DOI:
10.1039/C8RA04578J
(Paper)
RSC Adv., 2018,
8, 28810-28824
Anticancer activities of a β-amino alcohol ligand and nanoparticles of its copper(II) and zinc(II) complexes evaluated by experimental and theoretical methods†
Received
29th May 2018
, Accepted 7th August 2018
First published on 13th August 2018
Abstract
2-(2-(2-Hydroxyethylamino)ethylamino)cyclohexanol (HEAC) and copper and zinc complexes, [Cu(HEAC)Cl]Cl (1), [Cu(HEAC)Br]Br (2), [Zn(HEAC)Cl2] (3), were prepared and identified by elemental analysis, FT-IR, UV-Vis, 1H NMR spectroscopy and single-crystal X-ray diffraction. Also nanoparticles of 1–3 were prepared for anticancer studies by ultrasonic irradiation. Particle size and morphology of the nano particles are investigated by PXRD and SEM, respectively. X-ray analysis revealed that the ionic complexes 1 and 2 are isostructural. In the structure of complexes 1 and 2, the metal atom has a CuN2O2X (X: Cl (1), Br (2)) environment with square-pyramidal geometry, containing the tetradentate N2O2-donor HEAC. The bond length of the axial position in the square-pyramidal geometry of 1 and 2 is elongated. Complex 3 has a ZnN2OCl2 environment with trigonal bipyramidal geometry around the zinc atom in which the HEAC acts as mer-N2O-donor. The ability of HEAC and nano particles 1–3 to interact with the nine biomacromolecules (BRAF kinase, CatB, DNA gyrase, HDAC7, rHA, RNR, TrxR, TS and Top II) are investigated by docking calculations. For examination of the docking results, the in vitro activities of four compounds against the human leukemia cell line K562 were investigated by evaluation of IC50 values and mode of cell death (apoptosis). The thermodynamic stability of the compounds along with the charge distribution pattern were studied by DFT and NBO analysis, respectively.
1. Introduction
β-amino alcohols exhibit a broad spectrum of biological activities such as being used as antibacterial and tuberculostatic agents.1 Some of them are used as active pharmaceutical ingredients (APIs) such as α- and/or β-adrenergic agonists,2,3 HIV protease inhibitors4 and anti-hypertensives by blocking the α- and/or β-adrenergic receptors.5,6 The γ-secretase inhibitory activity and notch-sparing effects were reported for some γ-amino alcohols.7 β-Amino alcohols have been used as intermediates and chiral auxiliaries in organic synthesis,8,9 as well as organocatalysts.10 Amino alcohols such as mono- and diethanolamine are known to fix CO2 and are used as absorbents for the chemical absorption of CO2.11–13 The interactions of this class of compounds toward DNA and proteins have been established by docking studies.14,15
As a result, a number of synthetic methods have been developed, including asymmetric reduction of α-amino carbonyl compounds16 or α-hydroxy carbonyl compounds,17 asymmetric amino hydroxylation of olefins18 asymmetric hydroboration of enamines19 and ring opening of epoxides by amines.20 The last method has been developed through the use of different heterogeneous catalysts.21 Also the tandem reduction and ring-opening reaction of nitroarenes and epoxides has been reported.22
Because of the interesting chemistry of β-amino alcohols, this work describes the preparation, characterization, crystal structure and theoretical study of three new complexes, [Cu(HEAC)Cl]Cl (1), [Cu(HEAC)Br]Br (2), [Zn(HEAC)Cl2] (3), with 2-((2-((2-hydroxyethyl)amino)ethyl)amino)cyclohexan-1-ol (HEAC, Scheme 1). In addition to the expected biological properties of the β-amino alcohols derivatives,1–4 binding the copper and zinc atoms to this unit make these complexes a good choice for biologically active compounds.23–25 For studying the biological activities of the title compounds, a docking calculation was run to investigate the possibility of interactions between HEAC and its copper and zinc complexes with nine protein targets26 including, BRAF kinase, cathepsin B (CatB), DNA gyrase, histone deacetylase (HDAC7), recombinant human albumin (rHA), ribonucleotide reductases (RNR), thioredoxin reductase (TrxR), thymidylate synthase (TS), topoisomerase II (Top II). These proteins are used in this project either due to their reported roles in cancer growth or as transport agents that affect drug pharmacokinetic properties (e.g., rHA). Also, DNA gyrase was included to study of the possibility of anticancer properties also acting as antimalarial agents.27
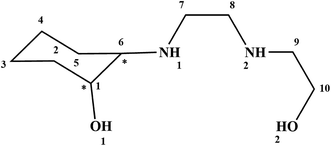 |
| | Scheme 1 The structure of the 2-(2-(2-hydroxyethylamino)ethylamino)cyclohexanol (HEAC). | |
The nano particles 1–3 were papered by ultrasonic irradiation to study the anticancer potential, mode of cell death and apoptosis on the K562 cell lines. Similar studies were performed on HEAC to determine the effect of coordination on the ligand anticancer activity. The K562 cell line is a pluripotent precursor cell that is positive for the Philadelphia (Ph) chromosome and was originally derived from a patient with human chronic myelogenous leukemia (CML) at the terminal stage of last crisis; this cell line is non-adherent and rounded, highly undifferentiated with an active proliferative capacity and the inhibition of apoptosis.28
2. Experimental
2.1. Materials and instrumentation
All starting chemicals and solvents were reagent or analytical grade and used as received. The HEAC ligand has been synthesized according to the literature.29 The infrared spectra of KBr pellets in the range 4000–400 cm−1 were recorded with a FT-IR TENSOR 27 spectrometer.1H NMR spectra were recorded on Bruker Aspect 3000 instrument. The carbon, hydrogen and nitrogen contents were determined by a Thermo Finnigan Flash Elemental Analyzer 1112 EA. The melting points were determined with a Barnsted Electrothermal 9200 electrically heated apparatus. The ultrasonic-assisted reaction was carried out using an ultrasonic bath – Sonica 2200ETH S3-Soltec. The electronic spectra were recorded in H2O using a Shimadzu model 2550 UV-Vis spectrophotometer (190–900 nm). The PXRD patterns were recorded using X'PertPro diffractometer (Holland) with CuKα1 radiation (k = 1.54056 Å). An accelerating voltage of 40 kV and an emission current of 30 mA were used. SEM images are recorded by scanning electron microscope (TSCAN, Czechoslovakia).
2.1.1. Synthesis of [Cu(HEAC)Cl]Cl (1). A solution of 0.20 g (1 mmol) of HEAC, dissolved in methanol (10 mL), was added to a stirring solution containing of CuCl2·2H2O (0.17 g, 1 mmol) in methanol (10 mL). The reaction mixture was stirred for five days at 60 °C and then filtered. Suitable blue prisms were obtained for X-ray diffraction studies by slow evaporation of the solution for a week and collection by filtration. Yield: 0.11 g, 33%; mp 189 °C. Anal. calcd for C10H22Cl2CuN2O2 (%): C, 35.67; H, 6.59; N, 8.32. Found: C, 35.42; H, 6.86; N, 8.41. IR (KBr, cm−1): 3255 (ν O–H), 3150 (ν N–H), 2935 (ν C–H and/or νas CH2), 2886 (νs CH2), 1451 (δas CH2), 1331 (δs CH2), 1249 and 1210 (ν C–O), 1103 (ν C–N). UV-Vis (H2O, λmax (nm)/ε): 679/80 (d → d).
2.1.1.1. Synthesis of nano particles of [Cu(HEAC)Cl]Cl (1). HEAC (0.69 g, 3.4 mmol) and CuCl2·2H2O (0.58 g, 3.4 mmol) were separately dissolved in ethanol (20 mL), and were irradiated inside an ultrasonic bath for 20 minutes at room temperature. The ligand solution was added to the metallic solution before being sonicated for 30 minutes and removing the solvent by rotary evaporation. Finally, the product was formed and collected. Yield: 0.52 g, 45%; mp 189 °C. Anal. calcd for C10H22Cl2CuN2O2 (%): C, 35.67; H, 6.59; N, 8.32. Found: C, 35.48; H, 6.72; N, 8.40. IR (KBr, cm−1): 3257 (ν O–H), 3151 (ν N–H), 2935 (ν C–H and/or νas CH2), 2886 (νs CH2), 1451 (δas CH2), 1331 (δs CH2), 1249 and 1211 (ν C–O), 1105 (ν C–N).
2.1.2. Synthesis of [Cu(HEAC)Br]Br (2). The procedure for the synthesis of 2 was similar to 1 except that the CuCl2·2H2O was replaced by CuBr2 (0.22 g, 1 mmol). Suitable blue prisms for X-ray diffraction studies were obtained by slow evaporation of the solution for a week and collected by filtration. Yield: 0.27 g, 63%; mp 180–184 °C. Anal. calcd for C10H22Br2CuN2O2 (%): C, 28.22; H, 5.21; N, 6.58. Found: C, 28.32; H, 5.20; N, 6.64. IR (KBr, cm−1): 3242 (ν O–H), 3200 (ν N–H), 2929 (ν C–H and/or νas CH2), 2881 (νs CH2), 1448 (δas CH2), 1328 (δs CH2), 1243 and 1204 (ν C–O), 1100 and 1050 (ν C–N). UV-Vis (H2O, λmax (nm)/ε): 680/76 (d → d).
2.1.2.1. Synthesis of nano particles of [Cu(HEAC)Br]Br (2). The procedure for the synthesis of nano particles 2 was similar to 1 except that the CuCl2·2H2O was replaced by CuBr2 (0.76 g, 3.4 mmol). Yield: 1.2 g, 83%; mp 180–184 °C. Anal. calcd for C10H22Br2CuN2O2 (%): C, 28.22; H, 5.21; N, 6.58. Found: C, 28.35; H, 5.26; N, 6.60. IR (KBr, cm−1): 3244 (ν O–H), 3200 (ν N–H), 2930 (ν C–H and/or νas CH2), 2881 (νs CH2), 1447 (δas CH2), 1328 (δs CH2), 1243 and 1205 (ν C–O), 1100 and 1050 (ν C–N).
2.1.3. Synthesis of [Zn(HEAC)Cl2] (3). The procedure for synthesis of 3 was similar to 1 except that the CuCl2·2H2O was replaced by ZnCl2 (0.14 g, 1 mmol). After evaporation of the solution, an oily product was formed which was dissolved in acetone (5 mL). Suitable colorless prisms for X-ray diffraction studies were obtained by evaporation of the solution and collection by filtration. Yield: 0.06 g, 18%; mp 170–173 °C. Anal. calcd for C10H21Cl2N2O2Zn (%): C, 35.58; H, 6.27; N, 8.30. Found: C, 35.59; H, 6.29; N, 8.43. IR (KBr, cm−1): 3429 (ν O–H), 3200 (ν N–H), 2941 (ν C–H and/or νas CH2), 2865 (νs CH2), 1458 (δas CH2), 1387 (δs CH2), 1202 (ν C–O), 1100 and 1046 (ν C–N). 1H NMR (300 MHz, DMSO-d6): (5.8)s, 1H, O1H, (4.8)s, 1H, O2H, (3.9)t, 2H, C10H2, (3.4)m, 1H, C1H, (2.6–3.3)m, 6H, C7H2, C8H2, C9H2, (2.5)m, 1H, C6H, (2.4)s, 2H, N1H, N2H, (1.9–2.0)m, 2H, C2H2, (1.7)m, 2H, C5H2, (1.2)m, 4H, C4H2, C3H2.
2.1.3.1. Synthesis of nano particles of [Zn(HEAC)Cl2] (3). The procedure for synthesis of nano particles 3 was similar to 1 except that the CuCl2·2H2O was replaced by ZnCl2 (0.46 g, 3.4 mmol). After rotary evaporation, an oily product was formed which was dissolved in acetone (15 mL). The final product was formed after evaporation of acetone. Yield: 0.53 g, 46%; mp 170–173 °C. Anal. calcd for C10H21Cl2N2O2Zn (%): C, 35.58; H, 6.27; N, 8.30. Found: C, 35.63; H, 6.30; N, 8.8.39.
2.2. Crystal structure determination
X-ray diffraction data for complexes 1 and 2 were collected at 93 K using a Rigaku FR-X Ultrahigh Brilliance Microfocus RA generator/confocal optics with XtaLAB P200 diffractometer. The data for complex 3 was collected at 173 K using a Rigaku SCXmini CCD diffractometer with a SHINE monochromator. Mo Kα radiation (λ = 0.71075 Å) was used for all the compounds and intensity data were collected using ω steps accumulating area detector images spanning at least a hemisphere of reciprocal space. All data were corrected for Lorentz polarization effects. A multiscan absorption correction was applied using CrystalClear.30 Structures were solved using dual space methods (SHELXT31) and refined by full-matrix least-squares against F2 (SHELXL-2013 (ref. 32)). Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were refined using a riding model or freely for N–H and O–H atoms, with the exception of the O–H groups on the disordered ethanolic groups of 3, these were assigned geometrically. All calculations were performed using the CrystalStructure interface. Selected crystallographic data are presented in Table 1. Diagrams of the molecular structure and unit cell were created using Ortep-III33,34 and Diamond.35 Selected bond lengths and angles are displayed in Table 2 and hydrogen bond geometries in Table 3.
Table 1 Crystal data and structure refinement for complexes 1–3
| |
1 |
2 |
3 |
| Empirical formula |
C10H22Cl2CuN2O2 |
C10H22Br2CuN2O2 |
C10H22Cl2N2O2Zn |
| Formula weight, g mol−1 |
336.75 |
425.65 |
338.58 |
| Crystal size, mm |
0.21 × 0.14 × 0.05 |
0.18 × 0.12 × 0.06 |
0.15 × 0.09 × 0.050 |
| Temperature, K |
93 |
93 |
173 |
| Crystal system |
Orthorhombic |
Orthorhombic |
Monoclinic |
| Space group |
Pbca |
Pbca |
P21/c |
| Unit cell dimensions |
| a, Å |
6.63584(15) |
6.68836(15) |
10.3713(8) |
| b, Å |
11.8221(3) |
12.3506(3) |
12.4965(9) |
| c, Å |
34.9929(8) |
35.1567(8) |
11.9806(9) |
| β, ° |
90 |
90 |
94.277(8) |
| Volume, Å3 |
2745.18(11) |
2904.13(12) |
1548.4(2) |
| Z |
8 |
8 |
4 |
| Calculated density, g cm−3 |
1.629 |
1.947 |
1.452 |
| Absorption coefficient, mm−1 |
1.972 |
7.017 |
1.924 |
| F(000), e |
1400.00 |
1688.00 |
704.00 |
| θ range for data collection, ° |
2.3–28.4 |
2.3–28.4 |
2.4–25.4 |
| h, k, l ranges |
−8 ≤ h ≤ 8, −15 ≤ k ≤ 15, −45 ≤ l ≤ 46 |
−8 ≤ h ≤ 8, −15 ≤ k ≤ 16, −44 ≤ l ≤ 46 |
−12 ≤ h ≤ 12, −15 ≤ k ≤ 15, −14 ≤ l ≤ 14 |
| Reflections collected/independent/Rint |
57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 562/3242/0.084 562/3242/0.084 |
61924/3452/0.056 |
12999/2830/0.187 |
| Data/restraints/parameters |
3242/4/170 |
3452/4/170 |
2830/2/188 |
| Goodness-of-fit on F2 |
1.25 |
1.06 |
0.834 |
| R1/wR2 (I ≥ 2σ(I)) |
0.0721/0.1834 |
0.0206/0.0535 |
0.0504/0.1154 |
| R1/wR2 (all data) |
0.0793/0.1862 |
0.0236/0.0543 |
0.0957/0.1293 |
| Largest diff. peak/hole, e Å−3 |
3.52/−0.93 |
0.68/−0.45 |
0.80/−0.55 |
Table 2 Selected bond length (Å) and angles (°) for complexes 1–3 with estimated standard deviations in parentheses
| 1 |
2 |
3 |
| Distances |
| Cu1–Cl1 |
2.2533(15) |
Cu1–Br1 |
2.3974(3) |
Zn1–Cl1 |
2.2431(14) |
| Cu1–Cl2 |
4.538(2) |
Cu1–Br2 |
4.6567(6) |
Zn1–Cl2 |
2.2953(14) |
| Cu1–N1 |
2.028(5) |
Cu1–N1 |
2.0194(16) |
Zn1–N1 |
2.151(5) |
| Cu1–N5 |
2.010(5) |
Cu1–N5 |
2.0098(17) |
Zn1–N5 |
2.069(4) |
| Cu1–O2 |
2.243(4) |
Cu1–O2 |
2.2384(14) |
Zn1–O10 |
2.303(4) |
| Cu1–O10 |
2.012(4) |
Cu1–O10 |
2.0039(14) |
|
|
|
| Angles |
| Cl1–Cu1–O2 |
97.74(12) |
Br1–Cu1–O2 |
98.53(4) |
Cl1–Zn1–Cl2 |
118.89(6) |
| Cl1–Cu1–N1 |
96.54(15) |
Br1–Cu1–N1 |
96.49(5) |
Cl1–Zn1–N1 |
105.26(13) |
| O2–Cu1–O10 |
92.52(17) |
O2–Cu1–O10 |
92.13(6) |
Cl2–Zn1–O10 |
89.10(11) |
| O2–Cu1–N5 |
102.09(18) |
O2–Cu1–N5 |
103.08(6) |
Cl2–Zn1–N5 |
119.09(12) |
| O10–Cu1–N5 |
82.16(19) |
O10–Cu1–N5 |
82.47(6) |
O10–Zn1–N5 |
75.91(15) |
| Cl1–Cu1–O10 |
98.58(13) |
Br1–Cu1–O10 |
97.97(4) |
Cl1–Zn1–O10 |
93.78(10) |
| Cl1–Cu1–N5 |
160.11(15) |
Br1–Cu1–N5 |
158.36(5) |
Cl1–Zn1–N5 |
120.72(12) |
| O2–Cu1–N1 |
81.27(18) |
O2–Cu1–N1 |
81.15(6) |
Cl2–Zn1–N1 |
92.84(13) |
| O10–Cu1–N1 |
164.31(19) |
O10–Cu1–N1 |
164.81(6) |
O10–Zn1–N1 |
156.94(16) |
| N1–Cu1–N5 |
85.1(2) |
N1–Cu1–N5 |
85.80(7) |
N1–Zn1–N5 |
83.13(17) |
Table 3 Hydrogen bonds dimensions (Å and °) in complexes 1–3
| D–H⋯A |
d(D–H) |
d(H⋯A) |
∠(DHA) |
d(D⋯A) |
| 1 |
| O(2)–H(2)⋯Cl(2) |
0.98(2) |
2.04(3) |
164(6) |
2.994(4) |
| O(10)–H(10)⋯Cl(2) |
0.97(2) |
2.18(5) |
145(6) |
3.025(4) |
| N(1)–H(1)⋯Cl(1) |
0.96(2) |
2.69(3) |
164(6) |
3.623(5) |
| N(5)–H(5)⋯Cl(2) |
0.99(2) |
2.35(3) |
165(6) |
3.314(5) |
|
| 2 |
| O(2)–H(2)⋯Br(2) |
0.932(18) |
2.230(19) |
172(3) |
3.1552(14) |
| O(10)–H(10)⋯Br(2) |
0.947(18) |
2.251(19) |
165(3) |
3.1759(14) |
| N(1)–H(1)⋯Br(1) |
0.920(16) |
2.840(17) |
165(2) |
3.7349(16) |
| N(5)–H(5)⋯Br(2) |
0.940(17) |
2.548(18) |
166(2) |
3.4675(17) |
|
| 3 |
| N(1)–H(1)⋯Cl(2) |
0.978(10) |
2.48(2) |
153(4) |
3.378(4) |
| N(5)–H(5)⋯Cl(2) |
0.979(10) |
2.338(17) |
164(4) |
3.288(4) |
| O(10)–H(10)⋯Cl(1) |
0.973(10) |
2.24(2) |
161(5) |
3.181(4) |
| O(2A)–H2E⋯Cl(2) |
0.98 |
2.73 |
139.7 |
3.539(9) |
2.3. Computational details
All structures were optimized with the Gaussian 09 software36 and calculated for an isolated molecule using Density Functional Theory (DFT)37 at the B3LYP/LanL2DZ level of theory, as well as for NBO analysis. The cif file of the complexes were used as input files for the theoretical calculations.
2.4. Docking details
The pdb files 4r5y, 3ai8, 5cdn, 3c0z, 2bx8, 1peo, 3qfa, 1njb, 4gfh for the nine receptors, BRAF kinase, cathepsin B (CatB), DNA gyrase, histone deacetylase (HDAC7), recombinant human albumin (rHA), ribonucleotide reductases (RNR), thioredoxin reductase (TrxR), thymidylate synthase (TS), topoisomerase II (Top II), respectively, used in this research were obtained from the Protein Data Bank (pdb).38 The full version of Genetic Optimisation for Ligand Docking (GOLD) 5.5 (ref. 39) was used for the docking studies. The Hermes visualizer in the GOLD Suite was used to further prepare the metal complexes and the receptors for docking. The optimized HEAC ligand and cif files of the complexes were used for docking studies. The region of interest used for Gold docking was defined as all the protein residues within 6 Å of the reference ligand “A” that accompanied the downloaded protein. All free water molecules in the structure of the proteins were deleted before docking. Default values for all other parameters were used and the complexes were submitted to 10 genetic algorithm runs using the GOLDScore fitness function.
2.5. Biological evaluation
2.5.1. Materials. Fetal bovine serum (FBS), propidium iodide (PI), acridine orange (AO) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were obtained from Sigma Aldrich. RPMI-1640 medium was obtained from Gibco (UK). Dimethyl sulfoxide (DMSO) was obtained from Merck. Fluorescent studies were done by fluorescent microscope (OLYMPUS, USA).
2.5.2. Cell culture and treatments. Human leukemia cell line K562 and its adriamycin-selected multidrug resistant subline K562/ADM were obtained from the Pasteur Institute of Iran. All the cell lines were grown in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and penicillin (100 U mL−1) and streptomycin (100 μg) (BIOCERA) at 37 °C in a humidified 5% CO2 incubator. To maintain MDR phenotype, adriamycin (1 mg L−1) was added to K562/ADM cultures and maintained in the drug-free medium for at least two weeks before being used. Compounds were dissolved in of DMSO (180 μL) for in vitro assays.
2.5.3. The MTT assay. Cells were cultured at a density of 1 × 104/100 μL in a 96-well plate and allowed to attach overnight. After the incubation with the testing chemicals for 48 h and 72 h, the cells were treated with the solution of MTT (5 mg mL−1) at 37 °C for 4 h. The absorbance was measured at 498 nm using a microplate reader (Multiskan Spectrum, Thermo Scientific). The inhibition rates were calculated on a plate-by-plate basis for the test wells. The IC50 values were calculated based on the inhibitory rate curves using Bliss' method.
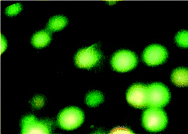
2.5.4. AO/PI staining assay. After treatment for 24 h with the compounds, the K562 cells were harvested, centrifuged at 400 rpm for 10 min and then washed in PBS. Then 1 mL aliquots were incubated in the dark with AO (10 μL) and PI (10 μL) for 15 min. After mixing the suspension with a solution of AO/PI, analysis was performed under a fluorescence microscope. Whilst a green fluorescence showed apoptosis, a red colored nucleus indicated necrotic cells.
3. Results and discussion
Reaction between HEAC and methanolic solution of the copper(II) chloride, copper(II) bromide and zinc(II) chloride provides complexes 1–3, respectively. The complexes are air-stable and soluble in DMF, DMSO and H2O. In all CSD searches which have been presented, for more precise results, the structures containing any error or disorder have been omitted.
3.1. Spectroscopic characterization
The frequency of IR bands for the free ligand are different from those of the corresponding complexes, providing significant indications of bonding sites of the HEAC. In the IR spectrum of HEAC, a broad peak at 3267 cm−1 can be assigned to the (ν O–H) which is shifted about 12 and 25 cm−1 to lower frequencies in complexes 1 and 2, respectively, whilst in 3 the peak is shifted to higher frequency than that of the free ligand (162 cm−1). Another peak above the 3000 cm−1 is corresponding to the stretching vibration of the amine groups of the ligand. Comparison of the IR spectra of the compounds revealed that the ν (N–H) is shifted to a higher frequency (53, 103 and 103 cm−1 in 1–3, respectively) which can be attributed to the coordination of these groups to the metal centers. Also the bands in the range of 2800–3000 cm−1, which can be assigned to the asymmetric and symmetric stretching vibrations of the C–H bonds, confirm the presence of the aliphatic moieties in the studied compounds.
In the 1H NMR spectrum of complex 3 (see Scheme 1 for numbering), the peaks corresponding to the hydrogen atoms of the two alcohol groups of HEAC are observed at the lowest magnetic field. The proton on the cyclohexane alcohol group shifts about 1 ppm to a lower magnetic field, with respect to the free ligand,40 revealing the coordination through this moiety whilst protons on the ethanolic arm do not undergo a significant shift. Another interesting part of this spectrum is the shifts of the amine groups to predict the coordination behavior of the HEAC. The peaks of amine protons are shifted by 0.3 ppm to a lower magnetic field after coordination. Based on this data we can conclude that the HEAC acts as N2O-donor toward the zinc atom.
UV-Vis spectra of the complexes 1 and 2 in aqueous solution exhibited a broad absorption which was assigned to the d–d transition of the copper(II) complexes. The energy for the d–d transition in the two complexes is very similar (679 and 680 nm for 1 and 2 respectively), showing that the HEAC ligand has a more significant effect on the ligand field strength than the coordinated halide ions.
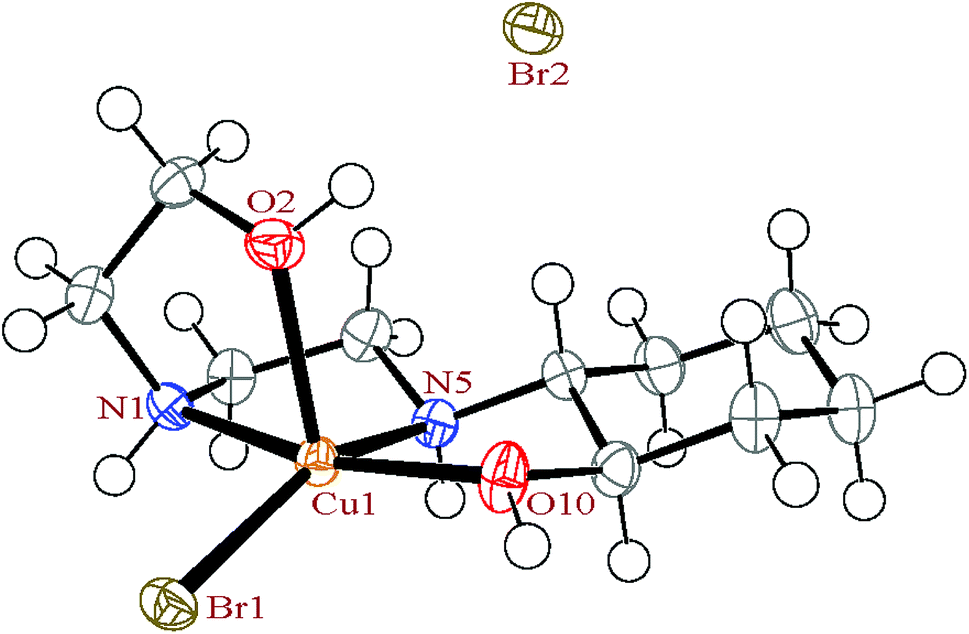
3.2. Description of the crystal structures
3.2.1. Crystal structure of [Cu(HEAC)Cl]Cl (1). X-ray analysis of complex 1 (Fig. 1) revealed a five coordinate Cu(II) center bound to the tetradentate HEAC ligand and one chloro ligand, with one chloride anion per copper center to balance charge. The nature of penta-coordinate geometry can be assessed by applying the formula proposed by Addison et al.41,42 The angular structural parameter, τ (τ = (β − α)/60, where α and β are the two largest angles at the copper atom with β ≥ α), was calculated to be 0.07 indicating a slightly distorted square-pyramidal geometry around the copper atom (Fig. 1 and 2). In this geometry, the oxygen atom of the ethanolic arm occupies the axial position whilst the cyclohexanolic oxygen and two amine nitrogen atoms lie on the equatorial plane along with the chloro ligand. The axial Cu–O bond distance is 0.231 (4) Å longer than the equatorial Cu–O, showing an elongated bond length down the z-axis, consistent with pseudo Jahn–Teller distortions observed in other Cu(II) square pyramidal structures.43 For comparing the geometrical parameters of 1 with the Cambridge Structural Database (CSD),44 a search was run on all analogues of 1 (all structures containing the base presented in Scheme 2; the macrocyclic ligands have been deleted for more precise results). Based on this study, a tetradentate ligand coordinated to a copper(II) center containing a chloro ligand commonly form with square-pyramidal geometry (81%) (Fig. 3). These square-pyramidal complexes can be further subdivided into two categories: where the chloro ligand can be either axial (Scheme 2(a)) or equatorial (Scheme 2(b)). The percentage of the structures containing the axial chloro ligand (59%) is higher than the equatorial variant. The average Cu–Cl distance in the structures containing axial chloro ligand is 0.271 Å longer than the equatorial variant (Scheme 2), confirming the presence of elongation of the bond in the axial direction. In complex 1, the chloro ligand is located in the equatorial plane with a bond length of 2.2533(15) Å which is comparable to the CSD average (Scheme 2(b)). The Cu–Cl bond length average of the complexes with trigonal bipyramidal geometry is similar to the square-pyramidal complexes containing a chloro ligand in the equatorial position.
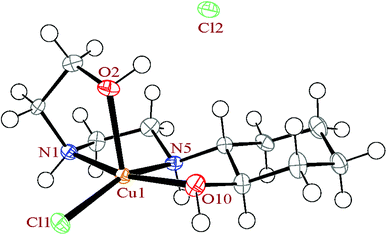 |
| | Fig. 1 The ortep diagram of the molecular structure of the complex 1. The ellipsoids are drawn at the 50% probability level. | |
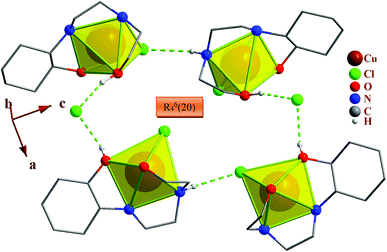 |
| | Fig. 2 Packing of the complex 1, showing the hydrogen bonds. Only the hydrogen atoms involved in hydrogen bonding are shown. Each CuN2O2Cl unit is shown as square-pyramid. Also R64(20) hydrogen bond motifs between four complexes 1 is shown. | |
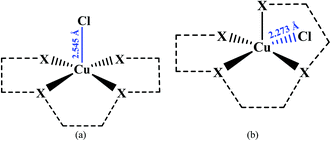 |
| | Scheme 2 Different types of the analogues of the complex 1. | |
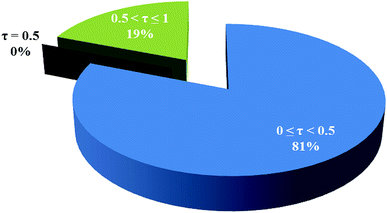 |
| | Fig. 3 Percentage of square-pyramidal and trigonal bi pyramidal geometries around the copper atom among the analogues of the complex 1. | |
In the crystal structure of 1, the geometry around the copper atom is square-pyramidal. Is this geometry common for the penta-coordinated copper atom? For answering this question, all CN = 5 copper complexes deposited in the CSD were analyzed by the Addison formula and results are shown in Fig. 4. Based on this study, common geometry around the copper atom in penta-coordinated complexes is square-pyramidal (88%), similarly in 1.
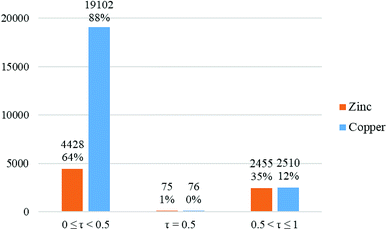 |
| | Fig. 4 Column chart of the percentage of different geometries around the penta-coordinated copper (blue columns) and zinc (orange columns) atoms in the CSD database. | |
In complex 1, the HEAC ligand acts as a tetradentate N2O2-donor and forms three five-membered, non-planar chelate rings. The angle between the mean planes through the two equatorial chelate rings is 7.5(3)°, showing these two chelate rings are almost coplanar and also are perpendicular to the axial chelate ring (with angle of 86.8(2)° between mean planes through the axial and equatorial chelate rings). The cyclohexane ring of the HEAC has a chair conformation and two of the carbon atoms are chiral with the same enantiomeric form. In addition, two new chiral centers are formed upon coordination (the nitrogen atoms) with different enantiomeric forms. Thus the complex has four chiral centers. However, the crystals overall contain a racemic mixture of R,R,R,S and S,S,S,R isomers in alternate layers.45,46
3.2.2. Crystal structure of [Cu(HEAC)Br]Br (2). X-ray analysis of complex 2 (Fig. 5) revealed that the complex is the isostructural bromine analogue of 1. It exhibits a five coordinate Cu(II) center bound to the tetradentate HEAC ligand and one bromo ligand, with one bromine anion per copper center to balance charge. For 2, τ = 0.11, which means that the geometry is still square-pyramidal, however the distortion is slightly greater than in 1. Similarly, to 1, the Cu–O bond length of the oxygen atom on the ethanolic arm in the axial position is by0.2345(14) Å longer than the equatorial one. A structural study of the CSD revealed that there are four examples47–50 for base presented in Scheme 2, in which the chloride ion is replaced by a bromide ion (the macrocyclic ligands have been deleted for more precise results). Three of the structures have square-pyramidal geometry around the metal atom (τ = 0.01–0.35 (ref. 47–49)) and one of them has tetragonal bi pyramidal (τ = 0.57 (ref. 50)). The averages of the axial and equatorial Cu–Br distances in the square-pyramidal examples are 2.543 and 2.430 Å, respectively, revealing a 0.113 Å elongation for axial bonds. In complex 1, the bromo ligand is located on the equatorial plane with a bond length of 2.3974(3) Å which is shorter than its analogues average.
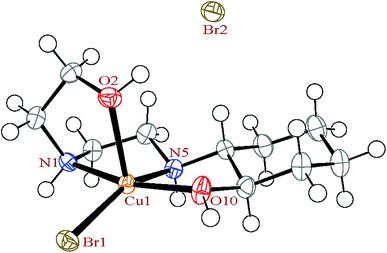 |
| | Fig. 5 The ortep diagram of the molecular structure of the complex 2. The ellipsoids are drawn at the 50% probability level. | |
In the crystal structure of 2, the N2O2-donor HEAC forms three five-membered non-planar chelate rings. The angle between mean planes through the two equatorial chelate rings and also between axial and equatorial chelate rings is 7.62(9) and 86.10(8)°, respectively, confirming a similar coordination geometry of HEAC in complexes 1 and 2. This complex has four chiral centers (two carbon and two nitrogen atoms) and contain a racemic mixture of R,R,R,S and S,S,S,R isomers in alternate layers.
3.2.3. Crystal structure of [Zn(HEAC)Cl2] (3). X-ray analysis of complex 3 (Fig. 6) revealed that the structure exhibits a five coordinate Zn(II) center bound to the HEAC ligand, which in this case is tridentate, along with two chloro ligands. The τ value is 0.61, confirming a trigonal bipyramidal geometry (Fig. 6 and 7). A structural study of the CSD database revealed that this geometry is not common among the zinc complexes with CN = 5 with the most common geometry for such complexes being square-pyramidal (64%), as was observed for copper complexes with the same coordination number. In this geometry two chloro ligands are located on the equatorial plane along with the N1 atom (Scheme 1) of the HEAC ligand, with the two axial positions being occupied by the O1 and N2 atoms (Scheme 1). The ethanolic arm of the HEAC does not participate in coordination and is parallel with the equatorial plane, modelled as disordered over two positions. Of the two Cu–N distances, the axial one is 0.082 (5) Å longer than the equatorial one, owing to the elongation down the z-axis.
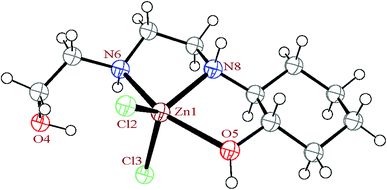 |
| | Fig. 6 The ortep diagram of the molecular structure of the complex 3. The ellipsoids are drawn at the 50% probability level. | |
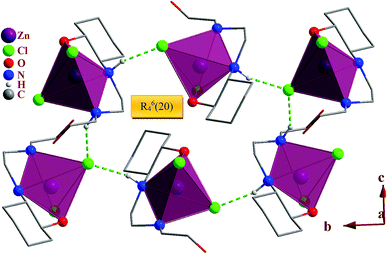 |
| | Fig. 7 Packing of the complex 3, showing the hydrogen bonds. Only the hydrogen atoms involved in hydrogen bonding are shown. Each ZnN2OCl2 unit is shown as trigonal bi pyramid. Also R64(20) hydrogen bond motifs between six complexes 3 is shown. | |
The HEAC ligand in 3 acts as N2O-donor to the zinc atom and forms two five-membered non-planar chelate rings in direction of the axial bonds. Each tridentate ligand can be coordinate to the metal in facial or meridional formations. In the mer form there are two 90° angles, one 180° angle, whereas in fac there are three 90°angles. In 3, two angles of coordinated HEAC are deviating from 90° due the chelating bite angle, while the third one is about 157.2°, confirming mer form (135°, exactly half way between fac and mer).51,52 The complex 3, has four chiral centers (two carbon and two nitrogen atoms) and contain a racemic mixture of R,R,R,S and S,S,S,R isomers in alternate layers.
3.2.4. Crystal network interactions. In the crystal networks of 1–3 (Fig. 2 and 7) intermolecular N–H⋯X (X: Cl (1, 3), Br (2)) and O–H⋯X (X: Cl (1, 3), Br (2)) hydrogen bonds appear between different moieties. In this way the halide ions act as proton acceptors and the nitrogen and oxygen atoms as proton donors. The free halide ions in 1 and 2 (Cl2 and Br2) play a more important role in the crystal network expansion than the coordinated halides (Cl1, Br1) owing to them forming intermolecular hydrogen bonds.In the crystal packing of the complexes, the N–H⋯Cl and O–H⋯Cl hydrogen bonds in 1, N–H⋯Br and O–H⋯Br in 2 and N–H⋯Cl in 3 participate in the formation of very different hydrogen bond motifs such as R43(14), R63(18) and R64(20) in 1 and 2 (Fig. 2 and 7) and R22(8), R64(20), R65(22) and R66(24) in 3 between adjacent complexes.51,53
The total intermolecular interaction energies of one molecule of 1 and 2 were calculated using Mercury54 and its CSD-materials tool.38,55 For this, the sum of the intermolecular interactions energies in a molecular packing shell containing 100 molecules around one molecule of 1 and 2 were calculated to be −129.76 (for cationic unit of 1), 58.35 (for anionic unit of 1), −139.709 (for cationic unit of 2) and 40.63 kJ mol−1 (for anionic unit of 2) (Fig. 8), confirming that one molecule of complex 2 is more stabilized in the solid state, by its network interactions, than 1. Also the cationic complex unit in 1 and 2 is more stabilized than the anionic species by intermolecular interactions. For the cationic unit of complex 1, 44% (Cu1 enantiomer) of the total energy is corresponding to the interactions with its 13 closest neighboring molecules (Fig. 8). This value for 2 with the same condition is 45%. The interactions between one molecule of the complexes 1 and 2 with three closest molecules in distance range of 3.5–5.5 Å, increase the energy component of the molecule by +68.35 and +51.51 kJ mol−1, respectively, whilst other interactions decrease the energy level of the studied units.
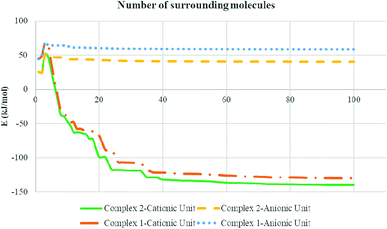 |
| | Fig. 8 Variation diagram of total intermolecular interactions energy (E) for complexes 1 and 2 with increasing the number of surrounding molecules. | |
3.3. PXRD and SEM studies of nano complexes
Fig. 9–11 shows both the simulated PXRD patterns obtained from the single crystal X-ray data of complexes 1–3 and that obtained experimentally from the nano complexes 1–3 prepared by the sonochemical process. The experimental data matched well, with slight variations in 2θ values. This indicates that the nano complexes 1–3 obtained by the sonochemical process possess an identical structure to the larger crystals. Estimated from the Sherrer formula for the calculation of particle sizes from the PXRD peaks (L = Kλ/(βcosθ), where L is the average crystallite size in nm, β is the breadth of the observed diffraction line at its half-maximum intensity, K is the so-called ‘shape factor’, and λ is the wavelength of the X-ray radiation (λ = 0.15406 nm)), the average sizes of the particles were calculated to be 42.85, 36.078 and 43.55 nm for complexes 1–3, which is in agreement with the values obtained from the SEM images (Fig. 9–11).
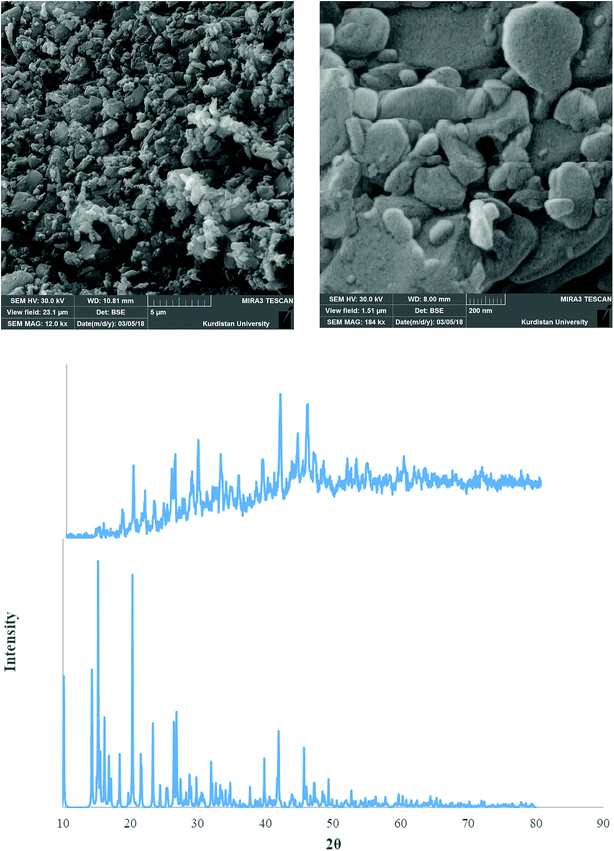 |
| | Fig. 9 PXRD pattern (middle) and SEM images (upper) of the nano complex 1. Also single crystal X-ray diffraction pattern of 1 (under) is presented. | |
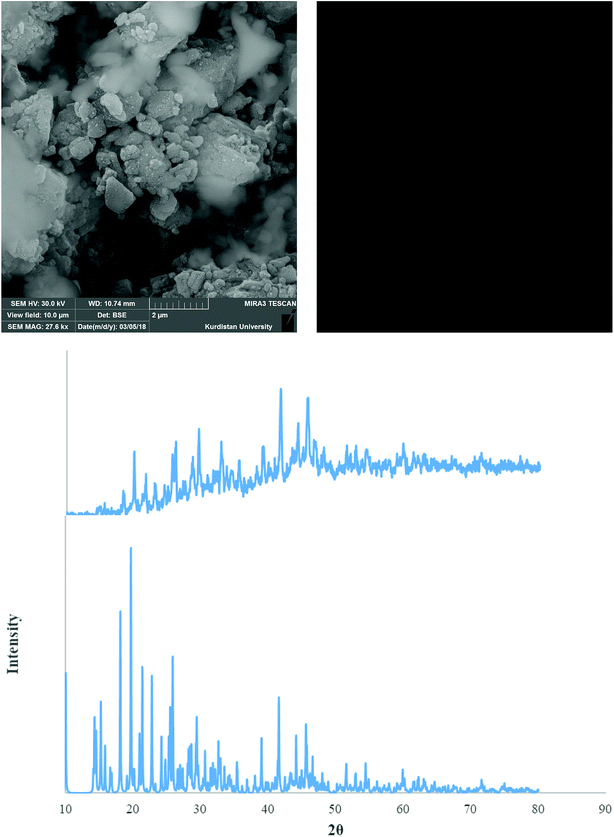 |
| | Fig. 10 PXRD pattern (middle) and SEM images (upper) of the nano complex 2. Also single crystal X-ray diffraction pattern of 2 (under) is presented. | |
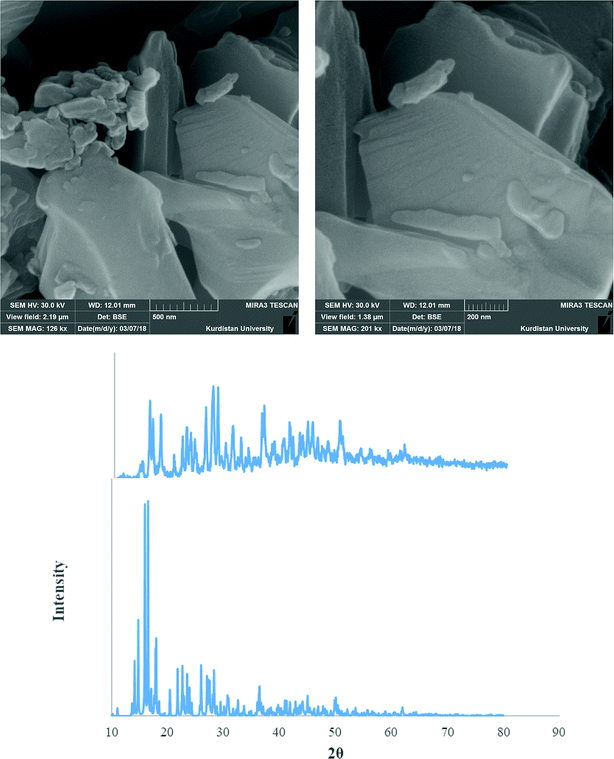 |
| | Fig. 11 PXRD pattern (middle) and SEM images (upper) of the nano complex 3. Also single crystal X-ray diffraction pattern of 3 (under) is presented. | |
3.4. Theoretical studies
For comparing the energy levels of the complexes for one isolated molecule, DFT calculations were performed. The data revealed that the order of thermodynamic stability of the optimized complexes is 1opt > 2opt > 3opt. The complex 1opt is −2235.98 and −81890.18 kcal mol−1 thermodynamically more stable than the 2opt and 3opt, respectively which shows that the chloro ligand of 1opt stabilizes the copper atom significantly more than the bromo ligand does.
For studying the charge distribution before and after complexation, a NBO analysis was done on the free HEAC ligand and its complexes (Table 4). The results reveal that the calculated charge on the metal atoms are in the range of 0.79–1.13 (Table 4) and lower than the formal charge (+2) owing to the electron donation of the ligand during complexation. Based on the calculated total charge values, the charge on the nitrogen, carbon and oxygen atoms in the optimized complexes does not have significant variation with respect to the free ligand, whilst the total charge on the hydrogen atoms is more positive than in the free ligand (Table 4). This observation reveals that the hydrogen atoms play an important role in electron donation toward metal atom, thus decreasing the charge of the copper and zinc atoms.
Table 4 The NBO analysis results for the complexes 1–3 and HEAC. The values are the total of charge on the similar atoms. The values of parentheses show the variation of charge on the atoms after coordination
| |
C |
H |
N |
O |
Bonded halide |
Non-bonded halide |
Metal |
| HEAC |
−2.27 |
5.45 |
−1.51 |
−1.67 |
— |
— |
— |
| Complex 1 |
−2.32 (−0.05) |
5.91 (+0.46) |
−1.48 (+0.03) |
−1.60 (+0.07) |
−0.55 |
−0.78 |
0.83 |
| Complex 2 |
−2.31 (−0.04) |
5.91 (+0.46) |
−1.49 (+0.02) |
−1.60 (+0.07) |
−0.50 |
−0.80 |
0.79 |
| Complex 3 |
−2.29 (−0.02) |
5.77 (+0.32) |
−1.61 (−0.10) |
−1.65 (+0.02) |
−1.34 (−0.67 per each chloride) |
— |
1.13 |
In the optimized HEAC, the HOMO is mostly delocalized on the ethylene diamine moiety while the LUMO is delocalized on the oxygen atom of the ethanolic arm of the ligand (Table 5). In the structure of the HOMO and LUMO of the complexes, the metal atom and halide ions play the main role. In 1opt and 2opt, the HOMO is mostly delocalized on the non-bonded halide ions whilst the LUMO is on the metal center. In 3opt, the metal atom is host to a significant portion of the LUMO and coordinated chloro ligand holds the majority of the HOMO.
Table 5 HOHO, LUMO orbitals and total energy for the optimized structures of HEAC and complexes 1–3opt
| |
HOMO |
LUMO |
Total energy (kcal mol−1) |
| HEAC |
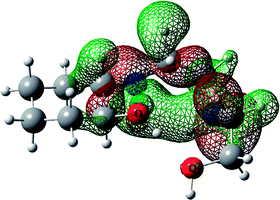 |
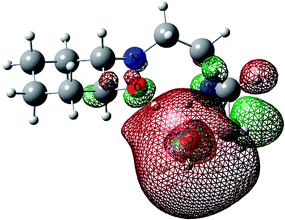 |
−410481.04 |
| Complex 1opt |
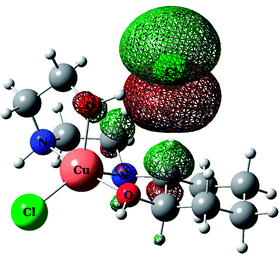 |
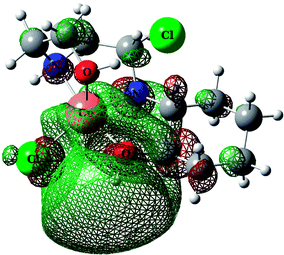 |
−552418.03 |
| Complex 2opt |
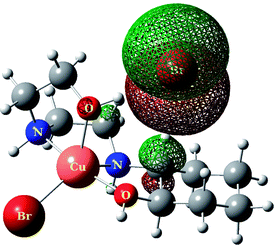 |
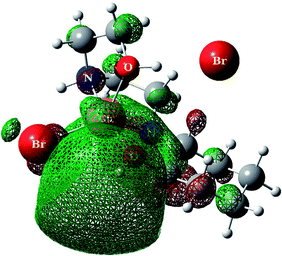 |
−550182.05 |
| Complex 3opt |
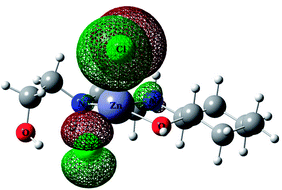 |
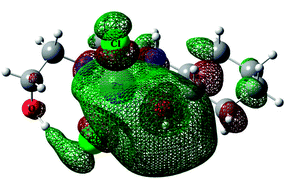 |
−470527.84 |
In the isolated molecules of 1opt, 2opt and 3opt, the metal atoms have τ values of 0.56, 0.57 and 0.10, respectively, revealing favored trigonal bi pyramidal geometry for the copper complexes and square-pyramidal geometry for zinc complex. These results are in contrast with the experimentally obtained solid state structures, suggesting that perhaps the intermolecular interactions affect the geometry around the metal center in solid phase.
3.5. Docking studies
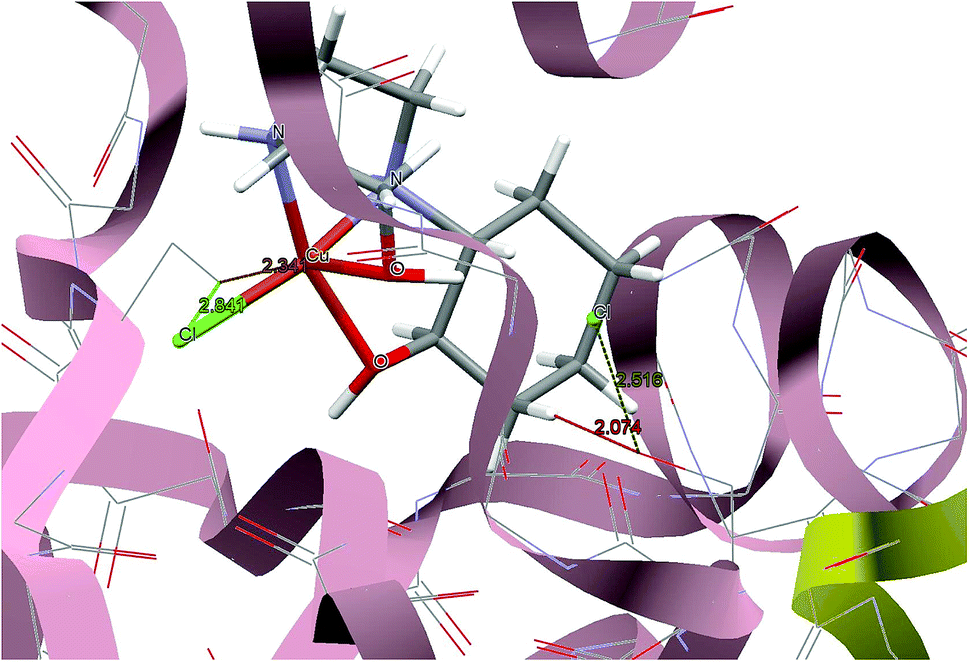
For predicting the biological activities of the HEAC ligand and complexes 1–3, interactions of these compounds with nine macromolecule receptors, using Gold39 docking software, were studied. The Gold docking results are reported in terms of the values of fitness which means that the higher fitness the better the docked interaction of the compounds.27 The results of the docking presented in this work are the best binding results out of the ten favorites predicted by Gold.
The general features from the Gold docking prediction (Table 6) confirm biologically active properties for all of the studied compounds. The best predicted target for HEAC is HDAC7, whilst for the studied complexes it was the TrxR target. Comparing the GOLDScore fitness values for the ligand and complexes showed that in some cases the ligand had a better interaction with the biomacromolecules (CatB, DNA-gyrase, HDAC7, TS) whilst the rest of the proteins (BRAF-kinase, rHA, RNR, TrxR, Top II) preferred the complexes. A fitness value comparison between studied complexes showed the general trend 3 > 2 > 1 in their binding ability towards proteins. Based on this result we can conclude that the zinc atom increases the binding ability of the HEAC to proteins better than the copper atom. Also the bromide ions of complex 2 are more efficient than chloride ions (1) in interaction toward biomacromolecules. The docking results of the interaction between the ligand and complex 1 with BRAF kinase protein are shown in the Fig. 12 and 13, respectively. In addition to the alcohol and amino moieties in the structures, the bonded and non-bonded chloride (1) and bromide (2) ions participate in hydrogen bonding with proteins.
Table 6 The calculated fitness values for the HEAC ligand and complexes 1–3
| |
BRAF-kinase |
CatB |
DNA-gyrase |
HDAC7 |
rHA |
RNR |
TrxR |
TS |
Top II |
| HEAC |
36.56 |
23.96 |
38.05 |
47.08 |
39.30 |
34.89 |
38.37 |
36.92 |
39.30 |
| Complex 1 |
36.89 |
21.89 |
34.57 |
38.52 |
38.30 |
33.80 |
44.26 |
34.24 |
34.06 |
| Complex 2 |
38.28 |
21.58 |
35.34 |
39.53 |
40.07 |
34.37 |
45.84 |
35.34 |
36.18 |
| Complex 3 |
37.80 |
22.70 |
37.00 |
40.06 |
37.06 |
35.18 |
43.16 |
35.38 |
41.24 |
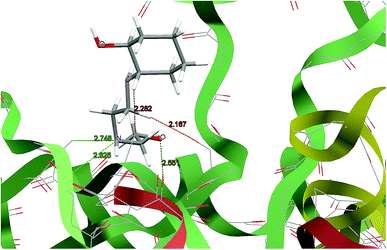 |
| | Fig. 12 Docking study results, showing the interaction between HEAC ligand and BRAF kinase protein. | |
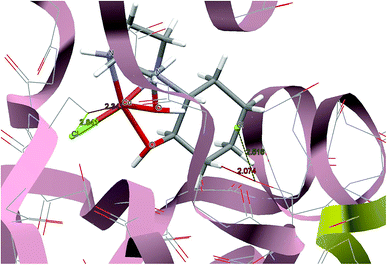 |
| | Fig. 13 Docking study results, showing the interaction between the complex 1 and BRAF kinase protein. | |
3.6. Biological screening
In addition to the docking studies, the in vitro cytotoxicities of HEAC and its complexes with copper (1, 2) and zinc (3) on the human cancer cell line K562 were evaluated experimentally. Results are expressed as IC50 values and are summarized in Table 7. The MTT assay showed that tested compounds exerted significant cytotoxic effects against K562 cell lines. For the studied compounds, the cytotoxic effects of the complex 1 is higher than the free ligand; showing that the biological activity of HEAC is increased upon binding to a copper atom containing a chloro ligand. This study also revealed that in addition to the nature of the coordinated metal atom, the type of the halide ions on the metal center affected the cytotoxicity of the compounds. Complexes 1 and 2 are isostructural, with the only difference being the halide atoms, however this resulted in the IC50 value of the complex containing the chloro ligand be about two times lower than the bromo ligand. The order of the cytotoxicity for all studied compounds is 1 > HEAC > 2 > 3. The IC50 values for different incubation time (24–72 h) are similar; showing that the cytotoxicity effect of all compounds is insensitive to the mentioned parameter.
Table 7 Concentrations of HEAC and its complexes 1–3 that induced a 50% decrease in K562 cell survival (expressed as IC50 (μg ml−1)). The compounds were incubated with cells for 24, 48 and 72 h. IC50 values are expressed as the mean ± SD determined from three independent experiments
| Compounds |
24 h |
48 h |
72 h |
| HEAC |
11.500 ± 0.430 |
11.750 ± 0.423 |
11.560 ± 0.541 |
| Complex 1 |
9.687 ± 0.174 |
9.471 ± 0.327 |
7.923 ± 0.031 |
| Complex 2 |
16.090 ± 0.194 |
16.612 ± 0.353 |
17.280 ± 0.211 |
| Complex 3 |
18.640 ± 0.323 |
18.510 ± 0.114 |
19.195 ± 0.141 |
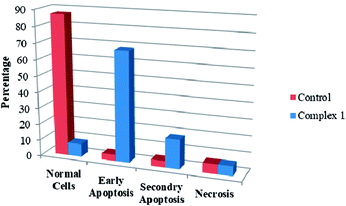
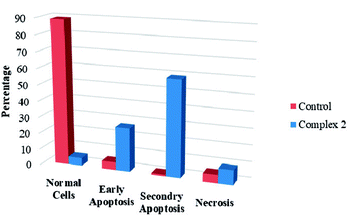
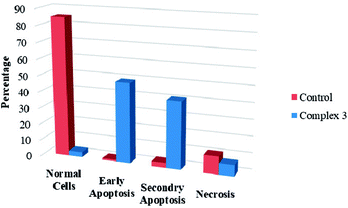
We also investigated the type of cell death induced by the compounds in K562 cells, based on the analysis of morphological characteristics of the cells after the double staining of with acridine orange (AO) and propidium iodide (PI) by fluorescence microscopy. Morphological features of apoptosis such as chromatin condensation, nuclear fragmentation and alterations in the size and the shape of cells were observed after 48 h treatment with IC50 concentrations of the compounds. Also, the features of late apoptosis were clearly expressed; the DNA was fragmented and stained orange and red. As shown in Table 8, the percentage of early apoptotic cells after treatment with complex 1 for 48 h is higher than the other compounds. Of the three tested compounds, the ability of complex 2 to induce secondary apoptosis in K562 cells is significant.
Table 8 Morphological changes of K562 cells after 48 h treatment with IC50 concentrations of complexes 1–3. The cells were stained with acridine orange and propidium iodide and examined by fluorescence microscope. The presence of early and late apoptosis along with the necrosis of K562 cells could be seen. (a): Complex 1, (b): Complex 2, (c): Complex 3
| Column graph |
Normal cells |
Cells after exposure to compounds |
 |
 |
 |
| (a) |
 |
 |
 |
| (b) |
 |
 |
 |
| (c) |
4. Conclusions
In this work, three complexes of copper and zinc, [Cu(HEAC)Cl]Cl (1), [Cu(HEAC)Br]Br (2), [Zn(HEAC)Cl2] (3); HEAC: 2-(2-(2-hydroxyethylamino)ethylamino)cyclohexanol, were synthesized and their spectral (IR, UV-Vis, 1H NMR) and structural (single crystal X-ray diffraction) properties were investigated. Also nanoparticles 1–3 for anticancer studies were prepared under ultrasonic irradiation and the particles size were analyzed by PXRD and SEM. In the isostructural complexes 1 and 2, the metal atom has square-pyramidal geometry which is formed by a tetradentate N2O2-donor (HEAC) and a chloro ligand. In this geometry, the axial Cu–O bond length is elongated with respect to the similar equatorial one. The zinc atom in complex 3 has the same coordination number to 1 and 2 but possesses a different geometry, trigonal bipyramidal. In this structure, the HEAC shows a different coordination manner (mer-N2O-donor) than the others. A structural study of the CSD database revealed that the common geometry around the penta-coordinated copper and zinc atoms is square-pyramidal (88% and 64%, respectively). In the crystal packing of the complexes, the halide ions participate in the formation of very different hydrogen bond motifs between adjacent complexes. The docking studies on all synthesized compounds showed that they are biologically active and can interact with the nine biomacromolecules (BRAF kinase, CatB, DNA gyrase, HDAC7, rHA, RNR, TrxR, TS and Top II). The best predicted targets for the free ligand is HDAC7, whilst for the complexes it is TrxR. The biological assay of the compounds on the human cancer cell line K562 confirmed the docking results. Among the complexes, the cytotoxic effects of complex 1 was higher than the free ligand with the order of cytotoxicity being 1 > HEAC > 2 > 3. Complexes 1 and 2 can induce early apoptosis (68%) and secondary apoptosis (59%) in K562 cells better than the others, respectively. The difference between theoretical (docking) and experimental (biological assay) results revealed that the anticancer activities of the studied compounds may be related to the other mechanisms than the interaction with these nine protein targets (for example DNA binding). The NBO analysis of the compounds revealed that among the hydrogen, carbon, nitrogen and oxygen atoms of the coordinated ligands, the hydrogen atoms act as electron donors and decrease the charge of the metal atom. The order of thermodynamic stability of the optimized complexes is determined as 1opt > 2opt > 3opt by DFT calculations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported financially by the Iran National Science Foundation (INSF).
References
- R. Yendapally and R. E. Lee, Bioorg. Med. Chem. Lett., 2008, 18, 1607–1611 CrossRef PubMed
 .
. - I. Declerck, B. Himpens, G. Droogmans and R. Casteels, Pflugers Arch., 1990, 417, 117–119 CrossRef PubMed .
- M. M. Weinberger, Pediatr. Clin. North Am., 1975, 22, 121–127 CrossRef PubMed .
- S. J. Kwon and S. Y. Ko, Tetrahedron Lett., 2002, 43, 639–641 CrossRef .
- W. H. Frishman, Circulation, 2003, 107, e117–e119 CrossRef PubMed .
- D. Nash, Clin. Cardiol., 1990, 13, 764–772 CrossRef PubMed .
- H.-X. Wei, D. Lu, V. Sun, J. Zhang, Y. Gu, P. Osenkowski, W. Ye, D. J. Selkoe, M. S. Wolfe and C. E. Augelli-Szafran, Bioorg. Med. Chem. Lett., 2016, 26, 2133–2137 CrossRef PubMed .
- D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–876 CrossRef PubMed .
- A. G. Myers, B. H. Yang, H. Chen, L. McKinstry, D. J. Kopecky and J. L. Gleason, J. Am. Chem. Soc., 1997, 119, 6496–6511 CrossRef .
- A. Russo and A. Lattanzi, Eur. J. Org. Chem., 2008, 2767–2773 CrossRef .
- A. J. Hunt, E. H. Sin, R. Marriott and J. H. Clark, ChemSusChem, 2010, 3, 306–322 CrossRef PubMed .
- J. E. Rainbolt, P. K. Koech, C. R. Yonker, F. Zheng, D. Main, M. L. Weaver, J. C. Linehan and D. J. Heldebrant, Energy Environ. Sci., 2011, 4, 480–484 RSC .
- T. Werner and N. Tenhumberg, J. CO2 Util., 2014, 7, 39–45 CrossRef .
- K. C. Rao, Y. Arun, K. Easwaramoorthi, C. Balachandran, T. Prakasam, T. E. Yuvaraj and P. Perumal, Bioorg. Med. Chem. Lett., 2014, 24, 3057–3063 CrossRef PubMed .
- S. Vanguru, L. Jilla, Y. Sajja, R. Bantu, L. Nagarapu, J. B. Nanubolu, B. Bhaskar, N. Jain, S. Sivan and V. Manga, Bioorg. Med. Chem. Lett., 2017, 27, 792–796 CrossRef PubMed .
- H. Takahashi, M. Hattori, M. Chiba, T. Morimoto and K. Achiwa, Tetrahedron Lett., 1986, 27, 4477–4480 CrossRef .
- F. A. Davis, M. S. Haque and R. M. Przeslawski, J. Org. Chem., 1989, 54, 2021–2024 CrossRef .
- G. Li, H. T. Chang and K. B. Sharpless, Angew. Chem., 1996, 108, 449–452 CrossRef .
- G. B. Fisher, C. T. Goralski, L. W. Nicholson, D. L. Hasha, D. Zakett and B. Singaram, J. Org. Chem., 1995, 60, 2026–2034 CrossRef .
- M. Hakimi, Z. Mardani, K. Moeini and M. A. Fernandes, J. Coord. Chem., 2012, 65, 2221–2233 CrossRef .
- T. Baskaran, A. Joshi, G. Kamalakar and A. Sakthivel, Appl. Catal., A, 2016, 524, 50–55 CrossRef .
- C. Shi, C. Ren, E. Zhang, H. Jin, X. Yu and S. Wang, Tetrahedron, 2016, 72, 3839–3843 CrossRef .
- M. Anjomshoa, M. Torkzadeh-Mahani, E. Dashtrazmi and M. Adeli-Sardou, J. Fluoresc., 2016, 26, 545–558 CrossRef PubMed .
- M. Sahlabadi, M. Daryanavard, H. Hadadzadeh and Z. Amirghofran, J. Mol. Struct., 2018, 1155, 450–456 CrossRef .
- M. Shebl, M. Saif, A. I. Nabeel and R. Shokry, J. Mol. Struct., 2016, 1118, 335–343 CrossRef .
- F. Marandi, K. Moeini, F. Alizadeh, Z. Mardani, C. K. Quah and W.-S. Loh, Z. Naturforsch., 2018, 73(6), 369–375 Search PubMed ,.
- A. A. Adeniyi and P. A. Ajibade, Molecules, 2013, 18, 3760–3778 CrossRef PubMed .
- C. B. Lozzio and B. B. Lozzio, Blood, 1975, 45, 321–334 Search PubMed .
- M. Hakimi, Z. Mardani, K. Moeini, F. Mohr and M. A. Fernandes, Polyhedron, 2014, 67, 27–35 CrossRef .
- A. K. Abu Al-Nasr and R. M. Ramadan, Spectrochim. Acta, Part A, 2013, 105, 14–19 CrossRef PubMed .
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed .
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed .
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef .
- M. N. Burnett and C. K. Johnson, Ortep-III, Report ORNL-6895, Oak Ridge National Laboratory, Oak Ridge, Tennessee, U.S., 1996 Search PubMed .
- G. Bergerhof, M. Berndt and K. Brandenburg, J. Res. Natl. Inst. Stand. Technol., 1996, 101, 221–225 CrossRef PubMed .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed .
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef .
- A. Gavezzotti, Acc. Chem. Res., 1994, 27, 309–314 CrossRef .
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef PubMed .
- M. Hakimi, Z. Mardani, K. Moeini, F. Mohr and M. A. Fernandes, Polyhedron, 2014, 67, 27–35 CrossRef .
- A. W. Addison, T. Rao, J. Reedjik, J. V. Rijn and G. Verschoor, Dalton Trans., 1984, 1349 RSC .
- M. Hakimi, Z. Mardani, K. Moeini and F. Mohr, Polyhedron, 2015, 102, 569–577 CrossRef .
- S. Roy, P. Mitra and A. K. Patra, Inorg. Chim. Acta, 2011, 370, 247–253 CrossRef .
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef .
- M. Hakimi, Z. Mardani, K. Moeini, E. Schuh and F. Mohr, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 272 Search PubMed .
- M. Hakimi, Z. Mardani, K. Moeini, E. Schuh and F. Mohr, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 267 Search PubMed .
- B. F. Hoskins and F. D. Whillans, J. Chem. Soc. A, 1970, 123 RSC .
- P. J. Toscano, K. J. Fordon, D. Macherone, S. Liu and J. Zubieta, Polyhedron, 1990, 9, 2375–2383 CrossRef .
- Y. M. Lee, E. S. Kim, H. J. Kim, H. J. Choi, Y. I. Kim, S. K. Kang and S. N. Choi, Dalton Trans., 2009, 126–133 RSC .
- S. Sarkar, S. Dey, T. Mukherjee, E. Zangrando, M. G. B. Drew and P. Chattopadhyay, J. Mol. Struct., 2010, 980, 94–100 CrossRef .
- M. Hakimi, Z. Mardani, K. Moeini, F. Mohr and M. A. Fernandes, Polyhedron, 2014, 67, 27–35 CrossRef .
- M. Hakimi, Z. Mardani, K. Moeini, N. Feizi and F. Mohr, J. Coord. Chem., 2017, 70, 1247–1259 CrossRef .
- M. Hakimi, K. Moeini, Z. Mardani and F. Mohr, Polyhedron, 2014, 70, 92–100 CrossRef .
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef .
- A. Gavezzotti and G. Filippini, J. Phys. Chem., 1994, 98, 4831–4837 CrossRef .
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Reza Kazemshoar-Duzduzania,
Keyvan Moeini
*a,
Reza Kazemshoar-Duzduzania,
Keyvan Moeini