
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTAK1 inhibition by natural cyclopeptide RA-V promotes apoptosis and inhibits protective autophagy in Kras-dependent non-small-cell lung carcinoma cells
Jianhong Yanga,
Tao Yanga,
Wei Yana,
Dan Lia,
Fang Wanga,
Zhe Wangb,
Yingjie Guoc,
Peng Baia,
Ninghua Tan *b and
Lijuan Chen*a
*b and
Lijuan Chen*a
aCancer Center, West China Hospital, Sichuan University, Collaborative Innovation Center for Biotherapy, Chengdu, China. E-mail: chenlijuan125@163.com
bSchool of Traditional Chinese Pharmacy, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing, China. E-mail: nhtan@cpu.edu.cn
cDepartment of Cardiology, The Second Affiliated Hospital of Guangdong Medical University, Zhanjiang, Guangdong, China
First published on 27th June 2018
Abstract
TAK1 kinase is required for the survival of Kras-dependent non-small-cell lung carcinoma (NSCLC) cells. Here, we report that the inhibition of TAK1 by a small natural cyclopeptide (RA-V) can promote apoptosis and inhibit protective autophagy in Kras-dependent NSCLC cells. Using short hairpin RNAs to deplete K-Ras, we identified H441 and H358 cells as Kras-dependent NSCLC cells which require protective basal autophagy for cell viability. We found that RA-V could selectively kill and induce apoptosis in H441 and H358 cells but had little effect on A549 and H460 (Kras-independent) cells. Furthermore, RA-V could inhibit basal autophagy in H441 and H358 cells. Mechanistic studies further showed that RA-V inhibits the level of TAK1 phosphorylation by binding directly to TAK1, resulting in the inhibition of the autophagy-related TAK1–AMPK–mTOR pathway. In addition, we found that RA-V could inhibit TAK1–P70S6K interaction, which may also inhibit basal autophagy. Our study shows that RA-V acts as an inducer of apoptosis and inhibitor of autophagy via the inhibition of TAK1 and provides the first example of TAK1 inhibition as a potential therapeutic strategy to promote apoptosis and inhibit protective autophagy in Kras-dependent NSCLC.
1. Introduction
Aberrant Kras activation frequently occurs in non-small-cell lung carcinoma. However, Kras mutation usually arises in the treatment of refractory tumors and remains a significant challenge in terms of treatment.1 For example, drug resistance often develops in cases of lung cancer featuring Kras mutation and treated with EGFR inhibitors.2 Thus, patients suffering from lung cancer with aberrant Kras activation will not benefit from targeted-EGFR treatment. TAK1 is activated to enhance Wnt-dependent transcription in Kras dependent cells, and is an absolute requirement for the survival of Kras-dependent cells. The inhibition of TAK1 is known to induce apoptosis in Kras-dependent cells.3 Consequently, inducing apoptosis-dependent cancer cell death in Kras-dependent cells by targeting TAK1 may represent an alternative option for the treatment of Kras-activated cancer.Autophagy is a catabolic pathway which is used by cells to remove damaged organelles, lipids and misfolded proteins. During starvation or other forms of stress, it regenerates energy from an internal source of nutrients to maintain energy homeostasis and thus ensure that cells survive.4 In Kras-activated cells, the level of basal autophagy is increased by mediating the TAK1–AMPK–mTOR pathway, which plays an important role in maintaining cell viability and tumorigenesis.5–7 In addition, TAK1 can activate autophagy by inhibiting the phosphorylation of p70S6 kinase1 (P70S6K) by blocking the interaction between raptor and P70S6K.8 These lines of evidence implied that the inhibition of protective autophagy must be taken into account in Kras-activated cancer treatment, and that targeting TAK1 may represent a reasonable treatment consideration.
TAK1, a serine/threonine kinase, plays an important role in the cascades of cellular responses regulated by a range of cytokines including TGF-β, IL-1 and TLR.9,10 Many intra-cellular kinases are activated by TAK1, including p38 MAPK, JNK and IKK. The transcription factor, AP-1, is controlled by p38 MAPK and JNK, while NF-kB is activated by IKK. Based on these functions, it is clear that TAK1 is a primary regulator of cell inflammatory responses, survival and differentiation. TAK1 also regulates the programmed cell death pathway, apoptosis and autophagy. TAK1 has been found to regulate the levels of apoptosis in a variety of cell lines and tissues.3,11,12 Furthermore, the activation of TAK1 can result in protective or cytotoxic autophagy in different types of cancer cells and tissues.5,8,13
RA-V (deoxybouvardin), a cyclopeptide, which was first isolated from Rubia cordifolia, shows strong anti-inflammatory and anti-cancer activities in various types of cell.14–18 Our previous study revealed that RA-V could bind directly to TAK1 and inhibit its kinase activity; it could then inhibit the downstream inflammation-related pathway indicating that RA-V is potent a TAK1 inhibitor.19 In the present study, we show that inhibition of TAK1 by RA-V can promote apoptosis and inhibit protective autophagy in Kras-dependent NSCLC cells.
2. Methods
2.1 Materials
RA-V (purity ≥ 99%) was prepared as described previously and identified by MS and NMR spectroscopies.15 RA-V was dissolved in DMSO to a concentration of 20 mM and was then preserved at −20 °C. RPMI1640, fetal bovine serum and lipofectamine 2000 were purchased from Life Technologies. Chloroquine (Sigma), rapamycin (Sigma), Z-VAD-FMK (Sigma) and acridine orange (Sigma) were dissolved in DMSO. The GFP-LC3 plasmid was obtained as a gift from Professor Canhua Huang (Sichuan University). Kras and P70S6K shRNA were purchased from ORIGENE. The following antibodies were used: caspase-3, Kras, BCL-2, BCL-XL, Beclin1 and LC3B (Cell Signaling Technology); Atg7, Beclin1, p-P70S6K, P70S6K, p-4E-BP1, 4E-BP1, p-Akt, Akt, p-mTOR, mTOR, p-AMPK and AMPK (ABCOM); Flag, Myc and β-actin (Santa Cruz).2.2 Cell lines
The NSCLC cells A549, H460, H441 and H358 were obtained from the American Type Culture Collection (ATCC). Cells were cultured in RPMI1640 1640 (10% fetal bovine serum (FBS), 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin) at 37 °C in an atmosphere of 5% CO2.2.3 MTT assay, annexin V-FITC/PI apoptosis assay, western blotting, GFP-LC3 plasmid transfection and TUNEL assay
These assays were performed in accordance with previously-published laboratory protocols.20,212.4 Characterization of Kras-dependent cell lines
NSCLC cells (A549, H460, H441 and H358) were seeded onto six-well plates for 24 h and then infected with increasing amounts of lentivirus expressing either a control or different doses of K-Ras shRNAs (control: NM_004985.3-570s1c1: CTCAGGACTTAGCAAGAAGTT; Kras: NM_033360.2-269s1c1: GACGAATATGATCCAACAATA) for a further period of 24 h. Cells were then treated with 1 μg ml−1 of puromycin and incubated for another 48 h. Cell viability was assessed by the MTT assay and the expression of Kras and caspase 3 were detected by western blotting.2.5 mRNA silencing
P70S6K trilence-27 siRNA (SR304164) was purchased from ORIGENE. Three specific siRNA constructs were tested for each target and the most effective was chosen. The siRNA constructs were transfected with Lipofectamine 2000 reagent (Invitrogen) for 48 h in H441 and H358 cells, in accordance with the manufacturer's instructions.2.6 Immunoprecipitation
The Flag-tagged TAK1 construct and the Myc-tagged S6K1 construct were transfected into HEK293T cells either alone or in combination. Twenty-four h after transfection, RA-V was added to cells and incubated for another 24 h. Cells were then lysed and incubated on ice for 30 min with lysis buffer (20 mM Tris (pH = 8.0), 137 mM NaCl, 10% glycerol, 1% NP-40 and 2 mM EDTA). Protein samples were then immunoprecipitated with Myc antibody overnight at 4 °C. Then, the immunoprecipitated proteins were pulled down by incubating with protein A-agarose beads (Cell Signaling Technology) for 2 h with rotary agitation. Finally, proteins were separated from the beads by boiling in 2× SDS loading buffer, and then detected by western blotting.2.7 Tumor xenograft model
All animal studies were approved by the Institutional Animal Care and Treatment Committee of Sichuan University. Female nude mice (BALB/c, 6–8 weeks of age, non-fertile and 18–20 g in weight) were obtained from Hufukang Biotech. Animals were kept on a cycle of 12 h light/darkness at 22 ± 1 °C and acclimated for at least one week prior to use. All experiments were carried out under controlled conditions and animals received appropriate humane care according to the National Institutes of Health Guidelines before and during experiments. Mice were injected subcutaneously with H460 or H441 cells (2 × 106 cells per mouse). Mice were then randomized into two groups until their tumors reached a size of approximately 100 mm3. Mice were then treated with or without 2.5 mg kg−1 RA-V via an injection into the tail vein every two days for two weeks. Tumor volumes were recorded every two days and measured using the following formula: tumor volume (mm3) = (length × width2) × 0.52.2.8 Statistical analysis
Data were analyzed using analysis of variance (ANOVA) and the unpaired Student's t-test. Values are expressed as mean ± standard deviation and P < 0.05 was considered to be statistically significant.3. Results
3.1 Identification of Kras-dependent NSCLC cells with high levels of basal protective autophagy
Lung cancers commonly harbor Kras mutations, but only a subset of lung cancers with Kras mutations rely upon Kras for survival (Kras-dependent cancer cells). In order to preferentially select Kras-dependent NSCLC cells, lentivirus shRNA was used to inhibit Kras, thereby allowing us to test the Kras dependency of four NSCLC cell lines: A549, H460, H441 and H358. As shown in Fig. 1A and B, the inhibition of Kras resulted in a reduction of cell viability and induced apoptosis in H441 and H358 cells, but not in A549 and H460 cells. These results implied that H441 and H358 were Kras-dependent cells while A549 and H460 were Kras-independent cells; these findings were in accordance with previously published research.3 To detect the level of basal autophagy in Kras-dependent cells, the GFP-LC3 plasmid was transfected into A549, H460, H441 and H358 cells; we then observed the clear aggregation of GFP-LC3 dots, a general marker of autophagy,22 in H441 and H358 cells but only a diffuse pattern of GFP-LC3 dots in A549 and H460 cells (Fig. 1C). This indicated that H441 and H358 cells showed high levels of basal autophagy. Furthermore, the strong LC3-II conversion, and relatively high expression of beclin1 in H441 and H358 cells detected by western blotting, was also indicative of high levels of basal autophagy in these cells (Fig. 1D). Then, blocking of autophagy by chloroquine (CQ) induced evident growth inhibition in H441 and H358 cells, but had no effect upon A549 and H460 cells (Fig. 1E). This implied that autophagy plays a protective role in Kras-dependent cells, which was in accordance with previous research.5,13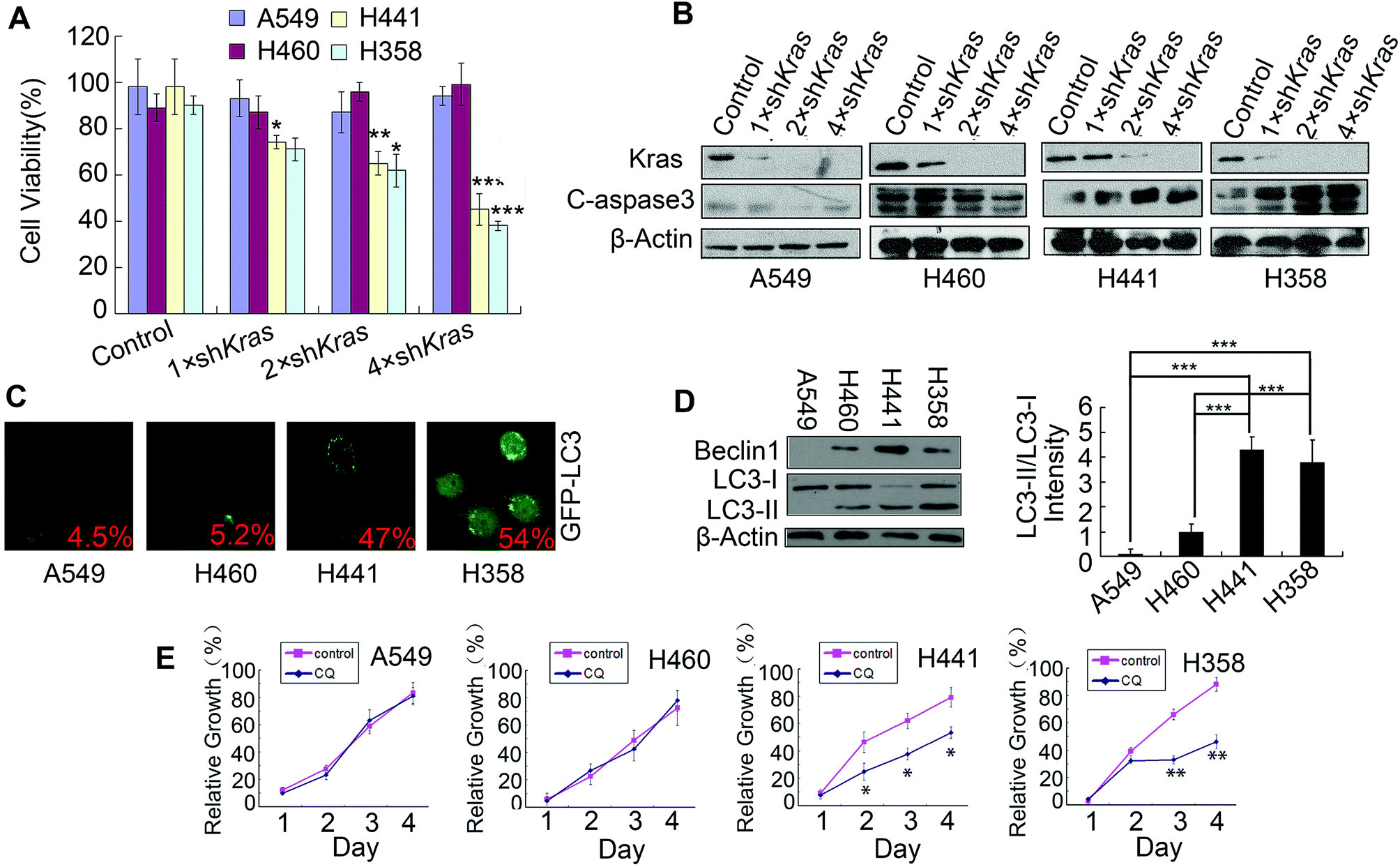 | ||
| Fig. 1 Identification of Kras-dependent NSCLC cells with high levels of basal protective autophagy. (A) NSCLC cells were infected with increasing amounts of lentivirus expressing control or different doses of K-Ras shRNAs for 24 h, then cells were treated with 1 μg ml−1 of puromycin and incubated for another 48 h. Cell viability was assessed by MTT assay. (B) NSCLC cells were infected with increasing amounts of lentivirus expressing control or different doses of K-Ras shRNAs for 24 h, then cells were treated with 1 μg ml−1 of puromycin and incubated for another 48 h. The expression of Kras and cleaved-caspase 3 were detected by western blotting. (C) NSCLC cells were transfected with GFP-LC3 plasmid and after 24 h transfection, RA-V was added to the cells. After incubation for 24 h, GFP-LC3-positive cells were detected and imaged under a fluorescence microscope. (D) Beclin1 and LC3B expression levels in NSCLC cells were detected by western blotting. (E) Cell growth detection of NSCLC cells treated with or without 25 μM of chloroquine (CQ). Data is presented as means ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.01, as compared to controls or the appointed group. | ||
3.2 RA-V selectively induces apoptosis-dependent cell death in Kras-dependent NSCLC cells
As RA-V (Fig. 2A) is a potential TAK1 inhibitor,19 we first investigated whether it also showed anticancer activity and induced apoptosis in Kras-dependent NSCLC cells. As shown in Fig. 2B, Kras-dependent cells (H441 and H358) were more sensitive to RA-V than Kras-independent cells (A549 and H460). Furthermore, we found that RA-V induced obvious apoptosis in Kras-dependent cells (H441 and H358) but had no effect on Kras-independent cells (A549 and H460) (Fig. 2C). Similar results were obtained by the detection of caspase 3, anti-apoptosis protein BCL-2 and BCL-XL by western blotting (Fig. 2D). To investigate whether RA-V induced cell death was apoptosis dependent, Z-VAD-FMK(zVAD), an apoptosis inhibitor was employed. As shown in Fig. 2E, zVAD clearly inhibited RA-V induced cell death in H441 and H358 cells, providing strong evidence that RA-V induced apoptosis-dependent cell death in Kras-dependent cells. Finally, we investigated the in vivo anticancer activity of RA-V in H460 (Kras-independent) and H441 (Kras-dependent) tumor models. Our results showed that treatment with 2.5 mg kg−1 of RA-V via the tail vein injection every other day caused tumor growth inhibition in the H441 model, but not the H460 model (Fig. 2F). This in vivo study showed that RA-V was more sensitive to the Kras-dependent tumor model, which was in accordance with results arising from our in vitro study.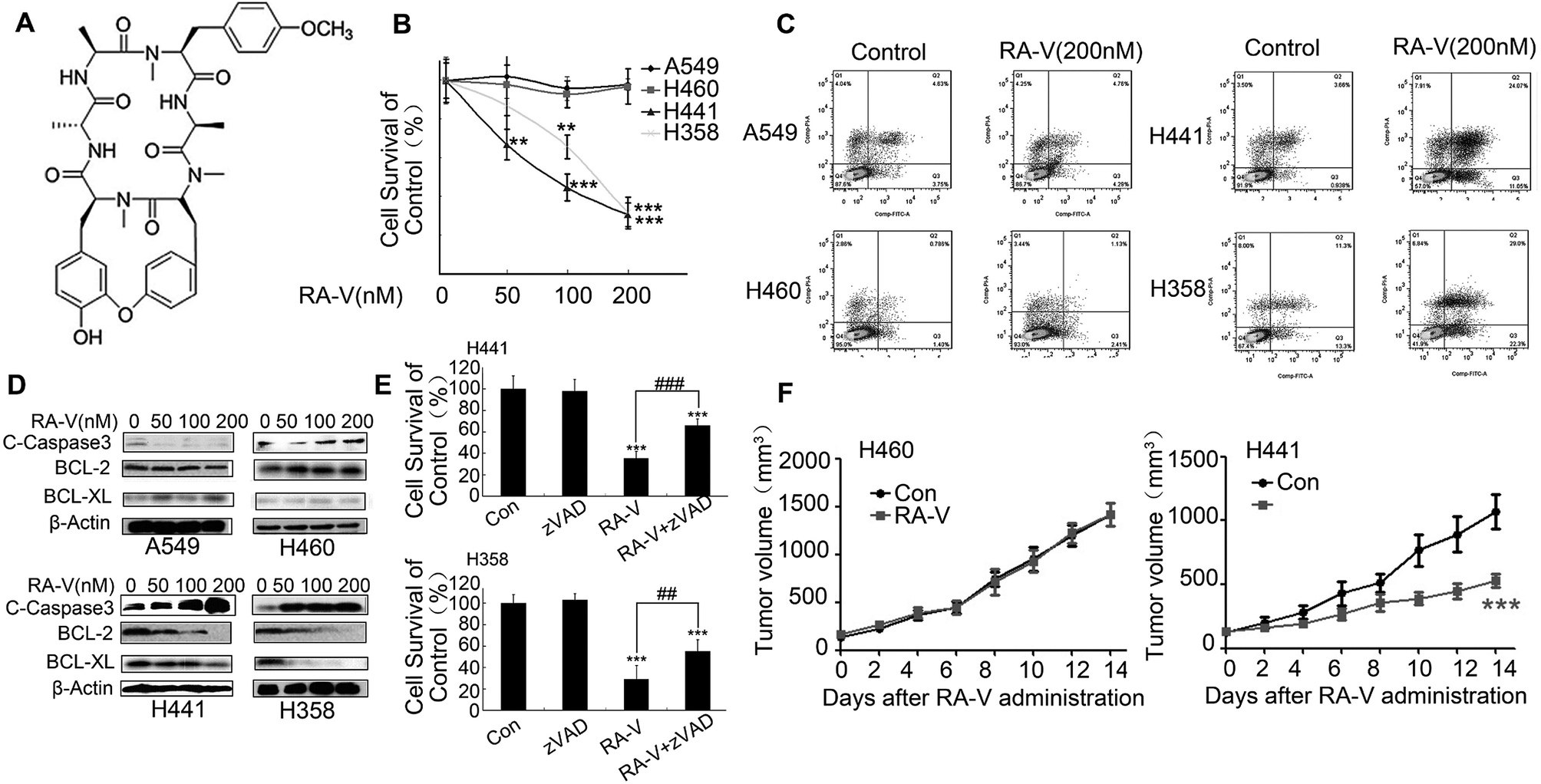 | ||
| Fig. 2 RA-V-induced apoptosis-dependent cell death in Kras-dependent NSCLCs. (A) The chemical structure of RA-V. (B) NSCLC cells were treated with 0, 50, 100 and 200 nM of RA-V for 48 h and cell viability was detected by MTT assay. (C) NSCLC cells were treated with 100 nM of RA-V for 48 h and then subjected to AnnexinV/PI double staining for apoptosis detection (D) NSCLC cells were treated with 0, 50, 100 and 200 nM of RA-V for 48 h and then subjected to western blotting to investigate the expression of cleaved-Caspase3, BCL-2 and BCL-XL. (E) H441 and H358 cells were treated with RA-V alone or in combination with Z-VAD-FMK (zVAD) for 48 h, and cell viability was assessed by MTT assay. (F) When tumors reached approximately 100 mm3, mice were treated with or without 2.5 mg kg−1 of RA-V via an injection to the tail vein every two days for two weeks. Tumor volumes were recorded every two days. Data are presents as means ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.01, as compared to controls or the appointed group. | ||
3.3 RA-V inhibits basal autophagy in Kras-dependent NSCLC cells by mediating the TAK1/AMPK/mTOR pathway
Next, we investigated whether RA-V could inhibit basal autophagy in Kras-dependent NSCLC cells by different methods. The aggregation of GFP-LC3 dots in H441 and H358 cells were clearly inhibited by RA-V (Fig. 3A), suggesting that RA-V can inhibit basal autophagy in Kras-dependent NSCLC cells. Numerous autophagy-related proteins will be up-regulated once autophagy is activated, such as LC3-II, beclin1 and ATG7.22,23 So, we next investigated whether RA-V inhibited the expression level of beclin1 and ATG7 in H441 and H358 cells. As shown in Fig. 3B, RA-V inhibited the expression levels of LC3-II, beclin1 and ATG7 in both H441 and H358 cells. Collectively, these results strongly suggested that RA-V inhibited basal autophagy in Kras-dependent NSCLC cells. In our attempt to study the mechanism of RA-V, we found that RA-V had no effect on the activity of AKT, but surprisingly that RA-V could inhibit the phosphorylation levels of both AMPK and TAK1 in a dose-dependent manner in both H441 and H358 cells (Fig. 3C). These observations suggested that RA-V inhibits the TAK1/AMPK/mTOR pathway in both H441 and H358 cells.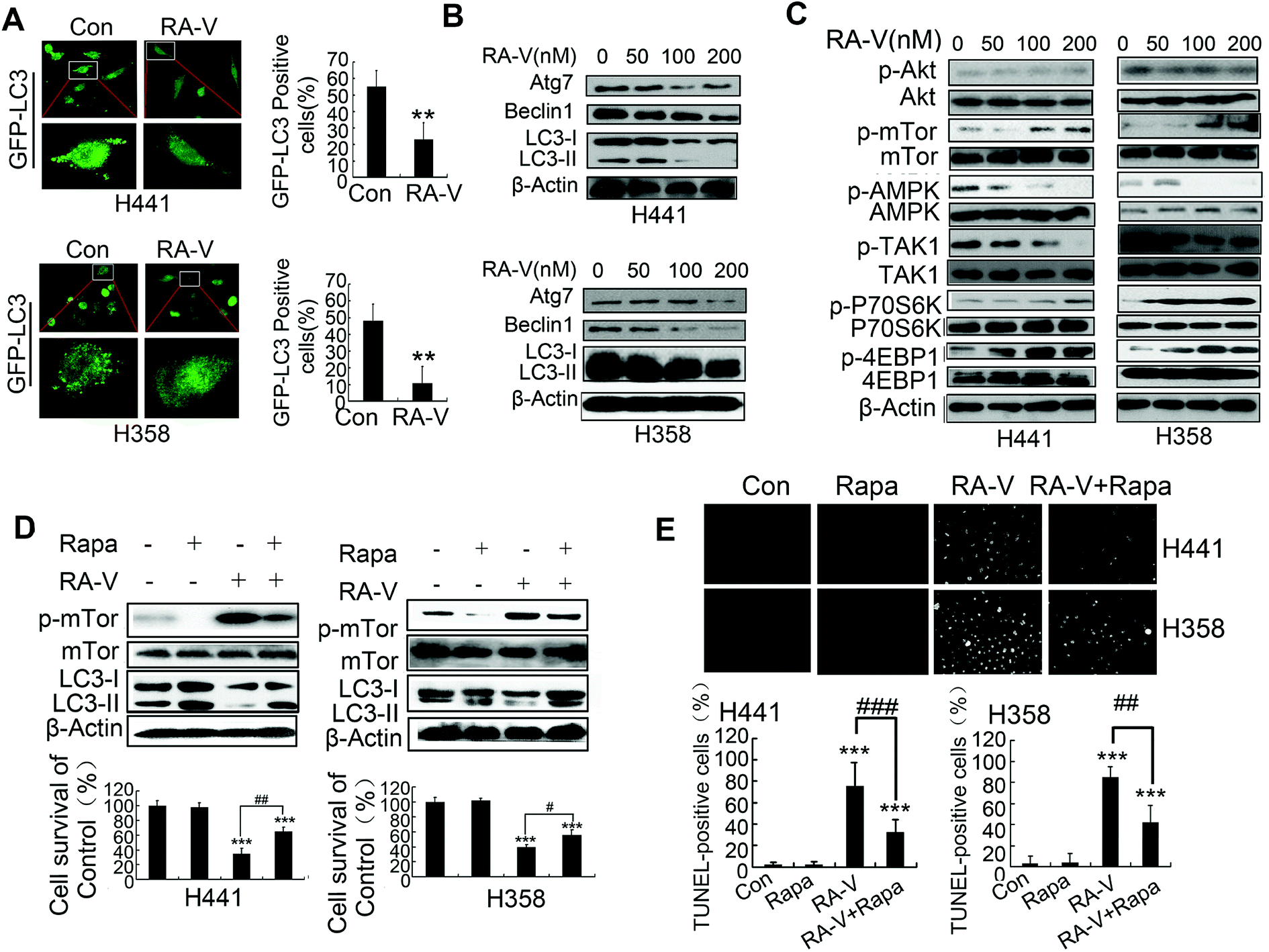 | ||
| Fig. 3 RA-V inhibits basal autophagy in Kras-dependent NSCLC cells by mediating the TAK1/AMPK/mTOR pathway. (A) Cells were transfected with GFP-LC3 plasmid for 24 h and then RA-V was added to the cells for another 24 h. GFP-LC3-positive cells were then detected and imaged under a fluorescence microscope. (B) Cells were treated with 0, 50, 100 and 200 nM RA-V for 48 h and then subjected to western blotting to determine the expression of Atg7, Beclin1 and LC3. (C) Cells were treated with 0, 50, 100 and 200 nM RA-V for 48 h and then subjected to western blotting to determine the expression of p-AKT, AKT, p-mTOR, mTOR, p-AMPK, AMPK, p-TAK1, TAK1, p-P70S6K, P70S6K, p-4E-BP1, 4E-BP1 and β-Actin. (D) Cells were treated with rapamycin (Rapa) or RA-V alone or in combination for 48 h, cell viability was assessed by MTT, and the expression of mTOR, p-mTOR and LC3 was detected by western blotting. (E) Cells were treated with Rapa or RA-V alone or in combination for 48 h, then cells were subjected to the TUNEL assay to monitor apoptosis. *P < 0.05, **P < 0.01, ***P < 0.01, as compared to controls or the appointed group. | ||
Further investigations showed that the inhibition of mTOR by rapamycin (Rapa) could increase autophagy levels and reduce cell death in H441 and H358 cells treated with RA-V (Fig. 3D), indicating that inhibition of the TAK1/AMPK/mTOR pathway contributes to the inhibition of autophagy by RA-V. We also used the TUNEL assay to monitor cell apoptosis levels and found that Rapa could reduce the level of apoptosis induced by RA-V in both H441 and H358 cells (Fig. 3E). Collectively, these data indicated that the inhibition of mTOR by Rapa could markedly increase basal protective autophagy and reduce apoptosis-dependent cell death in H441 and H358 cells treated with RA-V.
3.4 The TAK1/P70S6K pathway also contributes to the inhibition of protective basal autophagy by RA-V
TAK1 can activate autophagy by inhibiting the phosphorylation of p70S6 kinase1 (P70S6K) by blocking the interaction between raptor and P70S6K.8 We also found that RA-V could increase the phosphorylation level of both P70S6K and 4E-BP1 (Fig. 3C). This indicated that the TAK1/P70S6K pathway was also mediated by RA-V. Inhibition of P70S6K by P70S6K RNA interference (Fig. 4A) increased autophagy level and reduced cell death in H441 and H358 cells treated with RA-V (Fig. 4B), which indicated that the TAK1/P70S6K pathway contributes to the inhibition of autophagy by RA-V. Furthermore, the inhibition of P70S6K can reduce the level of apoptosis in RA-V treated H441 and H358 cells (Fig. 4C).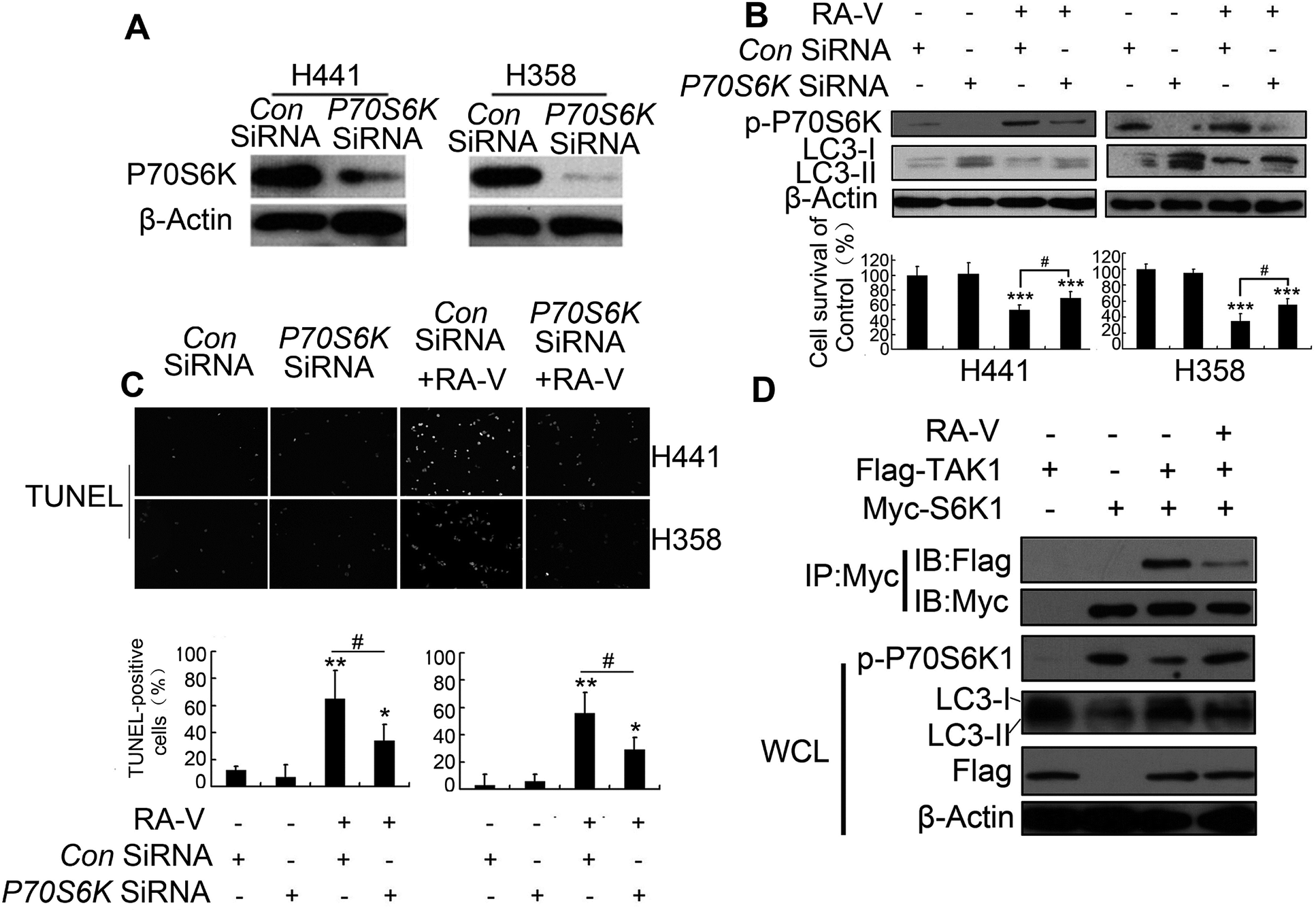 | ||
| Fig. 4 Inhibition of the TAK1/P70S6K pathways helps to protective basal autophagy inhibition by RA-V. (A) Detection of SiRNA effects: detection of P70S6K expression by western blotting in P70S6K-knocked-down cells. (B) RA-V were treated with or without control or P70S6K-silenced H358 or H441 cells for 48 h, cell viability was detected by MTT, and the expression of p-P70S6K and LC3 were detected by western blotting. (C) RA-V were treated with or without control or P70S6K-silenced H358 or H441 cells for 48 h, and apoptosis was detected by TUNEL assay. (D) H293T cells transfected with Flag-tagged TAK1 construct and Myc-tagged S6K1 construct alone or in combination, then treated with or without 100 nM of RA-V. Cell lysates were collected and immunoblotted with an anti-myc-antibody, followed by immunoblotting with flag or myc. The blots in the lower four panels were obtained from the same cell lysates using the indicated antibodies. Data are presented as means ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.01, as compared to controls or the appointed group. | ||
Given TAK1 can activate autophagy by inhibiting p70S6 kinase1 (P70S6K) phosphorylation by binding directly to P70S6K,8 we next investigated whether the binding of RA-V to TAK1 could affect the interaction between TAK1 and P70S6K. H293T cells, transfected with myc-P70S6K or/and flag-TAK1 constructs, were treated with or without RA-V; we then carried out myc immunoprecipitation and assayed for the binding of TAK1 to P70S6K using a flag antibody. As indicated in Fig. 4D, TAK1 could bind to P70S6K, inhibit the phosphorylation level of P70S6K and promote autophagy; furthermore, all of these observations could be inhibited by RA-V. These results revealed that the directly binding of RA-V to TAK1 could inhibit the interaction between TAK1 and P70S6K, and that mediation of the TAK1/P70S6K pathway contributes to the inhibition of autophagy by RA-V.
4. Discussion
Non-small-cell lung carcinoma accounts for 85% of lung cancers, which is the leading cause of death by cancer.24 Compared with small cell lung carcinoma (SCLC), NSCLC is relatively insensitive to chemotherapy. However, 10–35% of NSCLC patients (50% of Asians) harbor EGFR mutations and it is these patients who can specifically benefit from tyrosine kinase inhibitors (TKIs), such as erlotinib, gefitinib or afatinib.25–27 However, patients with EGFR mutations may also have Kras mutations, and these patients are insensitive to treatment with TKIs.27,28 In addition, other NSCLC patients with aberrant Kras activation also rely on Kras for survival. To overcome these Kras-dependent NSCLCs, specific Kras inhibitors are being investigated and developed.28,29 However, the direct inhibition of Kras has not yet led to the development of clinically-useful drugs. TAK1 is activated to enhance Wnt-dependent transcription in Kras-dependent cells, which is required for the survival of Kras-dependent cells. Inhibition of TAK1 will induce apoptosis in Kras-dependent cells.3 Consequently, inducing apoptosis-dependent cancer cell death in Kras-dependent cells by targeting TAK1 may represent an alternative option for the treatment of Kras-activated cancer.TAK1, transforming growth factor beta-activated kinase 1, is a Kras dependency-associated kinase.3 In Kras-dependent cells, the Wnt signal pathway is activated for cell survival; this occurs via the activation of TAK1.3,30,31 Thus, the inhibition of TAK1 may promote cell death in Kras-dependent cells. In our previous study, we found RA-V, a natural cyclopeptide, could inhibit the NF-κB signaling pathway by targeting TAK1.19 In the present study, we showed that RA-V could induce apoptosis in Kras-dependent NSCLCs. We identified that NSCLCs H441 and H358as as Kras-dependent cells and H460 and A549 as Kras-independent cells. RA-V was more sensitive to Kras-dependent cells and induced apoptosis in Kras-dependent cells. Also, we established a Kras-dependent tumor model (H441) and a Kras-independent tumor model (H460), and found that RA-V showed better anticancer activity in the H441 model but had no effect on the H460 model. Collectively, these results proved that inhibition of TAK1 by RA-V represents an effective treatment for Kras-dependent NSCLCs.
Autophagy is a process in which cells engulf their own cytoplasmic proteins or organelles and encapsulate them into vesicles that fuse with lysosomes to form autophagosomes. This leads the degradation of the contents of the packaged vesicles, thereby achieving the metabolic needs of the cells and allowing certain organelles to be renewed.4,22 In normal physiological circumstances, autophagy functions in a protective role to protect the cell from damage arising from starvation.23,32 In Kras-dependent cells, basal protective autophagy was increased by mediating the TAK1–AMPK–mTOR pathway, and thus plays an important role in maintaining cell viability and tumorigenesis. As expected, the inhibition of TAK1 by RA-V could inhibit the basal autophagy of H441 and H358 cells (Kras-dependent). Furthermore, our mechanistic investigations showed that RA-V inhibits the TAK1/AMPK/mTOR and TAK1/P70S6K pathways in H441 and H358 cells, and that the activation of TAK1/AMPK/mTOR pathway by rapamycin could inhibit cell death by RA-V. Furthermore, the knock down of P70S6K by P70S6K shRNA could also reduce RA-V-induced cell death. Importantly, RA-V could inhibit the interaction of TAK1 with P70S6K to inhibit autophagy in H358 cells. Collectively, these data show that the inhibition of TAK1 by RA-V can inhibit protective autophagy in Kras-dependent NSCLCs by inhibiting the TAK1/AMPK/mTOR and TAK1/P70S6K pathways.
Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (U1402222, 31470428) and National Key Programs of China during the 13th Five-Year Plan Period (2017ZX09201005-001-004), the National New Drug Innovation Major Project of China (2017ZX09309027), the Fund of Chinese Academy of Sciences (XDA09030301-4, Hundred Talents Program), the Fund for Introduction of High-level Talents from China Pharmaceutical University, the Program for Jiangsu Province Innovative Research Team, and “111” Project (B16046) from the Ministry of Education of China and the State Administration of Foreign Experts Affairs of China.References
- J. Downward, Targeting RAS signalling pathways in cancer therapy, Nat. Rev. Cancer, 2003, 3, 11–22 CrossRef PubMed.
- N. Normanno, S. Tejpar, F. Morgillo, L. A. De, C. E. Van and F. Ciardiello, Implications for KRAS status and EGFR-targeted therapies in metastatic CRC, Nat. Rev. Clin. Oncol., 2009, 6, 519 CrossRef PubMed.
- A. Singh, M. F. Sweeney, M. Yu, A. Burger, P. Greninger, C. Benes, D. A. Haber and J. Settleman, TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers, Cell, 2012, 148, 639–650 CrossRef PubMed.
- J. D. Rabinowitz and E. White, Autophagy and Metabolism, Science, 2010, 330, 1344–1348 CrossRef PubMed.
- J. Y. Guo, H. Y. Chen, R. Mathew, J. Fan, A. M. Strohecker, G. Karsliuzunbas, J. J. Kamphorst, G. Chen, J. M. S. Lemons and V. Karantza, Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis, Genes Dev., 2011, 25, 460–470 CrossRef PubMed.
- A. E. Lo Ré, M. G. Fernández-Barrena, L. L. Almada, L. D. Mills, S. F. Elsawa, G. Lund, A. Ropolo, M. I. Molejon, M. I. Vaccaro and M. E. Fernandez-Zapico, Novel AKT1-GLI3-VMP1 Pathway Mediates KRAS Oncogene-induced Autophagy in Cancer Cells, J. Biol. Chem., 2012, 287, 25325 CrossRef PubMed.
- G. Herrero-Martín, M. Høyer-Hansen, C. García-García, C. Fumarola, T. Farkas, A. López-Rivas and M. Jäättelä, TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells, EMBO J., 2009, 28, 677–685 CrossRef PubMed.
- S. J. Hyun, S. H. Min, K. Seong-Jin, K. Young-Il, P. Junsoo, L. H. Kyu and Y. O. Joon, TAK1 regulates autophagic cell death by suppressing the phosphorylation of p70 S6 kinase 1, Sci. Rep., 2013, 3, 1561 CrossRef PubMed.
- M. Landström, The TAK1-TRAF6 signalling pathway, Int. J. Biochem. Cell Biol., 2010, 42, 585–589 CrossRef PubMed.
- J. R. Delaney and M. Mlodzik, TGF-beta activated kinase-1: new insights into the diverse roles of TAK1 in development and immunity, Cell Cycle, 2006, 5, 2852 CrossRef PubMed.
- A. Sorrentino, N. Thakur, S. Grimsby, A. Marcusson, V. V. Bulow, N. Schuster, S. Zhang, C. H. Heldin and M. Landström, The type I TGF-|[beta]| receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner, Nat. Cell Biol., 2008, 10, 1199 CrossRef PubMed.
- K. Sayama, Y. Hanakawa, H. Nagai, Y. Shirakata, X. Dai, S. Hirakawa, S. Tokumaru, M. Tohyama, L. Yang and S. Sato, Transforming growth factor-beta-activated kinase 1 is essential for differentiation and the prevention of apoptosis in epidermis, J. Biol. Chem., 2006, 281, 22013 CrossRef PubMed.
- J. Y. Guo, G. Karsliuzunbas, R. Mathew, S. C. Aisner, J. J. Kamphorst, A. M. Strohecker, G. Chen, S. Price, W. Lu and X. Teng, Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis, Autophagy, 2013, 27, 1447–1461 Search PubMed.
- J. Zhou, Plant Cyclopeptides, Chem. Rev., 2006, 106, 840–895 CrossRef PubMed.
- J. T. Fan, J. Su, Y. M. Peng, Y. Li, J. Li, Y. B. Zhou, G. Z. Zeng, H. Yan and N. H. Tan, Rubiyunnanins C-H, cytotoxic cyclic hexapeptides from Rubia yunnanensis inhibiting nitric oxide production and NF-κB activation, Bioorg. Med. Chem., 2010, 18, 8226 CrossRef PubMed.
- G. G. Yue, J. T. Fan, J. K. Lee, G. Z. Zeng, T. W. Ho, K. P. Fung, P. C. Leung, N. H. Tan and C. B. Lau, Cyclopeptide RA-V inhibits angiogenesis by down-regulating ERK1/2 phosphorylation in HUVEC and HMEC-1 endothelial cells, Br. J. Pharmacol., 2011, 164, 1883–1898 CrossRef PubMed.
- X. Y. Fang, W. Chen, J. T. Fan, R. Song, L. Wang, Y. H. Gu, G. Z. Zeng, Y. Shen, X. F. Wu and N. H. Tan, Plant cyclopeptide RA-V kills human breast cancer cells by inducing mitochondria-mediated apoptosis through blocking PDK1-AKT interaction, Toxicol. Appl. Pharmacol., 2013, 267, 95–103 CrossRef PubMed.
- H. W. Leung, Z. Wang, G. G. Yue, S. M. Zhao, J. K. Lee, K. P. Fung, P. C. Leung, C. B. Lau and N. H. Tan, Cyclopeptide RA-V inhibits cell adhesion and invasion in both estrogen receptor positive and negative breast cancer cells via PI3K/AKT and NF-κB signaling pathways, Biochim. Biophys. Acta, 2015, 1853, 1827 CrossRef PubMed.
- Z. Wang and S. Zhao, et al., Natural cyclopeptide RA-V inhibits the NF-κB signaling pathway by targeting TAK1, Cell Death Dis., 2018, 9, 715 CrossRef PubMed.
- J. Yang, W. Wu, J. Wen, H. Ye, H. Luo, P. Bai, M. Tang, F. Wang, L. Zheng and S. Yang, Liposomal honokiol induced lysosomal degradation of Hsp90 client proteins and protective autophagy in both gefitinib-sensitive and gefitinib-resistant NSCLC cells, Biomaterials, 2017, 141, 188 CrossRef PubMed.
- J. Yang, Y. Zhou, X. Cheng, Y. Fan, S. He, S. Li, H. Ye, C. Xie, W. Wu and C. Li, Isogambogenic acid induces apoptosis-independent autophagic cell death in human non-small-cell lung carcinoma cells, Sci. Rep., 2015, 5, 7697 CrossRef PubMed.
- H. Abeliovich, Guidelines for the use and interpretation of assays for monitoring autophagy, Haematologica, 2012 Search PubMed.
- Z. Yang and D. J. Klionsky, An overview of the molecular mechanism of autophagy, Curr. Top. Microbiol. Immunol., 2009, 335, 1 Search PubMed.
- P. Goldstraw, D. Ball, J. R. Jett, T. L. Chevalier, E. Lim, A. G. Nicholson and F. A. Shepherd, Non-small-cell lung cancer, Lancet, 2011, 378, 1727–1740 CrossRef.
- D. Dupuy, Current status of thermal ablation treatments for lung malignancies, Seminars in Interventional Radiology, 2010, pp. 268–275 Search PubMed.
- F. R. Hirsch and P. A. Bunn, EGFR testing in lung cancer is ready for prime time, Lancet Oncol., 2009, 10, 522–527 CrossRef.
- M. G. Kris, How today's developments in the treatment of non-small cell lung cancer will change tomorrow's standards of care, Oncologist, 2005, 10(Suppl 2), 23 CrossRef PubMed.
- L. Ding, G. Getz, D. A. Wheeler, E. R. Mardis, M. D. Mclellan, K. Cibulskis, C. Sougnez, H. Greulich, D. M. Muzny and M. B. Morgan, Somatic mutations affect key pathways in lung adenocarcinoma, Nature, 2008, 455, 1069–1075 CrossRef PubMed.
- M. P. Patricelli, M. R. Janes, L. S. Li, R. Hansen, U. Peters, L. V. Kessler, Y. Chen, J. M. Kucharski, J. Feng and T. Ely, Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State, Cancer Discovery, 2016, 6, 316 CrossRef PubMed.
- R. Fodde and T. Brabletz, Wnt/beta-catenin signaling in cancer stemness and malignant behavior, Curr. Opin. Cell Biol., 2007, 19, 150 CrossRef PubMed.
- I. Wang, J. Snyder, Y. Zhang, J. Lander, Y. Nakafuku, J. Lin, G. Chen, T. V. Kalin, J. A. Whitsett and V. V. Kalinichenko, Foxm1 Mediates Cross Talk between Kras/Mitogen-Activated Protein Kinase and Canonical Wnt Pathways during Development of Respiratory Epithelium, Mol. Cell. Biol., 2012, 32, 3838 CrossRef PubMed.
- R. C. Scott, O. Schuldiner and T. P. Neufeld, Role and regulation of starvation-induced autophagy in the Drosophila fat body, Dev. Cell, 2004, 7, 167 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |