
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymorphism, phase transformation and energetic properties of 3-nitro-1,2,4-triazole†
Pengcheng Zhang a,
Xiuxiu Zhaoa,
Yao Dua,
Michael Gozinb,
Shenghua Li*a and
Siping Pang*a
a,
Xiuxiu Zhaoa,
Yao Dua,
Michael Gozinb,
Shenghua Li*a and
Siping Pang*a
aSchool of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: pangsp@bit.edu.cn; lishenghua@bit.edu.cn
bSchool of Chemistry, Faculty of Exact Science, Tel Aviv University, Tel Aviv, 69978, Israel
First published on 10th July 2018
Abstract
We report the preparation, analysis, and phase transformation behavior of polymorphs of 3-nitro-1,2,4-triazole. The compound crystallizes in two different polymorphic forms, Form I (tetragonal, P41212) and Form II (monoclinic, P21/c). Analysis of the polymorphs has been investigated using microscopy, differential scanning calorimetry, in situ variable-temperature powder X-ray diffraction, and single-crystal X-ray diffraction. On heating, Form II converts into Form I irreversibly, and on further heating, decomposition is observed. In situ powder X-ray diffraction studies revealed that Form II transforms to Form I above 98 °C, indicating that Form I is more stable than Form II at high temperature. Form II of 3-nitro-1,2,4-triazole has good detonation properties (Vdet = 8213 m s−1, PC–J = 27.45 GPa), and low sensitivity (IS > 40 J, FS = 360 N, ESD = 29 J), which make it a competitive candidate for use as a new insensitive explosive.
1. Introduction
Crystal polymorphism, the ability of a compound to exist in different crystalline forms that have different arrangements and/or conformations of molecules in the solid state, is a widespread phenomenon that has gained much interest in both academic and industrial research.1 In particular, the polymorphs of energetic materials have an important influence on energetic properties (insensitivity, higher density, higher energy).Energetic materials (explosives and propellants) are used extensively for both civilian and military applications. Some major considerations for successful weaponization of energetic materials include performance (e.g. energy density, rate of energy release), long term storage stability, and insensitivity to unwanted initiation. Therefore, a compromise or a balance of the energy-safety contradiction appears generally in practical applications.2 The term “Insensitive Munition (IM)” is used to describe munitions, which are safe to handle and difficult to initiate accidentally, but still have the power to fulfil the mission requirements to complete the mission.3 Selection of explosives that have inherently lower sensitivity to shock and/or thermal decomposition will reduce the vulnerability of the resulting formulation. TNT (2,4,6-trinitrotoluene, detonation velocity is 7363) and TATB (1,3,5-triamino-2,4,6-trinitrobenzen) (detonation velocity is 7541) are two well-known insensitive explosives; however, both of them have relatively low energy content.
Hence new insensitive compounds with high-energy-density are development targets of energetic material researchers. As an energetic nitrogen heterocyclic group, 1,2,4-triazole features high heats of formation, good thermal stabilities, big densities, which make it a highly efficient structural unit in the development of high-energy-density materials and many energetic triazole based derivatives have been synthesized.4 However, the research about polymorphs of energetic triazole compounds as well other organic compounds are rarely mentioned in the literature.5 In a solid state, the structural transformation was studied using the single-crystal-to-single-crystal (SCSC) and it was explained on the effect of solvent vapors, heat, light and sometimes by applying mechano-chemical forces.6
3-Nitro-1,2,4-triazole is a simple and easy to obtain intermediate, which is widely used in the field of medicine and organic semiconductor materials. We have known Form I of 3-nitro-1,2,4-triazole, it could be used as an explosive compound although there is no report about its energetic properties. In this paper, we report the two different forms of crystallization, analysis, and phase transformation behaviour of 3-nitro-1,2,4-triazole, as well as their energetic properties as insensitive explosives.
2. Results and discussion
Polymorphs screening
The crystallization of 3-nitro-1,2,4-triazole, depending on the conditions employed and solvents used (see the ESI, Tables S1 and S2†), leads to the formation of two different polymorphs. Form I (Fig. 1a) was reported in 1984.7 It has been recently demonstrated that the Form II has significant potential in energetic materials.8 Extensive experimental crystal polymorph screens were performed to identify the relevant polymorphs of 3-nitro-1,2,4-triazole. In numerous solvent-based crystallization trials only one crystal, Form II, was observed once at ambient pressure, and others are Form I (ESI, Tables S1 and S2†). However, both the Forms I and II were stable at ambient temperature and the Form I is more stable at high temperature up to 200 °C, where the Form II is unstable. Interestingly, the Form II undergoes irreversible phase transition to Form I at about 98 °C (ESI, Table S2†). The differential scanning calorimetric (DSC) and powder X-ray diffraction (PXRD) experiment results were clearly indicating this transformation. The crystal structures of Forms I and II are almost identical, except for Form I more disordering than Form II and also, the crystal packing as well as the bond interactions are quite different (ESI, Table S3†).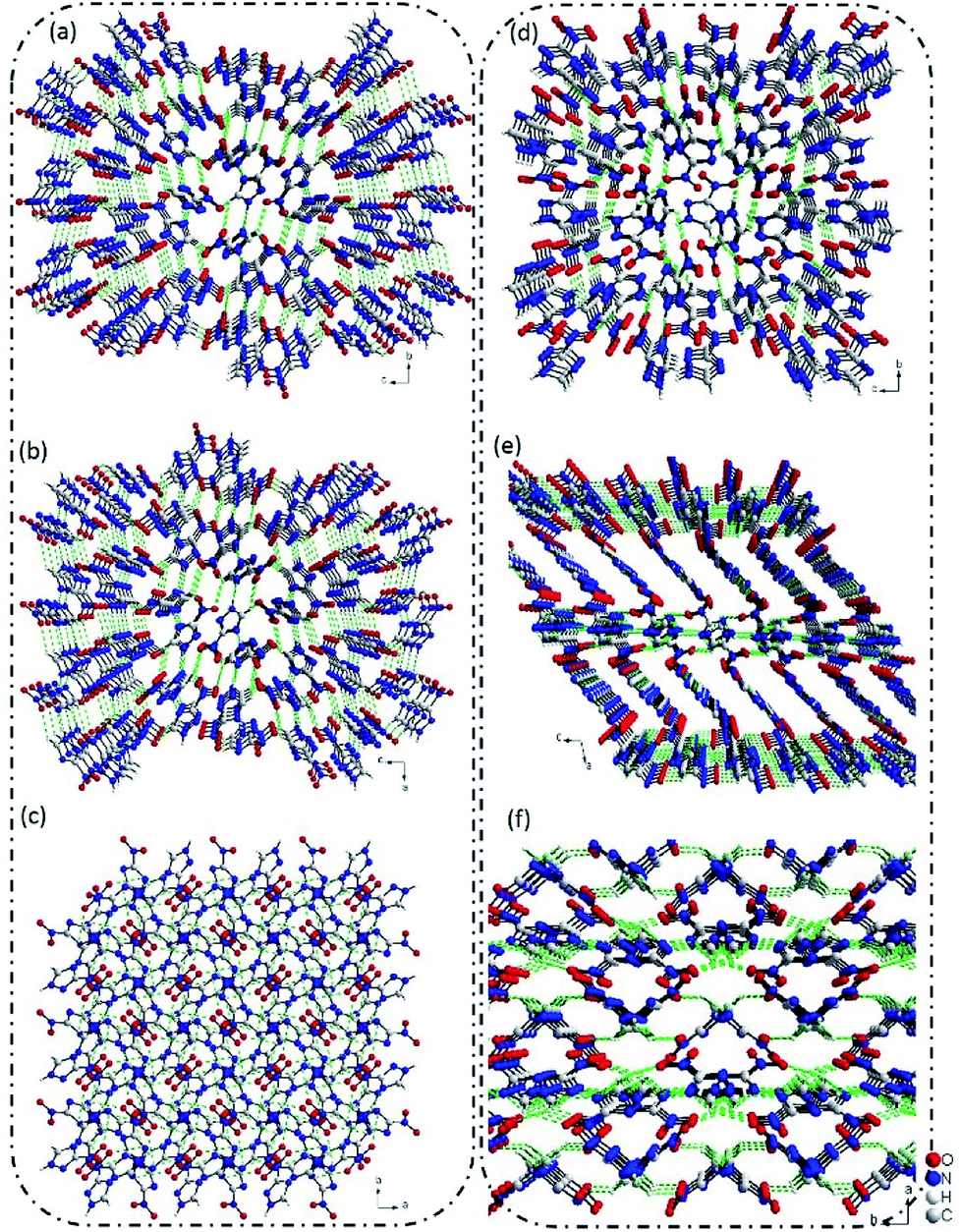 | ||
| Fig. 1 (a)–(c) The 3D supramolecular structure of Form I view along the a, b, c-axis, respectively; (d)–(f) the 3D supramolecular structure of Form II view along the a, b, c-axis, respectively. | ||
Crystal and molecular structure description of polymorphs
The single-crystal X-ray diffraction revealed that Form I crystallized in P41212 space group with a calculated density of 1.720 g cm−3 based on the diffraction data. In Form I, all of the atoms located in one plane, and each 3-nitro-1,2,4-triazole molecule connected with four adjacent molecules via hydrogen bonds of C–H⋯O and N–H⋯O. The distance of C⋯O and N⋯O are in the range of 3.1501–3.3945 Å and 3.2385–3.8411 Å. Through this connected mode, a 3D supramolecular structure was formed (Fig. 1a–c).Interestingly, when we re-crystal 3-nitro-1,2,4-triazole from methanol and ethyl acetate, a new crystal named Form II was obtained. Form II crystallized in P21/c space group with a density of 1.797 g cm−3. In Form II, each 3-nitro-1,2,4-triazole molecule interacted with four nearby molecules through hydrogen bonds and π–π stacking interactions to form a layer structure. The hydrogen bond length is in the range of 105.560–118.364. Then the layers stacked in an “AAAA” mode to build a 3D supramolecular structure (Fig. 1e and f). As shown in Fig. 1, the stacking mode of 3-nitro-1,2,4-triazole molecules contained in two crystal Forms are much different. In Form II, regular layer structures were observed along the b-axis. However, in Form 1, no layers could be observed along any direction. The supramolecular structure of Form II is similar to the high-energy-density material of 5-nitro-1H-tetrazole. This may explain the higher density of Form II than that of Form I in partly.
To gain more insights towards the different densities, calculated packing coefficient of two crystals, Hirshfeld surface and relative two-dimensional finger print spectra of two Form crystals were analyzed systematically. The calculated packing coefficients of both crystals are 71.9% and 75.3%, respectively. Generally, high packing coefficient ratio also imply high density. In this work, two Form of crystals are constructed by the same molecule. Therefore, the higher packing coefficient demonstrated the higher density. Hirshfeld surface of the molecules contained in different crystal Forms were carried out by Crystalexplorer17.5 software. From the picture of Hirshfeld surface, the red and blue spots demonstrate the high and low contact populations, respectively. As shown in Fig. 2, each 3-nitro-1,2,4-triazole molecule interacted with adjacent molecules through strong hydrogen (red spots) and π–π stacking interaction (white plane). The ratio of hydrogen bond interactions contained in Form I and Form II are 56.1% and 50.9%, respectively. From the figure print spots of the Forms, it is shown that the longest distance between two interaction molecules contained in Form I and Form II were appearance at the spots of (2.3, 2.3) and (2.1, 2.1), respectively, demonstrated the closer interaction were existence in Form II.
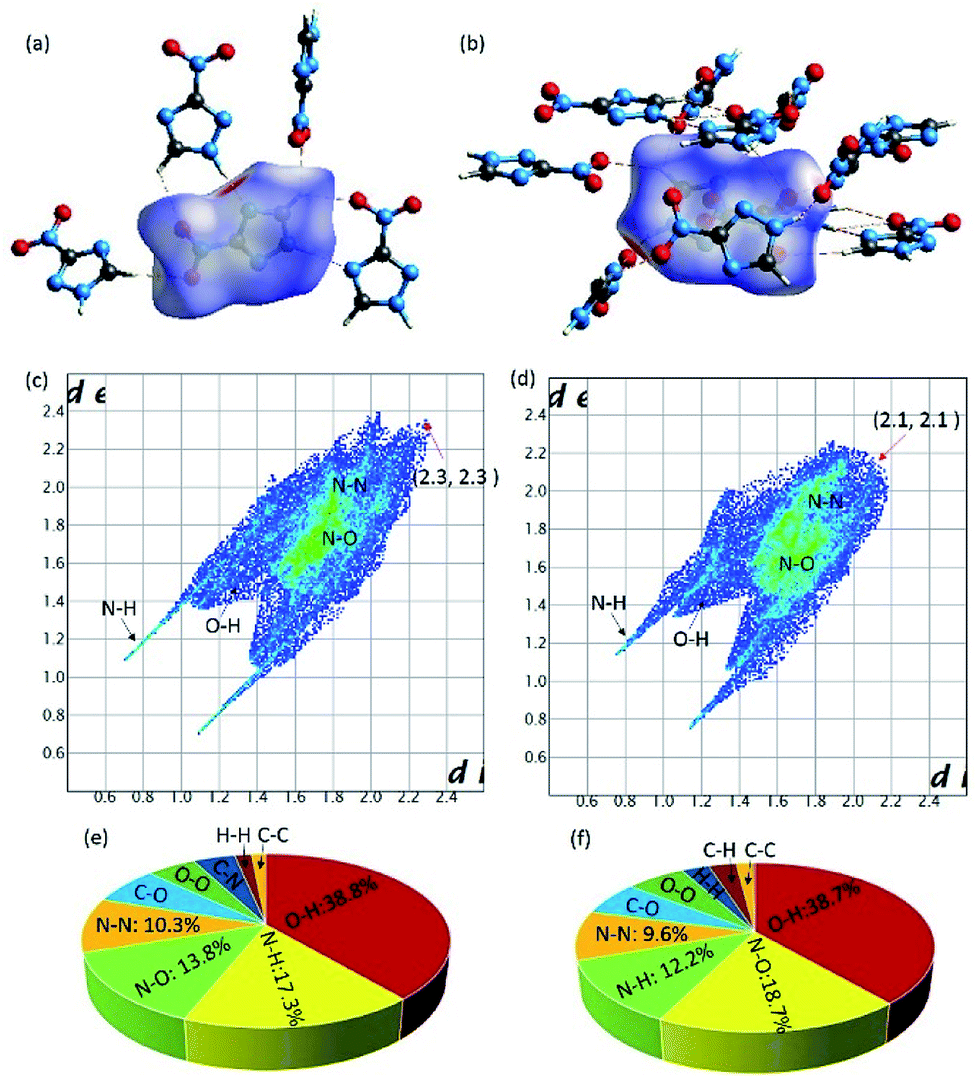 | ||
| Fig. 2 Hirshfeld surface calculations of Form I (a) and Form II (b); finger print plots of Form I (c) and Form II (d); the individual atomic contact percentage contribution to the Hirshfeld surface of Form I (e) and Form II (f). | ||
Furthermore, it is to be note that intensive O–N interactions were observed from the finger print spots of Form II, which is reflected the existence of strong π–π stacking interaction, as shown in Fig. 2. The ratio of O–N interactions from Form II and Form I are 18.7% and 13.8%, respectively, demonstrating the existence of higher percentage of interlayer contact in Form II than that contained in Form I. The higher calculated packing coefficient, more hydrogen bonds, and stronger π–π stacking interaction in Form II may the main reasons, which promote the higher density.
Investigation of stability and transformation behavior of the forms
Differential scanning calorimetry (DSC) and in situ variable-temperature powder X-ray diffraction (PXRD). Moreover, only a few single crystals of Form II could be obtained from the method discussed above, leading to a question regarding the large-scale preparation of this polymorph, as a pure phase.To know the nature of phase transition, DSC measurement was carried out from room temperature to 300 °C. It is evident from Fig. 3 that no changes were observed in the energy profile for Form I, except for a sharp exothermic peak at 217 °C, which is due to the decomposition of Form I. However, for Form II (Fig. 3), two exothermic peaks were observed (98 and 217 °C). Since the latter peak corresponds to the decomposition point of Form I, it suggests that a phase transition of Form II → Form I might have occurred at 98 °C, and it subsequently melted at 217 °C.
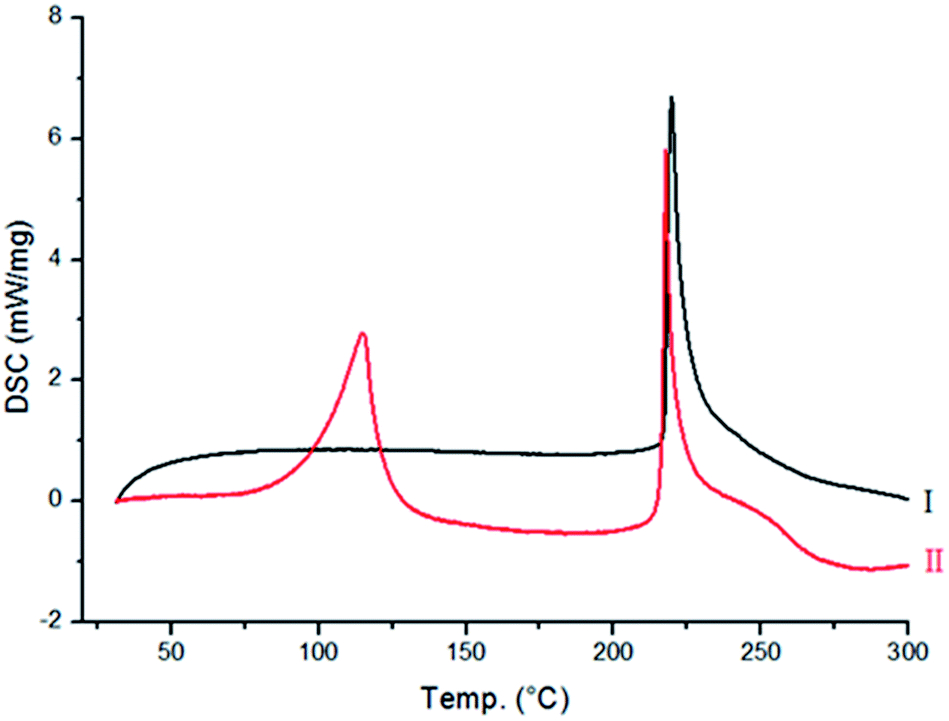 | ||
| Fig. 3 DSC traces of Forms I–II, heating up to decomposition point. | ||
Furthermore, the evidence for the phase transition from Form II into Form I was also obtained in situ variable-temperature powder diffraction experiments. It is evident from Fig. 4a that Form I did not show any phase transitions as the position and intensity of the peaks remain intact throughout the process, irrespective of the temperature variations. However, phase transitions of Forms II to Form I are evident from the appearance of new peaks during heating. The transformations are shown in blue and purple in Fig. 4. The patterns in blue and purple are identical to the patterns shown in Fig. 4a for Form I. This unequivocally confirms the transition of Forms II into I before the decomposition of the Forms II. In Form II, the transition is observed between 98 and 115 °C (with the appearance of the new peaks at 2θ 20, and 23° and disappearance of peaks at 2θ 9, 24, 27, and 35°) (Fig. 4b).
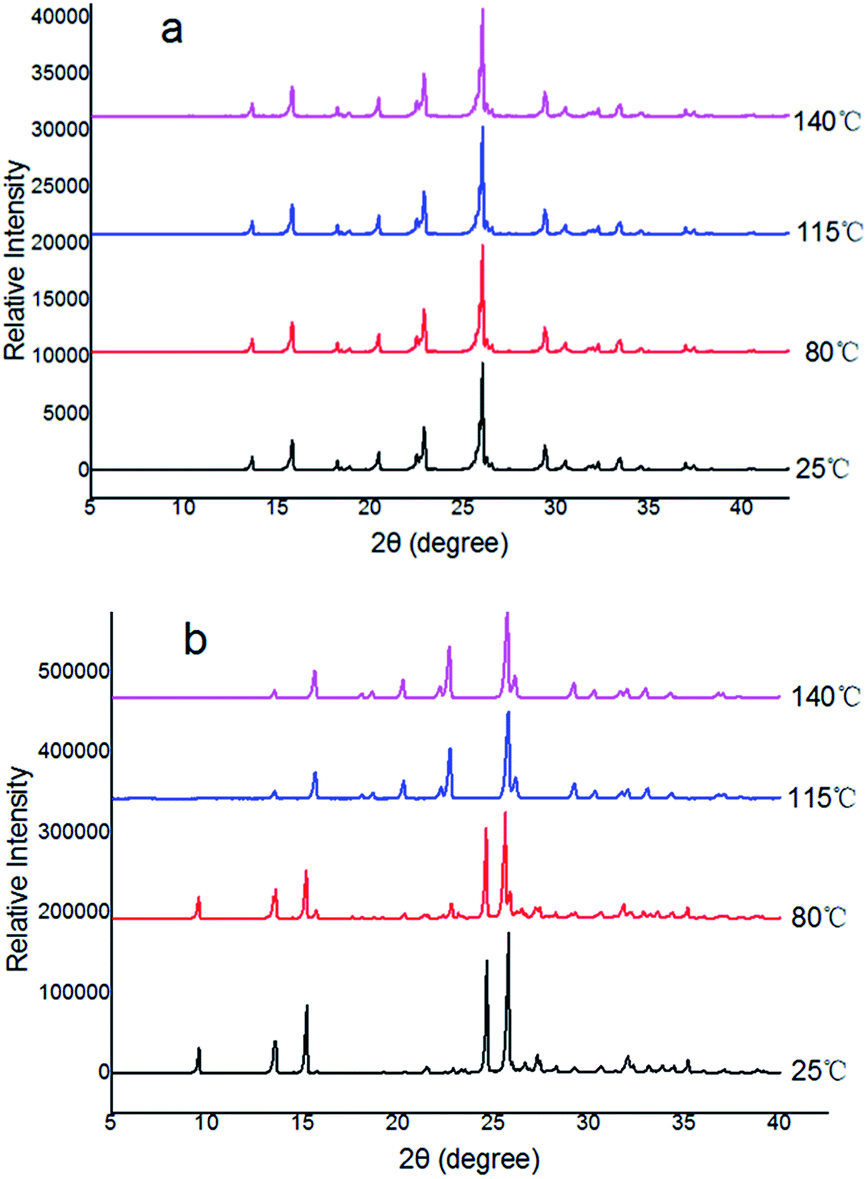 | ||
| Fig. 4 In situ variable-temperature powder X-ray diffraction patterns for the polymorphs: (a) Form I, (b) conversion of Form II → Form I. | ||
Phase to phase transformation
Phase to phase transformation, occurring due to cooperative movement of atoms in the solid state, is an interesting and challenging phenomenon in terms of obtaining good-quality single crystals even after phase transformations, which gives more insight about the mechanism of transformation from one solid form to another.9 Single crystals of Form I did not show any changes in the unit cell dimensions during in situ heating even up to 140 °C, demonstrating the stability of Form I, and no unit cell parameters could be obtained above this temperature. However, single crystals of Form II showed a change in the unit cell parameters, and the phase transition occurred at 98 °C, which corresponds to Form II to I transformation. As per our knowledge, the temperature may play a major role to result the phase transition.10 Also, it may fact that the two solid forms are isostructural in terms of the arrangement of molecules in both two and three dimensions, making easier movement of atoms in the solid-state.Physicochemical properties: heat of formation, detonation parameters, and sensitivities
Density is one of the significant physical characteristics of energetic materials.11 The densities of Form I and II are 1.72 and 1.79 g cm−3, respectively, which are higher that of TNT. Compared to the Form I, the density of the Form II increases by 0.07 g cm−3, which may contribute to the relatively regular molecular arrangement of Form II in the supramolecular structure.The calculated heats of formation of Form I and II was 146.57 kJ mol−1, which were higher than those of both TNT and TATB. Based on calculated heats of formation and experimental, the detonation performances of Form I and Form II, were calculated with EXPLO5 (version 5.05)12 in Table 2. Both Form I and II exhibited better detonation performance than TNT and TATB, as expected, the Form II exhibits the highest value (Vdet = 8213 m s−1, PC–J = 27.45 GPa), compared to Form I, the detonation velocity increases by 266m s−1, highlight the importance of improving detonation properties of energetic compounds through polymorphs screening. To further evaluate their safer properties, the sensitivities of Form I and II toward impact, friction and electrostatic discharge were tested by using standard procedures (Table 2). Both of them exhibited insensitivities to an electrostatic discharge of approximately 29.24 J. Its impact sensitivities and friction sensitivities were above 40 J and 360 N respectively, which are the same as TATB.
| Form I (CCDC 1848407) | Form II (CCDC 1848408) | |
|---|---|---|
| Empirical formula | C2H2N4O2 | C2H2N4O2 |
| Formula weight | 114.08 | 114.08 |
| Crystal habit | Irregular blocks | Needles |
| Crystal color | Yellow | White |
| Crystal system | Tetragonal | Monoclinic |
| Space group | P41212 | P21/c |
| a (Å) | 6.8413(3) | 8.7895(6) |
| b (Å) | 6.8413(3) | 10.0707(8) |
| c (Å) | 18.8196(12) | 9.9645(5) |
| α (deg) | 90 | 90 |
| β (deg) | 90 | 107.082(6) |
| γ (deg) | 90 | 90 |
| V (Å3) | 880.82(8) | 843.10(10) |
| Z | 8 | 8 |
| Dcalc. (g cm−3) | 1.720 | 1.797 |
| T (K) | 101.8 | 101.8 |
| μ (mm−1) | 0.152 | 0.159 |
| 2θ range (deg) | 7.36 to 52 | 6.78 to 52 |
| F(000) | 464 | 464 |
| GOF on F2 | 1.098 | 1.078 |
| Final R indexes [I > 2σ(I) i.e. Fo > 4σ(Fo)] | R1 = 0.0375, wR2 = 0.0860 | R1 = 0.0318, wR2 = 0.0734 |
| Final R indexes [all data] | R1 = 0.0410, wR2 = 0.0883 | R1 = 0.0378, wR2 = 0.0772 |
| Form I | Form II | TATB | 2,4,6-TNT | |
|---|---|---|---|---|
| a BAM drop hammer.b BAM friction tester.c Nitrogen content.d Oxygen balance.e Temperature of decomposition by DSC (β = 5 °C, onset values).f Density derived from X-ray structure measurement at 25 °C.g Molar enthalpy of formation.h Energy of explosion.i Explosion temperature.j Detonation pressure.k Detonation velocity. | ||||
| Molecular formula | C2H2N4O2 | C2H2N4O2 | C6H6N6O6 | C7H5N3O6 |
| Molecular mass [g mol−1] | 114.08 | 114.08 | 258.2 | 227.13 |
| Impact sensitivitya [J] | >40 | >40 | >40 | 15 (ref. 13) |
| Friction sensitivityb [N] | 360 | 360 | 360 | 353 |
| ESD test [J] | 29.24 | 29.24 | 17.75 | — |
| Nc [%] | 49.1 | 49.1 | 33 | 18.50 |
| Ωd [%] | −24.1 | −24.1 | −55.8 | −73.96 |
| Tdec.e [°C] | 217 | 217 | 330 | 290 |
| ρf [g cm−3] | 1.72 | 1.79 | 1.858 | 1.648 (ref. 14) |
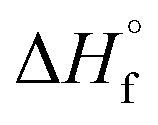 g [kJ mol−1] g [kJ mol−1] |
146.57 | 146.57 | 139.8 | −55.5 |
| EXPLO5 values, V5.05 | ||||
| −ΔEU°h [kJ kg−1] | 4786 | 4822 | 4520 | 5258 |
| TEi [K] | 3561 | 3530 | — | 3663 |
| PC–Jj [GPa] | 24.81 | 27.45 | 25.9 | 23.5 |
| Vdetk [m s−1] | 7947 | 8213 | 7541 | 7459 |
Since 3-nitro-1,2,4-triazole-Form II has better detonation performance than TNT and TATB, while retain low sensitivity, it could be used as new insensitive explosive, replacing TNT and TATB.
3. Experimental
Chemicals and crystal growth
3-Nitro-1,2,4-triazole was purchased from Sigma-Aldrich. HPLC grade solvents were used for crystallization. From PXRD, it is confirmed that the commercially available material 3-nitro-1,2,4-triazole is found to be Form II. Crystallization was carried out from a wide variety of solvents, solvent mixtures, and other crystallization methods (see the ESI†), which resulted in two different solid forms labeled as Forms I, II.Crystallization
The sample of 3-nitro-1,2,4-triazole (≥98%) used in this work was purchased from Maya Reagent. For crystallization experiments, 3-nitro-1,2,4-triazole was dissolved in a solvent or mixture of solvents and left at ambient temperature (for few days) to allow slow evaporation of solvent until crystals appeared. The solvents used were ethanol (Aldrich, >99.8%), methanol (Fisher, >99.5%), acetone (Fisher, >99%), acetone (Aldrich, 99.5% atom% D), dichloromethane (Fisher, >99%), and glacial acetic acid (Fisher, >99%). In addition, the following mixed solvent systems (∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) were used: methanol/ethyl acetate. Powder XRD studies of the crystallization products were carried out at ambient temperature on a Bruker D8 instrument operating in transmission mode (CuKα1 radiation, Ge monochromated).
:1 ratio) were used: methanol/ethyl acetate. Powder XRD studies of the crystallization products were carried out at ambient temperature on a Bruker D8 instrument operating in transmission mode (CuKα1 radiation, Ge monochromated).
Powder XRD analysis of the crystallization products indicated that, with the exception of crystallization from hydroxylamine, all crystallization experiments gave the same product, which we designate as α. Crystallization from hydroxylamine, on the other hand, which we designate as β.
Single-crystal X-ray diffraction
Details of the single-crystal X-ray diffraction intensity measurements and refinements are given in Table 1. All operations for Forms I, II, I′, I′′, and II′ were performed on a Bruker-Nonius Kappa Apex2 diffractometer, using graphite-monochromated MoKα radiation (λ = 0.7107 Å). All diffractometer manipulations, including data collection, integration, scaling, and absorption corrections, for these forms were carried out using the Bruker Apex2 software.15 Data collection for Form I and II was carried out at 120 K, using a frame time of 20 s and a detector distance of 60 mm. The optimized strategy used for data collection consisted of four φ and six ω scan sets, with 0.5° steps in φ or ω. The structures were solved by using SHELXS and refined by least-squares methods using SHELXL.16 From the systematic absences, the observed metric constants, and intensity statistics, space groups P41212 and P21/c were chosen for Forms I, II, respectively, at the beginning; subsequent solution and refinement confirmed the correctness of the choice. The structures (Forms I, II, I′, I′′, and II′) were solved using SIR92 and subsequent electron-density difference syntheses17 and refined (full-matrix least-squares) using the Oxford University Crystals for Windows program.18,19 All non-hydrogen atoms were refined using anisotropic displacement parameters.PXRD
Powder X-ray diffraction measurements were carried out with a Rigaku Dmax 2500 diffractometer using Cu Kα radiation (λ = 1.5418 Å). The sample holder was a copper block, and a very thin layer of powder sample was pressed on this block. The patterns were collected in the 2θ range of 5–90° with a step size of 0.02° and 1.0 s counting per step. The copper block with the sample was heated at the rate of 10 °C min−1. The diffraction pattern was collected while the sample temperature was held constant within 1 °C of the set temperature, and the data were acquired in 16 min. The diffraction data were collected at room temperature and subsequently after every 10 °C to monitor the change in structure during heating. The positions of the peaks were fixed by deconvoluting the peaks using Rigaku multipeak separation software available with the diffractometer system.Thermal analysis
The DSC experiments were performed using a DSC 1 Star System with STARe Excellence Software from Mettler-Toledo AG. Samples in the range of 3–4 mg were placed in Al crucibles after surface-drying on filter paper. The sample was heated from 30 to 300 °C at a rate of 5 °C min−1.4. Conclusions
To summarize, preparation and rational analysis of two polymorphs of 3-nitro-1,2,4-triazole have been reported and thoroughly characterized. In the two forms, the –NH group presents that between the –NO2 groups forms intramolecular N–H⋯O hydrogen bonding. In Forms I and II, molecules interact through N–H⋯O hydrogen bond formation. In the phase transition of Form II to I has also been demonstrated, Form II is more stable at ambient conditions, whereas Form I is more stable at higher temperatures. Compared to Form I, the density of Form II increases by 0.07 g cm−3, the detonation velocity increases by 266 m s−1, highlight the importance of improving detonation properties of energetic compounds through polymorphs screening. The Form II of 3-nitro-1,2,4-triazole have good detonation properties (Vdet = 8213 m s−1, PC–J = 27.45 GPa), and low sensitivity (IS > 40 J, FS > 360 N, ESD = 29 J), which make it a competitive candidate for using as new insensitive explosives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from NSAF (U153062), the National Natural Science Foundation of China (21576026) and the Fundamental Research Funds for the Central Universities.References
- (a) R. Hilfiker, Org. Process Res. Dev., 2007, 11, 649–650 Search PubMed; (b) R. J. Davey, Cryst. Growth Des., 2002, 2, 675–676 CrossRef; (c) D. Singhal and W. Curatolo, Adv. Drug Delivery Rev., 2004, 56, 335–347 CrossRef PubMed; (d) A. B. Park, L. J. chyall, J. Dunlap, C. Schertz, D. Jonaitis, B. C. Stahly, S. Bates, R. Shipplett and S. Childs, Expert Opin. Drug Discovery, 2007, 2, 145–154 CrossRef PubMed.
- (a) Q. L. Yan, A. Cohen, N. Petrutik, A. Shlomovich, L. Burstein, S. P. Pang and M. Gozin, J. Mater. Chem. A, 2016, 4, 9941–9948 RSC; (b) A. Cohen, Y. Z. Yang, Q. L. Yan, A. Shlomovich, N. Petrutik, L. Burstein, S. P Pang and M. Gozin, Chem. Mater., 2016, 28, 6118–6126 CrossRef; (c) W. Liu, Q. H. Lin, Y. Z. Yang, X. J. Zhang, Y. C. Li, Z. H. Lin and S. P. Pang, Chem.–Asian J., 2014, 9, 479–486 CrossRef PubMed.
- I. J. Powell, Propellants, Explos., Pyrotech., 2016, 41, 409–413 CrossRef.
- (a) W. Liu, S. h. Li, Y. C. Li, Y. Z. Yang, Y. Yu and S. P. Pang, J. Mater. Chem. A, 2014, 2, 15978–15986 RSC; (b) C. Qi, S. H. Li, Y. C. Li, Y. Wang, X. X. Zhao and S. P. Pang, Chem.–Eur. J., 2012, 18, 16562–16570 CrossRef PubMed.
- (a) L. Mazur, A. E. Koziol, K. N. Jarzembska, R. Paprocka and B. Modzelewska-Banachiewicz, Cryst. Growth Des., 2017, 17, 2104–2115 CrossRef; (b) J. Z. Huo, X. M. Su, X. X. Wu, Y. Y. Liu and B. Ding, CrystEngComm, 2016, 18, 6640–6652 RSC; (c) C.-P. Li, J.-M. Wu and M. Du, Inorg. Chem., 2011, 50, 9284–9289 CrossRef PubMed; (d) J. P. Reddy, D. Swain and V. R. Pedireddi, Cryst. Growth Des., 2014, 14, 5064–5071 CrossRef; (e) T. S. Thakur, R. Sathishkumar, A. G. Dikundwar, T. N. Guru Row and G. R. Desiraju, Cryst. Growth Des., 2010, 10, 4246–4249 CrossRef.
- A. Chaudhary, A. Mohammad and S. M. Mobin, Cryst. Growth Des., 2017, 17, 2893–2910 CrossRef.
- G. Evrard, F. Durant and A. Michel, Bull. Soc. Chim. Belg., 1984, 93, 233–234 CrossRef.
- M. Hemamalini and H.-K. Fun, Acta Crystallogr., 2011, E67, o15 Search PubMed.
- (a) E. N. Duesler, K. E. Wiegers, D. Y. Curtin and I. C. Paul, Mol. Cryst. Liq. Cryst., 1980, 59, 289–298 CrossRef; (b) H. Nakanishi, W. Jones and J. M. Thomas, Chem. Phys. Lett., 1980, 71, 44–48 CrossRef; (c) V. Enkelmann, G. Wegner, K. Novak and K. B. Wagener, J. Am. Chem. Soc., 1993, 115, 10390–10391 CrossRef; (d) J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000–1002 CrossRef PubMed; (e) A. Natarajan, C. K. Tsai, S. I. Khan, P. McCarren, K. N. Houk and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2007, 129, 9846–9847 CrossRef PubMed; (f) J. A. G. D. Ripmeester Enright, C. I. Ratcliffe, K. A. Udachin and I. L. Moudrakovski, Chem. Commun., 2006, 4986–4996 RSC; (g) J. Tian, P. K. Thallapally, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2009, 131, 13216–13217 CrossRef PubMed; (h) E. Y. Lee and M. P. Suh, Angew. Chem., 2004, 116, 2858–2861 CrossRef; (i) J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef; (j) O. Ohmori, M. Kawano and M. Fujita, J. Am. Chem. Soc., 2004, 126, 16292–16293 CrossRef PubMed; (k) E. Deiters, V. Bulach and M. W. Hosseini, Chem. Commun., 2005, 3906–3908 RSC; (l) G. J. Halder and C. J. Kepert, Aust. J. Chem., 2006, 59, 597–604 CrossRef; (m) K. L. Gurunatha, S. Mohapatra, P. A.Suchetan and T. K. Maji, Cryst. Growth Des., 2009, 9, 3844–3847 CrossRef; (n) D. Bradshaw, J. E. Warren and M. Rosseinsky, Science, 2007, 315, 977–979 CrossRef PubMed; (o) C. Q. li, D. C. Swenson and L. R. MacGillivray, Angew. Chem., Int. Ed., 2005, 44, 3569–3572 CrossRef PubMed; (p) S. Supriya and S. K. Das, J. Am. Chem. Soc., 2007, 129, 3464–3465 CrossRef PubMed; (q) I. Halasz, Cryst. Growth Des., 2010, 10, 2817–2823 CrossRef.
- (a) E. Lee, Y. Kim, J. Heo and K.-M. Park, Cryst. Growth Des., 2015, 15, 3556–3560 CrossRef; (b) T. T. Zhang, Y. Lu, Z. M. Zhang, Q. Lan, D. Liu and E. B. Wang, Inorg. Chim. Acta, 2014, 411, 128–133 CrossRef; (c) S. Uchida, E. Takahashi and N. Mizuno, Inorg. Chem., 2013, 52, 9320–9326 CrossRef PubMed.
- X. X. Zhao, S. H. Li, Y. Wang, Y. C. Li, F. Q. Zhao and S. P. Pang, J. Mater. Chem. A, 2016, 4, 5495–5504 RSC.
- M. SućeskaEXPLO5.5 program, Zagreb, Croatia, 2010 Search PubMed.
- R. Mayer, J. Köhler and A. Homburg, Explosives, Wiley-VCH, Weinheim, 5th edn, 2002 Search PubMed.
- N. I. Golovina, A. N. Titkov, A. V. Raevskii and L. O. Atovmyan, J. Solid State Chem., 1994, 113, 229–238 CrossRef.
- Apex2, Version 2 User Manual, M86–E01078, Bruker Analytical X-ray Systems, Madison, WI, 2006 Search PubMed.
- G. M. Sheldrick, SHELXTL-PLUS Program for Crystal structure Solution and Refinement, University of Gottingen, Gottingen, Germany, 1997 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef.
- C. K. Prout and L. J. Pearce, CAMERON, Chemical Crystallography Laboratory, Oxford, U.K., 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For ESI and crystallographic data in CIF. CCDC 1848407 and 1848408. See DOI: 10.1039/c8ra04116d |
| This journal is © The Royal Society of Chemistry 2018 |