DOI:
10.1039/C8RA04186E
(Paper)
RSC Adv., 2018,
8, 24617-24626
Halogen bond triggered aggregation induced emission in an iodinated cyanine dye for ultra sensitive detection of Ag nanoparticles in tap water and agricultural wastewater
Received
16th May 2018
, Accepted 19th June 2018
First published on 10th July 2018
Abstract
Aggregation induced emission (AIE) has emerged as a powerful method for sensing applications. Based on AIE triggered by halogen bond (XB) formation, an ultrasensitive and selective sensor for picomolar detection of Ag nanoparticles (Ag NPs) is reported. The dye (CyI) has an iodine atom in its skeleton which functions as a halogen bond acceptor, and aggregates on the Ag NP plasmonic surfaces as a halogen bond donor or forms halogen bonds with the vacant π orbitals of silver ions (Ag+). Formation of XB leads to fluorescence enhancement, which forms the basis of the Ag NPs or Ag+ sensor. The sensor response is linearly dependent on the Ag NP concentration over the range 1.0–8.2 pM with an LOD of 6.21 pM (σ = 3), while for Ag+ it was linear over the 1.0–10 μM range (LOD = 2.36 μM). The sensor shows a remarkable sensitivity for Ag NPs (pM), compared to that for Ag+ (μM). The sensor did not show any interference from different metal ions with 10-fold higher concentrations. This result indicates that the proposed sensor is inexpensive, simple, sensitive, and selective for the detection of Ag NPs in both tap and wastewater samples.
1. Introduction
The photophysical properties of organic and inorganic molecules have motivated researchers’ interest throughout time. In particular, luminescence, which is the radiative state of molecules absorbing light, has been used extensively in different applications such as molecular logic gates,1,2 sensors,3–5 organic thermometers,6 and cellular imaging.2,7 However, luminescence has some disadvantage such as stability and aggregation induced quenching (AIQ). The latter dominates at high concentrations or in the solid state. On the other hand, in some molecules luminescence increases upon aggregation, which is known as aggregation induced emission (AIE). AIE-based materials have been used in different applications such as pharmaceutical analysis,8 biomedical applications,9,10 and biological imaging.11–13 The molecular forces behind AIE were mainly ascribed to the restriction of intramolecular rotation (RIR), twisted intramolecular charge transfer (TICT), J-aggregate dyes, and hydrogen bonds (HB).14 However, halogen bond (XB) induced emission enhancement is scarce.8 XB is a parallel realm to HB, which is comparable in terms of energy strength and directionality.15,16 XB forms between halogen atoms X (X = F, Cl, Br, and I) and electron donor atoms Y (N, S, and O). The formed XB (C–Y⋯X) has an electrostatic nature as it formed between the electron deficient part of X and the high electron density cap of the Y atom (Type I). In addition, the XB might form between the electron density belt on the equatorial axis of the X atom and the electron deficient part of the Y atom (Type II).15 XB has proven itself as a competitive binding motif with HB in different applications such as supramolecular architecture and anion recognition.15 XB could be formed between the sigma hole of a halogen atom with an organic substrate,17,18 a macrocycle,19 or a metal center,20,21 and the negative belt on the nanostructure.22,23 XB was used as a binding motif in the inhibition of gold-based bimetallic catalysis by aryliodides.24 In addition, the interaction between the Au NPs and the 1,4-diiodobenzene was used in single molecular junctions.25
Recently, silver nanoparticles (Ag NPs) have been widely used in different human activities. The potent antimicrobial effect of Ag NPs is widely used in different applications.26 For example, Ag NPs are currently used in food packaging processes, wound healing fibers, and nanosilver toothpaste. However, Ag NPs are toxic when there is leakage into water drains and food supplies. Therefore, the development of sensors for detecting small traces of Ag NPs is in high demand. Several sensors for Ag NPs have been developed based on different methodologies such as fluorogenic oligonucleotide,27 ICP-based techniques,28 electrothermal atomic absorption spectroscopy (ETAAS),29,30 and X-ray fluorescence spectrometry combined with cloud point extraction (TXRF).31 On the other hand, colorimetric32,33 as well as chemiluminescence34–36 based methods were used for Ag NP determination. The proposed techniques are sensitive to Ag NP traces, but overcoming requirements for expensive techniques for the extraction and an increased number of separation steps is another challenge.
Here we present XB as a new motif for the aggregation of dyes (cyanine) on metal nanoparticle surfaces. Such aggregation induced emission enhancement of the cyanine dye derivative was used as a ratiometric sensor for both Ag+ and Ag NPs in aqueous solutions. The molecular interactions between the XB donor (I atom) and the halogen bond acceptor induces the fluorescence enhancement of the dye. In addition, the sensitivity of the Ag NP plasmonic surfaces to the XB formation increases the sensitivity by 106 times compared to the silver ions. The sensor principle was successfully applied for the in situ detection of Ag NPs in both tap and wastewater real samples.
2. Materials and methods
2.1 Material preparation
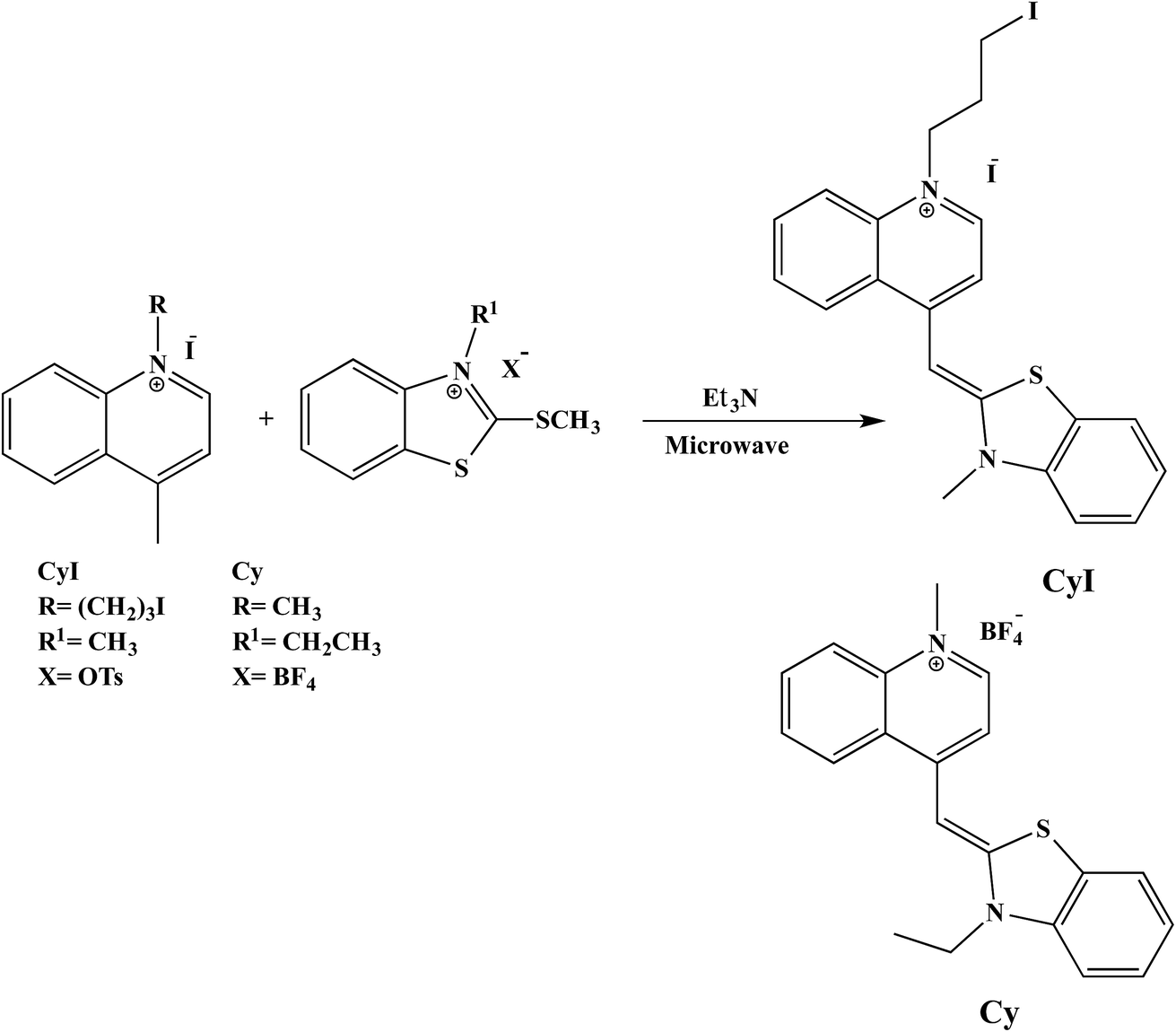
2.1.1 Cyanine dyes. Substituted cyanine dyes were prepared according to the literature using a microwave assisted solvent-free synthesis.37 In brief, benzothiazolium salt (2 mmol) and 4-methylquinolinium salt (2 mmol) with a few drops of triethylamine were mixed in a glass conical flask. The mixture was subjected to microwave irradiation at 320 or 260 W for 8 or 5 min for CyI and Cy respectively. After cooling and washing with diethyl ether, an orange to yellowish orange precipitate was obtained.
1-Methyl-4-((3-methylbenzo[d]thiazol-2(3H)-ylidene)methyl)quinolin-1-ium (Cy). Yellowish orange crystals, yield: 87%; mp: 272–276 °C; IR (KBr): ν = 1466 (SH), 1520, 1616 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN); 1H NMR (DMSO-d6): δ = 1.39 (t, J = 7.0 Hz, 3H, CH3), 4.16 (s, 3H, CH3), 4.63 (q, J = 6.8 Hz, 2H, CH2), 6.89 (s, 1H, CH), 7.33–8.76 (m, 10H, Ar-H); 13C NMR: δ = 12.3 (CH3), 40.9 (CH2), 42.3 (CH3), 87.2 (CH), 107.8, 112.5, 118.1, 122.7, 123.9, 124.5, 125.4, 126.9, 128.4, 133.2, 137.9, 139.3, 144.7, 148.7 (Ar-C), 158.6 (NCS).
1-(3-Iodopropyl)-4-((3-methylbenzo[d]thiazol-2(3H)-ylidene)methyl)quinolin-1-ium (CyI). Orange crystals, yield: 90%; mp: 230–235 °C; IR (KBr): ν = 1469 (SH), 1500, 1606 cm−1 (CC, CN); 1H NMR (DMSO-d6): δ = 2.37 (m, 2H, CH2), 3.39 (t, J = 7.0 Hz, 2H, CH2), 3.96 (s, 3H, CH3), 4.53 (t, J = 7.3 Hz, 2H, CH2), 6.73 (s, 1H, CH), 7.12–8.72 (m, 10H, Ar-H); 13C NMR: δ = 2.3 (CH2), 32.1 (CH2), 33.9 (CH3), 54.3 (CH2), 88.5 (CH), 107.6, 112.7, 117.1, 122.6, 123.7, 123.8, 124.3, 125.6, 126.4, 127.8, 133.2, 136.1, 140.0, 143.3, 148.0 (Ar-C), 159.7 (NCS).
C, CN); 1H NMR (DMSO-d6): δ = 1.39 (t, J = 7.0 Hz, 3H, CH3), 4.16 (s, 3H, CH3), 4.63 (q, J = 6.8 Hz, 2H, CH2), 6.89 (s, 1H, CH), 7.33–8.76 (m, 10H, Ar-H); 13C NMR: δ = 12.3 (CH3), 40.9 (CH2), 42.3 (CH3), 87.2 (CH), 107.8, 112.5, 118.1, 122.7, 123.9, 124.5, 125.4, 126.9, 128.4, 133.2, 137.9, 139.3, 144.7, 148.7 (Ar-C), 158.6 (NCS).
1-(3-Iodopropyl)-4-((3-methylbenzo[d]thiazol-2(3H)-ylidene)methyl)quinolin-1-ium (CyI). Orange crystals, yield: 90%; mp: 230–235 °C; IR (KBr): ν = 1469 (SH), 1500, 1606 cm−1 (CC, CN); 1H NMR (DMSO-d6): δ = 2.37 (m, 2H, CH2), 3.39 (t, J = 7.0 Hz, 2H, CH2), 3.96 (s, 3H, CH3), 4.53 (t, J = 7.3 Hz, 2H, CH2), 6.73 (s, 1H, CH), 7.12–8.72 (m, 10H, Ar-H); 13C NMR: δ = 2.3 (CH2), 32.1 (CH2), 33.9 (CH3), 54.3 (CH2), 88.5 (CH), 107.6, 112.7, 117.1, 122.6, 123.7, 123.8, 124.3, 125.6, 126.4, 127.8, 133.2, 136.1, 140.0, 143.3, 148.0 (Ar-C), 159.7 (NCS).
2.1.2 Ag NP preparation. Ag nanoparticles were prepared using the citrate reduction of AgNO3 according to a reported method.38 A calculated amount of AgNO3 (17 mg, 0.1 mmol) was dissolved in 100 ml DW and the solution was heated to 95 °C. Then, the appropriate amount of trisodium citrate (100 mg, 0.38 mmol) was gently added under continuous stirring. The Ag NP concentration was calculated according to the literature.38 For the metal ions interference experiment, 1 mM of different inorganic salts was dissolved in double-distilled water and titrated against 3 ml of cyanine dye (10 μM) using a micropipette. Double-distilled water was used throughout the course of the investigations.
2.2 Sample preparation for detection
For Ag NP sensing in both tap water and wastewater, the samples were prepared using the standard addition method. The appropriate concentration of AgNO3 was dissolved in tap water or the wastewater from Kitchener Drain, Kafrelsheikh, Egypt. The solution was then heated to 95 °C and the required trisodium citrate was slowly added under continuous stirring. Then, the solution was irradiated under a UV lamp for 30 min in the presence of trisodium citrate as the reducing agent in order to completely convert the possibly formed AgCl to Ag NPs. The required volume of the Ag NP solution was added to 10 μM of CyI in a 3 ml cuvette.
2.3 Characterization
The morphology and average size of the Ag NPs were measured using a transmission electron microscope with an acceleration voltage up to 200 kV (TEM, JEOL-JEM-2100, Tokyo, Japan). A UV-vis spectrophotometer (Shimadzu UV-2450) was used to measure the optical properties over a range of 200–700 nm. A spectrofluorometer (Shimadzu, RF-5301PC) was used for fluorescence measurements.
2.4 Theoretical calculations
The structures were optimized using DFT calculations implemented in Gaussian09.39 The B3LYP/LanL2DZ basis set was employed for the optimization of iodine and silver atoms. The stability of the structures was confirmed by performing the frequency calculations. All of the optimized structures gave positive frequencies for the stable adducts on the potential energy scan. Visualization of the data was performed using the VMD program (version 1.9).
3. Results and discussion
3.1 Photophysical properties of CyI
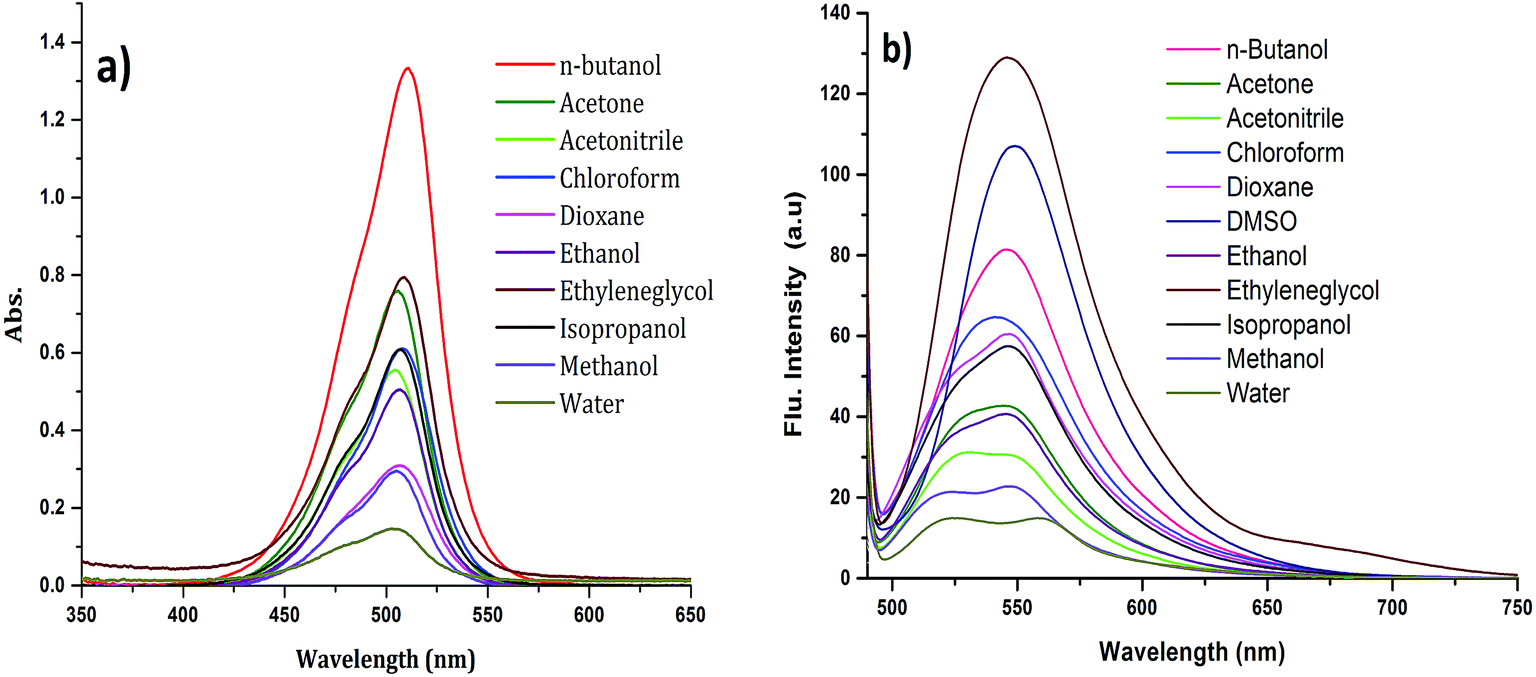
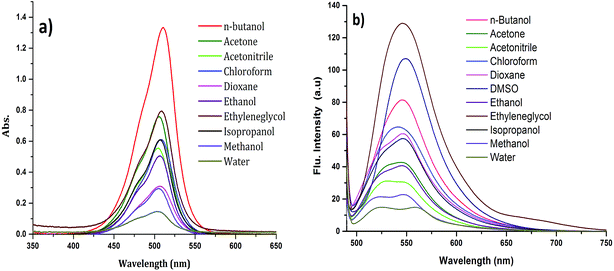
The absorption spectrum of CyI is shown in Fig. 1. The dye possesses a characteristic peak at ca. 270 nm, which was attributed to the π–π* transition for the conjugated aromatic system while a strong and narrow absorption peak observed at 505 nm was ascribed to an intramolecular charge transfer (ICT) transition,40 which is sensitive to the solvent polarity (Fig. 1a). On the other hand, CyI gives a characteristic fluorescence peak at 520 nm upon excitation (λex = 470 nm). The fluorescence quantum yield of CyI was measured using coumarin 153 (Φ = 0.38 in ethanol)41 as the reference dye. The results show a low quantum yield (Φ = 0.013 in water) of CyI due to the presence of the heavy I atom, and this significantly increases upon the addition of Ag NPs (Φ = 0.048 in water, 50 μm Ag NPs). The fluorescence peak is sensitive to the solvent environment, which is blue shifted in non-polar solvents and red shifted in polar solvents, suggesting the polar nature of the excited state (Fig. 1b and Table 1).
 |
| | Fig. 1 Absorption (a) and emission (λex = 420 nm) (b) spectra of CyI in different solvents at room temperature. | |
Table 1 UV-Vis absorption and fluorescence of CyI in different solvents
| Solvent |
ET(30) |
λmaxa |
Wavenumbera |
εb |
λema |
Wavenumbera |
| Wavenumber in cm−1. ε in M−1 cm−1. |
| Water |
63.1 |
504 |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 841 841 |
2940 |
524, 558 |
17921.15 |
| MeOH |
55.4 |
505 |
19802 |
5920 |
523, 547 |
18281.54 |
| EtOH |
51.9 |
507 |
19724 |
10120 |
529, 545 |
18348.62 |
| CH3CN |
45.6 |
504 |
19841 |
11120 |
530, 548 |
18248.18 |
| Acetone |
42.2 |
506 |
19763 |
15200 |
525, 545 |
18348.62 |
| CHCl3 |
39.1 |
508 |
19685 |
12220 |
542 |
18450.18 |
| n-Butanol |
49.7 |
511 |
19570 |
26680 |
546 |
18315.02 |
| DMSO |
45.1 |
511 |
19570 |
26680 |
548 |
18248.18 |
| CH2Cl2 |
40.7 |
512 |
19531.25 |
89600 |
544 |
18382.35 |
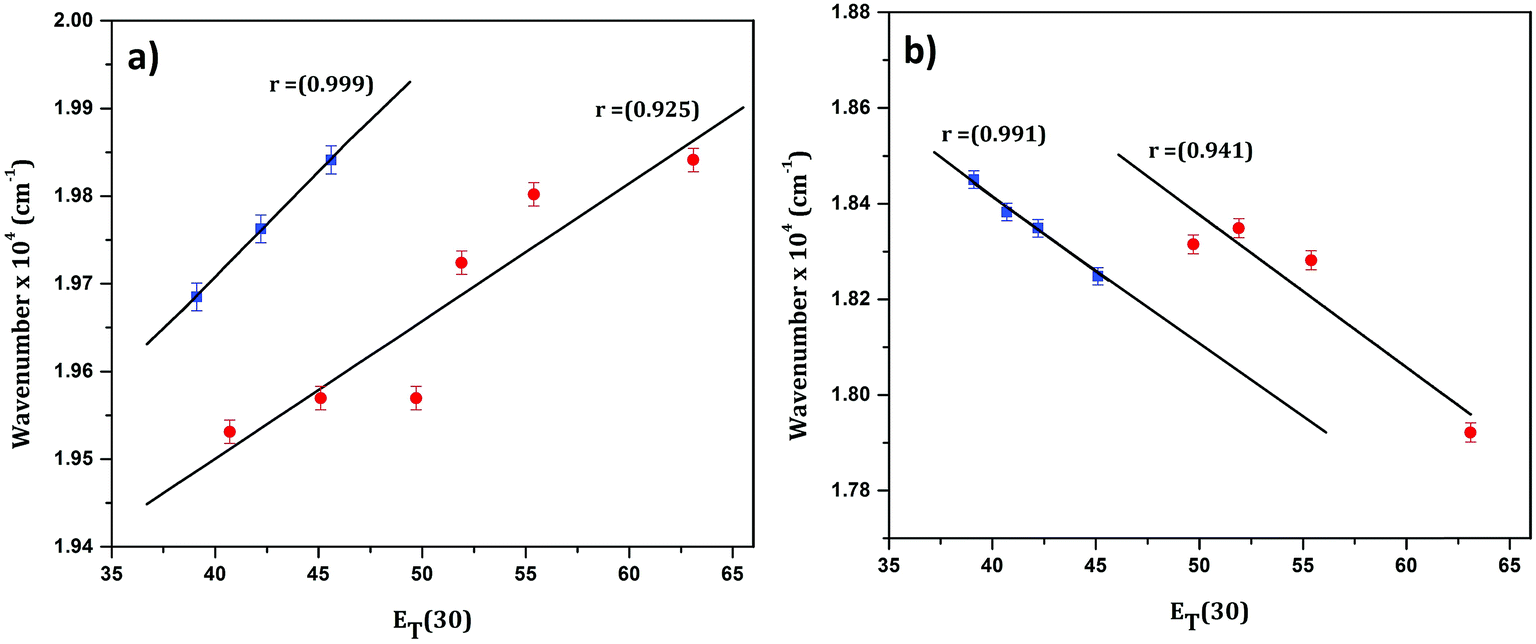
In order to confirm the polar nature of both the ground and excited states of CyI, a plot of the absorption maximum and emission maximum versus the Dimroth–Reichardt (ET(30))42 solvent parameter in different solvents was created. Fig. 2 shows a significant solvatochromic effect for CyI in the selected solvents with the emergence of two data sets, one for the aprotic solvents (CH2Cl2, DMSO and CH3CN) and the other for protic solvents (water, ethanol, and methanol). This result demonstrates the role of proton donating ability in forming hydrogen bonds between the solvent and the solute in both ground and excited states. In addition, absorption energies increase with the increase in ET(30), while the emission energies decrease.
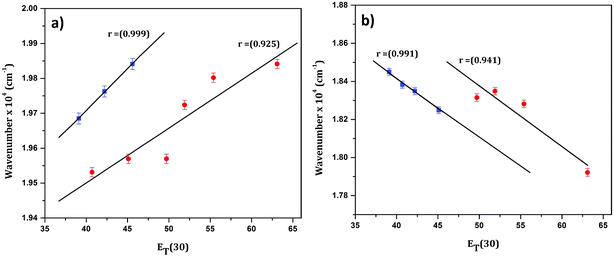 |
| | Fig. 2 Linear plot of (a) absorption and (b) emission maximum versus the ET(30) solvent parameter for CyI. | |
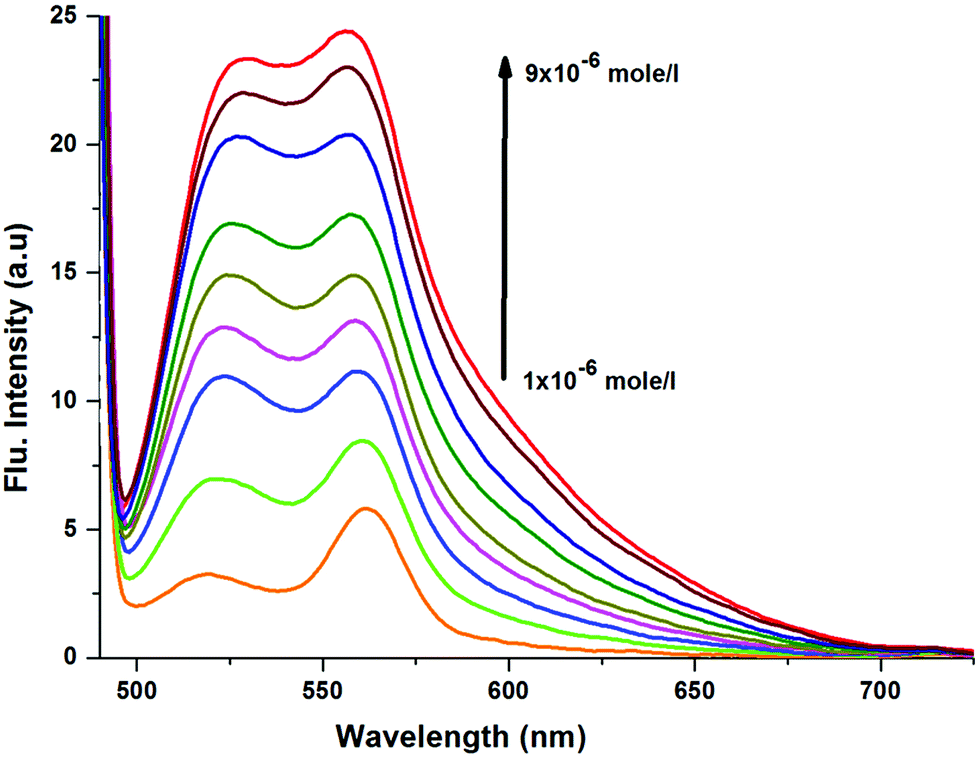
Notably, CyI gives two emission peaks at 524 and 558 nm in polar solvents, and these could be attributed to aggregation processes. In order to confirm the aggregation process, emissions of different concentrations of CyI were recorded (Fig. 3). The results show that the aqueous solution of CyI gives two emission peaks, attributed to the monomer (short wavelength) and the aggregated form (longer wavelength). The ratio between the longer and shorter wavelengths increases with the increase in the dye concentration, which confirms the aggregation process. Interestingly, there is an emission shoulder which appears at 600 nm and could be attributed to the aggregated form of CyI.
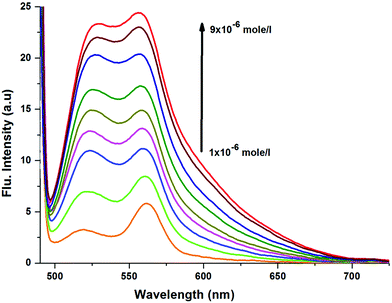 |
| | Fig. 3 Emission spectra of CyI at different concentrations. | |
3.2 Ag NP preparation and their interactions with CyI
Ag NPs were prepared using the direct reduction of Ag+ with trisodium citrate under heating. The obtained NPs have a spherical shape and a diameter of 20–30 nm (Fig. 4a). To investigate the effect of the dye (CyI) on Ag+, an aqueous solution of Ag+ with CyI (0.1 mM) was left for two days in air at room temperature. Interestingly, aggregated Ag NPs were observed under TEM (Fig. 4b). Electron transfer from the dye (CyI) to Ag+ is responsible for the reduction process. This process is thought to occur through halogen bond (XB) formation, and this is confirmed using theoretical calculations (DFT) as described in a later section.
 |
| | Fig. 4 TEM of Ag NPs prepared using (a) trisodium citrate as the reducing agent, (b) Ag+ and CyI as the reducing agent, and (c) CyI + Ag NPs (after 2 days). | |
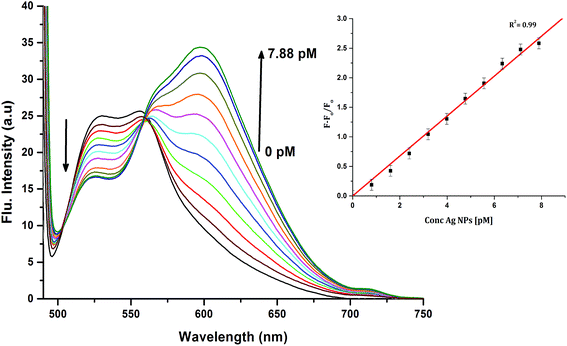
In order to investigate the interactions between the cyanine dye derivatives with Ag+ and Ag NPs, the absorption and fluorescence spectra of the dyes were recorded at different concentrations. The absorption spectra of CyI show characteristic maximum absorption peaks at 504 nm, which decrease upon the addition of both Ag+ and Ag NPs aqueous solutions with an isosbestic point at 455 nm that gives an indication of complex formation. The fluorescence of CyI demonstrates two characteristic peaks with maxima at 535 and 600 nm which were ascribed to the presence of the monomer (535 nm) and the aggregated form (600 nm peak). Upon addition of different concentrations of Ag+ and Ag NPs, a subsequent decrease in the monomer peak is observed, and a successive increase in the peak at longer wavelengths is observed (Fig. 5 and 6). The new peak (600 nm) is attributed to the aggregation of CyI by the complexation with both Ag+ and Ag NPs. To confirm the aggregation process, an aqueous solution of CyI was subjected to Ag NPs, which have a uniform spherical shape as in Fig. 4a. The TEM image shows the aggregation of Ag NPs by CyI (Fig. 4c).
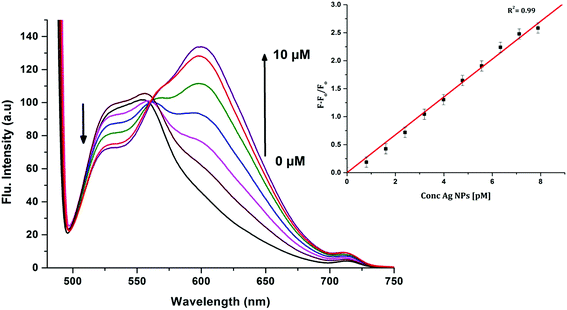 |
| | Fig. 5 Emission spectra of 10 μM CyI and increasing concentration of Ag+ cations in aqueous media (pH = 7) at room temperature. The inset is the change in the emission spectra (600 nm) upon addition of different Ag+ concentrations. | |
 |
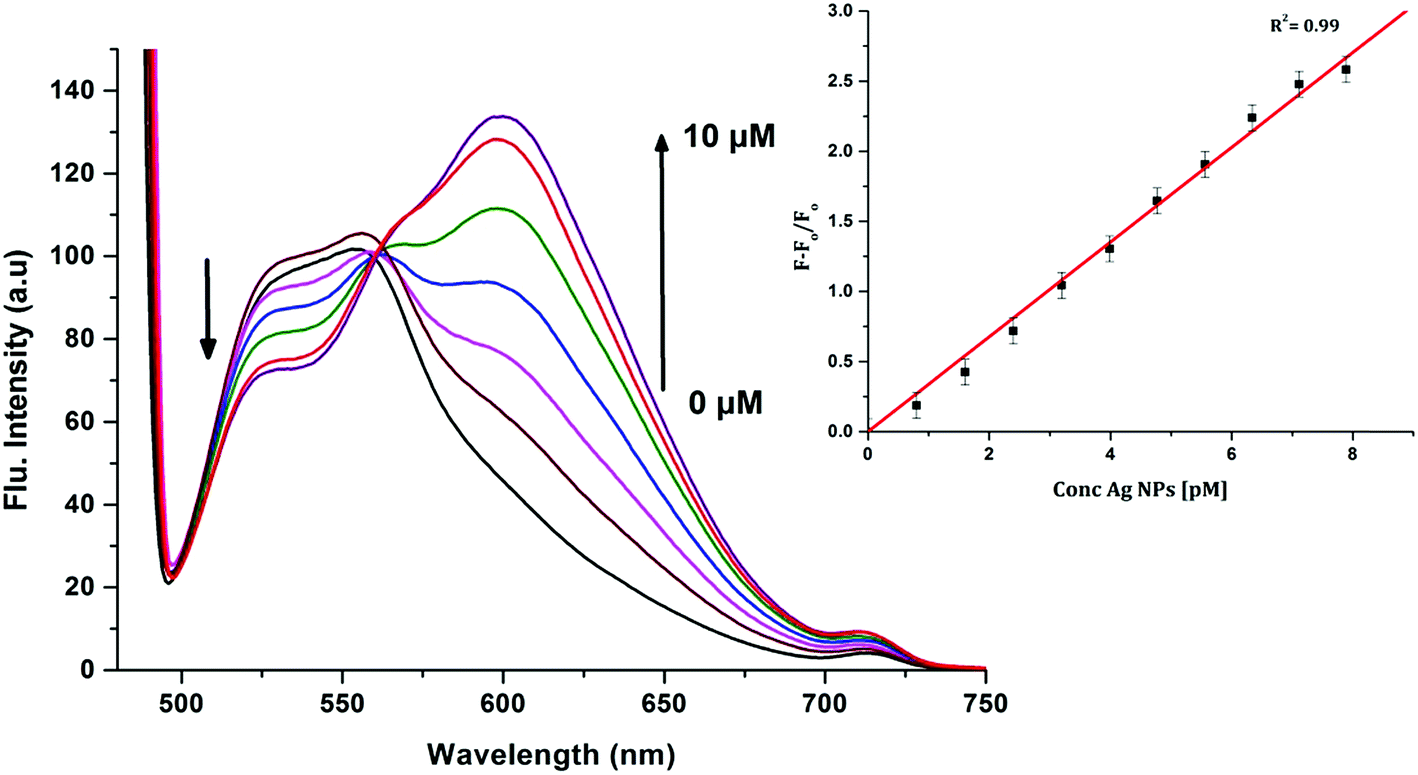
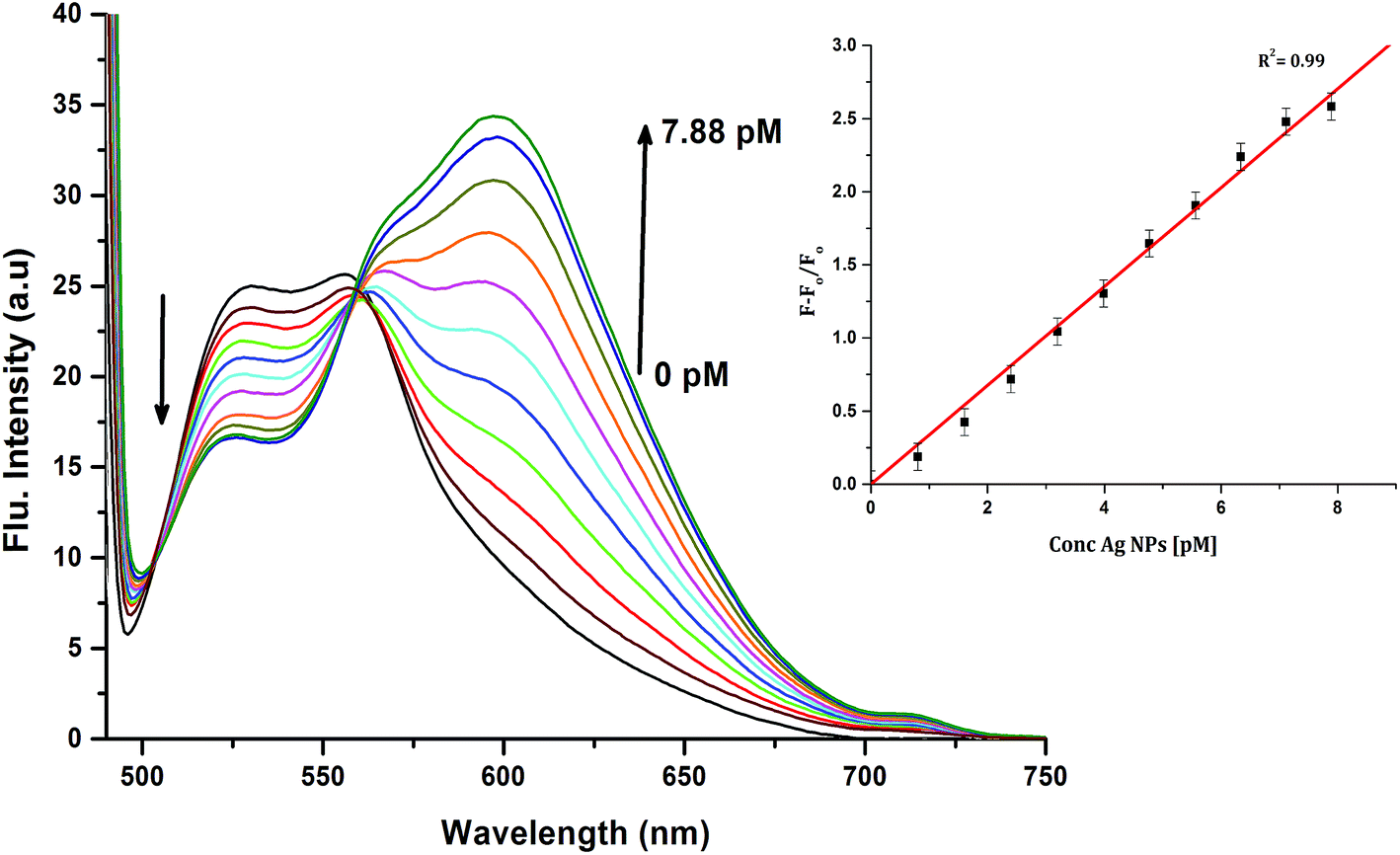
| | Fig. 6 Emission spectra of 10 μM CyI and increasing concentration of Ag NPs in aqueous media (pH = 7) at room temperature. The inset is the change in the emission spectra (600 nm) upon addition of different Ag NP concentrations. | |
3.3 Proposed mechanism for the interaction of CyI with both Ag+ and Ag NPs
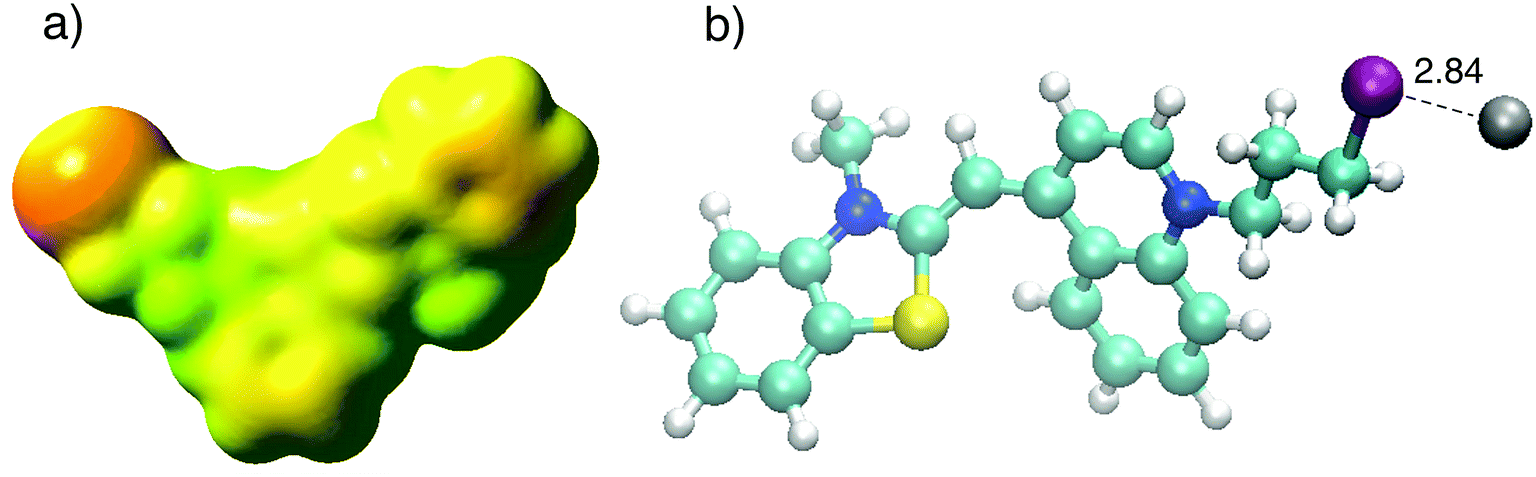
In order to investigate the binding mechanism between CyI with both Ag+ and Ag NPs, theoretical calculations (B3LYP/LanL2DZ) were used to corroborate the experimental work. The optimized structure of CyI with Ag+ shows that the stable structure had a 1:1 ratio of CyI and Ag+. The molecular electrostatic potential of CyI shows the distribution of the electron density over the whole molecule and the presence of a negative belt on the iodine atom, with a positive cap on the I atom linear (C–I) bond axes (Fig. 7a). These results show the motivation for the XB to form through the negative belt on the iodine atom and the positive charge on the Ag+ cation. The binding between the Ag+ and I atoms matches the XB in terms of bond length shortening and directionality. The Ag⋯I bond length is 2.84 Å which is 23.2% less than the sum of the van der Waals radii of Ag (1.72 Å) and I (1.98 Å), while the bond angle is 105.8° which is in accordance with Type II XB (Fig. 7b).15 NBO analysis for the optimized structure was performed to investigate the nature of the bond stabilization.17,18 The data analysis reveals strong interactions between the lone pair from the iodine atom to the vacant Ag orbital (LP (3) I 40 → LP*(6) Ag (41)) with a stabilization energy of 40.01 kcal mol−1. The high ECT value indicates strong charge transfer through XB formation, which is responsible for the reduction of Ag+ by CyI.
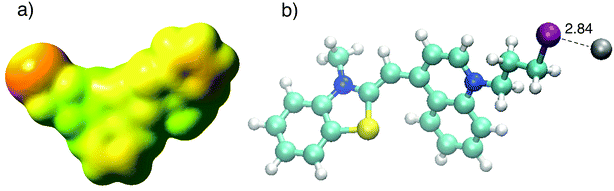 |
| | Fig. 7 (a) Calculated MEP of CyI and (b) optimized structure of CyI–Ag at the B3LYP/LanL2DZ level of theory. | |
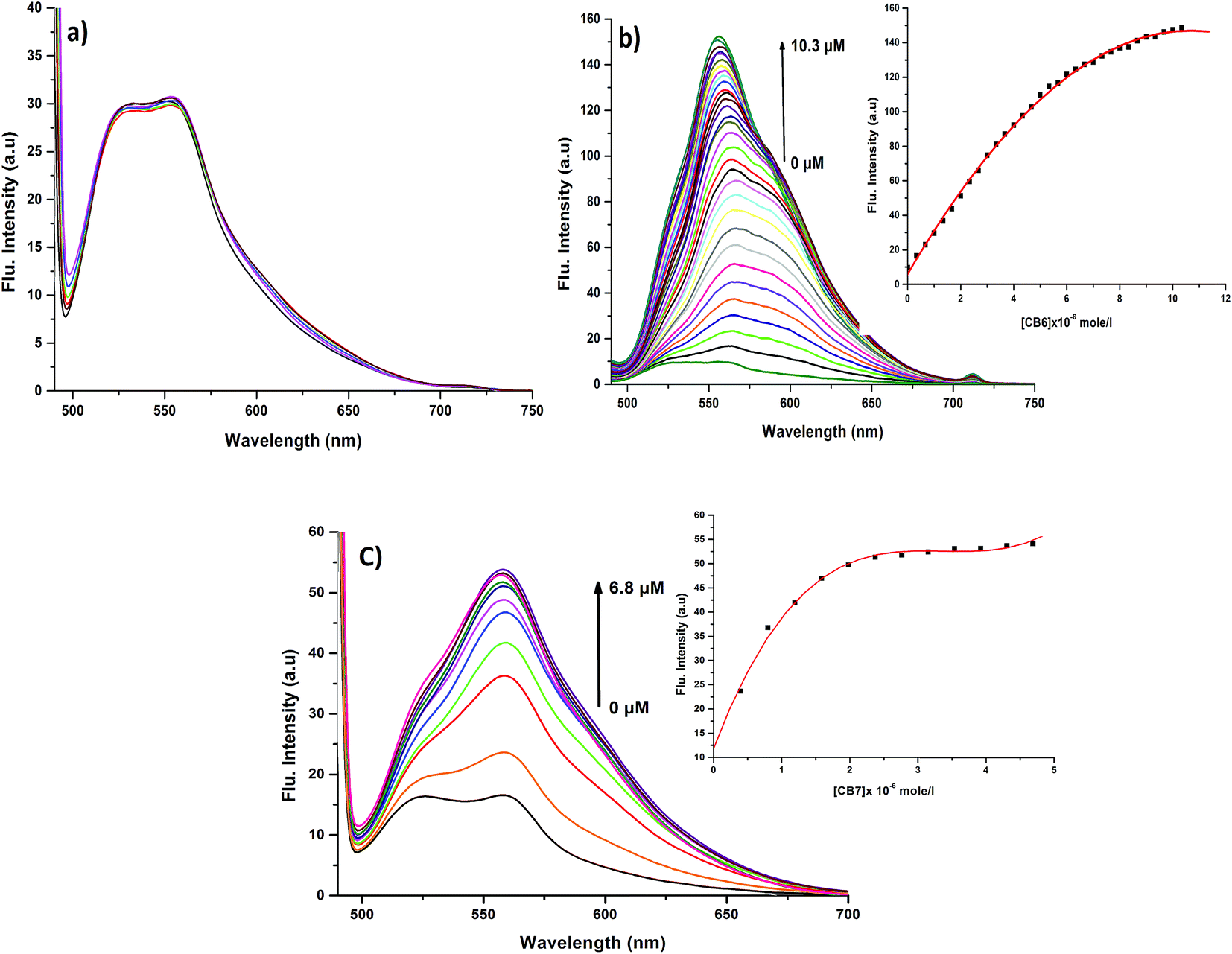
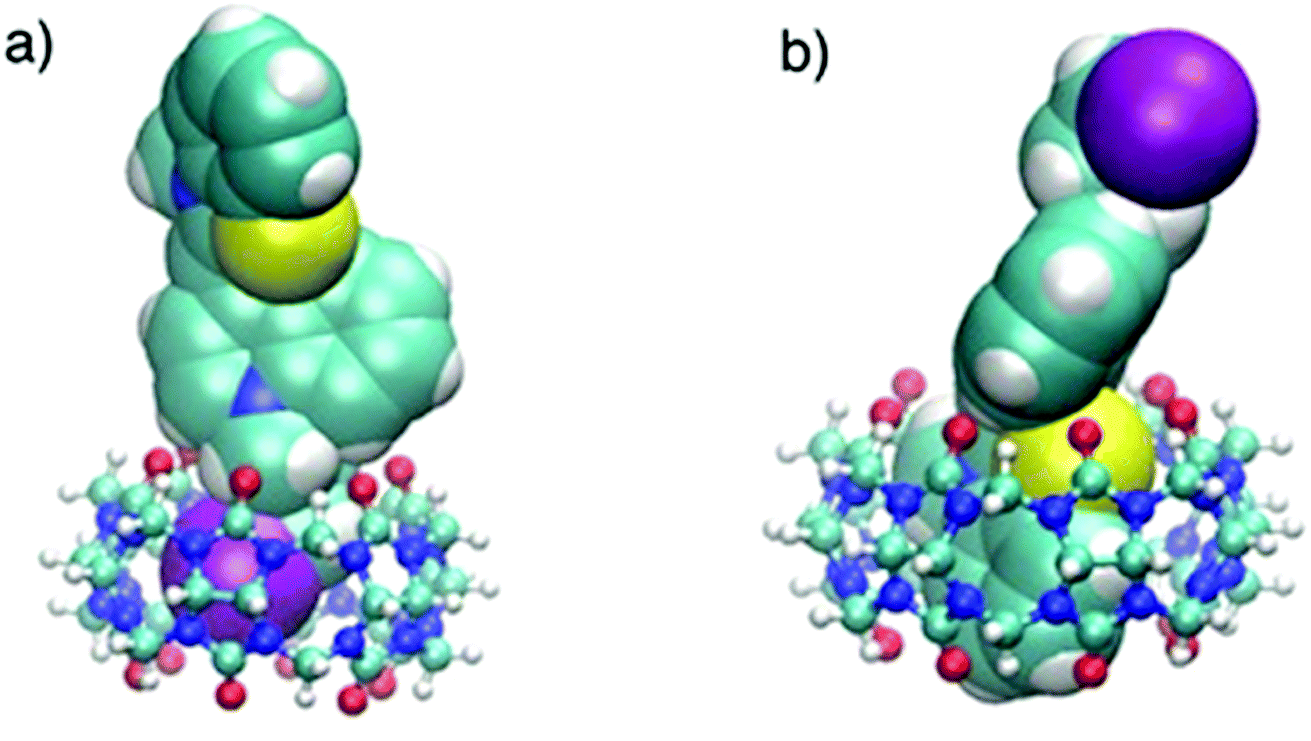
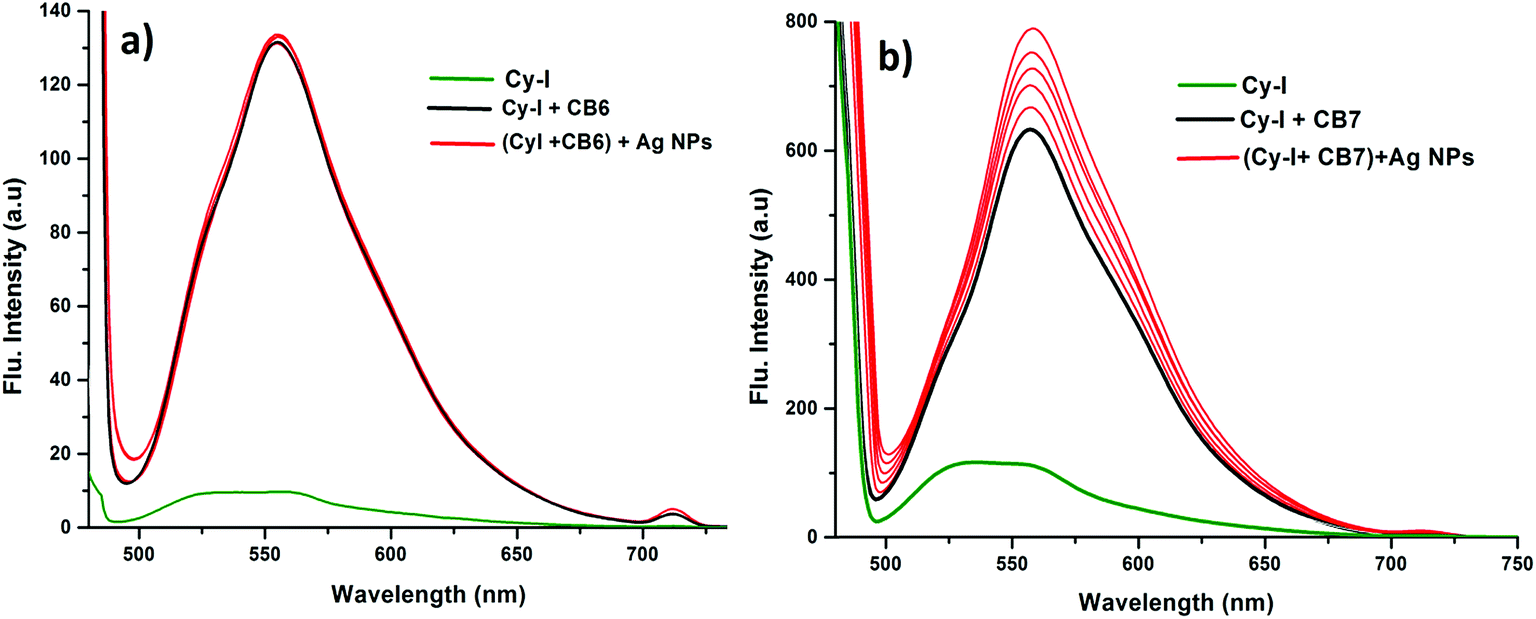
In order to prove the complexation mode, we used Cy (Scheme 1) in which there is no I atom in the dye skeleton. There is no significant increase in emission for the 600 nm peak (Fig. 8a), which is direct evidence that the complexation occurs through the binding of the iodine with Ag+ and Ag NPs. Another piece of evidence comes from the supramolecular binding of CyI with cucurbit[n]uril (CBn). CBn is a macrocyclic compound with n glycouril units. The cavity of the CBn is variable according to the number of glycouril units, with a size of 68, 142, and 242 Å for CB5, CB6, and CB7 respectively.43 CB5 is very small and could only bind with some inorganic anions such as Cl− and NO3−.43 On the other hand, CB6 is big enough to accommodate aliphatic amines, while the volume of CB7 is big enough to accommodate aromatic rings. Successive addition of different concentrations of either CB6 or CB7 to CyI enhances the emission peak at 600 nm (Fig. 8b and c). The fluorescence enhancement in both cases could be attributed to the disturbance of the charge separation in CyI. This effect was mainly due to the binding of CB6 to the iodine atom (k = 1.34 × 104 M−1) or CB7 to the benzothiazole moiety (k = 2.64 × 105 M−1).
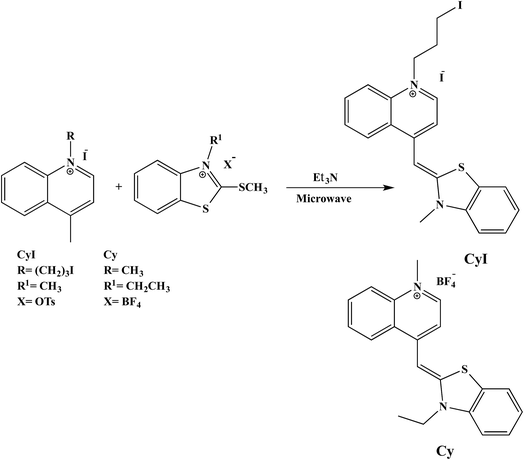 |
| | Scheme 1 Microwave synthesis of Cy and CyI. | |
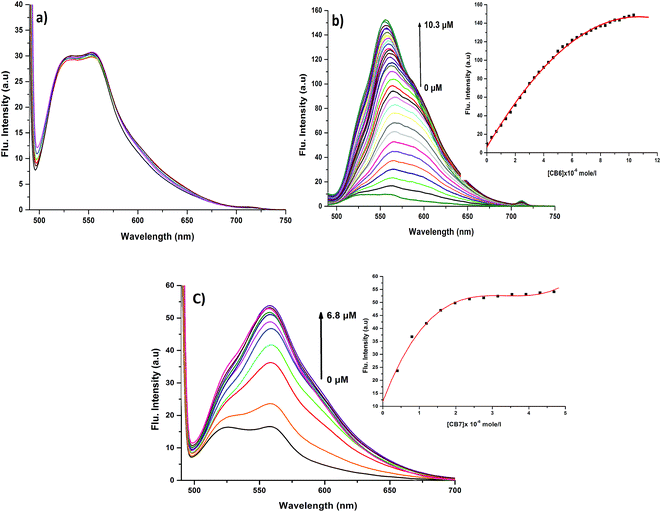 |
| | Fig. 8 Emission spectra of (a) Cy (10 μM) at different concentrations from Ag NPs, (b) CyI (10 μM) under different concentrations of CB6 and (c) CyI (10 μM) under different concentrations of CB7 in aqueous solution. | |
This was supported by the DFT calculations (B3LYP/LanL2DZ), which showed that the structure for CB6 was stabilized through iodine (Fig. 9a), while CB7 was stabilized by the complete inclusion of the benzothiazole moiety (Fig. 9b). To further investigate the binding mode of CyI with Ag+ and Ag NPs, different concentrations of CB6 were added to CyI until the saturation point was reached, then Ag+ or Ag NPs were added. There was no fluorescence enhancement upon the addition of Ag+ or Ag NPs (Fig. 10a), which was attributed to blocking of the binding sites (I atoms) by complete complexation with CB6, and hence the binding site is no longer accessible. However, when using CB7, which has a bigger volume accessible to benzothiazole, addition of Ag+ or Ag NPs results in a further increase in the emission at the maximum wavelength (Fig. 10b). This result shows that after complexation of CB7 with benzothiazole, the iodine atom is still accessible to the Ag+ or Ag NPs for binding.
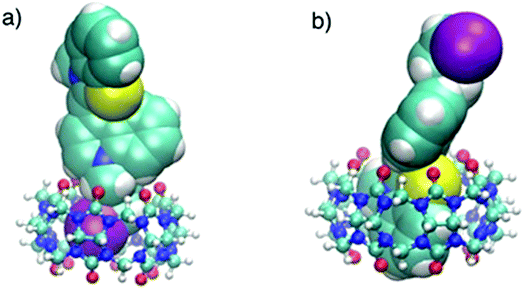 |
| | Fig. 9 Optimized structures (B3LYP/LanL2DZ) of (a) CyI–CB6, and (b) CyI–CB7 molecular adducts. | |
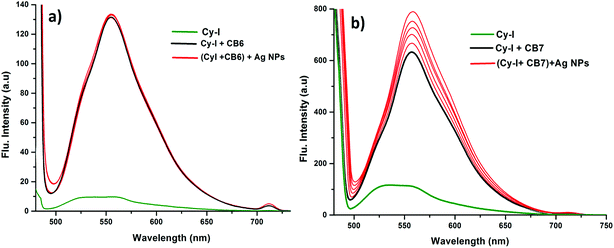 |
| | Fig. 10 Emission spectra of (a) 10 μM CyI (green), CyI + 10.3 μM CB6 (black) and CyI + 10.3 μM CB6 + 7.88 μM Ag NPs (red); (b) 10 μM CyI (green), CyI + 6.8 μM CB7 (black) and CyI + 6.8 μM CB6 + 7.88 μM Ag (red). | |
3.4 Selectivity
The response of the investigated optical nanosensor to different metal ions was investigated using emission of CyI at 600 nm. Among metal ions, Mn2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Sn2+, Cd2+, Hg2+, Cr3+, Pb2+, K+, Ag+ and Ag NPs were investigated. These metals were chosen due to their widespread occurrence in the environment and their toxic effects. Among different metal ions, Ag NPs, Ag+, and Hg2+ revealed stronger abilities to bind with CyI. However, the sensor response was highly selective to Ag NPs compared to Ag+ and Hg2+ ions (selectivity was 106 times greater). According to their ionic radii, Ag+ and Hg2+ have larger values (0.129, 0.116 nm) respectively compared with the other studied ions, so the probability of halogen bond formation with iodine centers increases.
3.5 Sensor sensitivity
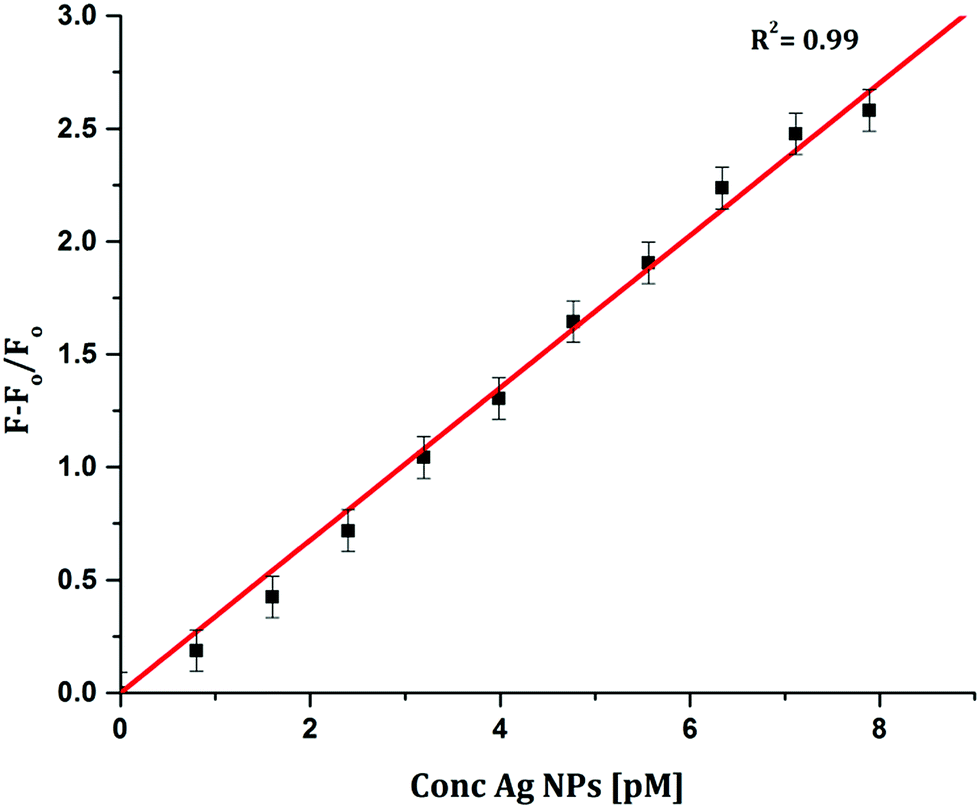
To determine the sensitivity of the sensor, different concentrations of Ag+ or Ag NPs were added to CyI. Upon addition of Ag NPs in the concentration range between 1 and 7.88 pM, the emission peak of CyI at 600 nm increased. The saturation point was reached at the ratio of 1:1 CyI to Ag NPs. Plotting the relation between the concentration of the Ag NPs and the emission response (F − F0)/F0 gives a linear relationship with a correlation coefficient (R2) of 0.99 (Fig. 11). The sensor exhibits a limit of detection (LOD) of 6.21 pM. Meanwhile the sensor response to Ag+ was linear in the 1.0–10 μM range (LOD = 2.36 μM). The sensitivity of the sensor to Ag NPs is compared to the reported methods (Table 2). Moreover, the fluorescence enhancement triggered by XB is easy to perform and did not need any complicated pre-preparation time or sophisticated instruments such as ETAAS (Table 2).
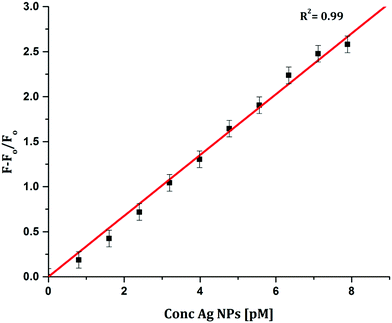 |
| | Fig. 11 Calibration curve for CyI emission with increasing concentrations of Ag NPs. | |
Table 2 Methods for Ag+ and Ag NP detection
| Analytical method |
Ag+ |
Ag NPs |
Reference |
| Linear range |
LOD |
Linear range |
LOD |
| Fluorometric |
66.7–667 nM |
|
64.2–241 nM |
|
27 |
| Fluorometric |
1–100 nmol l−1 |
50 pmol l−1 |
|
|
44 |
| ICPMS |
|
|
0.1–146 μg l−1 |
0.006 μg l−1 |
28 |
| ICPMS |
|
0.07 μg l−1 |
0.025–99.4 μg l−1 |
0.13 μg l−1 |
45 |
| ETAAS |
|
|
0.02–0.3 μg l−1 |
0.0007 μg l−1 |
29 |
| ETAAS |
|
|
0.1–0.4 μg l−1 |
0.0007 μg l−1 |
30 |
| FIA-CL |
|
|
0.0003–0.0009 |
0.0002 μg l−1 |
36 |
| UV spectroscopy |
|
|
17–425 μg l−1 |
1 μg l−1 |
46 |
| TXRF |
|
|
5–1000 μg l−1 |
0.7 μg l−1 |
31 |
| Fluorescence |
0.1–5.0 μM |
14 ppb |
|
|
34 |
| Colorimetric |
1.0–5.0 μM |
0.5 μM |
|
|
47 |
| Chemiluminescence |
|
|
2.5–80 pM |
1.9 pM |
36 |
| Fluorescence |
1.0–10.0 μM |
2.36 μM |
1.0–7.9 pM |
6.21 pM |
This work |
4. Real sample application
To validate our method for detection of Ag NPs in real samples, a standard addition method was used. We prepared in situ Ag NPs in samples from tap water and wastewater. The Ag NPs were prepared using the chemical reduction of Ag+ by trisodium citrate. Then the samples were exposed to UV light (365 nm) for 10 min in the presence of sodium citrate to verify the complete reduction and conversion of the possible formation of AgCl to Ag NPs.48 Tap water was used from the Kafrelsheikh University water supply, while wastewater was used from Kitchener Drain, Kafrelsheikh, Egypt. Kitchener Drain is located 20 km north of Cairo and extends more than 69 km through three Egyptian governorates. It is considered to be the most heavily polluted water drain in Egypt, and this is mainly from industrial and agricultural activities. The most abundant heavy metals in Kitchener Drain were Cd, Ni, and Pb.49
The experiment was performed in triplicate (n = 3). Using the standard calibration curve (Fig. 11), the proposed sensor was able to recover the in situ Ag NPs with high sensitivity, with recoveries between 105.2 and 96.9% (Table 3). This result indicates that the proposed sensor is sensitive and selective for the detection of Ag NPs under real conditions.
Table 3 Determination of Ag NPs in tap and wastewater real samples
| Sample type |
Added Ag NPs (pM) |
Ag NPs found (pM) |
Recovery % |
| Tap water |
0.96 |
1.01 |
105.2 |
| 1.28 |
1.28 |
100 |
| 1.60 |
1.57 |
98.1 |
| Wastewater |
1.50 |
1.47 |
98 |
| 1.87 |
1.87 |
100 |
| 2.25 |
2.18 |
96.9 |
5. Conclusions
An inexpensive and selective sensor for Ag NPs was developed. The mechanism of detection was based on the emission enhancement of the dye (CyI) upon the formation of halogen bonds with the Ag NP plasmonic surfaces. The detection limit was 6.21 pM in the range from 5.0 to 50 pM (n = 3). The method is selective for Ag NPs with a concentration 106 times lower than Ag+ and Hg2+ ions. The method is applicable to in situ determination of the concentration of Ag NPs in both tap and wastewater in real samples.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Mostafa Abdelbar, Kamel Shoueir, Maged El-Kemary, and Hamdy S. El-Sheshtawy acknowledge the financial support from Kafrelsheikh University, Egypt. Hamdy S. El-Sheshtawy acknowledges the Computational Laboratory for Analysis, Modeling, and Visualization (CLAMV) at the Jacobs University, Bremen (Germany) for computation time.
References
- V. F. Pais, M. Lineros, R. Lopez-Rodriguez, H. S. El-Sheshtawy, R. Fernandez, J. M. Lassaletta, A. Ros and U. Pischel, J. Org. Chem., 2013, 78, 7949–7961 CrossRef PubMed.
- F. F. Du, X. J. Gong, W. J. Lu, Y. Liu, Y. F. Gao, S. M. Shuang, M. Xian and C. Dong, Talanta, 2018, 179, 554–562 CrossRef PubMed.
- H. S. El-Sheshtawy, M. S. Abd El Sadek and I. S. Yahia, Nanosci. Nanotechnol. Lett., 2014, 6, 18–25 CrossRef.
- Z. Xie, X. Kong, L. Feng, J. Ma, Y. Li, X. Wang, W. Bao, W. Shi and Y. Hui, Sens. Actuators, B, 2018, 257, 154–165 CrossRef.
- T. Tao, Y. Gan, J. Yu and W. Huang, Sens. Actuators, B, 2018, 257, 303–311 CrossRef.
- V. F. Pais, J. M. Lassaletta, R. Fernandez, H. S. El-Sheshtawy, A. Ros and U. Pischel, Chem.–Eur. J., 2014, 20, 7638–7645 CrossRef PubMed.
- J. Qi, C. Sun, A. Zebibula, H. Zhang, R. T. K. Kwok, X. Zhao, W. Xi, J. W. Y. Lam, J. Qian and B. Z. Tang, Adv. Mater., 2018, 30, 1706856 CrossRef PubMed.
- W.-S. Zou, S. Lin, J.-Y. Li, H.-Q. Wei, X.-Q. Zhang, D.-X. Shen, J.-Q. Qiao, H.-Z. Lian, D.-Q. Xie and X. Ge, New J. Chem., 2015, 39, 262–272 RSC.
- Q. Wan, Q. Huang, M. Liu, D. Xu, H. Huang, X. Zhang and Y. Wei, Appl. Mater. Today, 2017, 9, 145–160 CrossRef.
- X. Zhang, K. Wang, M. Liu, X. Zhang, L. Tao, Y. Chen and Y. Wei, Nanoscale, 2015, 7, 11486–11508 RSC.
- L. Mao, M. Liu, D. Xu, Q. Wan, Q. Huang, R. Jiang, Y. Shi, F. Deng, X. Zhang and Y. Wei, Appl. Surf. Sci., 2017, 423, 469–475 CrossRef.
- R. Jiang, M. Liu, C. Li, Q. Huang, H. Huang, Q. Wan, Y. Wen, Q.-y. Cao, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 80, 708–714 CrossRef PubMed.
- R. Jiang, H. Liu, M. Liu, J. Tian, Q. Huang, H. Huang, Y. Wen, Q.-y. Cao, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 81, 416–421 CrossRef PubMed.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef PubMed.
- H. Wang, W. Z. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef PubMed.
- H. S. El-Sheshtawy, M. M. Ibrahim, I. El-Mehasseb and M. El-Kemary, Spectrochim. Acta, Part A, 2015, 143, 120–127 CrossRef PubMed.
- H. S. El-Sheshtawy, H. M. A. Salman and M. El-Kemary, Spectrochim. Acta, Part A, 2015, 137, 442–449 CrossRef PubMed.
- H. S. El-Sheshtawy, B. S. Bassil, K. I. Assaf, U. Kortz and W. M. Nau, J. Am. Chem. Soc., 2012, 134, 19935–19941 CrossRef PubMed.
- A. S. D. Ivanov, I. V. A. Novikov, Y. V. Kirina and V. Y. Kukushkin, Chem. Commun., 2016, 52, 5565–5568 RSC.
- R. Bertani, P. Sgarbossa, A. Venzo, F. Lelj, M. Amati, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677–695 CrossRef.
- Q. Zhao, J. Mol. Model., 2014, 20, 2133–2138 CrossRef PubMed.
- I. Blakey, Z. Merican, L. Rintoul, Y.-M. Chuang, K. S. Jack and A. S. Micallef, Phys. Chem. Chem. Phys., 2012, 14, 3604–3611 RSC.
- R. N. Dhital, C. Kamonsatikul, E. Somsook, Y. Sato and H. Sakurai, Chem. Commun., 2013, 49, 2542–2544 RSC.
- Y. Komoto, S. Fujii, K. Hara and M. Kiguchi, J. Phys. Chem. C, 2013, 117, 24277–24282 CrossRef.
- A. U. Khan, N. Malik, M. Khan, M. Cho and M. M. M. Khan, Bioprocess Biosyst. Eng., 2018, 41, 1–20 CrossRef PubMed.
- Y.-H. Lin and W.-L. Tseng, Chem. Commun., 2009, 6619–6621 RSC.
- J.-f. Liu, J.-b. Chao, R. Liu, Z.-q. Tan, Y.-g. Yin, Y. Wu and G.-b. Jiang, Anal. Chem., 2009, 81, 6496–6502 CrossRef.
- G. Hartmann, C. Hutterer and M. Schuster, J. Anal. At. Spectrom., 2013, 28, 567–572 RSC.
- G. Hartmann, T. Baumgartner and M. Schuster, Anal. Chem., 2014, 86, 790–796 CrossRef PubMed.
- L. Torrent, M. Iglesias, M. Hidalgo and E. Margui, J. Anal. At. Spectrom., 2018, 33, 383–394 RSC.
- R. A. González-Fuenzalida, Y. Moliner-Martínez, M. González-Béjar, C. Molins-Legua, J. Verdú-Andres, J. Pérez-Prieto and P. Campins-Falcó, Anal. Chem., 2013, 85, 10013–10016 CrossRef PubMed.
- P. Lodeiro, E. P. Achterberg and M. S. El-Shahawi, Talanta, 2017, 164, 257–260 CrossRef PubMed.
- A. Chatterjee, M. Santra, N. Won, S. Kim, J. K. Kim, S. B. Kim and K. H. Ahn, J. Am. Chem. Soc., 2009, 131, 2040–2041 CrossRef PubMed.
- S. Jang, P. Thirupathi, L. N. Neupane, J. Seong, H. Lee, W. I. Lee and K.-H. Lee, Org. Lett., 2012, 14, 4746–4749 CrossRef PubMed.
- G. Z. Tsogas, D. L. Giokas and A. G. Vlessidis, Anal. Chem., 2014, 86, 3484–3492 CrossRef PubMed.
- H. H. Alganzory, M. M. H. Arief, M. S. Amine and E.-Z. M. Ebeid, J. Chem. Pharm. Res., 2014, 6, 143–161 Search PubMed.
- A. M. Al-Soliemy, O. I. Osman, M. A. Hussein, A. M. Asiri and S. A. El-Daly, J. Fluoresc., 2016, 26, 1199–1209 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, K. N. Kudin, T. Vreven, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03 Revision E.01, 2004 Search PubMed.
- H. S. El-Sheshtawy and A. M. Abou Baker, J. Mol. Struct., 2014, 1067, 225–232 CrossRef.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 Search PubMed.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 2nd edn, 1990 Search PubMed.
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559–577 CrossRef.
- L. Bian, X. Ji, W. Hu and J. Agric, Food Chem., 2014, 62, 4870–4877 CrossRef PubMed.
- J.-b. Chao, J.-f. Liu, S.-j. Yu, Y.-d. Feng, Z.-q. Tan, R. Liu and Y.-g. Yin, Anal. Chem., 2011, 83, 6875–6882 CrossRef PubMed.
- Z.-H. Wu and W.-L. Tseng, Anal. Methods, 2011, 3, 2915–2920 RSC.
- C. Qin, W.-Y. Wong and L. Wang, Macromolecules, 2011, 44, 483–489 CrossRef.
- G. Wang, T. Nishio, M. Sato, A. Ishikawa, K. Nambara, K. Nagakawa, Y. Matsuo, K. Niikura and K. Ijiro, Chem. Commun., 2011, 47, 9426–9428 RSC.
- H. B. El-Habet, E. S. Naeem, T. M. Abel-Meeged and S. Sedeek, International Journal of Plant & Soil Science, 2014, 3, 911–933 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Kamel R. Shoueira,
Ibrahim El-Mehassebb,
El-Zeiny M. Ebeidcd and
Maged El-Kemaryab
*ab,
Kamel R. Shoueira,
Ibrahim El-Mehassebb,
El-Zeiny M. Ebeidcd and
Maged El-Kemaryab