
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptically active distorted cyclic triptycenes: chiral stationary phases for HPLC†
Tomoyuki Ikai *a,
Naoya Nagataa,
Seiya Awataa,
Yuya Wadaa,
Katsuhiro Maedaa,
Motohiro Mizunoa and
Timothy M. Swager*b
*a,
Naoya Nagataa,
Seiya Awataa,
Yuya Wadaa,
Katsuhiro Maedaa,
Motohiro Mizunoa and
Timothy M. Swager*b
aGraduate School of Natural Science and Technology, Kanazawa University, Kakuma-machi, Kanazawa 920-1192, Japan. E-mail: ikai@se.kanazawa-u.ac.jp
bDepartment of Chemistry, Massachusetts Institute of Technology (MIT), 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: tswager@mit.edu
First published on 5th June 2018
Abstract
A pair of optically active triptycene derivatives ((R,R)- and (S,S)-8) with a distorted cyclic structure were synthesized via an intramolecular etherification and evaluated as a novel chiral selector for high-performance liquid chromatography. The (R,R)- and (S,S)-8-based chiral stationary phases (CSPs) were found to be particularly effective in the resolution of axially chiral biaryl compounds. The importance of the distorted cyclic structure present in 8 for chiral recognition was demonstrated by comparisons with the corresponding non-cyclic model compound ((R,R)-9), which did not display enantioselectivity in the separation of the test racemates.
Enantiodiscrimination is present in all living organisms, and different enantiomeric compounds often show different physiological activities in a biological system.1–4 The systematic investigation of biological properties of individual enantiomers is essential, particularly in the development of new chiral drugs. Therefore, the efficient preparation of both enantiopure isomers and the precise determination of the enantiomeric excess (ee) of chiral compounds is of ever increasingly importance.5–9 Direct resolution by high-performance liquid chromatography (HPLC) using a chiral stationary phase (CSP) is a powerful technique for this purpose as a result of its simplicity, reproducibility and scalability.10–15 A wide variety of CSPs consisting of optically active molecules or polymers have been developed to date.16–19 The complexity and diversity of chiral pharmaceuticals and related compounds is increasing daily and correspondingly current methods do not always provide adequate methods for the separation on CSPs, as a result the parallel development of novel chiral selectors is required to meet this expanding diversity.
Triptycene is a paddle-wheel shaped aromatic hydrocarbon with D3h-symmetry, consisting of three benzene rings in a [2.2.2] ring structure with two bridgehead carbons.20–25 Although the unsubstituted triptycene scaffold is achiral, many derivatives can be inherently chiral depending on the substitution pattern.26–29 In such cases, the bridgehead carbons in triptycene units become asymmetric centers. Recently, chiral triptycenes showing enantioselective recognition30 and asymmetric catalytic activities31 have been developed. We have reported that optically active triptycene-based supramolecular systems32 and polymers33 can be designed as circularly polarized luminescence materials. Considering the unique structural features of triptycene, including a three-dimensional framework with very limited conformational freedom, we anticipated that promising chiral recognition materials can be developed through appropriate modification of triptycene.
To create novel functional materials using a chiral triptycene framework, we designed and synthesized a pair of enantiomeric chiral selectors ((R,R)- and (S,S)-8, Scheme 1), which have a cyclic structure distorted by the chirality of triptycene unit. The chiral recognition abilities of optically active triptycenes were evaluated as CSPs for HPLC separations. We demonstrate that a distorted cyclic structure in (R,R)- and (S,S)-8 is particularly important in the efficient enantioseparation of axially chiral biaryl compounds.
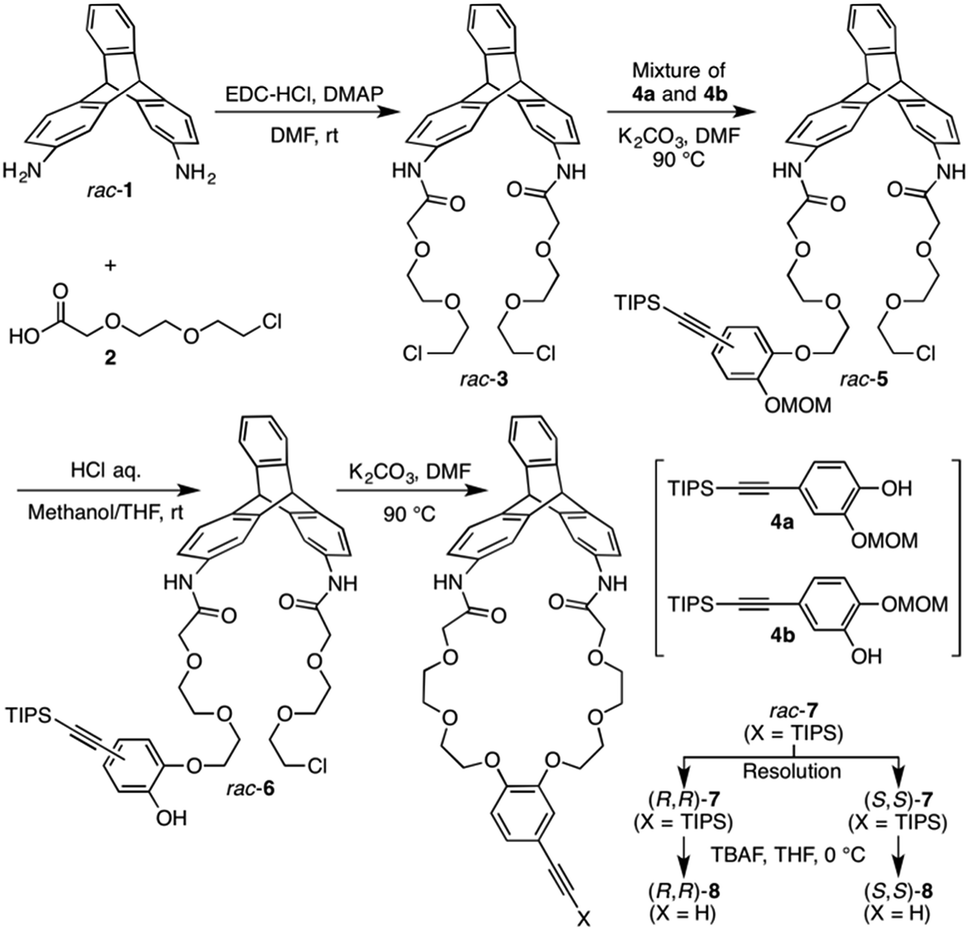 | ||
| Scheme 1 Synthesis of optically active triptycene derivatives ((R,R)- and (S,S)-8) with a distorted cyclic structure. | ||
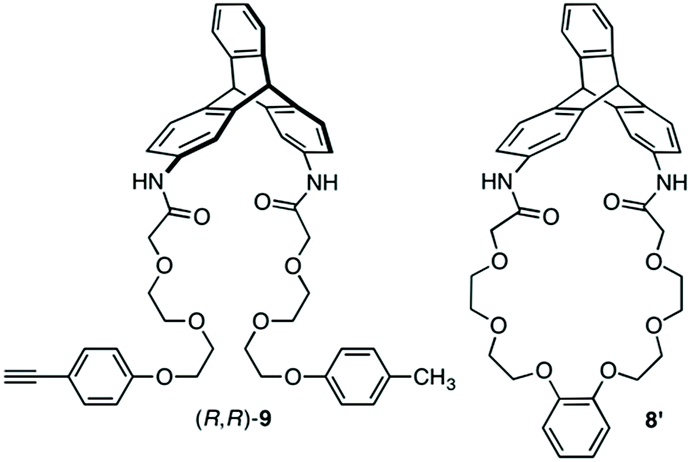
The synthetic route to the target optically active compound 8 is shown in Scheme 1. Key intermediate rac-2,6-diaminotriptycene (1) was amidated with 2-[2-(2-chloroethoxy)ethoxy]acetic acid (2), then reacted with mono-protected catechol derivatives (4a and 4b) to give rac-5, containing two regioisomers differing in the substitution position of the protected ethynyl group. Because these isomers are converted into an identical product (rac-7) in the next two reactions, the resulting regioisomeric mixture was carried forward to the next step without separation. After deprotecting the methoxymethyl (MOM) ether of rac-5 with HCl, an intramolecular etherification of rac-6 was conducted under dilute conditions, affording cyclic compound rac-7 in 78% yield. (R,R)- and (S,S)-7 were successfully obtained by resolution of rac-7 with preparative HPLC on a chiral column (Daicel Chiralpak IG) (Fig. 1A). We confirmed that the two fractionated components had enantiomeric relationships with each other by circular dichroism (CD) spectroscopy (Fig. 1D) and that they are pure with ee values > 96% (Fig. 1B and C). The absolute configurations of the first- and second-eluted components in Fig. 1A were assigned as 9R,10R and 9S,10S, respectively. This assignment is based on the comparison of the CD spectral patterns of optically active 1, prepared from the first-eluted components in Fig. 1A and a reported triptycene derivative with a 9R,10R configuration ((R,R)-1a in Fig. S1, ESI†).34 The subsequent deprotection of the triisopropylsilyl group of (R,R)- or (S,S)-7 gave the corresponding optically active 8 bearing an ethynyl cross-linkable group. As expected, the resulting 8 maintained a high enantiopurity of >96% ee (Fig. S2, ESI†). We also prepared a corresponding non-cyclic compound ((R,R)-9, Chart 1) bearing diethylene glycol moieties for comparative studies (Scheme S1C and Fig. S3, ESI†).
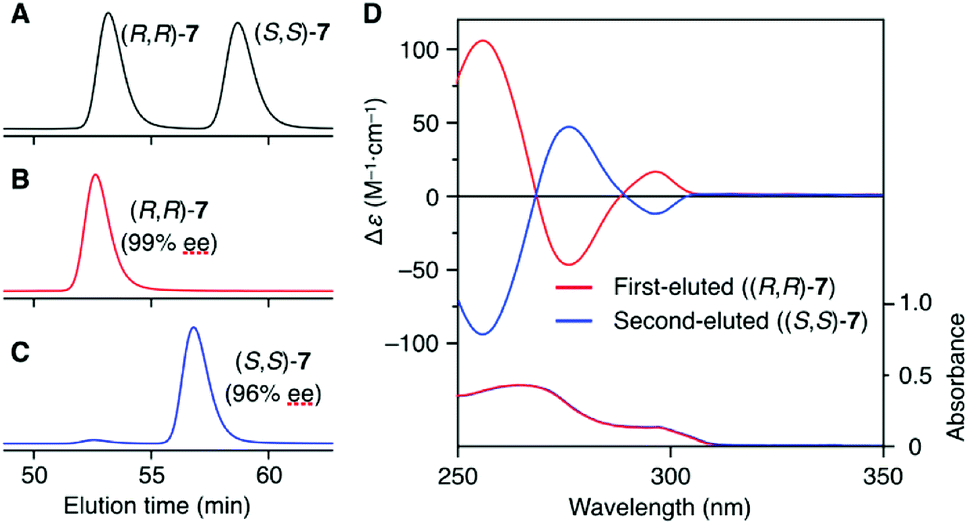 | ||
| Fig. 1 Elution profiles of rac-7 (A), (R,R)-7 (B) and (S,S)-7 (C) on Chiralpak IG (column, 25 cm × 0.46 cm (i.d.); eluent, hexane/ethyl acetate (3/2, v/v); flow rate, 0.4 mL min−1; temperature, ca. 20 °C). The chromatograms depict UV traces recorded at 254 nm. (D) CD and absorption spectra of the first- (red line) and second-eluted (blue line) components in chloroform at 25 °C. [7] = 1.0 × 10−4 M. | ||
 | ||
| Chart 1 | ||
Scheme 2 outlines the immobilization scheme on silica. The Huisgen 1,3-dipolar cycloaddition reaction between the azide-functionalized silica gel (A-silica) and the ethynyl group of 8 or 9 was affected using copper(I) iodide as the catalyst in a mixture of N,N-dimethylformamide and triethylamine.35 This afforded a chiral packing material containing a triptycene derivative immobilized onto the silica surface through the 1,2,3-triazole linker. The presence of triptycene derivatives immobilized on the silica gel was confirmed by infrared spectroscopy (Fig. S4–6, ESI†), and their contents in the modified silica gel were determined to be approximately 7 wt% by thermogravimetric analysis, from which the number of chiral selectors per unit weight of the modified silica gel was calculated to be ca. 6 × 1019 g−1. Based on the crystal structure of an analogue of 8 (8′, Chart 1), which did not contain an ethynyl group, the area occupied per single chiral selector is estimated to be ca. 1.5 × 10−18 m2 (Fig. S7A, ESI†). Thus, the area covered with chiral selectors per unit weight of the modified silica gel was determined to be ca. 90 m2 g−1 (Fig. S7B, ESI†). Considering that the specific surface area of the silica gel used is ca. 100 m2 g−1, most of the silica surface is covered with the chiral selectors. This reasoning is predicated on the assumption that the chiral selectors are immobilized onto the silica surface as a monolayer. To investigate the chiral recognition ability as a CSP for HPLC, the resultant materials were packed into stainless-steel columns using the slurry method.36 Their resolution abilities for six racemic compounds (10 and 11a–e, Chart 2) were evaluated using a hexane/ethanol mixture as an eluent. The resolution results are summarized in Table 1, and Fig. 2A shows the representative chromatograms for the resolution of rac-10 on the (R,R)-8-based CSP, in which the enantiomers were eluted at retention times of t1 and t2. The hold-up time (t0) was estimated to be 6.43 min and the retention factors, k1 [= (t1 − t0)/t0] and k2 [= (t2 − t0)/t0], were calculated to be 1.54 and 1.76, respectively, leading to a separation factor α (= k2/k1) of 1.14.
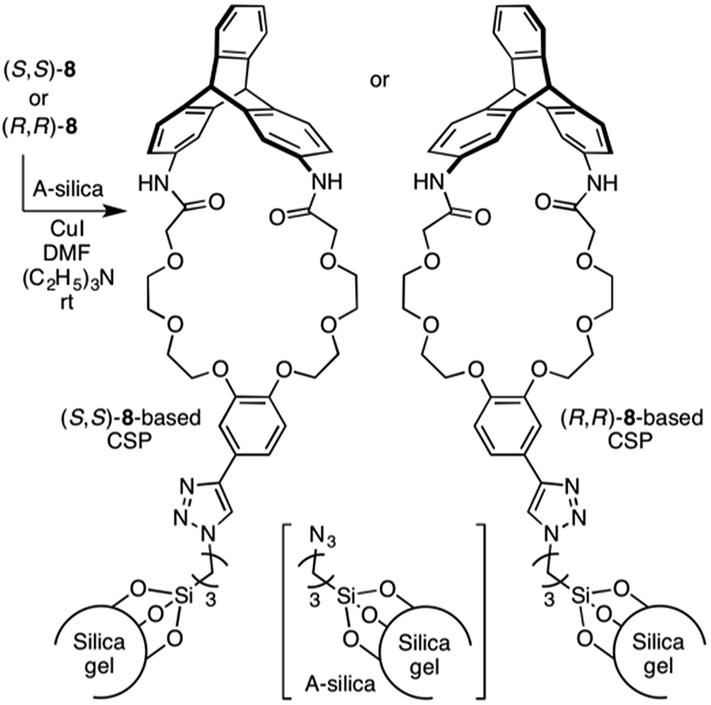 | ||
| Scheme 2 Immobilization of (R,R)- and (S,S)-8 onto A-silica. | ||
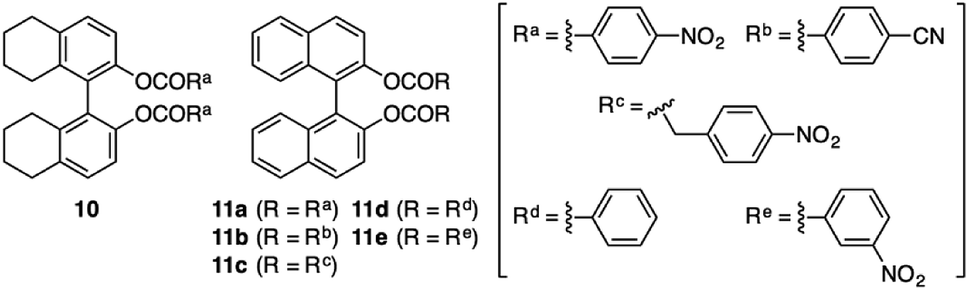 | ||
| Chart 2 | ||
| CSP | (R,R)-8 | (S,S)-8 | (R,R)-9 | |||
|---|---|---|---|---|---|---|
| Racemate | k1 | α | k1 | α | k1 | α |
| a Column: 25 cm × 0.20 cm (i.d.). Eluent: hexane/ethanol (97/3, v/v). Flow rate: 0.20 mL min−1. Temperature: 20 °C. The characters in parentheses represent the absolute configuration of the first-eluted enantiomer. | ||||||
| 10 | 1.54 (S) | 1.14 | 1.40 (R) | 1.13 | 1.14 | 1.00 |
| 11a | 5.82 (S) | 1.09 | 5.87 (R) | 1.08 | 4.87 | 1.00 |
| 11b | 7.49 (S) | 1.14 | 7.24 (R) | 1.11 | 6.74 | 1.00 |
| 11c | 8.09 | 1.00 | 7.48 | 1.00 | 6.56 | 1.00 |
| 11d | 1.27 | 1.00 | 1.22 | 1.00 | 0.71 | 1.00 |
| 11e | 9.66 | 1.00 | 9.48 | 1.00 | 7.22 | 1.00 |
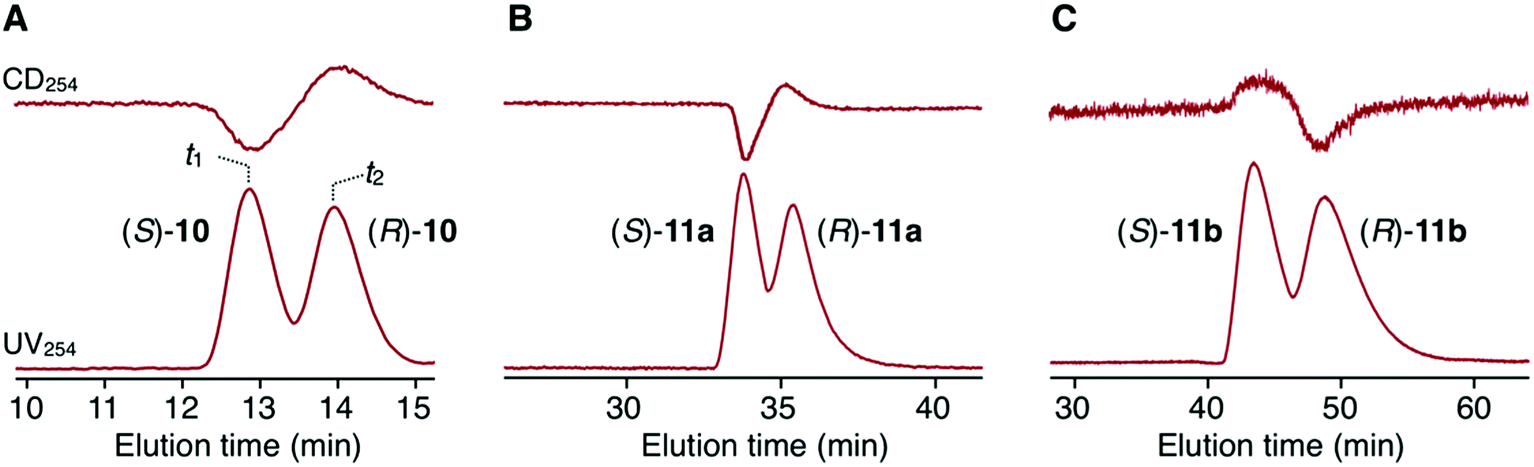 | ||
| Fig. 2 Chromatograms for the resolution of 10 (A), 11a (B) and 11b (C) on the (R,R)-8-based CSP (column dimensions, 25 × 0.20 cm (i.d.); eluent, hexane/2-propanol (97/3, v/v); flow rate, 0.2 mL min−1; temperature, 20 °C). The lower and upper chromatograms depict the UV and CD traces recorded at 254 nm, respectively. | ||
The (R,R)-8-based CSP displayed an efficient chiral recognition ability toward axially chiral biaryl compounds (10, 11a and 11b; Fig. 2). In all cases, the (S)-isomers eluted first, followed by the (R)-isomers, indicating that the (R,R)-8-based CSP interacts preferentially with the (R)-isomers rather than the corresponding antipodes. Conversely, 11d and 11e, which did not have a substituent at the para-position of the phenyl ring, were not resolved on the (R,R)-8-based CSP. It appears that the para-substituted nitro and cyano groups in 10, 11a and 11b act as key interaction sites with (R,R)-8. The k1 values for 10, 11a and 11b gradually decreased with an increase in the eluent polarity accompanied by an increase in the ethanol content (Table S1, ESI†), and electrostatic interactions, including hydrogen bonding with the amide groups and dipole–dipole interaction with the ether groups, appear to have a dominant role in the chiral recognition on (R,R)-8. When the NMR spectrum of (R,R)-8 in CDCl3 (30 mM) was measured in the presence of an equimolar amount of rac-10 (Fig. S8, ESI†), the methylene proton resonances (ca. 3.5–3.9 ppm) derived from the ethylene glycol residues were slightly shifted to downfield compared with those of pure (R,R)-8. Alternatively, no clear difference was observed in the NH resonance region around 8.7 ppm. These observations indicate that the ether groups arranged along the distorted cyclic structure in (R,R)-8 may be more important enantiodiscrimination sites than the amide groups. Fig. 3 displays the X-ray crystal structure of 8′ (Chart 1).37 The X-ray crystallographic analysis revealed that the NH hydrogens are interacting with the ether oxygens through intramolecular hydrogen bonding. This suggests a minimal contribution of the NH group in (R,R)-8 to the molecular recognition. Our understanding of the basis for separation is limited at present. The exact chiral discrimination mechanism for (R,R)-8 will require additional investigations, including 2D NMR and computational simulation studies, which are ongoing. No resolution was observed for racemic 11c, which has a flexible methylene spacer between the ester and nitrophenyl groups, with a (R,R)-8-based CSP. Here the increased conformational freedom of the analyte probably makes it difficult for (R,R)-8 to discriminate between the chirality of (R)- and (S)-11c. We also observed that resolution with the CSP is dependent on the column temperature, with more efficient resolutions being achieved at lower temperatures (Table S2 and Fig. S9, ESI†). Particularly, the enantiomers of 10 were almost completely resolved at 0 °C, with an α value of 1.17 (Fig. S9A, ESI†). Fig. S10† shows the CD spectra of (R,R)-8 measured in chloroform at various temperatures suggesting that the molecular conformations change. (R,R)-8 displayed more intense cotton effects at lower temperatures, consistent with the suppression of thermal fluctuations. Consistent with this CD analysis, the temperature-dependent resolution ability of the (R,R)-8-based CSP is likely the result of structural variations in the chiral selector. As expected based on the enantiomeric relationship, the (S,S)-8-based CSP showed virtually identical resolution ability to (R,R)-8-based CSP, except for the elution order (Table 1 and S3, ESI†). In sharp contrast to these results, the non-cyclic (R,R)-9-based CSP did not show any recognition ability for the racemates studied (Table 1 and S4, ESI†), even though the k1 values were comparable to those of the (R,R)-8-based CSP. These results demonstrate that the asymmetric orientation of the ether groups within the distorted cyclic structure plays a critical role in the resolution of 10, 11a and 11b (to observe the conformation clearly see Movie S1, ESI†). As was the case with the previously reported chiral macrocyclic receptors,38–40 a favorable enantiodiscrimination site is afforded to 8 through the cyclic formation and hence the performance of this motif is an important lead for future CSP development.
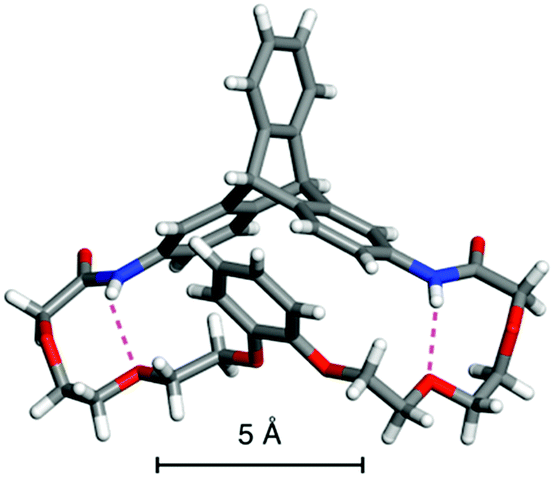 | ||
| Fig. 3 X-ray crystal structure of 8′ represented by a stick model. The hydrogen bonds are indicated by purple dashed lines. | ||
In summary, we have demonstrated new triptycene-based CSPs, which display apparent chiral recognition abilities toward a series of axially chiral biaryl compounds. A comparative study using a non-cyclic model compound ((R,R)-9) revealed that the cyclic structure of (R,R)-8 is critical for discriminating chiral compounds. Further investigations to elucidate the chiral recognition mechanism of a triptycene-based CSP and to create improved materials for chiral resolution are in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (R2702) and the Grants-in-Aid for Scientific Research (C), Grant No. 17K05875. Work at MIT was supported in part by the National Science Foundation, DMR-1410718. The authors thank Prof. Shigehisa Akine and Dr Yoko Sakata (Kanazawa University) for the single crystal X-ray structure analysis.Notes and references
- E. J. Ariëns, Med. Res. Rev., 1986, 6, 451–466 CrossRef.
- B. Kasprzyk-Hordern, Chem. Soc. Rev., 2010, 39, 4466–4503 RSC.
- W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760–770 CrossRef PubMed.
- Q. Zhou, L.-S. Yu and S. Zeng, Drug Metab. Rev., 2014, 46, 283–290 CrossRef PubMed.
- O. McConnell, A. Bach, C. Balibar, N. Byrne, Y. Cai, G. Carter, M. Chlenov, L. Di, K. Fan, I. Goljer, Y. He, D. Herold, M. Kagan, E. Kerns, F. Koehn, C. Kraml, V. Marathias, B. Marquez, L. McDonald, L. Nogle, C. Petucci, G. Schlingmann, G. Tawa, M. Tischler, R. T. Williamson, A. Sutherland, W. Watts, M. Young, M.-Y. Zhang, Y. Zhang, D. Zhou and D. Ho, Chirality, 2007, 19, 658–682 CrossRef PubMed.
- K. De Klerck, D. Mangelings and Y. Vander Heyden, J. Pharm. Biomed. Anal., 2012, 69, 77–92 CrossRef PubMed.
- D. Speybrouck and E. Lipka, J. Chromatogr. A, 2016, 1467, 33–55 CrossRef PubMed.
- D. C. Patel, M. F. Wahab, D. W. Armstrong and Z. S. Breitbach, J. Chromatogr. A, 2016, 1467, 2–18 CrossRef PubMed.
- H. Leek, L. Thunberg, A. C. Jonson, K. Ohlen and M. Klarqvist, Drug Discovery Today, 2017, 22, 133–139 CrossRef PubMed.
- W. H. Pirkle and T. C. Pochapsky, Chem. Rev., 1989, 89, 347–362 CrossRef.
- D. R. Taylor and K. Maher, J. Chromatogr. Sci., 1992, 30, 67–85 Search PubMed.
- E. R. Francotte, J. Chromatogr. A, 2001, 906, 379–397 CrossRef PubMed.
- T. Nakano, J. Chromatogr. A, 2001, 906, 205–225 CrossRef PubMed.
- C. Yamamoto and Y. Okamoto, Bull. Chem. Soc. Jpn., 2004, 77, 227–257 CrossRef.
- D. C. Patel, M. F. Wahab, D. W. Armstrong and Z. S. Breitbach, J. Chromatogr. A, 2016, 1467, 2–18 CrossRef PubMed.
- Y. Okamoto and T. Ikai, Chem. Soc. Rev., 2008, 37, 2593–2608 RSC.
- T. J. Ward and K. D. Ward, Anal. Chem., 2012, 84, 626–635 CrossRef PubMed.
- G. K. E. Scriba, J. Chromatogr. A, 2016, 1467, 56–78 CrossRef PubMed.
- R. Sardella, F. Ianni, M. Marinozzi, A. Macchiarulo and B. Natalini, Curr. Med. Chem., 2017, 24, 796–817 CrossRef PubMed.
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649–2653 CrossRef.
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1189 CrossRef PubMed.
- J.-S. Yang and J.-L. Yan, Chem. Commun., 2008, 1501–1512 RSC.
- J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301–3315 RSC.
- L. Zhao, Z. Li and T. Wirth, Chem. Lett., 2010, 39, 658–667 CrossRef.
- Y. X. Ma, Z. Meng and C. F. Chen, Synlett, 2015, 26, 6–30 CrossRef.
- N. Harada, Y. Tamai and H. Uda, J. Am. Chem. Soc., 1980, 102, 506–511 CrossRef.
- N. Harada, Y. Tamai and H. Uda, J. Org. Chem., 1984, 49, 4266–4271 CrossRef.
- J. Daub, L. Jakob and J. Salbeck, Chem. Ber., 1988, 121, 2187–2194 CrossRef.
- K. Shahlai, H. Hart and A. Bashirhashemi, J. Org. Chem., 1991, 56, 6912–6916 CrossRef.
- G.-W. Zhang, P.-F. Li, Z. Meng, H.-X. Wang, Y. Han and C.-F. Chen, Angew. Chem., Int. Ed., 2016, 55, 5304–5308 CrossRef PubMed.
- F. K.-C. Leung, F. Ishiwari, Y. Shoji, T. Nishikawa, R. Takeda, Y. Nagata, M. Suginome, Y. Uozumi, Y. M. A. Yamada and T. Fukushima, ACS Omega, 2017, 2, 1930–1937 CrossRef.
- T. Ikai, Y. Wada, S. Awata, C. Yun, K. Maeda, M. Mizuno and T. M. Swager, Org. Biomol. Chem., 2017, 15, 8440–8447 Search PubMed.
- T. Ikai, T. Yoshida, S. Awata, Y. Wada, K. Maeda, M. Mizuno and T. M. Swager, ACS Macro Lett., 2018, 7, 364–369 CrossRef.
- The absolute configuration of (R,R)-1a had been determined by the single crystal X-ray structure analysis..
- T. Ikai, S. Awata, T. Kudo, R. Ishidate, K. Maeda and S. Kanoh, Polym. Chem., 2017, 8, 4190–4198 RSC.
- Y. Okamoto, M. Kawashima and K. Hatada, J. Chromatogr. A, 1986, 363, 173–186 CrossRef.
- Because the single crystal preparation of (R,R)-8 could not be achieved, we prepared the racemic crystal from rac-8′ instead. Only the crystal structure of the (R,R)-isomer is shown in Fig. 3..
- X. X. Zhang, J. S. Bradshaw and R. M. Izatt, Chem. Rev., 1997, 97, 3313–3362 CrossRef PubMed.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef PubMed.
- M. H. Hyun, J. Chromatogr. A, 2016, 1467, 19–32 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization of triptycene derivatives and additional spectroscopic, chromatographic and computational data. CCDC 1839125. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra04434a |
| This journal is © The Royal Society of Chemistry 2018 |