DOI:
10.1039/C8RA05757E
(Paper)
RSC Adv., 2018,
8, 29338-29343
Rational design of sequestered DNAzyme beacons to enable flexible control of catalytic activities†
Received
6th July 2018
, Accepted 8th August 2018
First published on 17th August 2018
Abstract
DNAzymes as functional units play increasingly important roles for DNA nanotechnology, and fine control of the catalytic activities of DNAzymes is a crucial element in the design and construction of functional and dynamic devices. So far, attempts to control cleavage kinetics can be mainly achieved through varying the concentrations of the specific metal ions. Here we present a facile sequestered DNAzyme beacon strategy based on precisely blocking the catalytic core of the DNAzyme, which can flexibly regulate the DNAzyme cleavage kinetics without changing the concentrations of metal ions. This strategy can be extended to couple with a large number of other RNA-cleaving DNAzymes and was successfully applied in designing a dual stem-loop structure probe for arbitrary sequence biosensing, which provides the possibility of scaling up versatile and dynamic DNA devices that use DNAzymes as functional modules.
Introduction
RNA-cleaving DNAzymes, screened through in vitro selection, are a class of single-stranded nucleic acids with protease-like catalytic activities.1–5 Compared with proteases, DNAzymes present low cost and high stability against chemical and nuclease degradations. DNAzymes as functional units play increasingly important roles for DNA nanotechnology, such as biosensors,6–11 DNA nanostructures,12,13 logic gates,14–18 DNA nanomachines,19–22 DNA-based hydrogels,23,24 drug deliverers,25–28 and theranostics.29,30 Since DNAzymes enable stimuli-responsive catalytic functions, they can therefore provide additional versatility over base-pair hybridization based nanodevices and, because DNAzymes often catalyze multiple turnover reactions, such devices can have amplification effects. Thus, fine control of the catalytic activities of DNAzymes is a crucial element in the design and construction of functional and autonomous devices. Most DNAzymes consist of two binding arms and one catalytic core, activated via binding with unique metal ions such as Pb2+,31,32 UO22+,33 Zn2+,34,35 Na+,36 and Cr3+.37 Attempts to control cleavage kinetics can be mainly achieved through varying the concentrations of the specific metal ions. However, it remains a major challenge to develop facile strategies for fine control of the catalytic activities of DNAzymes without changing the concentrations of metal ions, which limits the possibility of scaling up versatile and dynamic DNA devices that use DNAzymes as functional modules.
Herein, we propose a novel sequestered DNAzyme beacon strategy based on precisely blocking the catalytic cores of DNAzymes, which can flexibly control of the catalytic activities without changing the concentrations of metal ions. We explore the effects of the binding strength and the concentration of triggering stimulator on the cleavage kinetics. Furthermore, by applying the sequestered DNAzyme beacon strategy, we design a dual stem-loop structure probe to construct general biosensing systems for arbitrary target sequence. Taken together, these results demonstrate the possibility of flexibly designing more dynamic and controllable DNAzyme-based nanodevices.
Materials and methods
Reagents and chemicals
All HPLC-purified DNA oligonucleotides were purchased from Sangon Inc. (Shanghai, China). The DNA sequences and modifications are listed in Table S1.† The secondary structures of all oligonucleotides, including the thermodynamic parameters, were calculated using NUPACK software (http://www.nupack.org/). DNA stock solutions were prepared by dissolving oligonucleotides in TE buffer (50 mM Tris(hydroxymethyl)methyl aminomethane (Tris), 1 mM ethylene diamine tetraacetic acid (EDTA); pH 8.0) and stored in the dark at 4 °C before use. Zinc chloride (ZnCl2) and all other chemicals employed were of analytical grade and used without further purification. All solutions were prepared with ultrapure water, which was obtained through a Millipore Milli-Q water purification system (Molsheim, France) and had an electrical resistance of ≥18.2 MΩ.
Time-dependent fluorescence measurements and kinetic analysis
Sequestered DNAzyme beacons (SDB) were annealed in 50 mM HEPES (pH 7.0) and 100 mM NaCl buffer by heating at 95 °C for 5 min and then slowly cooling to room temperature over 2 hours. Fluorescence measurements were performed by using a QM-40 fluorescence spectrometry (PTI, Canada) at 25 °C. Sample solutions were excited at 522 nm (slit 2 nm), and the emission signal was recorded with wavelength of 539 nm (slit 2 nm). For all time-dependent kinetic measurements, 20 μL of 10 mM Zn2+ (for 10-23 DNAZyme based system, 1 mM Pb2+ was used), 2 μL of 50 μM DNAzyme substrate (S), and 2 μL of 10 μM SDB were added one after another to buffer solution with a total volume of 200 μL. After thoroughly mixing the components, the fluorescence change were monitored in 60s by fluorescence spectrometry. In all graphs, time t = 0 indicates the time of SDB being added to the solutions. The enzyme-catalyzed reactions were assumed to follow the rate-law of pseudo-first order kinetics. The observed rate constant kobs was obtained by curve fitting using OriginPro 8 based on the equation y = a + be−kx, where k is the observed rate constant.
Nucleic acids analysis
To detect target DNA, 2 μL of 50 μM S, 2 μL of 10 μM Probe sequence and 5 μL of specific concentration of target DNA were added one after another to buffer solution with a total volume of 180 μL. After thoroughly mixing, the components were incubated at room temperature for 20 min. A 20 μL of 10 mM Zn2+ was added to induce the DNAzyme catalysed cleavage reaction. After vortexing to mix the components thoroughly, the fluorescence change was monitored in 60s by fluorescence spectrometry. In all graphs, time t = 0 indicates the time of Zn2+ being added to the solutions.
Results and discussion
The mechanism of the sequestered DNAzyme beacons
The mechanism of the controlled catalytic activities by designing sequestered DNAzyme beacons (SDBs) is illustrated in Fig. 1. The 17E Zn2+-dependent DNAzyme is applied as the model biocatalyst in our systems.35 Similar to most of other in vitro selected DNAzymes, 17E is composed of a 14 nt catalytic core and two binding arms. An extended blocking sequence with an overhung stick end (red domain) is introduced to the 3′ end of 17E (blue domain), which can hybridized partially with the catalytic core of 17E. Thus, the catalytic core is controllably sequestered in the stem of the hairpin structure, resulting in the reducing of the cleavage reaction rate of 17E. By modulating the binding strength of the stem, the cleavage rates can be tuned continuously. It is noteworthy that the sequestered DNAzyme beacon strategy presented here (1) is facile and do not need to change the component of the DNAzyme, (2) can be extended to couple with a large number of other RNA-cleaving DNAzymes, and (3) provides the possibility of scaling up DNA devices that use DNAzymes as functional modules, which could be applied to many field while metal ions cannot be readily changed, such as in vivo applications.
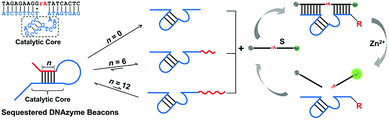 |
| | Fig. 1 Mechanism proposed for the sequestered DNAzyme beacons (SDBs) with controllable catalytic activities. Inset shows the secondary structure of Zn(II)-dependent deoxyribozyme–substrate complex 17E/DS. SDB is constructed by 17E (blue domain) with an extended blocking sequence (red domain) attached at the 3′ end. The catalytic core of 17E is controllably sequestered in the stem of the hairpin structure. The hairpin structure of SDB can be reversibly unfolded. The 17E in SDB can be then liberated and hybridize with a fluorophore and quencher labelled substrate to form a catalytic system. The nucleotide sequences of all strands are provided in Table S1 (ESI†). | |
Control of cleavage rate by modulating the binding strength and hybridization state of the catalytic core
The relationship between the cleavage rate and the binding strength of the catalytic core was investigated. The 18 nt substrate strand (S) was synthesized with a FAM fluorophore and a BHQ1 quencher at either end, serving as the signal reporter. The progress of the cleavage reaction can therefore be observed by monitoring the donor fluorescence intensity. In this work, the number of the hybridized bases in catalytic core is annotated by n. As shown in Fig. 2A, the reaction rate (kobs) is the highest when 17E is free (n = 0, kobs = 0.18 min−1), and that is reduced by over one order of magnitude when 12 bases of the catalytic core are hybridized (n = 12, kobs = 0.013 min−1). The results reveals that by modulating the binding length n, the cleavage rates can be changed by over one order of magnitude.
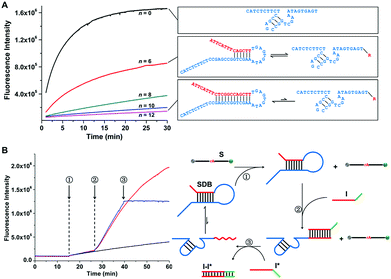 |
| | Fig. 2 Control of cleavage rates through control of binding strength of the catalytic core. (A) Catalytic cleavage reactions of SDBs with various binding length n. Initial concentrations: 1 mM Zn2+, 100 nM SDB, and 500 nM S. (B) Dynamic control of cleavage reaction. All traces start from the addition of inactive SDBs (n = 12) into S solutions. For the middle trace, a complementary initiator (I) was added. For the top trace, after the addition of I, an anti-I sequence I* was added to displace I off from the SDB. Initial concentrations: 1 mM Zn2+, 100 nM SDB, 1000 nM S, 400 nM I, and 400 nM I*. | |
Changing the hybridization state of the catalytic core allows dynamic and allosteric control of the cleavage rate (Fig. 2B). First, an inactive SDB (n = 12) is added in S solution (step 1), and the cleavage rate is very slow since the catalytic core is almost completely sequestered by the blocking sequence. By adding complementary initiator (I), the blocking sequence is displaced from the catalytic core (step 2), which resulted in dramatically increasing of the cleavage rate. Also, when the complementary strand I* is added, the cleavage reaction can be effectively stopped by removing I from the blocking sequence through forming I–I* duplex. Therefore, the above results confirm that the cleavage kinetics of proposed SDB can be tuned by modulating the blocking strength and hybridization state of the catalytic core of 17E.
Fine adjustment of the cleavage rate
Fine adjustment of the cleavage rate can be made by changing the blocking site. Serials of SDBs with catalytic cores sequestered at 3′, 5′, and center were designed respectively. As shown in Fig. 3A, lengthening n from 0 to 14 nt slows the cleavage rates from 0.19 to 0.0093 min−1. Notably, when the same number of bases of the catalytic core are hybridized, different cleavage rates are observed for SDBs with catalytic cores sequestered at 3′, 5′, and center. Take 5 bases blocking as example, the cleavage rate of SDB sequestered at 3′ end of the catalytic core is the fastest (kobs = 0.18 min−1), and that is slowest at center (kobs = 0.059 min−1). Earlier studies pointed out that the unpaired T and three G/C pairs in the catalytic core play crucial roles in 17E.38 First, the T acts as spacer between the neighboring C and the short G/C paired stem to ensure that C is at the most adequate position to carry out the catalytic function.39 In addition, the three G/C pairs promote activity due to higher stem stability.40 Hence, comparing with SDBs sequestered at 3′ and 5′, center blocking (CCGGT) inhibits both the unpaired T and the three-G/C paired stem (Fig. 3B), which results in the slowest cleavage rate. We demonstrate that the proposed SDBs enable flexible control of the cleavage kinetics.
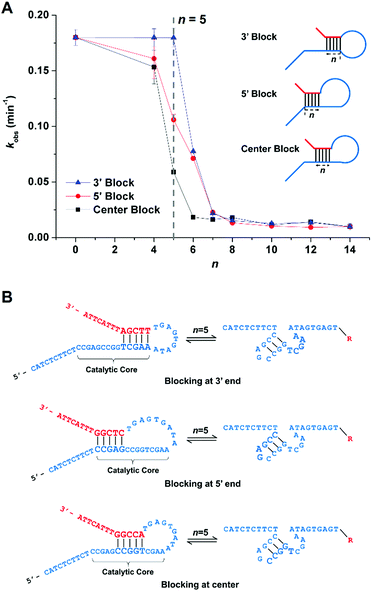 |
| | Fig. 3 (A) The effect of binding lengths and blocking sites on cleavage kinetics. Inset shows proposed secondary structures of SDBs. The error bars represent the standard deviation of three measurements. (B) The proposed second structures of SDBs with 5 bases blocking at 3′ end, 5′ end, and center. | |
To further demonstrate the power of our design strategy, we applied the sequestered DNAzyme beacon strategy to couple with a divalent metal-dependent DNAzyme, 10-23 DNAzyme.41,42 Serials of SDBs with catalytic cores sequestered at center were designed respectively. As shown in Fig. 4, in a fixed concentration of Pb2+ (100 μM), lengthening n from 1 to 15 nt slows the cleavage rates from 0.217 to 0.0006 min−1. These results demonstrate that the sequestered DNAzyme beacon strategy presented can be extended to couple with other RNA-cleaving DNAzymes to enable flexible control of the cleavage kinetics without changing the concentrations of mental ions.
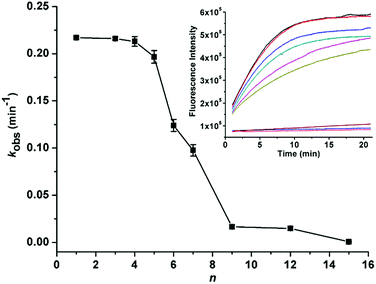 |
| | Fig. 4 The effect of binding lengths on 10-23 DNAzyme cleavage kinetics. The error bars represent the standard deviation of three measurements. Inset shows the change in fluorescence in the presence of different length blocking: 0, 1, 3, 4, 5, 6, 7, 9, 12, and 15 nt from bottom to top, respectively. Initial concentrations: 100 μM Pb2+, 100 nM SDB, and 500 nM S. | |
Control of cleavage rate by tuning the addition of triggering stimulator
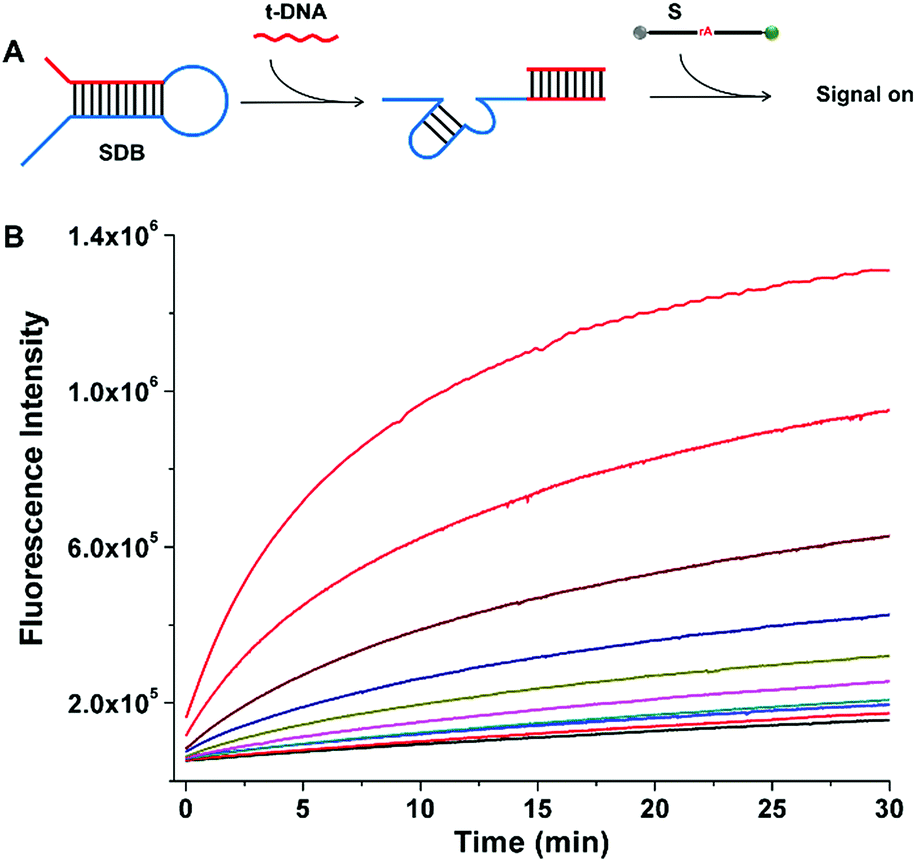
The cleavage kinetics of SDBs can also be finely regulated by tuning the concentration of additional triggering stimulator, which can be applied to target DNA detection. As shown in Fig. 5A, a completely sequestered DNAzyme beacon was designed. The DNA trigger (t-DNA) with specific sequence is complementary to the sequence of the red domain with an overhang toehold in SDB. In the absence of t-DNA, the 17E DNAzyme (blue domain) is sequestered by the hybridization of catalytic core, which provides a low background fluorescence. In the presence of t-DNA, the 17E DNAzyme is liberated from the caged structure with catalytic activity being restored. The activate 17E DNAzyme can catalyze the cleavage of the substrate, which resulting in an obvious increase of fluorescent signal. As the concentration of t-DNA increases from 0.5 to 100 nM, the fluorescence intensity generated by the cleavage of substrate also increases (Fig. 5B), which indicates that the cleavage reaction can be enhanced in a t-DNA-concentration-dependent way (Fig. S1†). Therefore, it is demonstrated that the presence of t-DNA can enhance the catalytic activity of the DNAzyme, and the kinetics of the cleaving reaction can be regulated by tuning the addition of triggering stimulator.
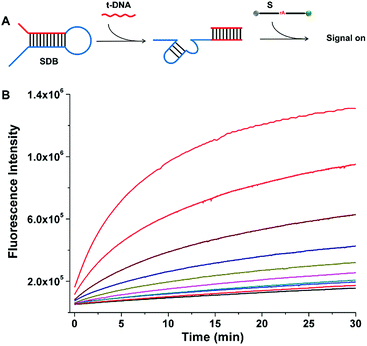 |
| | Fig. 5 Control of cleavage rates through control of stimulator concentration. (A) Schematic representation of the SDB activated by the addition of DNA trigger. (B) The effect of t-DNA concentrations on the catalytic reaction rates. The curves show the change in fluorescence in the presence of different concentrations of t-DNA (nM): 0, 0.5, 1, 2, 5, 10, 15, 20, 50, and 100 from bottom to top, respectively. | |
SDB-based dual stem-loop structure probe for arbitrary target sequence biosensing
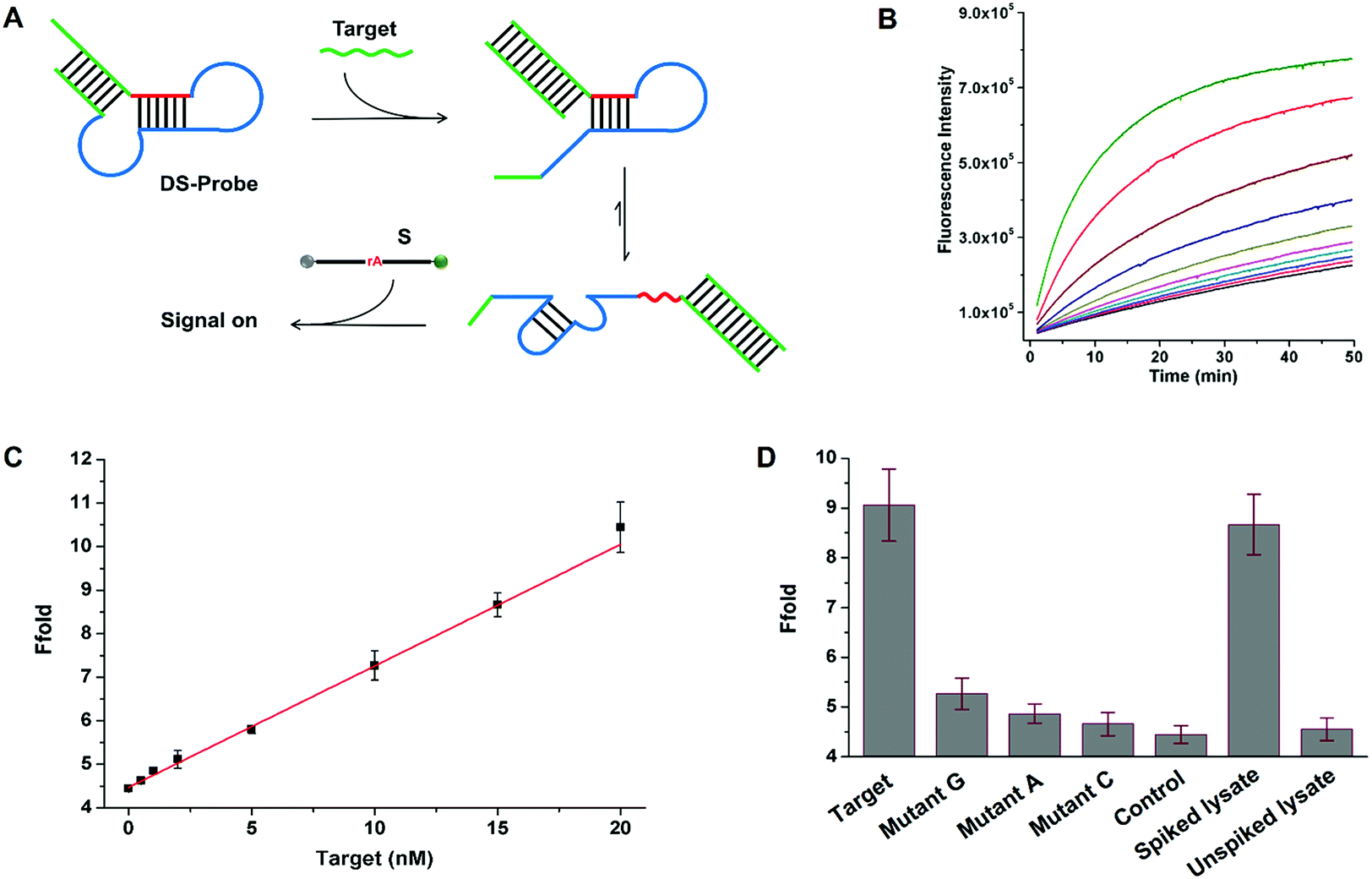
To further demonstrate the universality of SDB in the applications of nucleic acid analysis. We designed a general sensing strategy for the detection of arbitrary target sequence (Fig. 6A). A general probe (DS-probe) was designed using a metastable dual stem-loop structure. DS-probe comprises several domains that make up recognition and signalling modules (i.e. SDB). The inner 6 bp stem (red domain) weakly sequesters the 17E DNAzyme by forming a SDB through hybridizing with the catalytic core. The outer stem (green domain) stabilizes the structure and is available for arbitrary target recognition. In the absence of target DNA (target), the weakly sequestered SDB is stabilized by the synergistic dual stem-loop structure, and is inactive to substrate reporter. In the presence of target, the outer stem is opened through toehold-mediated strand-displacement reaction with target. The relatively weak SDB freed by target recognition allows it to interconvert between hairpin and catalyst forms, which resulting in an obvious signal increase arised from substrate cleavage. Fig. 6B shows the time-dependent fluorescence changes upon analyzing different concentrations of target. As the concentration of the analyte increases from 0.5 to 100 nM, the fluorescence intensity generated by the cleavage of substrate also increases. A linear correlation is obtained in the range of 0–20 nM with a detection limit of 0.13 nM based on 3σ/slope (σ, standard deviation of the blank samples) (Fig. 6C).
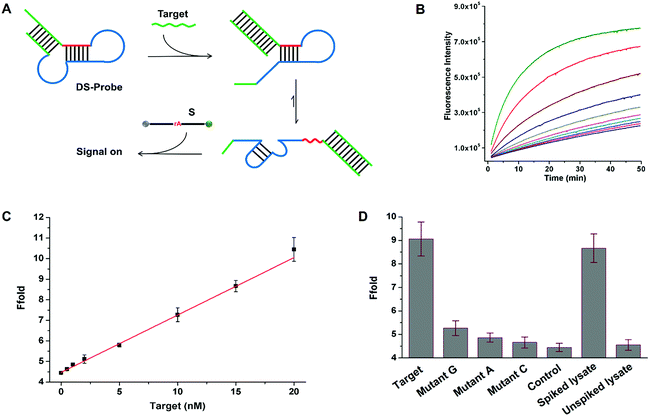 |
| | Fig. 6 Sensing system for the detection of arbitrary target sequence. (A) Schematic representation of the general target sensing strategy. A metastable dual stem-loop structure probe was designed by combining recognition and DNAzyme modules. (B) Time-dependent fluorescence response on varying concentrations of target. The curves show the change in fluorescence in the presence of different concentrations of the target (nM): 0, 0.5, 1, 2, 5, 10, 15, 20, 50, and 100 from bottom to top, respectively. (C) The linear responses of fluorescence enhancement at low target concentrations. (D) Fluorescence enhancement of the sensing system for a series of targets (15 nM): target sequence, single-base mutant sequences, control sample, and target sequence spiked or unspiked in 5-fold diluted HeLa cell lysates, respectively. The error bars represent the standard deviation of three measurements. | |
To quantitatively evaluate the specificity of the proposed sensing system, a series of single-base mismatch sequences of target were tested to obtain the corresponding discrimination factors. The discrimination factor is defined as the ratio of the signal gain obtained with the complementary target DNA to that obtained with the mismatch sequence under the same conditions. As shown in Fig. 6D, the discrimination factors are 5, 9, and 17 for T > G, T > A, and T > C substitutions, respectively, clearly demonstrating the good selectivity of the sensing system. To evaluate the robustness of the sensing system in the physiological medium, a 5-fold diluted HeLa cell lysate was spiked with the target DNA. The spiked recovery of 15 nM target DNA is 95% with a RSD of 7% (n = 3; Fig. 6D). Meanwhile, no evident fluorescence enhancement is observed using an unspiked lysate sample, indicating the possibility for biological sample applications. Therefore, the above results together demonstrate the universality of SDB in the applications of nucleic acid biosensing. By utilizing these strategies, DNAzymes with desired catalytic activity can be applied to biosensors and DNAzyme-based nanodevices.
Conclusions
In conclusion, we have developed a sequestered DNAzyme beacon strategy that allows flexible control of DNAzyme catalytic activities in DNA structures. The crucial element of the proposed strategy is that the catalytic cores of DNAzymes are precisely blocked by designed hybridization. As a result, the cleavage kinetics of proposed SDB can be finely tuned by over one order of magnitude via modulating the blocking strength and the concentration of triggering stimulator. The sequestered DNAzyme beacon strategy was further successfully employed to construct a dual stem-loop structure probe for the detection of arbitrary target sequence. DNAzyme is an important basis of many DNA nanodevices. We expect the additional design flexibility provided by the sequestered DNAzyme beacons to contribute to the development of DNAzyme-based nanodevices, such as biosensors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21605134).
Notes and references
- S. K. Silverman, Trends Biochem. Sci., 2016, 41, 595 CrossRef PubMed.
- R. R. Breaker, Nat. Biotechnol., 1997, 15, 427 CrossRef PubMed.
- Y. Lu, Chem.–Eur. J., 2002, 8, 4588 CrossRef.
- G. F. Joyce, Annu. Rev. Biochem., 2004, 73, 791 CrossRef PubMed.
- J. C. Achenbach, W. Chiuman, R. P. G. Cruz and Y. Li, Curr. Pharm. Biotechnol., 2004, 5, 312 Search PubMed.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2007, 46, 7587 CrossRef PubMed.
- L. M. Lu, X. B. Zhang, R. M. Kong, B. Yang and W. Tan, J. Am. Chem. Soc., 2011, 133, 11686 CrossRef PubMed.
- X. H. Zhao, L. Gong, X. B. Zhang, B. Yang, T. Fu, R. Hu, W. Tan and R. Yu, Anal. Chem., 2013, 85, 3614 CrossRef PubMed.
- L. Freage, A. Trifonov, R. Tel-Vered, E. Golub, F. Wang, J. S. McCaskill and I. Willner, Chem. Sci., 2015, 6, 3544 RSC.
- L. Gong, Z. Zhao, Y. F. Lv, S. Y. Huang, T. Fu, X. B. Zhang, G. L. Shen and R. Q. Yu, Chem. Commun., 2015, 51, 979 RSC.
- W. Zhou, R. Saran and J. Liu, Chem. Rev., 2017, 117, 8272 CrossRef PubMed.
- A. V. Garibotti, S. M. Knudsen, A. D. Ellington and N. C. Seeman, Nano Lett., 2006, 6, 1505 CrossRef PubMed.
- X. Mao, A. J. Simon, H. Pei, J. Shi, J. Li, Q. Huang, K. W. Plaxco and C. Fan, Chem. Sci., 2016, 7, 1200 RSC.
- M. N. Stojanovic, S. Semova, D. Kolpashchikov, J. Macdonald, C. Morgan and D. Stefanovic, J. Am. Chem. Soc., 2005, 127, 6914 CrossRef PubMed.
- J. Elbaz, O. Lioubashevski, F. Wang, F. Remacle, R. D. Levine and I. Willner, Nat. Nanotechnol., 2010, 5, 417 CrossRef PubMed.
- J. Elbaz, F. Wang, F. Remacle and I. Willner, Nano Lett., 2012, 12, 6049 CrossRef PubMed.
- R. Orbach, B. Willner and I. Willner, Chem. Commun., 2015, 51, 4144 RSC.
- R. Orbach, F. Remacle, R. D. Levine and I. Willner, Chem. Sci., 2014, 5, 1074 RSC.
- Y. Tian, Y. He, Y. Chen, P. Yin and C. Mao, Angew. Chem., Int. Ed., 2005, 44, 4355 CrossRef PubMed.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206 CrossRef PubMed.
- R. Pei, S. K. Taylor, D. Stefanovic, S. Rudchenko, T. E. Mitchell and M. N. Stojanovic, J. Am. Chem. Soc., 2006, 128, 12693 CrossRef PubMed.
- Y. Tang, B. Ge, D. Sen and H. Z. Yu, Chem. Soc. Rev., 2014, 43, 518 RSC.
- H. Lin, Y. Zou, Y. Huang, J. Chen, W. Y. Zhang, Z. Zhuang, G. Jenkins and C. J. Yang, Chem. Commun., 2011, 47, 9312 RSC.
- Y. Huang, L. Fang, Z. Zhu, Y. Ma, L. Zhou, X. Chen, D. Xu and C. Yang, Biosens. Bioelectron., 2016, 85, 496 CrossRef PubMed.
- Z. Zhang, D. Balogh, F. Wang and I. Willner, J. Am. Chem. Soc., 2013, 135, 1934 CrossRef PubMed.
- G. Zhu, J. Zhang, E. Song, M. Donovan, K. Zhang, C. Liu and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7998 CrossRef PubMed.
- H. M. Meng, X. Zhang, Y. Lv, Z. Zhao, N. N. Wang, T. Fu, H. Fan, H. Liang, L. Qiu and G. Zhu, ACS Nano, 2014, 8, 6171 CrossRef PubMed.
- W. H. Chen, X. Yu, A. Cecconello, Y. S. Sohn, R. Nechushtai and I. Willner, Chem. Sci., 2017, 8, 5769 RSC.
- H. Fan, X. Zhang and Y. Lu, Sci. China: Chem., 2017, 60, 591 CrossRef.
- W. Zhou, J. Ding and J. Liu, Theranostics, 2017, 7, 1010 CrossRef PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223 CrossRef PubMed.
- J. Li and Y. Lu, J. Am. Chem. Soc., 2000, 122, 10466 CrossRef.
- J. Liu, A. K. Brown, X. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056 CrossRef PubMed.
- S. W. Santoro, G. F. Joyce, K. Sakthivel, S. Gramatikova and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2433 CrossRef PubMed.
- J. Li, W. Zheng, A. H. Kwon and Y. Lu, Nucleic Acids Res., 2000, 28, 481 CrossRef PubMed.
- S.-F. Torabi, P. Wu, C. E. McGhee, L. Chen, K. Hwang, N. Zheng, J. Cheng and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5903 CrossRef PubMed.
- W. Zhou, M. Vazin, T. Yu, J. Ding and J. Liu, Chem.–Eur. J., 2016, 22, 9835 CrossRef PubMed.
- W. Zhou, Y. Zhang, J. Ding and J. Liu, ACS Sens., 2016, 1, 600 CrossRef.
- B. Wang, L. Cao, W. Chiuman, Y. Li and Z. Xi, Biochemistry, 2010, 49, 7553 CrossRef PubMed.
- A. Peracchi, M. Bonaccio and M. Clerici, J. Mol. Biol., 2005, 352, 783 CrossRef PubMed.
- S. W. Santoro and G. F. Joyce, Biochemistry, 1998, 37, 13330 CrossRef PubMed.
- Q. Wang, D. Zhang, M. Cheng, J. He and K. Liu, Acta Pharm. Sin. B, 2012, 2, 28 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting tables and figures. See DOI: 10.1039/c8ra05757e |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mengxi Chenga,
Danling Daia,
Zhonghua Xionga and
Feng Liu
*a,
Mengxi Chenga,
Danling Daia,
Zhonghua Xionga and
Feng Liu