradiation, the dispersion was stirred for 60 min in dark to achieve adsorption–desorption equilibrium. During the photocatalytic process, 3 mL aliquots of the reaction suspension were withdrawn every 15 minutes and centrifuged at 10000 rpm for 6 min to remove the suspended catalyst particles. The collected supernatant solutions were then analyzed using a spectrometer (TU-1901 UV-vis) at the characteristic absorption peak for MB at 664 nm. HPLC was adopted for analysis of phenol concentration and the distribution of its degradation products. The mobile phase was composed of methanol and water (volume ratio: 60/40), and the elution time was 6 min at a flow rate of 1 mL min−1. The detector was set at the wavelength of 270 nm, and C18 reversed phase column (Agilent 1100, 4.6 nm × 200 nm) was used for chromatographic analysis.
The degradation efficiency (%) was calculated as follows:
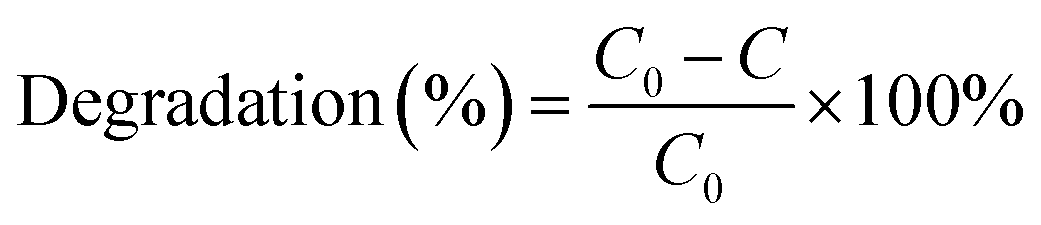 | (1) |
3. Results and discussion
Fig. 1a shows the XRD spectra of K4Nb6O17, Ag@AgBr, AgBr/K4Nb6O17 and Ag@AgBr/K4Nb6O17 composites. In the spectrum of pure K4Nb6O17, characteristic diffraction peaks were detected at 2θ angles of 9.30°, 23.45°, 25.28°, 27.59°, 28.97°, 31.41°, 46.48° and 51.42°, attributed to the (040), (200), (180), (0100), (032), (132), (0160) and (422) crystal planes, respectively, which can be ascribed to the monoclinic K4Nb6O17 (JCPDS 31-1064). The sharper diffraction peaks in the spectrum of K4Nb6O17 indicate its good crystallinity. In the spectrum of AgBr, characteristic diffraction peaks were detected at 2θ angles of 26.73°, 30.96°, 44.35°, 55.04°, 66.48°, and 73.12°, attributed to the (111), (200), (220), (222), (400) and (640) crystal planes, respectively, which can be ascribed to AgBr (JCPDS 06-0438). In the patterns of AgBr/K4Nb6O17 and Ag@AgBr/K4Nb6O17 composites, the characteristic diffraction peaks belonging to both K4Nb6O17 and AgBr were detected, indicating that the AgBr/K4Nb6O17 and Ag@AgBr/K4Nb6O17 composites possessed two-phase composition. However, the diffraction peaks attributed to Ag could not be easily observed in the reduced Ag@AgBr/K4Nb6O17 composite, which may be because the amount and the size of Ag nanoparticles were too low to be detected in the partially reduced Ag@AgBr photocatalysts. As shown in Fig. 1b, it was found that as the amount of AgBr increased, the characteristic peaks of the compound, such as those corresponding to the (222), (400) and (640) crystal planes belonging to AgBr, gradually increased, particularly the peak ascribed to the (640) crystal plane, indicating that AgBr nanoparticles successfully adsorbed on the surface of K4Nb6O17. However, it is difficult to draw conclusions concerning the existence of Ag according to the XRD patterns, which is similar to the cases of Ag/AgBr/TiO2 and Ag-AgBr-WO3 nanocomposites.28,29 In order to examine the elemental composition and valence state of Ag in the Ag@AgBr/K4Nb6O17 composite, XPS was performed. | ||
| Fig. 1 (a) XRD spectra of K4Nb6O17, AgBr, AgBr/K4Nb6O17 and Ag@AgBr/K4Nb6O17, (b) XRD spectra of K4Nb6O17, AgBr and Ag@AgBr/K4Nb6O17 with various contents of AgBr. | ||
Fig. 2 shows the XPS spectra of the composite of Ag@AgBr/K4Nb6O17. Strong peaks of Ag 3d, Br 3d, and O 1s are observed in the full XPS spectrum of Ag@AgBr/K4Nb6O17 (Fig. 2a) and the corresponding high-resolution spectra, which are noted in Fig. 2b–d. As shown in Fig. 2b, two typical peaks of Ag 3d located at about 373.63 eV and 367.65 eV can be attributed to the Ag 3d3/2 and Ag 3d5/2 binding energies, respectively. The Ag 3d3/2 peak and 3d5/2 peak can be further divided into two different peaks. In particular, the peaks at 373.25 eV and 367.28 eV are attributed to Ag(I) of AgBr, while those at 374.12 eV and 367.99 eV are attributed to metallic Ag (0),28 confirming that some Ag+ ions were successfully reduced to metallic Ag via irradiation. The XPS spectrum of Br 3d (as seen in Fig. 2c) shows a binding energy peak at 68.52 eV and comprises two individual Br 3d3/2 and Br 3d5/2 peaks with binding energies of 69.38 eV and 68.53 eV, respectively, indicating that Br was mainly present in the form of Br−.29 The O 1s spectrum for the surface of the sample is shown in Fig. 2d. A broad peak was observed and deconvoluted to three peaks at 529.5 eV, 529.97 eV and 532.05 eV, which were assigned to the adsorbed molecular H2O, hydroxyl oxygen and lattice oxygen, respectively.30 Based on the XPS analysis, the atomic contents of silver and bromide ions on the surface of Ag@AgBr/K4Nb6O17 were calculated to be 4.93 and 3.64 mol%, respectively. This indicates that the Ag+ ions had been reduced to metallic Ag. Therefore, the joint characterization from XRD and XPS confirms the coexistence of Ag (0) and AgBr on the surface of the K4Nb6O17 support, which is in agreement with previous research studies,28,29,31 where the same results were obtained by other characterization analysis.
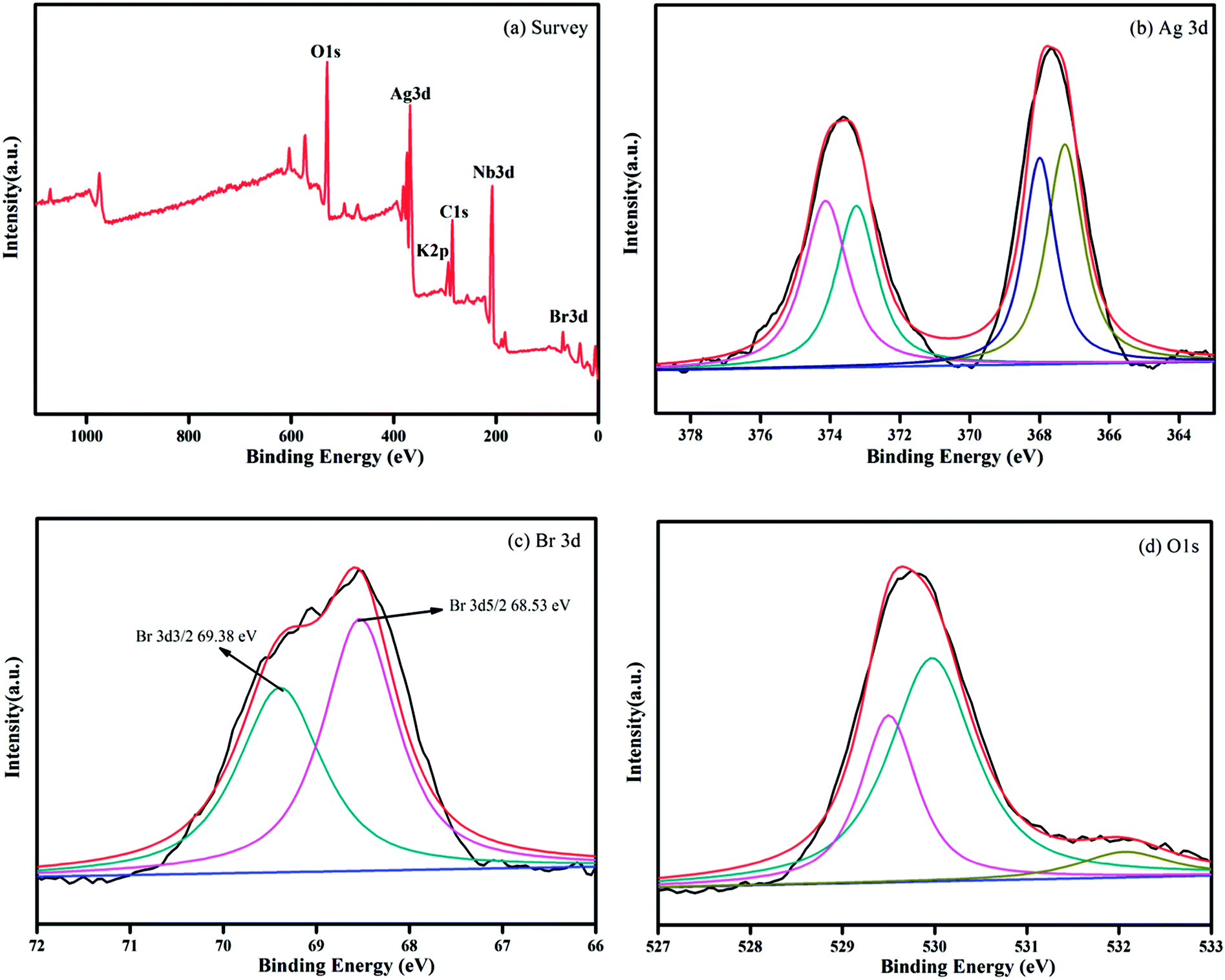 | ||
| Fig. 2 XPS spectra of the Ag@AgBr/K4Nb6O17 sample: (a) survey of the sample and (b) Ag 3d, (c) Br 3d, and (d) O 1s spectra of the sample. | ||
In order to further understand the changes in Ag content during photo-reduction, we performed ICP analysis. AgBr/K4Nb6O17 and Ag@AgBr/K4Nb6O17 (80 mg) were added to 25 mL of nitric acid and then, 3 mL aliquots were sampled from the reactor at given intervals to analyze the concentrations of Ag+ in the aqueous solution. The results showed that Ag content in the AgBr/K4Nb6O17 and Ag@AgBr/K4Nb6O17 composites was 147 g kg−1 and 168 g kg−1, respectively, indicating that a certain amount of AgBr was reduced to Ag, which was consistent with the analysis results of XPS.
In order to clarify the morphology and microstructure of the composites and to ensure that the Ag@AgBr nanoparticles were successfully attached to the surface of K4Nb6O17, the composites were characterized by SEM, TEM, SAED and EDX. As shown in Fig. 3a, K4Nb6O17 exhibited uniformly layered structures and Ag@AgBr (Fig. 3b) showed regular spherical particles. The average particle size was about 1 μm, but there was an observable degree of agglomeration. In the SEM image of Ag@AgBr deposited on K4Nb6O17, as shown in Fig. 3c, it could be clearly seen that the Ag@AgBr nanoparticles were uniformly distributed on the surface of the K4Nb6O17 with a size range of 10–20 nm; also, the particles did not affect the layered structures of the K4Nb6O17. Interestingly, the particle size of the Ag@AgBr particles deposited onto the K4Nb6O17 surface was much smaller than that of the pure Ag@AgBr particle, which was attributed to the existence of K4Nb6O17 that could effectively inhibit the agglomeration of Ag@AgBr nanoparticles. Moreover, the deposition of Ag@AgBr nanoparticles may greatly improve the light absorption and the specific surface area, which was beneficial to the photocatalysis activity, as seen from the UV-vis and BET analysis.
 | ||
| Fig. 3 SEM images of (a) K4Nb6O17; (b) Ag@AgBr; (c) Ag@AgBr/K4Nb6O17; (d–f) TEM, SAED and EDX images of Ag@AgBr/K4Nb6O17 composites. | ||
From the TEM images, it was found that K4Nb6O17 presents a uniform two-dimensional lamellar structure and Ag@AgBr nanoparticles were tightly adsorbed on the surface of K4Nb6O17 with average diameters of 20 nm (Fig. 3d). Moreover, as shown in Fig. 3e, the SAED patterns exhibited rectangular patterns, consistent with the XRD results and indexed to the monoclinic phase of K4Nb6O17. According to the SAED pattern, the Ag@AgBr nanoparticles do not affect the crystallinity of K4Nb6O17 due to the strong diffraction of K4Nb6O17. It should be noted that the brightest central spot in the SAED was blocked in order to obtain better contrast. The EDX spectrum shown in Fig. 3f contains peaks associated with O, Br, K, Ag and Nb, where the K, Nb, and O peaks originated from to K4Nb6O17 and Ag and Br peaks originated from to Ag@AgBr.
In addition, the associated EDX elemental maps were obtained to evaluate the chemical uniformity within individual particles, as shown in Fig. 4. Fig. 4a shows the SEM of the Ag@AgBr/K4Nb6O17. The results of elemental mappings shown in Fig. 4(b–d) evidenced that K, Nb and O are from K4Nb6O17 and the Br and Ag elements (Fig. 4(e and f)) were evenly distributed on the obtained Ag@AgBr/K4Nb6O17. Moreover, the associated elemental mapping images were obtained to evaluate the chemical uniformity within individual particles, which clearly confirmed that the Ag@AgBr had been successfully deposited on the surface of the K4Nb6O17 composite.
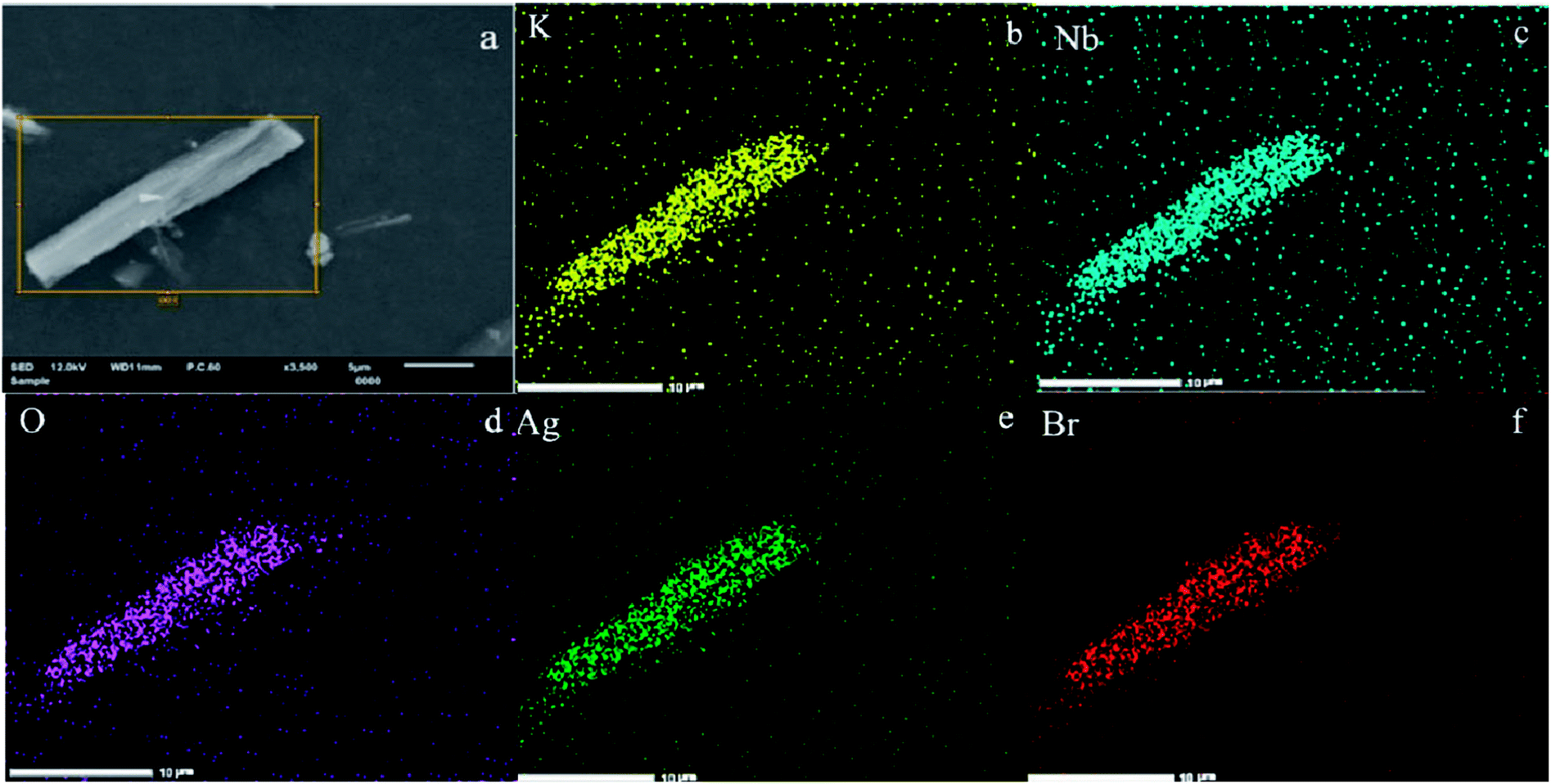 | ||
| Fig. 4 (a) SEM image of Ag@AgBr/K4Nb6O17. (b–f) Elemental maps of different particles in Ag@AgBr/K4Nb6O17. | ||
Fig. 5 shows the UV-vis diffuse reflectance spectra of the as-prepared photocatalysts. In the absorption spectrum of pristine K4Nb6O17, it can be seen that the absorption band edge is about 400 nm and the calculated band gap is 3.1 eV, indicating that there is no absorption of visible light. Due to the SPR effect of noble metal Ag particles,32 Ag@AgBr shows strong absorption in the visible light region. Therefore, the modifications of Ag@AgBr nanoparticles on the surface of K4Nb6O17 can also bring an improvement in the strong absorption and utilization of visible light. Moreover, the Ag@AgBr/K4Nb6O17 composite exhibits better visible light absorption than the AgBr/K4Nb6O17 composite as increased visible light absorption indicates that more photogenerated carriers can be excited and thus promote the photocatalytic performance of the composite.
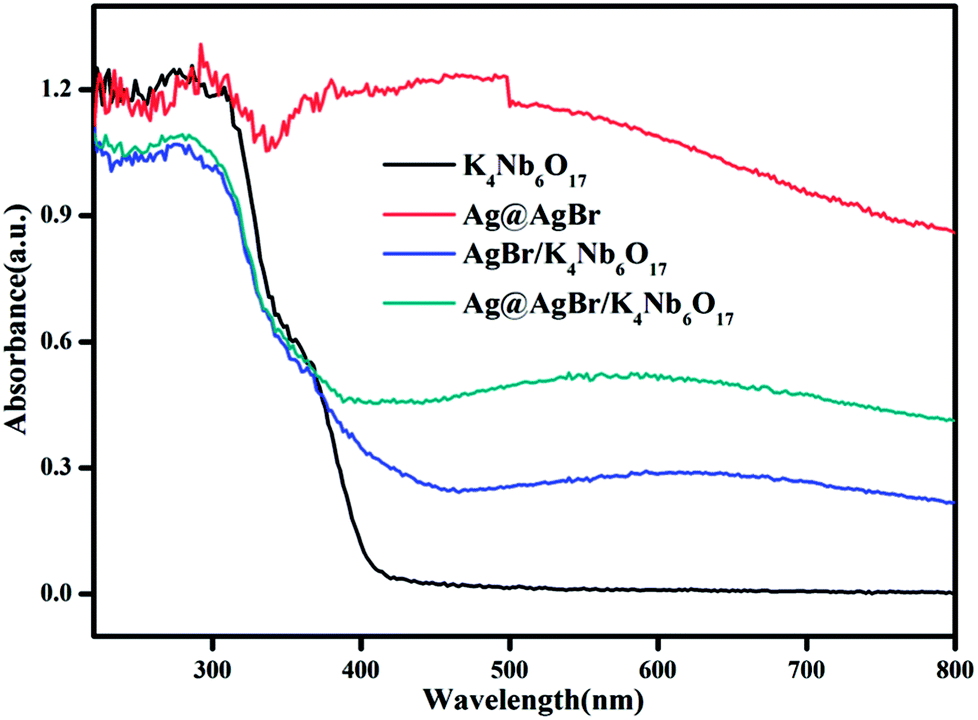 | ||
| Fig. 5 UV-vis diffuse reflectance spectra of the as-prepared photocatalysts. | ||
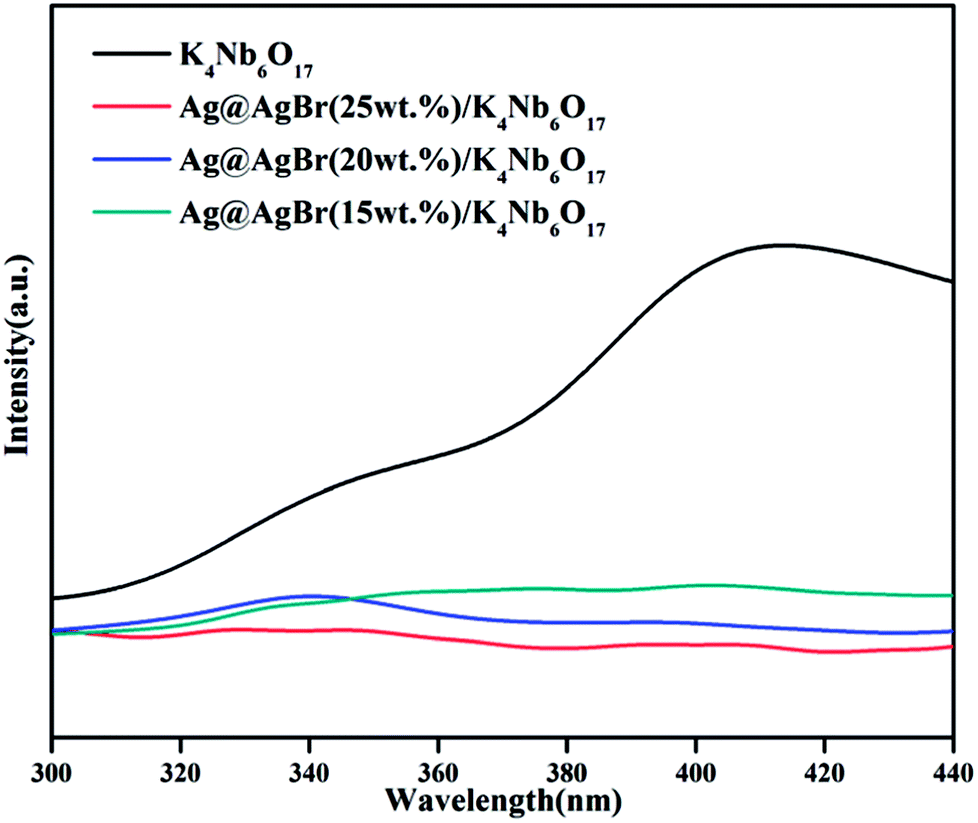
PL spectra can be recorded to assess the photogenerated electron–hole pair transfer and recombination processes in semiconductors. The lower fluorescence emission intensity corresponds to lower recombination rate of photogenerated electron–hole pairs and higher photocatalytic performance.33 Fig. 6 shows the PL spectra of pure K4Nb6O17 and various Ag@AgBr content Ag@AgBr/K4Nb6O17 composites. It can be seen that in the PL spectrum of the K4Nb6O17 sample, the main emission peak is centered at about 420 nm, mainly due to electron–hole recombination. After the Ag@AgBr nanoparticles were modified on the surface of K4Nb6O17, the fluorescence intensity dropped sharply, indicating that the introduction of Ag@AgBr effectively inhibits the recombination of photogenerated electron–hole pairs. The presence of Ag@AgBr effectively enhances the separation efficiency of photogenerated charges and the lifetime of photo-generated electrons and holes, thus increasing the activity of the composites.
 | ||
| Fig. 6 Fluorescence spectra of photocatalysts (λex = 250 nm). | ||
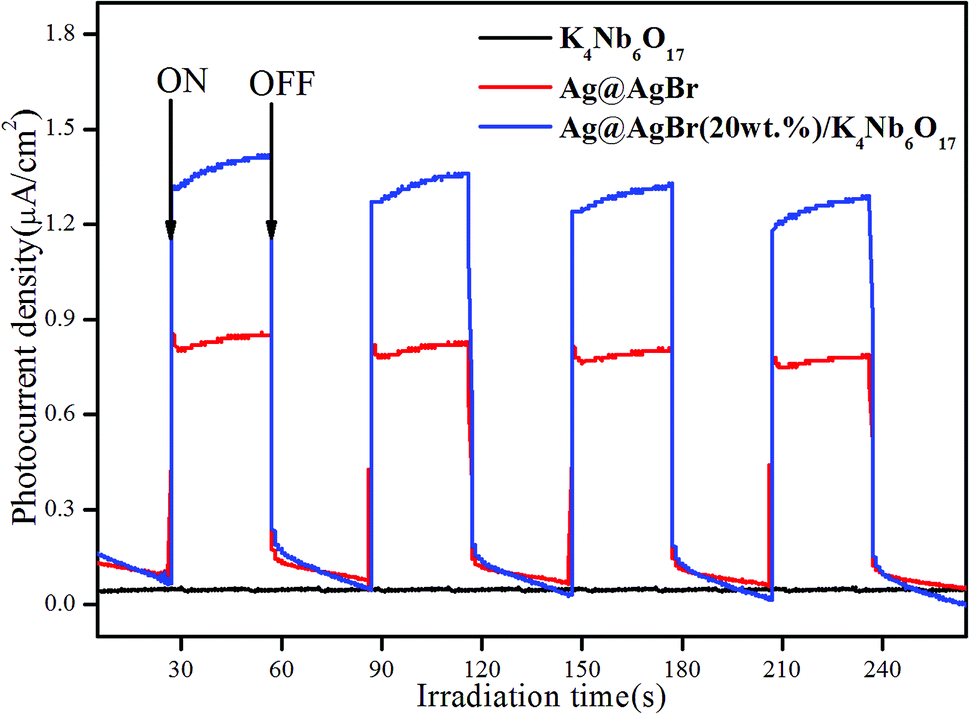
In order to verify the electron transfer rate, the transient photoelectric response of the photocatalyst was tested, and the results are shown in Fig. 7. The photocurrent response intensity of the catalyst was measured under the conditions of xenon lamp-irradiation with ON–OFF intervals of 30 s. A significant photoelectric response was observed from the ON/OFF photoperiod. It is generally believed that the higher the photocurrent response intensity, the faster the transfer rate of photogenerated charges.34 Clearly, pure K4Nb6O17 showed almost no photocurrent response due to the large band gap with no visible light response, while Ag@AgBr possessed a higher photocurrent response. In addition, the Ag@AgBr/K4Nb6O17 composite exhibited the highest photocurrent intensity, which was 1.7-times higher than that of pure Ag@AgBr. These results demonstrate that Ag@AgBr nanoparticles modified on the surface of K4Nb6O17 could greatly promote the rapid separation of charges, which was conducive to promoting the photocatalytic activity of the composite.
 | ||
| Fig. 7 Transient photocurrent response for K4Nb6O17, Ag@AgBr and Ag@AgBr/K4Nb6O17 composites. | ||
In order to further study the charge transfer efficiency, EIS was also recorded and the response of K4Nb6O17, Ag@AgBr and Ag@AgBr/K4Nb6O17 composites under visible light was analyzed. EIS is a useful tool to investigate charge transfer processes on the electrode and at the contact interface between electrode and electrolyte, which could be used to probe the separation efficiency of charge carriers. The smaller the arc radius of an EIS Nyquist plot, the higher would be the efficiency of interfacial charge transfer and more effective would be the separation of photogenerated electron–hole pairs.35 As shown in Fig. 8, the diameter of the semicircle became shorter after the Ag@AgBr and K4Nb6O17 materials were coupled, which indicated that the Ag@AgBr/K4Nb6O17 composites showed superior charge transport efficiency than K4Nb6O17 and Ag@AgBr. The combined results of EIS, photocurrent and PL verify that the Ag@AgBr nanoparticle-decorated K4Nb6O17 could be more conducive to the separation of photo-generated carriers and improvement of the photocatalytic activity.
 | ||
| Fig. 8 Electrochemical impedance spectroscopy for K4Nb6O17, Ag@AgBr and Ag@AgBr/K4Nb6O17 composites. | ||
The catalytic activity of the photocatalyst was analyzed by degradation of the MB solution under visible light. As shown in Fig. 9a, the characteristic peak intensity of MB gradually decreases with the progress of photodegradation, and the final degradation is 96% after 60 min in the presence of the Ag@AgBr (20 wt%)/K4Nb6O17 composite. In order to more clearly compare the activity of the catalyst, we set up a blank experiment and a control experiment. It can be seen from Fig. 9b that the photocatalyst has almost no adsorption or degradation of MB under dark reaction conditions, while there was almost no self-degradation of MB without catalyst or light. K4Nb6O17 displayed no visible light catalytic activity due to the insufficient visible light response. Moreover, MB degradation was only 43% with the same amount of Ag@AgBr under the same conditions. In comparison, the Ag@AgBr (20 wt%)/K4Nb6O17 photocatalyst exhibited the highest photocatalytic activity, which was about 2.2-times higher than that of Ag@AgBr, indicating that the combination of Ag@AgBr nanoparticles with K4Nb6O17 could effectively improve the activity due to the surface plasmon resonance as well as the rapid separation of photogenerated electron–hole pairs. To further investigate the synergistic effect of the present composites, the 20 wt% Ag@AgBr + 80 wt% K4Nb6O17 sample was prepared by mechanical mixing and used to degrade MB solution. The result indicated that the activity of this sample was much lower than that of Ag@AgBr (20 wt%)/K4Nb6O17 composites, thus providing further evidence to explain that the main factor for the increase in activity was the inhibition of photogenerated charge recombination, which was observed only in case of Ag@AgBr (20 wt%)/K4Nb6O17 photocatalyst. In addition, the P–Ag@AgBr/K4Nb6O17 composite photocatalyst could degrade 76% of MB, which may be due to the smaller specific surface area of the P–Ag@AgBr/K4Nb6O17 composites.
 | ||
| Fig. 9 (a) UV-vis spectral changes of MB solution in the presence of Ag@AgBr (20 wt%)/K4Nb6O17; (b) the activity of different catalysts to degrade MB in visible light. | ||
In order to investigate the effect of Ag@AgBr nanoparticles modification on the activity of the composite catalyst, the photocatalytic activity of Ag@AgBr/K4Nb6O17 with various concentrations of Ag@AgBr were tested. It is generally accepted that photocatalytic degradation of organic pollutants follows the first-order kinetics equation and the relevant equation is listed below36
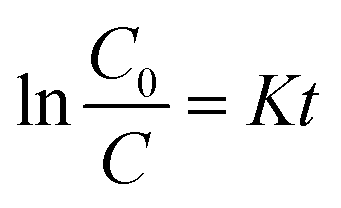
In this formula, K (Fig. 10a) represents the apparent first order rate constant, which was calculated by plotting ln(C0/C) versus time (t). In fact, the amount of Ag@AgBr nanoparticles modified on K4Nb6O17 has a great influence on the photocatalytic activity as the photocatalytic activity increased gradually with the increase in Ag@AgBr nanoparticle modification. When the modification amount was 20 wt%, the best photocatalytic activity was obtained and the apparent rate constant was about 0.047 min−1. In contrast, the photocatalytic activity decreased when the content of Ag@AgBr was higher than 20 wt%. These results indicate that the introduction of Ag@AgBr nanoparticle could promote visible light absorption and improve the efficiency of charge separation and also, there exists an optimum ratio of Ag@AgBr nanoparticles modification. Furthermore, the higher content of Ag@AgBr nanoparticles can create an unsuitable ratio between Ag@AgBr and K4Nb6O17 and decrease the interface contact, thus suppressing the interfacial charge transfer. The excess Ag@AgBr particles act as a new charge recombination center,37 thus inhibiting the charge transfer efficiency and reducing the photocatalytic performance of the composite.
 | ||
| Fig. 10 (a) Apparent rate constants for the photocatalytic degradation of MB over various Ag@AgBr/K4Nb6O17 catalysts. (b) Degradation of MB under visible light irradiation with various initial dye concentrations. | ||
The effect of initial MB concentration on the photocatalytic degradation rate was investigated over the concentration range of 5–25 ppm and the results are shown in Fig. 10c. The photodegradation efficiency of MB was found to decrease with the increase in initial concentration of dye and the degradation efficiency observed was nearly 100% after 45 min irradiation when the initial concentration was 5 mg L−1. The observed difference in activity upon changing the initial solution concentration may have been due to the visible light screening effect of the dye. The transmittance in the solution decreased with the increase in the initial concentration of MB, resulting in fewer photons reaching the surface of the catalyst; hence, the photocatalytic degradation rate decreased. Moreover, the active surface on the catalyst is crucial for the reaction and degradation to take place. As the initial concentration of dye was increased while all other parameters were maintained constant, fewer active sites became available for the degradation reaction. As a consequence, the rate of reaction decreased with the increase in the initial dye concentration.
The stability of a photocatalyst is important for its assessment and application. The photocatalytic activity of the Ag@AgBr (20 wt%)/K4Nb6O17 composites in the degradation of MB were studied in consecutive cycles under the same conditions. As shown in Fig. 11a, the photocatalytic activity decreases slightly over five consecutive cycles and the degradation in the final run was 86%. This decrease was thought to be due to some catalyst washout during the recovery steps, which could be minimized through the use of centrifugation between runs. To assess the structural stability, the crystalline structure and morphology of Ag@AgBr/K4Nb6O17 composites before and after the experiment were examined. As shown in Fig. 11b, in XRD spectra of Ag@AgBr/K4Nb6O17, there was almost no change in the intensities of the characteristic diffraction peaks after the photocatalytic reaction, which implied the stability of crystal structure of the composite photocatalyst. Moreover, the morphology of the composite before and after the reaction was examined, and the results showed that there were no significant changes in the morphology of the Ag@AgBr/K4Nb6O17 composite; only a small amount of the Ag@AgBr nanoparticles were slightly agglomerated (Fig. 11c and d). All these results indicate that this composite could be used as a stable and high-performance photocatalyst for environment protection.
 | ||
| Fig. 11 (a) Cycling runs for photocatalytic degradation of MB in the presence of Ag@AgBr/K4Nb6O17. (b) XRD patterns of Ag@AgBr/K4Nb6O17 before and after the photocatalytic reaction. (c and d) SEM images of photocatalysts before and after photocatalytic degradation of MB solution. | ||
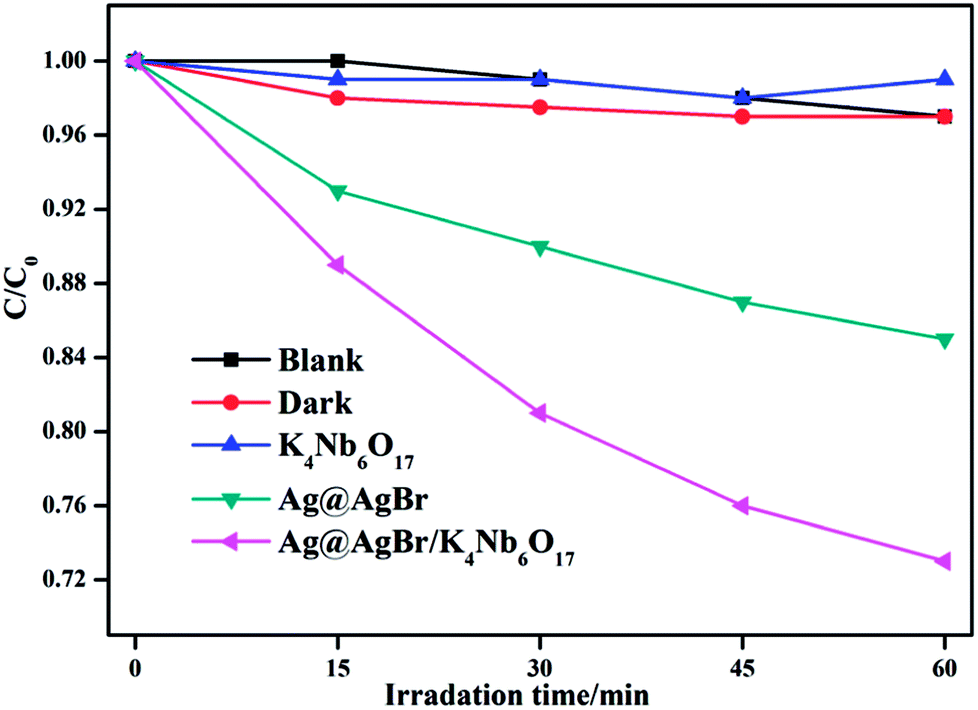
In order to further investigate the activity of the as-prepared photocatalyst, we prepared a phenol, a typical colorless organic pollutant, as another contaminant to study the activity under the same conditions. As displayed in Fig. 12, the photolysis of the phenol solution in the blank condition is negligible and the adsorption capacity of the composite material to the phenol in the dark environment is also very limited. In contrast, the Ag@AgBr/K4Nb6O17 photocatalysts exhibit higher catalytic degradation activities for phenol solution than pure Ag@AgBr, which was ascribed to the introduction of Ag@AgBr nanomaterial, higher visible light absorption and utilization, more photo-generated charge excitation and faster photo-generated carrier separation.
 | ||
| Fig. 12 The activity of K4Nb6O17, Ag@AgBr and Ag@AgBr/K4Nb6O17 photoacatalyst to degrade phenol in visible light. | ||
It is well known that the photocatalytic process involves the excitation of semiconductors by light to create electron and hole pairs, which can be used in redox reactions to degrade organic pollutants or produce hydrogen. In fact, the electron and hole pairs are not directly involved in the degradation process.38 Instead these pairs first generate free radicals (such as ·OH and ·O2−) with adsorbed H2O or O2 and then, the free radicals participate in the photodegradation process.39 For different photocatalytic degradation processes, the specific active species are different due to different energy levels, structure, and phase composition. In the photocatalytic degradation process, the addition of the appropriate free radical quencher will result in considerable reduction in the photocatalytic activity. We can imply the photocatalytic reaction mechanism by determining the free radical species. In this system, isopropanol (IPA) and EDTA-2Na were used as capture agents for ·OH and h+, respectively,13 and the results are shown in Fig. 13. The result was clearly obtained on comparing the photocatalytic activities of Ag@AgBr/K4Nb6O17 composite with and without addition of any scavenger (Fig. 13a–c). After the addition of EDTA-2Na and IPA, the photocatalytic activity decreased significantly, indicating that the photolysis process over Ag@AgBr/K4Nb6O17 composites was controlled by the photogenerated holes and radicals. Fig. 13d clearly shows the changes in K with various scavengers, suggesting that addition of IPA and EDTA-2Na have a negative effect on the photocatalytic activity.
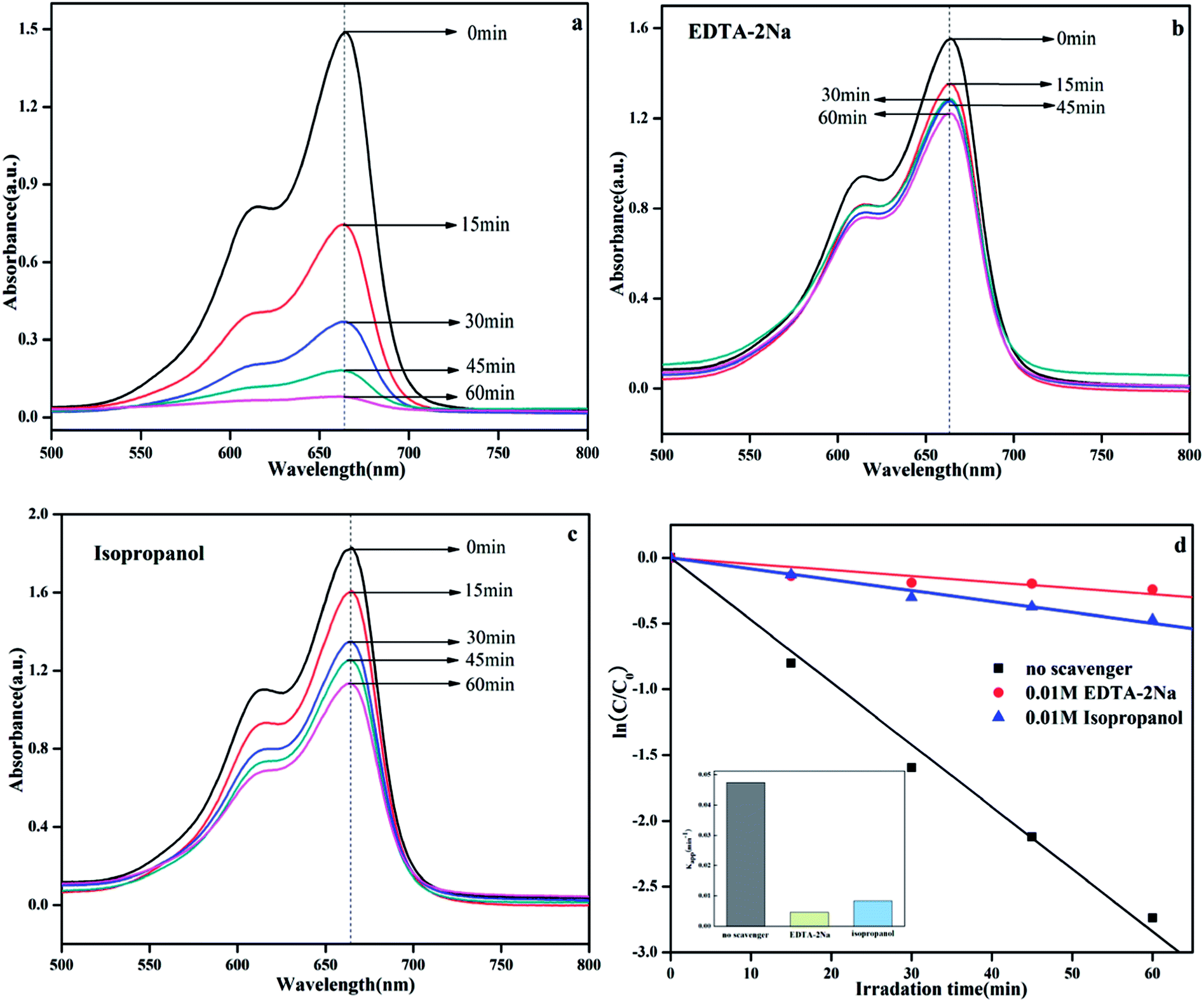 | ||
| Fig. 13 UV-vis spectral changes of MB solution over Ag@AgBr (20 wt%)/K4Nb6O17 with various scavengers: (a) no scavenger, (b) EDTA-2Na, and (c) IPA. (d) Kinetic curves for the photocatalytic degradation of MB; inset: apparent rate constants for the photocatalytic degradation of MB. | ||
The specific surface area is also an important factor affecting photocatalytic activity. According to the results of the nitrogen sorption tests, the specific surface area of pure K4Nb6O17, Ag@AgBr (20 wt%)/K4Nb6O17 and P–Ag@AgBr (20 wt%)/K4Nb6O17 was calculated to be 0.62, 3.65, 2.82 m2 g−1, respectively. Clearly, the specific surface area of the Ag@AgBr/K4Nb6O17 composite modified with Ag@AgBr nanoparticles significantly increased. The larger specific surface areas could provide more active sites for reactant adsorption, which is beneficial for the photocatalytic activity. Moreover, the Ag@AgBr/K4Nb6O17 composite prepared by the oil–water self-assembly method exhibits larger surface area than that prepared by the precipitation method. This is because the simultaneous presence of oil and water could effectively inhibit the agglomeration of Ag@AgBr nanoparticles, thus providing larger specific surface area and higher photocatalytic activity.
The key factor affecting the photocatalytic activity is the rapid separation of photogenerated electron–hole pairs. According to the literature on the mechanisms of Ag@AgX,4,40 it is widely accepted that the Ag nanoparticles could not only produce SPR effect, but also realize the rapid separation of photogenerated charges. Based on the experimental results and literature reports, the mechanism of photodegradation of MB was analyzed over Ag@AgBr/K4Nb6O17 composites system, as shown in Fig. 14.
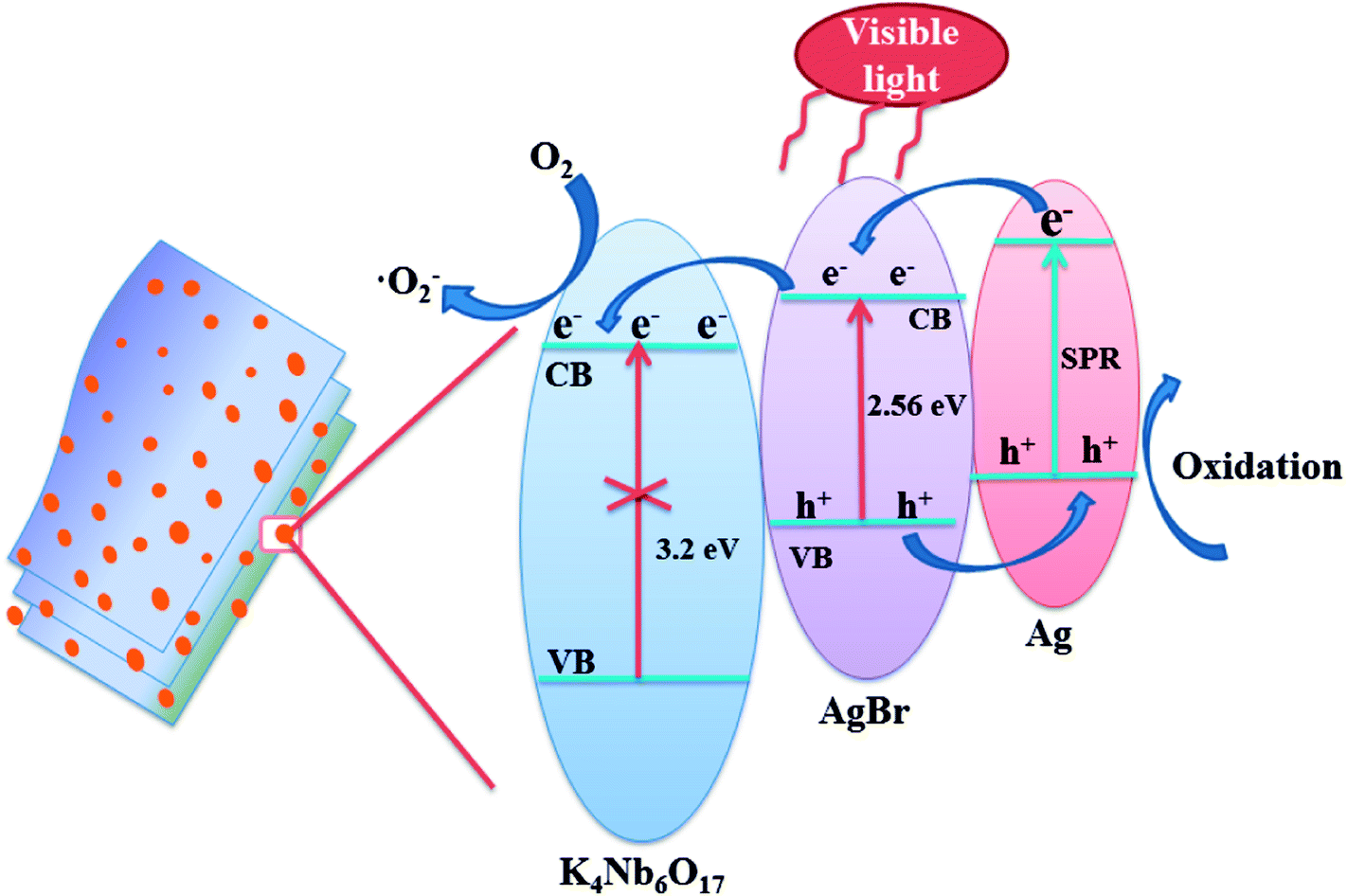 | ||
| Fig. 14 Schematic of photo-generated carriers' transportation for the Ag@AgBr/K4Nb6O17 composite. | ||
According to the literature,25,26 Ag@AgBr and K4Nb6O17 in Ag@AgBr/K4Nb6O17 composites have matched energy level structure, which is conducive in separating charge pairs. A schematic of photo-generated carriers' transportation for Ag@AgBr/K4Nb6O17 composite is shown in Fig. 14. Under visible irradiation, both AgBr and Ag nanoparticles could stimulate the generation of photogenerated electron–hole pairs. Interestingly, the electrons on the Ag could quickly migrate to the conduction band of AgBr and the holes of AgBr would move to the Ag nanoparticles at the interface of the composites. Moreover, since the conduction band of K4Nb6O17 is more negative than the conduction band of AgBr, the electrons on the CB of AgBr could rapidly transfer to the surface of K4Nb6O17, where the pure K4Nb6O17 could not be excited by visible light irradiation; such a transfer method could promote the separation of the photogenerated charges, which is in accordance with the results of the PL, photocurrent and EIS measurements. Moreover, the electrons could be captured by adsorbed O2 to generate reactive ·O2− radicals, which then break down the organic contaminants.41 In contrast, the holes could participate in the photocatalytic oxidation of organic pollutants directly, which is consistent with the results of the radical scavenger experiments.
4. Conclusions
In summary, the Ag@AgBr nanoparticle-decorated K4Nb6O17 composites were successfully synthesized via the oil-in-water self-assembly method. The Ag@AgBr (20 wt%)/K4Nb6O17 composite displayed the highest photocatalytic activity, degrading 96% MB solution under visible light irradiation for 60 min, which was 2.3-times higher than that of bulk Ag@AgBr. In addition, the Ag@AgBr/K4Nb6O17 composite could degrade phenol to some extent. The improved photocatalytic performance was attributed to the introduction of Ag@AgBr nanoparticles, where the Ag@AgBr nanoparticles not only enhanced visible light absorption efficiency due to the Ag nanoparticles' SPR, but also greatly accelerated the separation of the photogenerated electron–hole pairs. In addition, the roles of the radical species were investigated in which the holes and ·O2− radicals were thought to dominate the photocatalytic process. In conclusion, Ag@AgBr/K4Nb6O17 composites used as highly efficient and stable photocatalysts can effectively degrade organic pollutants, which is beneficial for the environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 51672081), Hebei Natural Science Funds for the Joint Research of Iron and Steel (B2016209348), Natural Science Foundation of Hebei Province, China (No. B2018209356).References
- K. Mondal and A. Sharma, RSC Adv., 2016, 6, 83589–83612 RSC.
- C. Gao, J. Wang and H. X. Xu, et al., Chem. Soc. Rev., 2017, 46, 2799–2823 RSC.
- M. J. Kale and T. Avanesian, ACS Catal., 2014, 4, 116–128 CrossRef.
- L. Ma, X. Yang and Z. Q. Zhou, et al., RSC Adv., 2016, 6, 97808–97817 RSC.
- L. Xu, F. Y. Zhang and X. Y. Song, et al., J. Mater. Chem. A, 2015, 3, 5923–5933 RSC.
- R. M. Cobley, C. M. Zeng and J. Li, et al., Chem. Rev., 2011, 111, 3669–3712 CrossRef PubMed.
- S. U. Lee, H. Jung and D. H. Wi, et al., J. Mater. Chem. A, 2018, 6, 4068–4078 RSC.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef PubMed.
- Y. P. Bi, S. X. Ouyang and N. Umezawa, et al., J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef PubMed.
- J. Wang, P. Guo and M. F. Dou, et al., RSC Adv., 2014, 4, 51008–51015 RSC.
- H. Xu, J. X. Zhu and Y. X. Song, et al., RSC Adv., 2014, 4, 9139–9147 RSC.
- G. Wang, X. C. Ma and B. B. Huang, et al., J. Mater. Chem., 2012, 22, 21189–21194 RSC.
- C. Hu, T. W. Peng and X. X. Hu, et al., J. Am. Chem. Soc., 2010, 132, 857–862 CrossRef PubMed.
- M. S. Zhu, P. L. Chen and M. H. Liu, ACS Nano, 2011, 5, 4529–4536 CrossRef PubMed.
- S. Wang, C. Liu and L. Z. Zheng, et al., New J. Chem., 2016, 40, 1323–1329 RSC.
- L. L. Sun, W. Wu and Q. Y. Tian, et al., ACS Sustainable Chem. Eng., 2016, 4, 1521–1530 CrossRef.
- H. A. Ghaly, A. S. El-Kalliny and T. A. Gad-Allah, et al., RSC Adv., 2017, 7, 12726–12736 RSC.
- J. J. Li, Y. L. Xie and Y. J. Zhong, et al., J. Mater. Chem. A, 2015, 3, 5474–5481 RSC.
- J. Q. Li, H. J. Hao and J. Zhou, et al., Appl. Surf. Sci., 2017, 422, 626–637 CrossRef.
- S. W. Zhang, J. X. Li and X. K. Wang, et al., ACS Appl. Mater. Interfaces, 2014, 6, 22116–22125 CrossRef PubMed.
- Y. Y. Bai, F. R. Wang and J. K. Liu, Ind. Eng. Chem. Res., 2016, 55, 9873–9879 CrossRef.
- G. W. Geng, B. Guan and P. L. Chen, et al., RSC Adv., 2017, 7, 9948–9957 RSC.
- F. Y. Chen, W. J. An and L. Liu, et al., Appl. Catal., B, 2017, 217, 65–80 CrossRef.
- L. Liu, S. L. Lin and J. S. Hu, et al., RSC Adv., 2015, 5, 62306–62313 RSC.
- W. Q. Cui, H. Wang and Y. H. Liang, et al., Chem. Eng. J., 2013, 230, 10–18 CrossRef.
- S. L. Lin, L. Liu and J. S. Hu, et al., Appl. Surf. Sci., 2015, 324, 20–29 CrossRef.
- Y. H. Liang, S. L. Lin and L. Liu, et al., Appl. Catal., B, 2015, 164, 192–203 CrossRef.
- Y. H. Zhang, Z. R. Tang and X. Z. Fu, et al., Appl. Catal., B, 2011, 106, 445–452 CrossRef.
- P. Wang, B. B. Huang and X. Y. Qin, et al., Inorg. Chem., 2009, 48, 10697–10702 CrossRef PubMed.
- C. L. Yu, K. Yang and Y. Xie, et al., Nanoscale, 2013, 5, 2142–2151 RSC.
- M. R. Elahifard, S. Rahimnejad and S. Haghighi, et al., J. Am. Chem. Soc., 2007, 129, 9552–9553 CrossRef PubMed.
- Z. C. Wang, J. H. Liu and W. Chen, Dalton Trans., 2012, 41, 4866–4870 RSC.
- W. J. An, W. Q. Cui and Y. H. Liang, et al., Appl. Surf. Sci., 2015, 351, 1131–1139 CrossRef.
- Y. J. Wang, R. Shi and J. Lin, et al., Energy Environ. Sci., 2011, 4, 2922–2929 RSC.
- Y. F. Liu, W. Q. Yao and D. Liu, Appl. Catal., B, 2015, 163, 547–553 CrossRef.
- J. Di, J. X. Xia and S. Yin, et al., J. Mater. Chem. A, 2014, 2, 5340–5351 RSC.
- L. Jin, G. Q. Zhu and M. Hojamberdiev, et al., Ind. Eng. Chem. Res., 2014, 53, 13718–13727 CrossRef.
- L. Liu, Y. H. Qi and J. R. Lu, et al., Appl. Catal., B, 2016, 183, 133–141 CrossRef.
- Y. L. Li, Y. M. Liu and J. S. Wang, et al., J. Mater. Chem. A, 2013, 1, 7949–7956 RSC.
- L. Q. Ye, J. Y. Liu and C. Q. Gong, et al., ACS Catal., 2012, 2, 1677–1683 CrossRef.
- D. Chen, H. Zhang and Y. Liu, et al., Energy Environ. Sci., 2013, 6, 1362–1387 RSC.
| This journal is © The Royal Society of Chemistry 2018 |