
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,2,3-Triazole based bisphosphine, 5-(diphenylphosphanyl)-1-(2-(diphenylphosphanyl)-phenyl)-4-phenyl-1H-1,2,3-triazole: an ambidentate ligand with switchable coordination modes†
Latchupatula Radhakrishna,
Madhusudan K. Pandey and
Maravanji S. Balakrishna *
*
Phosphorus Laboratory, Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: krishna@chem.iitb.ac.in; msb_krishna@iitb.ac.in; Fax: +91-22-5172-3480/2576-7152
First published on 18th July 2018
Abstract
The reaction of 1-(2-bromophenyl)-4-phenyl-1H-1,2,3-triazole (1) with Ph2PCl yielded bisphosphine, 5-(diphenylphosphanyl)-1-(2-(diphenylphosphanyl)phenyl)-4-phenyl-1H-1,2,3-triazole (2). Bisphosphine 2 exhibits ambidentate character in either the κ2-P,N or κ2-P,P coordination mode. Treatment of 2 with [M(CO)4(piperidine)2] (M = Mo and W) yielded κ2-P,N and κ2-P,P coordinated Mo0 and W0 complexes [M(CO)4(2)] [M = W-κ2-P,N (4); Mo-κ2-P,P (5); W-κ2-P,P (6)] depending on the reaction conditions. Formation and stability of κ2-P,P coordinated Mo0 and W0 complexes were assessed by time dependent 31P{1H} NMR experiments and DFT studies. The complex 4 on treatment with [AuCl(SMe2)] afforded the hetero-bimetallic complex [μ-PN,P-{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2AuCl)}-κ2-P,N}W(CO)4] (7). The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction between 2 and [CpRu(PPh3)2Cl] yielded [(η5-C5H5)RuCl{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-κ2-P,P] (8), whereas the similar reaction with [Ru(η6-p-cymene)Cl2]2 in a 2:1 molar ratio produced a cationic complex [(η6-p-cymene)RuCl{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-κ2-P,N]Cl (9). Similarly, treatment of 2 with [M(COD)(Cl)2] (M = Pd and Pt) in a 1:1 molar ratio yielded PdII and PtII complexes [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}PdCl2] (10) and [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}PtCl2] (11). The reaction of 2 with 2 equiv. of [AuCl(SMe2)] afforded [Au2Cl2{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-μ-P,P] (12). Most of the complexes have been structurally characterized. Palladium complex 10 shows excellent catalytic activity towards Cu-free Sonogashira alkynylation/cyclization reactions.
:1 reaction between 2 and [CpRu(PPh3)2Cl] yielded [(η5-C5H5)RuCl{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-κ2-P,P] (8), whereas the similar reaction with [Ru(η6-p-cymene)Cl2]2 in a 2:1 molar ratio produced a cationic complex [(η6-p-cymene)RuCl{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-κ2-P,N]Cl (9). Similarly, treatment of 2 with [M(COD)(Cl)2] (M = Pd and Pt) in a 1:1 molar ratio yielded PdII and PtII complexes [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}PdCl2] (10) and [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}PtCl2] (11). The reaction of 2 with 2 equiv. of [AuCl(SMe2)] afforded [Au2Cl2{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-μ-P,P] (12). Most of the complexes have been structurally characterized. Palladium complex 10 shows excellent catalytic activity towards Cu-free Sonogashira alkynylation/cyclization reactions.
Introduction
Chelating ligands having both soft- and hard-donor atoms have drawn considerable interest owing to their ability to stabilize the metal atoms before and after the oxidative addition and reductive elimination reactions, two important steps in metal mediated homogeneous catalysis.1 If the ligands possess more than two donor atoms, and unlike pincer complexes, show flexibility in their coordination behavior to vary the donor atoms under the influence of temperature,2 light3 or redox reagents,4 they can also find application in molecular switches, logic gates, sensors etc.5 Therefore incorporating heteroaryl groups having donor atoms such as nitrogen, oxygen or sulphur in close proximity to phosphine moieties can generate interesting ligand systems with rich coordination chemistry.6 In this context, mono- or bisphosphines containing pyridine,7 imidazole,8 pyrazole9 or furan10 moieties have attracted more attention, whereas triazole based systems have been less explored,11 though they exhibit ambidentate behavior and find application in catalysis.2,12Recently we reported the synthesis of two triazole based mono phosphines by exploiting the temperature controlled Li/H exchange phenomenon and studied their group 11 metals chemistry.13 As a continuation of our work, herein, we describe the synthesis of C1-symmetric triazole based bisphosphine 2. We also demonstrate the ambidentate behavior of ligand 2 with Mo0 and W0 metal centers using extensive NMR studies and DFT calculations. The coordination chemistry of ligand 2 with PdII, PtII, RuII and AuI metal precursors and the catalytic activity of palladium complex 10 in tandem Cu-free Sonagashira coupling/cyclization reactions are also investigated.
Results and discussion
Synthesis of 5-(diphenylphosphanyl)-1-(2-(diphenylphosphanyl)phenyl)-4-phenyl-1H-1,2,3-triazole (2)
The temperature-controlled lithiation of 1-(2-bromophenyl)-4-phenyl-1H-1,2,3-triazole (1), followed by the treatment of Ph2PCl resulted in two different 1,2,3-triazole functionalized monophosphines [o-Ph2PC6H4{1,2,3-N3C(Ph)C(H)}] and [C6H5{1,2,3-N3C(Ph)C(PPh2)}] depending on the reaction conditions.13 This method was employed to synthesize triazole based new bisphosphine in one or two step reactions. Treatment of 1-(2-bromophenyl)-4-phenyl-1H-1,2,3-triazole (1) with two equivalents of nBuLi, followed by the addition of two equivalents of Ph2PCl yielded bisphosphine [o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}] (2) (Scheme 1). Alternately, 1-(2-bromophenyl)-4-phenyl-1H-1,2,3-triazole (1) on treatment with one equivalent of nBuLi followed by the addition of Ph2PCl at −78 °C afforded kinetically stable triazole based mono phosphine A, which on further treatment with nBuLi followed by the addition of Ph2PCl at room temperature produced bisphosphine [o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}] (2) as shown in Scheme 1. The 31P{1H} NMR spectrum of 2 showed two doublets at −30.4 and −17.5 ppm with a JPP coupling of 22.3 Hz. The existence of coupling between the two phosphorus atoms, which are five bonds apart, was confirmed by 31P–31P COSY experiment. Although, the origin of the coupling is not clear, the large 5JPP value indicates the least possibility of through bond coupling and does not rule out the possibility of through-space coupling. C1-symmetric biaryl-bisphosphines14 and imidazole based bisphosphines (Table 1) have shown through-space coupling with JPP values of similar magnitude.8b Bischalcogenide derivatives of 2 (2a and 2b; See ESI Scheme S1†) did not show phosphorus–phosphorus coupling probably due to the non-availability of phosphorus lone pairs for through space interaction, thus suggesting the coupling to be of through-space in nature. The assignment of chemical shifts in 2 are based on previously reported triazole based mono phosphines.13 The triazole bonded phosphorus is significantly shielded compared to the ortho-carbon bound phosphorus atom. | ||
| Scheme 1 Synthesis of triazole based phosphorus ligand 2. | ||
| Compounda | R | δPb | JPP (Hz) | Reference |
|---|---|---|---|---|
| a In all these compounds phosphorus atoms are five bonds apart.b δ in ppm.c Not observed. | ||||
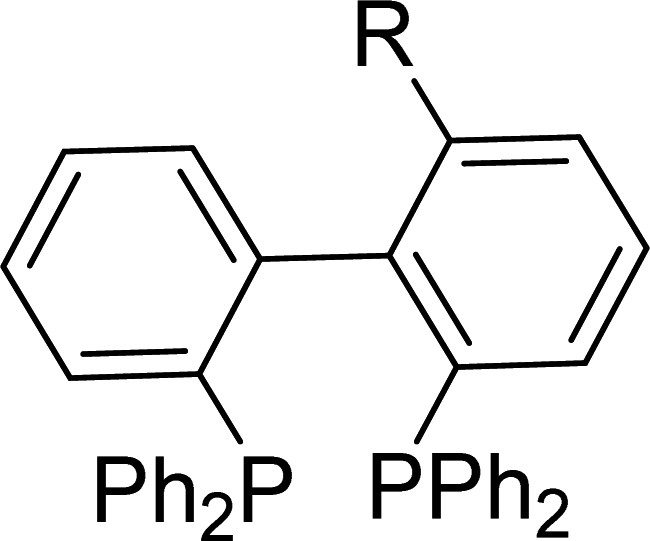 |
OCH3 | −13.6 (d), −14.0 (d) | 18.3 | 14 |
| CH3 | −12.8 (d), −14.6 (d) | 27.1 | 14 | |
| Cl | −12.0 (d), −14.1 (d) | 22.5 | 14 | |
| C6H5 | −13.9 (d), −15.1 (d) | 11.4 | 14 | |
| N(CH3)2 | −14.4 (d), −14.5 (d) | 5.3 | 14 | |
| [o-Ph2P(C6H4){1-N(CH)2-4-NC(PPh2)}] | −18.0 (d), −30.3 (d) | 24.3 | 8b | |
| [o-Ph2P(C6H4){1-N(CH)2-4-N(CH3)C(PPh2)}]OTf | −19.6 (d), −22.6 (d) | 27.9 | 8b | |
| [o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}] (2) | −17.5 (d), −30.4 (d) | 22.3 | This paper | |
| [o-Ph2P(O)(C6H4){1,2,3-N3C(Ph)C((O)PPh2)}] (2a) | 28.5 (s), 12.7 (s) | c | This paper | |
| [o-Ph2P(Se)(C6H4){1,2,3-N3C(Ph)C((Se)PPh2)}] (2b) | 31.6 (s) 1JPSe = 754 (Hz), 18.5 (s) 1JPSe = 752 (Hz) | c | This paper | |
Single crystals of 2 suitable for X-ray diffraction analysis were obtained from an ethanol solution of 2 kept at 0 °C for 24 h. The phosphorus centers exhibit distorted trigonal pyramidal geometry with both the phosphorus atoms being orthogonal to each other as shown in Fig. 1. The P2–C7 [1.830(3) Å] bond distance is little shorter compared to P1–C2 (1.859(3) Å) bond distances due to the better electron withdrawing nature of the triazole ring.
 | ||
| Fig. 1 Molecular structure of 2 with 50% ellipsoid probability. Hydrogen atoms were omitted for clarity. Selected bond distances (Å) and bond angles (°) of 2: P1–C2 1.859(3), P1–C15 1.835(3), P2–C7 1.830(3), P2–C27 1.827(3), N1–N2 1.352(3), N2–N3 1.312(3), C2–P1–C15 101.42(12), C2–P1–C21 101.18(12), C15–P1–C21 103.93(12), C7–P2–C27 103.14(12), C7–P2–C33 101.64(12), C27–P2–C33 103.12(12), N1–N2–N3 106.7(2). | ||
Coordination chemistry of bisphosphine 2
Recently, we demonstrated the coordination behavior of triazole based monophoshines towards group 11 metals, in which ligand showed mono-, di- as well as tridentate coordinating modes.13 Although, bidentate mode is common with the triazole based phosphines, the reports on the participation of two nitrogen atoms in coordination is rare. Bisphosphine 2 can act as a potential tetradentate ligand. The plausible coordinating modes for the ligand 2 are listed in Chart 1. Although, simple κ2-P,P (I) and κ-P, κ-P-coordination (IV) via phosphorus atoms are the most common and preferred modes for 2, it can also show chelating [κ2-P,N] (II), as well as chelating-cum-bridging [κ2-P,N and μ-PN,P] (III) modes to perform as a tridentate ligand. However the possibility of 2 showing other coordinating modes V–IX is less likely (Chart 1), but are not precluded, as appropriate metal precursors might enable these coordination modes. Therefore, it would be interesting to study the coordination chemistry of 2 with various transition metal precursors.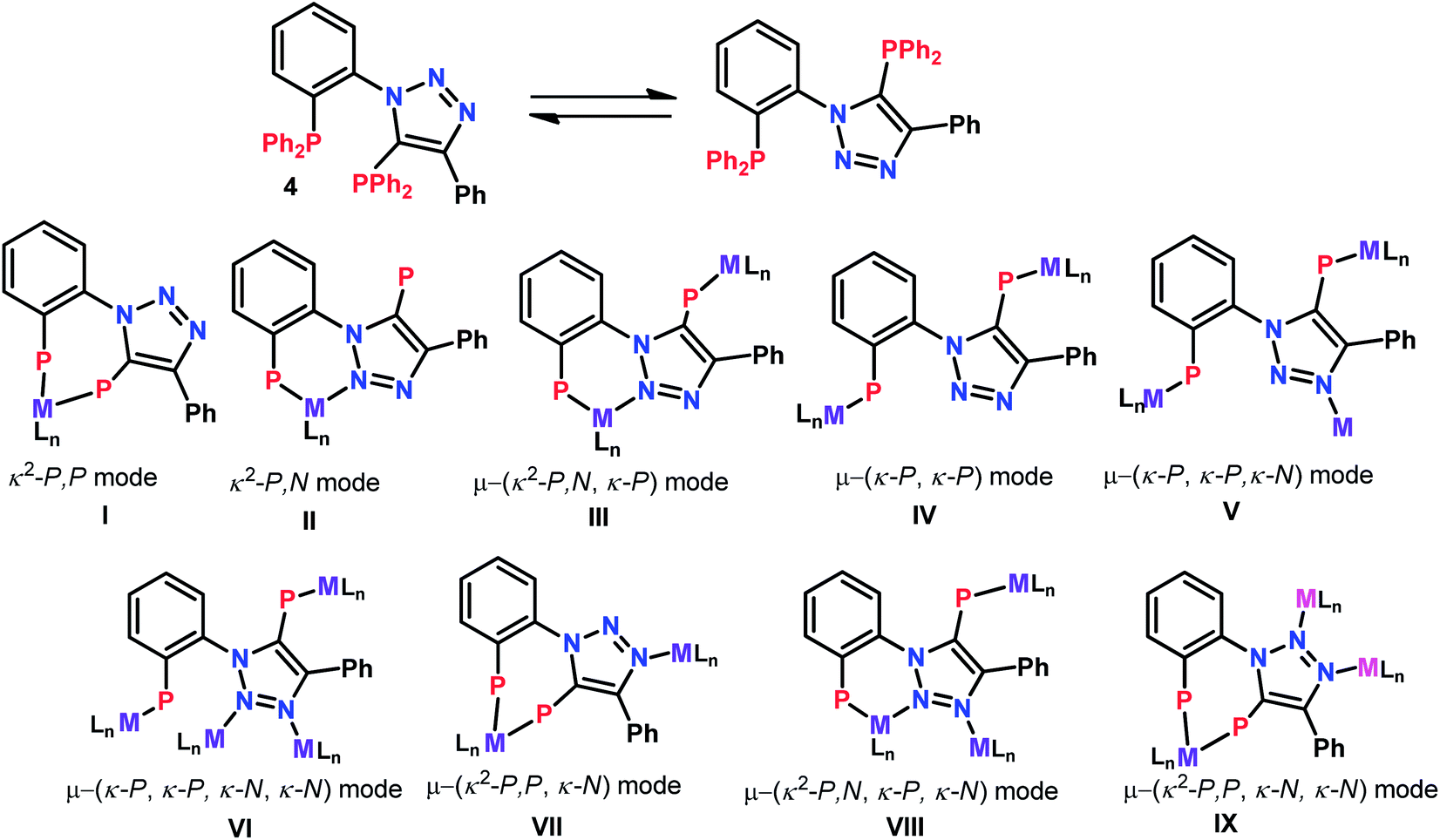 | ||
| Chart 1 Possible coordinating modes for bisphosphine 2. | ||
Treatment of bisphosphine 2 with [Mo(piperidine)2(CO)4] in CH2Cl2 at room temperature resulted in [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}Mo(CO)4] (5) with ligand showing κ2-P,P coordination. The same reaction with [W(piperidine)2(CO)4] afforded initially κ2-P,N coordinated complex, [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,N}W(CO)4] (4) which on prolonging the reaction time, yielded κ2-P,P coordinated complex [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}W(CO)4] (6) as shown in Scheme 2. The complexes 4 and 5 were characterized by 31P{1H} NMR spectroscopy and their structures were confirmed by single crystal X-ray analysis. The 31P{1H} NMR spectrum of complex 4 shows two singlets at 23 (1JWP ≈ 239.2 Hz) and −23.9 ppm, clearly indicating the coordination of aryl group bound phosphorus atom, probably along with one of the triazole nitrogen atoms and triazolyl phosphorus atom being left uncoordinated. The 31P{1H} NMR spectrum of complex 5 consists of two doublets at 35.6 and 23.7 ppm with a 2JPP ≈ 19 Hz indicating κ2-P,P coordination to molybdenum.
 | ||
| Scheme 2 Synthesis of Mo0 and W0 complexes 4–6. | ||
To gain some insight into the stability of complexes 3 and 4, we have examined the ΔG values obtained from DFT calculations. The energy difference (ΔG) between the κ2-P,N coordinated W0 complex 4 and the κ2-P,N coordinated Mo0 intermediate 3 is −175.1 kcal mol−1, which clearly indicates relatively more stability of 4. Similarly, the energy difference (ΔG) between the complexes 3 and 5 is −6.6 kcal mol−1 and the same between 4 and 6 is −6.4 kcal mol−1. Therefore, the kinetically controlled κ2-P,N-complex isomerizes to κ2-P,P-complex. A dichloromethane solution of κ2-P,N coordinated complex 4 on stirring at room temperature for 72 h or the reaction of 2 with [W(piperidine)2(CO)4] in toluene under reflux conditions for 12 h yielded κ2-P,P-complex 6 in quantitative yield. The 31P{1H} NMR spectrum of 6 shows two doublets at 18.9 ppm (1JWP ≈ 238.3 Hz, 2JPP ≈ 12.8 Hz) and 6.9 ppm (1JWP ≈ 247.3 Hz, 2JPP ≈ 12.8 Hz). The course of the reaction was also monitored by time dependent 31P{1H} NMR spectroscopy. Initially in the case of molybdenum, two resonances were observed at 34.1 and −24.9 ppm, corresponding to coordinated and uncoordinated phosphorus atoms indicating the formation of κ2-P,N coordinated complex 3. However, within 30 minutes 50% of the κ2-P,N complex transformed into κ2-P,P complex, and the isomerization was complete within 2 h (Fig. 2). In the case of tungsten, isomerization starts after 12 h and 50% conversion was observed after 40 h and the isomerization was complete after 72 h (Fig. 3). These NMR studies clearly show the formation of thermodynamically favored κ2-P,P coordination complexes via isomerization of initially formed κ2-P,N coordinated complexes.
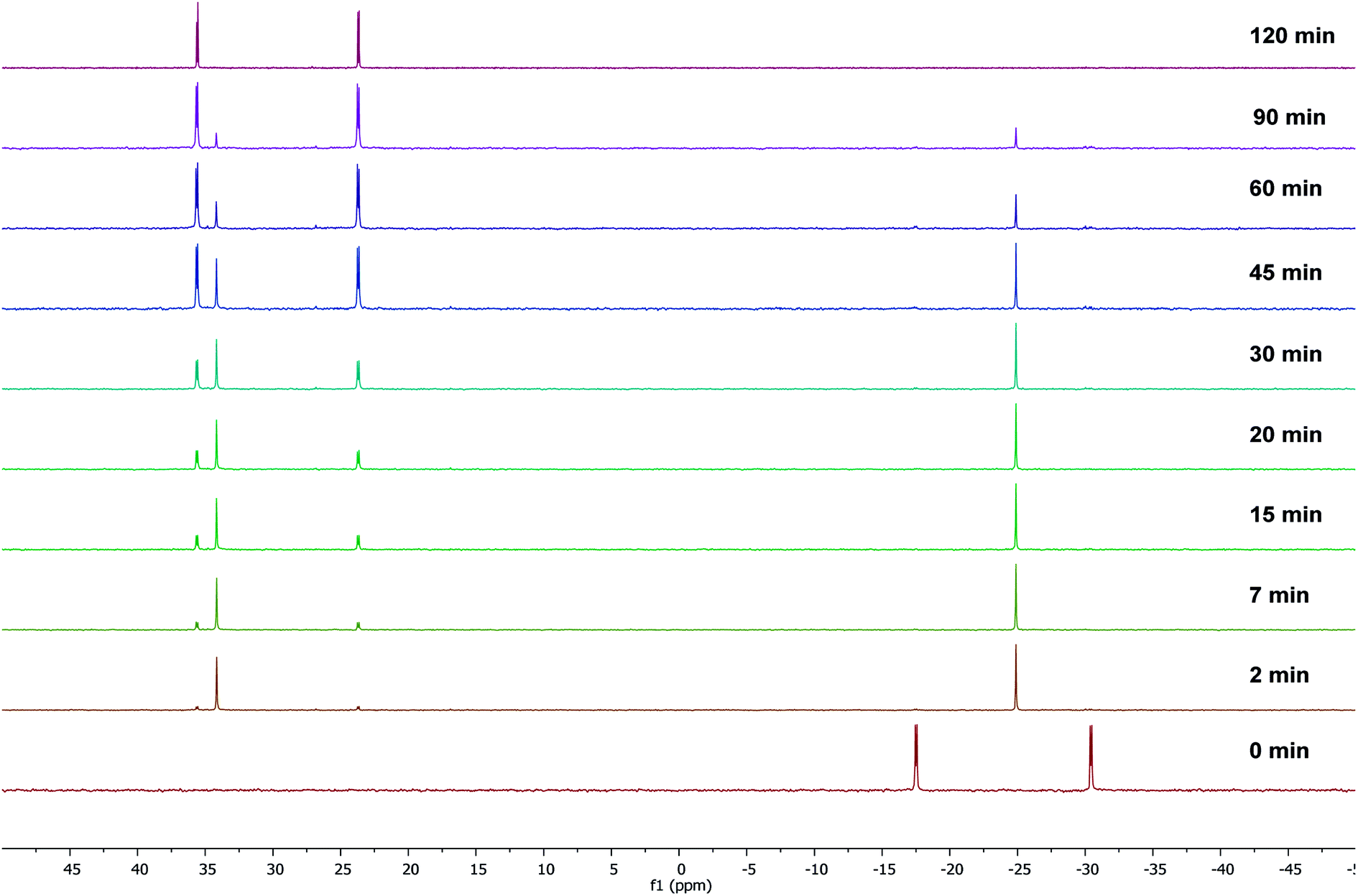 | ||
| Fig. 2 Time dependent 31P{1H} NMR spectral data for the isomerization of complex 3 to 5. | ||
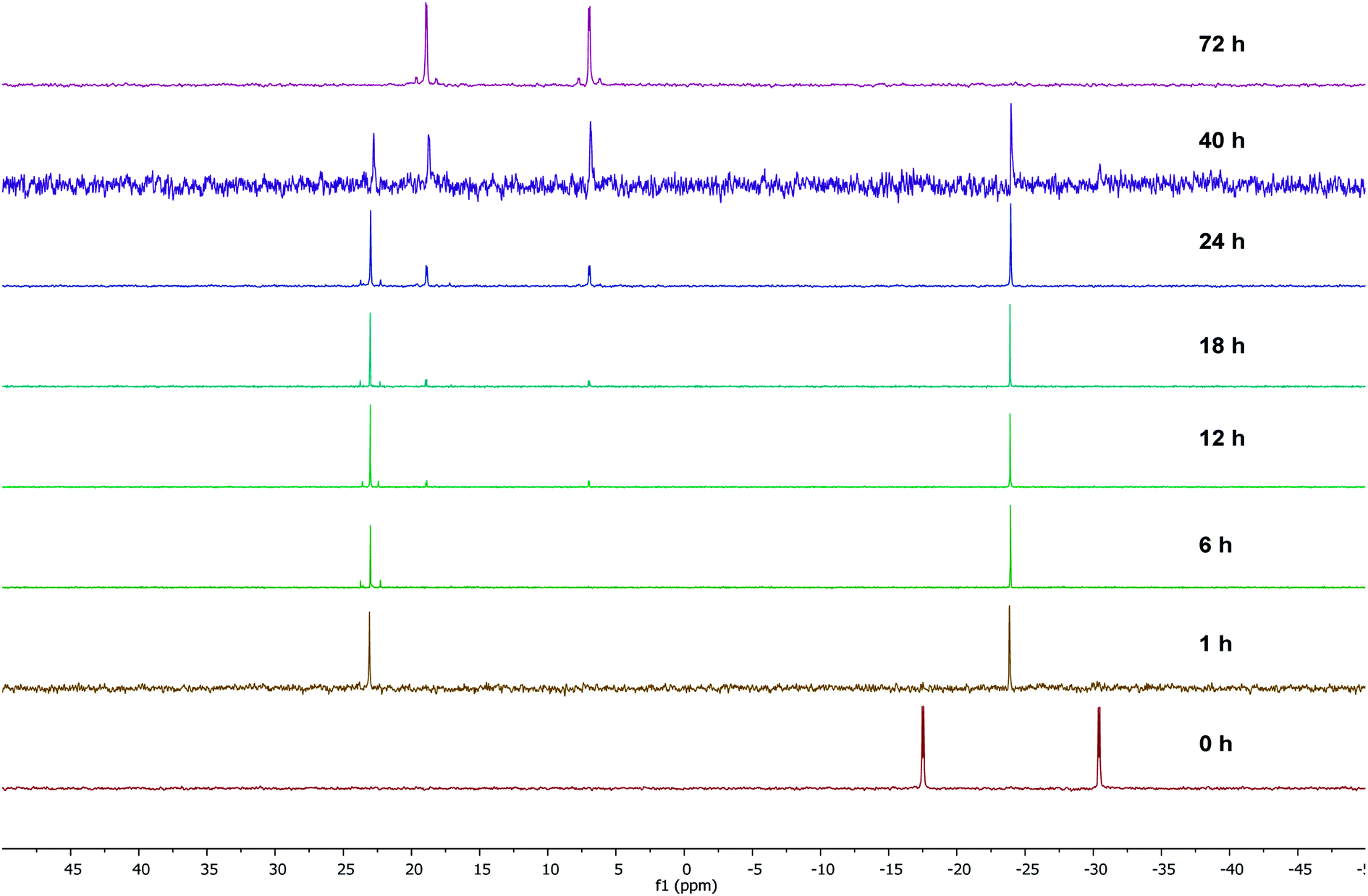 | ||
| Fig. 3 Time dependent 31P{1H} NMR spectral data for the isomerization of complex 4 to 6. | ||
The IR spectrum of complex 4 showed three νCO bands at 1850, 1887 and 2013 cm−1, whereas those of 5 and 6 are appended at 1901, 2023 cm−1 and 1892, 2018 cm−1, respectively. The νCO features are similar to those of analogous complexes of the type cis-[M(CO)4(LL)] (M = Mo and W; LL = dppm and dppe).15
The X-ray quality crystals of complexes 4–6 were obtained by solvent diffusion methods. The molecular structures of complexes 4–6 are shown in Fig. 4. The crystal structure of complex 4 reveals distorted octahedral geometry around tungsten center flanked by one of the triazolyl nitrogens (N2), phosphorus atom and four carbonyl groups. The P1–W1–N2 (75.10(9)°) angle is considerably smaller despite the formation of six-membered chelate ring which may be due to the relative orientation of N and P lone pairs. All P–Mo–C and N–Mo–C bond angles are greater than 90°, with the largest deviations being 97.74(15)° and 96.93(18)° for P1–W1–C40 and N2–W1–C39, respectively. The W–C bond distances of mutually trans carbonyls [W1–C41, 2.039(5) Å and W1–C42, 2.021(5) Å] are slightly longer than the W–C bond distances for the carbonyls trans to phosphorus and nitrogen (W1–C39 (1.972(5) Å) and W1–C40 (1.978(5) Å)), due to the poor π acceptor ability of phosphorus and triazolyl nitrogen moieties compared to the carbonyls. The P1–M–P2 bond angles in 5 and 6 are 87.76(3)° and 87.81(4)°, respectively. In 5 and 6, the M–C bond distances for the carbonyl trans to the phosphorus [Mo (1.997(4) and 1.998(4) Å) and W (2.000(6) and 2.006(6))] is shorter than the M–C bond distances of mutually trans carbonyls [Mo (2.030(4) and 2.044(4) Å) and W (2.027(5) and 2.039(5) Å)].
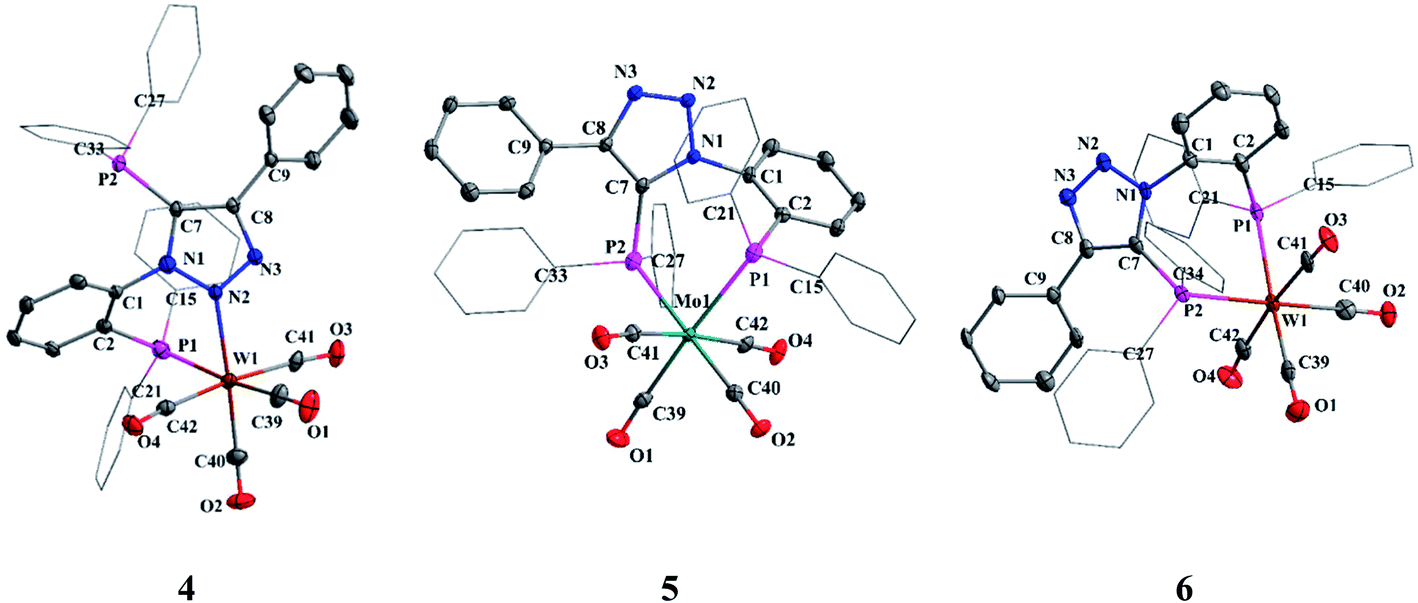 | ||
| Fig. 4 Molecular structures of 4, 5 and 6. All hydrogen atoms and solvent molecules were omitted for clarity. | ||
In order to gain some insight into the preference between the κ2-P,N and the κ2-P,P coordinating modes in Mo and W–carbonyl complexes, calculations were performed using the M06/6-31G**,16 lanl2dz level of theory17 for complexes 3–6. Crystal structure data were used for coordinates while performing the DFT calculations.18 The computed geometric parameters of complexes 4–6 are in good agreement with the single crystal X-ray crystallographic structures (see ESI: Tables S1 and S2†). The HOMO and LUMO energy values are tabulated in Table 2.19 In all these complexes, the HOMO level is majorly contributed by the metal center and carbonyl groups, whereas the LUMO level is majorly contributed by the ligand (Fig. 5).20 The HOMO of κ2-P,P coordinated complexes 5 and 6 are slightly higher in energy as compared to the κ2-P,N coordinated complexes 3 and 4.21 The HOMO–LUMO energy differences between κ2-P,P in 5 and 6 (4.4 eV and 4.29 eV) are even more compared to the κ2-P,N coordinated complexes 3 and 4 (3.63 eV and 3.44 eV). It clearly suggests that the complexes 5 and 6 are more stable compared to the corresponding κ2-P,N coordinated complexes 3 and 4.
| HOMO (eV) | LUMO (eV) | Contribution of HOMO (%) | Contribution of LUMO (%) | |||||
|---|---|---|---|---|---|---|---|---|
| M | CO | 2 | M | CO | 2 | |||
| a M = Mo (3 and 5), W (4 and 6). | ||||||||
| 3 | −5.15 | −1.52 | 60 | 27 | 13 | 5 | 4 | 92 |
| 4 | −5.05 | −1.61 | 56 | 30 | 14 | 5 | 4 | 91 |
| 5 | −5.67 | −1.27 | 64 | 24 | 12 | 15 | 4 | 80 |
| 6 | −5.61 | −1.32 | 60 | 26 | 14 | 14 | 5 | 81 |
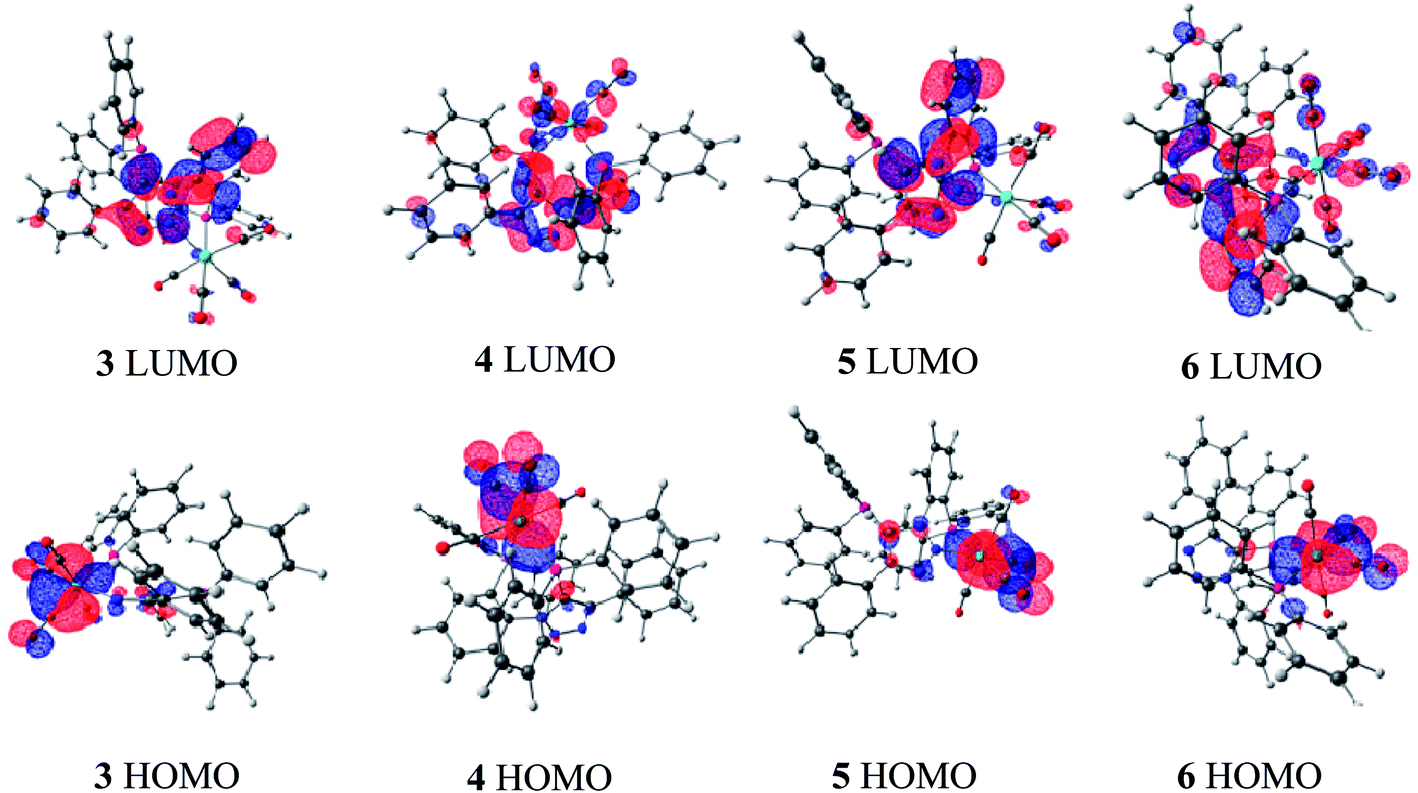 | ||
| Fig. 5 Frontier molecular orbitals of compounds 3–6. | ||
The complex 4 can function as a metalloligand due to the presence of one uncoordinated phosphorus atom. The reaction of 4 with [AuCl(SMe2)] in dichloromethane resulted in hetero-bimetallic complex 7 as shown in Scheme 3. The 31P{1H} NMR spectrum of 7 shows two singlets at 22.9 (1JWP ≈ 237.5 Hz) and 12.8 ppm. A shift in the 31P{1H} NMR signal of the triazole bound phosphorus (ΔδP = 33) indicates its coordination to gold. The νCO of complex 7 is similar to that of 4 and shows four νCO bands at 1857, 1888 and 1901 cm−1 and 2010 cm−1.
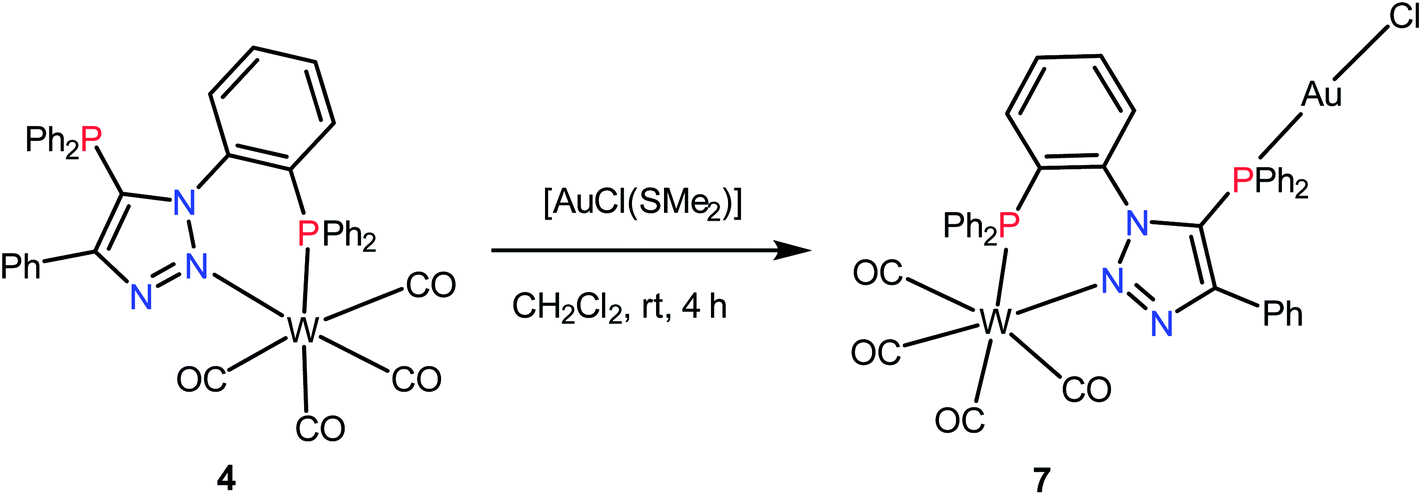 | ||
| Scheme 3 Synthesis of W, Au complex 7. | ||
The molecular structure of complex 7 is shown in Fig. 6. The crystal structure of complex 7 reveals distorted octahedral geometry around tungsten and linear geometry around gold atom. The P1–W1–N2 and P1–Au1–Cl1 bond angels are 76.14(9)° and 175.86(5)°, respectively. The W1–N2 and W2–P1 bond distances are 2.244(5) Å and 2.5080(15) Å, whereas W–C bond distances vary from 1.964(6) to 2.035(6) Å. Further, complex 7 shows weak Au⋯Carene interaction between gold and triazole (N1) attached phenyl group. The Au⋯Carene separation is 3.40 Å for ipso C1 and 3.480 Å for ortho C6, which lies within the range of Au–Carene interactions (3.07–3.55 Å) reported in the literature.22
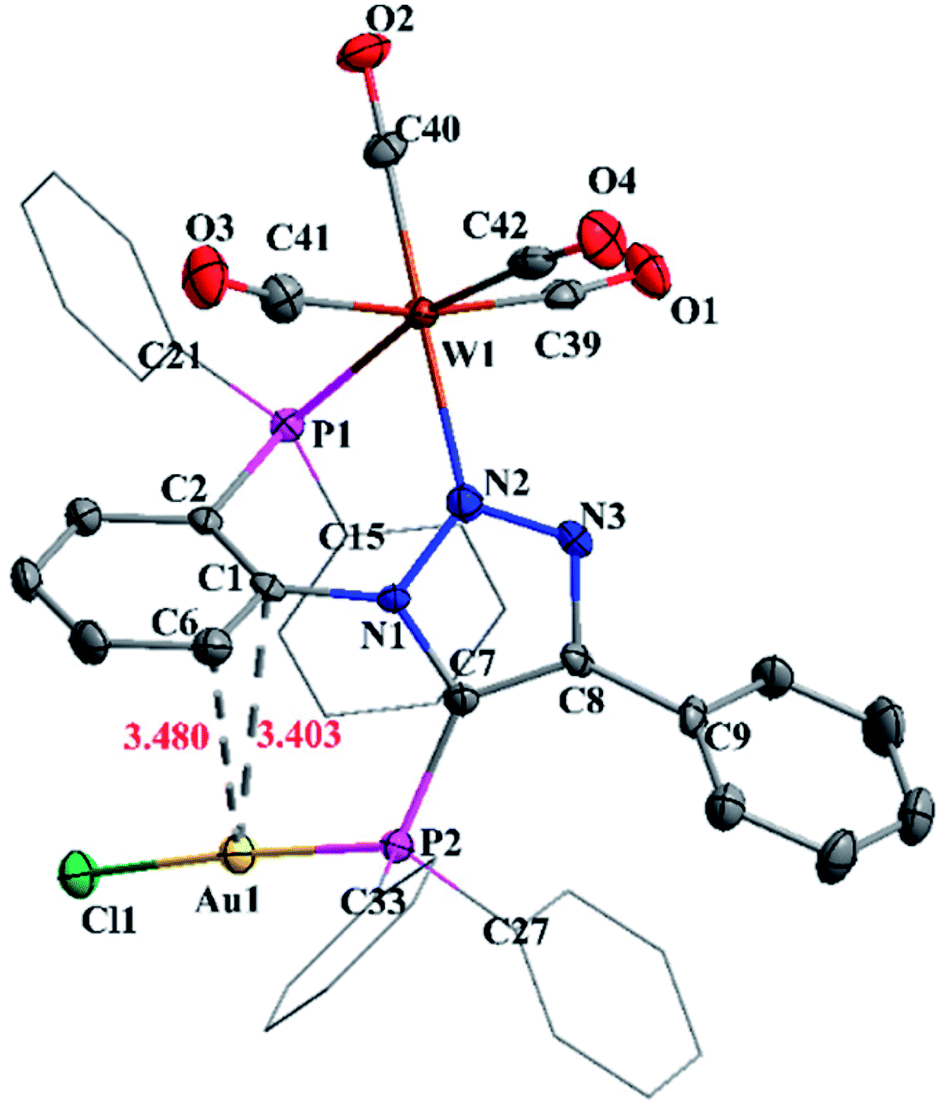 | ||
| Fig. 6 Molecular structure of 7 with 50% ellipsoid probability. Hydrogen atoms were omitted for clarity. | ||
The reaction of bisphosphine 2 with one equvivalent of [(η5-C5H5)Ru(PPh3)2Cl] in toluene under reflux conditions afforded mononuclear complex [(η5-C5H5)RuCl{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-κ2-P,P] (8). The 31P{1H} NMR spectrum of 8 consists of two doublets at 54.3 and 36.3 ppm with a 2JPP = 51.9 Hz, indicating κ2-P,P coordination. The 1H NMR spectrum of 8 shows a singlet at 4.25 ppm for cyclopentadienyl protons. Reaction of 2 with 0.5 [Ru(η6-p-cymene)Cl2]2 in dichloromethane yielded a red colored cationic complex [(η6-p-cymene)RuCl{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-κ2-P,N]Cl (9) as shown in Scheme 4. Complex 9 showed two singlets in its 31P{1H} NMR spectrum at 35.5 and −21.3 ppm, indicating κ2-P,N coordination keeping the triazolyl phosphorus (−21.3 ppm) uncoordinated. The 1H NMR spectrum of 9 showed four doublets at 6.53, 6.42, 5.92 and 5.25 ppm for the cymene group. The aliphatic protons of the cymene group showed septet at 3.10 ppm and two doublets at 1.07 and 1.24 ppm with a 3JHH = 6.5 Hz for the isopropyl group and a singlet at 1.71 ppm for methyl group.
 | ||
| Scheme 4 Synthesis of Ru(II) complexes 8 and 9. | ||
The crystals of complex 8 were obtained by diffusion of petroleum ether into a saturated solution of 8 in chloroform. The unit cell contains two independent molecules with identical bond parameters. In 8, ruthenium atom adopts a pseudo-octahedral geometry (Fig. 7). The P1–Ru1–P2, P1–Ru–Cl1 and P7–Ru–Cl1 bond angels are 91.86(4), 88.72(4) and 93.59(4)°, respectively. It specifies slight deviation from the ideal octahedral angle. The Ru–P, Ru–Cl and Ru–C bond distances are in the range expected for typical half-sandwich complexes.8c The phosphorus (P2) attached phenyl hydrogen (H34) is involved in weak intermolecular hydrogen bonding with triazole nitrogen (N3) with a N3⋯H34 distance of 2.50 Å. This interaction plays a major role in bulk crystal packing (see ESI Fig. S1†).
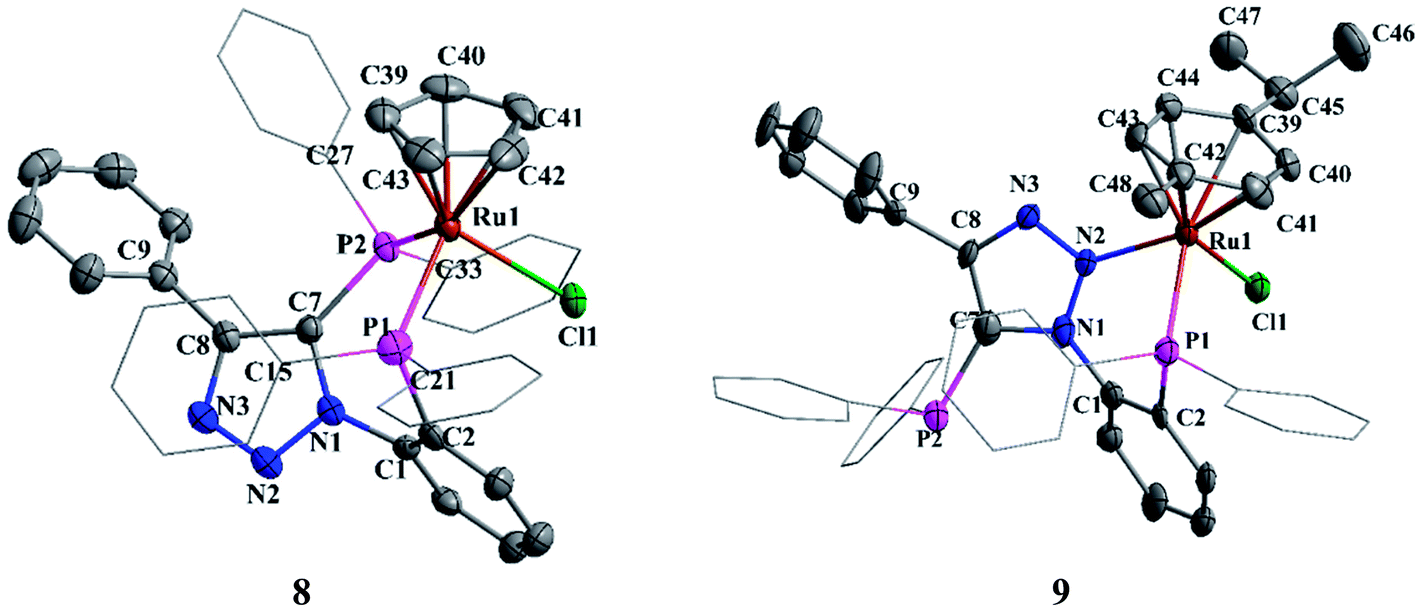 | ||
| Fig. 7 Molecular structures of 8 and 9. Hydrogen atoms and solvent molecule were omitted for clarity. | ||
In complex 9, the ruthenium center shows pseudo-octahedral geometry with p-cymene, one of the nitrogen (N2) of triazole group, phosphorus atom (P2) and chloride ion occupying the coordinating sites (Fig. 7). The P1–Ru1–N2 (80.42(16)°), P1–Ru1–Cl1 (89.85(7)°) and N2–M–Cl1 (88.30(16)°) bond angles clearly indicate the distorted octahedral geometry around the ruthenium atom. The Ru1–P1, Ru1–N1 and Ru1–Cl1 bond distances are 2.3060(19), 2.136(6) and 2.4074(19) Å, respectively. The Ru1–C (2.237 Å) mean distance is comparable to the same in analogues complexes such as: [(η6-p-cymene)RuCl{2-NMe2-3-iPr2P-indene}-κ2-P,N]BF4 (2.226 Å)23 and [(η6-p-cymene)RuCl{PR1R2}-κ2-P,N]ClO4 R1 = (O–C10H6–C10H6–O) R2 = [O–C6H4{2-C(NN(Me)CH)CH}] (2.242 Å).24
The reactions of [M(COD)Cl2] (M = Pd, Pt) with 2 in 1:1 molar ratio in dichloromethane afforded [MCl2(2)] (M = Pd (10); Pt (11)) in good yield. The 31P{1H} NMR spectra of 10 and 11 confirm the κ2-P,P coordination. Interestingly palladium complex did not show 2JPP coupling, whereas platinum complex showed 2JPP and 1JPtP couplings of 17.52 Hz and 3582 & 3721 Hz, respectively.
The suitable crystals of complexes 10 and 11 were obtained from chloroform solution layered with petroleum ether. Both palladium and platinum atoms adopt square planar geometry (Fig. 8). The P1–M–P2 bond angles in 10 and 11 are 94.72(3)° and 94.11(16)°, respectively. Triazole nitrogen atoms N3 and N2 show weak intermolecular hydrogen bonding with phenylene hydrogen atoms H25 and H14 of two other molecules (See ESI Fig. S2†) which is evident from the N3⋯H25 and N2⋯H14 bond distances of 2.570 Å and 2.553 Å, respectively. No such interactions were observed in platinum complex 11.
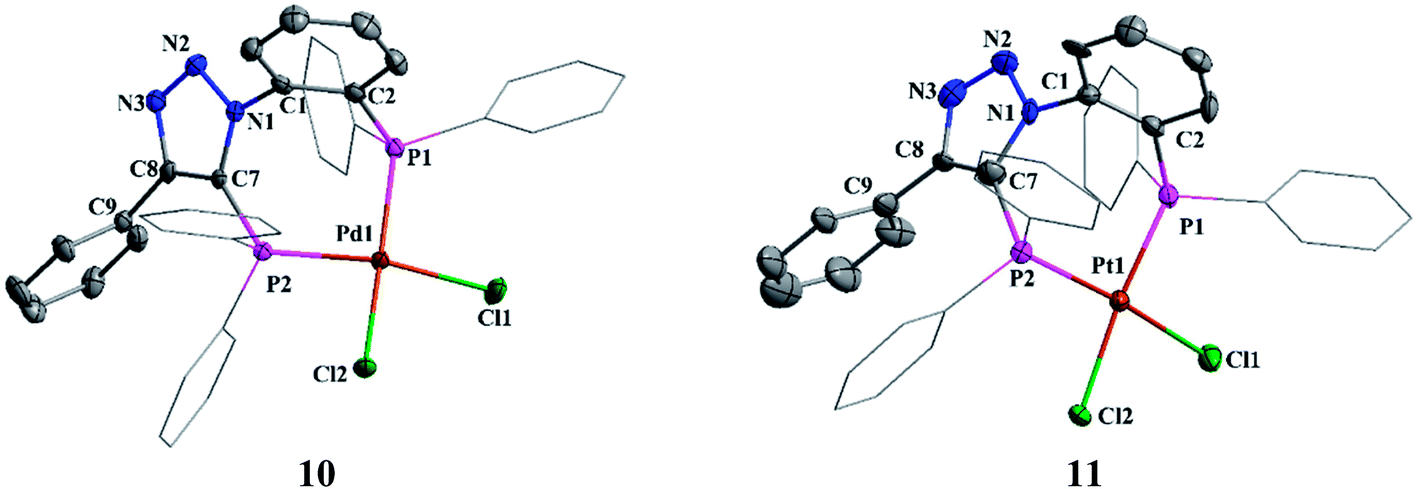 | ||
| Fig. 8 Molecular structures of 10 and 11. All hydrogen atoms and solvent molecule were omitted for clarity. | ||
The reaction of 2 with two equivalents of [AuCl(SMe2)] in dichloromethane yielded digold complex, [Au2Cl2{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-μ-P,P] (12). The 31P{1H} NMR spectrum of 12 exhibits two resonances at 24.5 and 12.2 ppm. The mass spectrum of complex 12 shows a peak at 1018.0853 corresponding to [M − Cl]+.
A perspective view of the molecular structure of 12 is shown in Fig. 9. The bond angels P1–Au1–Cl1 (174.94(5)°) and P2–Au2–Cl2 (174.27(4)°) are almost linear but orthogonal to each other. AuI centers display linear geometry with an Au⋯Au separation of 3.3406(3) Å, indicating intramolecular aurophilic interaction. The Au⋯Au distance is within the range (ca. 2.50–3.50 Å) and less than the sum of the van der Waals radii (3.8 Å) reported in the literature.25
 | ||
| Fig. 9 Molecular structure of 12 with 50% ellipsoid probability. Hydrogen atoms and solvent molecule were omitted for clarity. Selected bond distances (Å) and bond angles (°): P1–Au1 2.2224(11), P2–Au2 2.2101(11), P1–C2 1.814(4), P2–C7 1.806(4), Au1–Cl1 2.2834(11), Au2–Cl2 2.2703(11), Au1–Au2 3.3406(3), P1–Au1–Cl1 174.27(4), P2–Au2–Cl2 174.94(5), C2–P1–Au1 115.02(15), C7–P2–Au2 111.46(14), N1–N2–N3 107.6(3). | ||
Sonogashira coupling and sequential cyclization of alkyne and 2-halophenols to form synthetically useful 2-substituted benzofuran-type frameworks is an important methodology as these compounds constitute ubiquitous core structure in natural products and pharmaceuticals.26 For the preparation of benzofurans, a palladium catalyst in the presence copper salt as co-catalyst is generally employed for tandem alkynylation/cyclization reactions of terminal alkynes with o-hydroxy aryl halides.27 However, Cu-free Sonogashira alkynylation and sequential cyclization methods are scant in the literature.28 In order to assess the catalytic efficiency of palladium complex 10, tandem Cu-free Sonogashira alkynylation/cyclization reactions were performed.
Initially, the reaction of 2-iodophenol with phenyl acetylene was tested with 1 mol% of palladium catalyst 10 in various solvents. An excellent yield of 2-phenylbenzofuran was observed when Cs2CO3 was used as a base in dimethyl sulfoxide at 80 °C for 12 h. The yield was lowered considerably in other solvents (Table 3, entries 1–6). The coupling reaction was strongly influenced by the base as well as catalyst loading. Replacing Cs2CO3 with K2CO3 gave only moderate conversion (Table 3, entry 7). Catalyst loading of 0.5 mol% resulted in lower conversion rate (Table 3, entry 9). On decreasing the reaction time to 6 h, the conversion was not significantly affected (Table 3, entry 8). Under the optimized reaction conditions, various combinations of substituted o-iodophenols and alkynes were subjected to the tandem coupling/cyclization reaction to produce 2-substituted benzofurans in good to excellent yields. The results of the tandem Cu-free Sonogashira alkynylation/cyclization reaction are summarized in Chart 2. Both aromatic and aliphatic acetylene were tolerated in this conversion. In contrast, the coupling reaction of 2-iodophenol with 2-ethenylpyridine was not successful and resulted in negligible conversion.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Time (h) | Conversion (%)a |
| a Conversion determined by GC-MS using 2-iodophenol as internal standard.b Isolated yield.c 0.5 mol% catalyst used. | ||||
| 1 | Dioxane | Cs2CO3 | 12 | 55 |
| 2 | Dioxane/H2O(1:1) |
Cs2CO3 | 12 | 68 |
| 3 | CH3CN | Cs2CO3 | 12 | 60 |
| 4 | CH3CN/H2O(1:1) |
Cs2CO3 | 12 | 76 |
| 5 | DMF | Cs2CO3 | 12 | 85 |
| 6 | Toluene | Cs2CO3 | 12 | 48 |
| 7 | DMSO | K2CO3 | 12 | 74 |
| 8 | DMSO | Cs2CO3 | 6 | 99 (95b) |
| 9 | DMSO | Cs2CO3 | 12 | 78c |
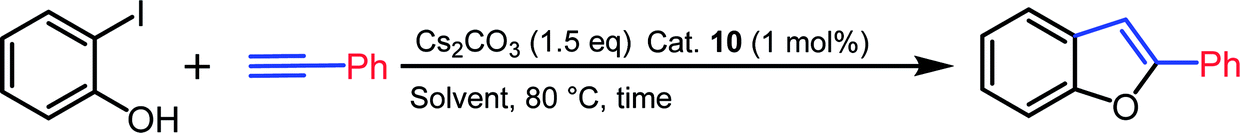
 | ||
| Chart 2 Substrate scope for tandem Cu-free Sonagashira alkynylation/cyclization reaction. Conditions: 2-iodophenol 0.5 mmol, alkynes 0.6 mmol, DMSO 3 mL, Cs2CO3 0.75 mmol, 80 °C, catalyst (1 mol%). Isolated yield. | ||
Conclusions
A new triazole-based bisphosphine 2 has been synthesized. The ambidentate bisphosphine 2 shows κ2-P,P, κ2-P,N-chelation as well as μ-(κ2-P,N, κ-N) and μ-(κ-P, κ-P) bridging coordination modes to act as di- or tridentate ligand. The kinetically controlled κ2-P,N coordination modes in Mo0 and W0 complexes “switched” to the thermodynamically preferred κ2-P,P coordination modes and the isomerization process was monitored by time dependent 31P{1H} NMR studies and further supported by DFT studies. The palladium complex 10 is found to be an efficient catalyst for tandem Cu-free Sonagashira alkynylation/cyclization reaction. Further catalytic investigation are under active investigation in our laboratory.Experimental section
General procedures
All the air sensitive compounds were handled and stored in the MBRAUN Glove box. All reactions were performed under an inert atmosphere of dry nitrogen or argon, using standard Schlenk techniques. All the solvents were dried by conventional methods and distilled prior to use. The compounds [M(piperidine)2(CO)4] (M = Mo and W),33 [(η5-C5H5)Ru(PPh3)2Cl],34 [Ru(η6-p-cymene)Cl2]2,35 [M(COD)Cl2] (M = Pd or Pt),36 [AuCl(SMe2)],37 5-(diphenylphosphino)-1,4-diphenyl-1H-1,2,3-triazole,13 1-(2-(diphenylphosphanyl)phenyl)-4-phenyl-1H-1,2,3-triazole (A)13 and 2-iodo-4,6-dimethylphenol38 were prepared according to the published procedures. Other reagents were obtained from commercial sources and used after purification.Instrumentation methods
The solution NMR spectra were recorded on a Bruker FT spectrometers (Advance-400 or 500) MHz at ambient probe temperatures. 13C{1H} and 31P{1H} NMR spectra were acquired using broad band decoupling method. The spectra were recorded in CDCl3 solutions with CDCl3 as an internal lock; chemical shifts of 1H and 13C NMR spectra are reported in ppm downfield from TMS, used as an internal standard. The chemical shifts of 31P{1H} NMR spectra are referred to 85% H3PO4 as an external standard. Positive values indicate downfield shifts. Infrared spectra were recorded on a PerkinElmer 400 FTIR instrument in KBr disk. The microanalyses were performed using a Carlo Erba Model 1112 elemental analyzer. Mass spectra were recorded using Waters Q-Tof micro (YA-105). Melting points of all compounds were determined on a Veego melting point apparatus and are uncorrected. GC-MS analyses were carried out in Agilent 7890A GC system.Synthesis of [o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}] (2)
Mp: 167–170 °C. Anal. calcd for C38H29N3P2: C, 77.41; H, 4.96; N, 7.13. Found C, 77.26; H, 5.38; N, 7.22% 1H NMR (500 MHz, CDCl3): δ 7.39 (td, J = 7.6, 1.3 Hz, 2H), 7.34–7.29 (m, 7H), 7.28–7.24 (m, 5H), 7.24–7.18 (m, 9H), 7.13 (dd, J = 7.3, 4.1 Hz, 4H), 7.11–7.06 (m, 1H), 7.01–6.97 (m, 2H).13C NMR (126 MHz, CDCl3) δ 152.69, 135.38, 133.98, 133.83, 132.92, 131.00, 130.51, 129.68, 129.27, 129.03, 128.95, 128.67, 128.61, 128.46, 127.73, 127.59. 31P{1H} NMR (202 MHz, CDCl3): δ −17.52 (d, JPP = 22.3 Hz), −30.43 (d, JPP = 22.3 Hz). HRMS (ES) m/z calcd for C38H30N3P2 ([M + H]+) 590.1907; found 590.1909.
Synthesis of [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,N}W(CO)4] (4)
A solution of 2 (0.03 g, 0.051 mmol) and [W(CO)4(piperidine)2] (0.024 g, 0.051 mmol) in dichloromethane (8 mL) was stirred for 4 h at room temperature. Then, the solvent was evaporated under reduced pressure, washed with petroleum ether (3 × 5 mL) and dried under vacuo to give analytically pure product of 4 as a yellow solid. Yield: 80% (0.036 g). Mp: 212–215 °C. IR (KBr disk): νCO = 2013, 1887, 1850 cm−1. Anal. calcd for C42H29N3O4P2W: C, 56.97; H, 3.36; N, 4.75. Found C, 56.43; H, 3.68; N, 5.13%. 1H NMR (400 MHz, CDCl3): δ 7.76–7.68 (m, 2H), 7.63–7.50 (m, 10H), 7.46 (dd, J = 13.4, 5.9 Hz, 4H), 7.21 (t, J = 7.2 Hz, 1H), 7.03–6.97 (m, 1H), 6.96–6.81 (m, 7H), 6.71 (td, J = 7.8, 1.9 Hz, 2H), 6.22 (t, J = 7.5 Hz, 2H). 31P{1H} NMR (162 MHz, CDCl3): δ 23.01 (1JWP = 239.2), −23.94. HRMS (ES) m/z calcd for C42H30N3O4P2W ([M + H])+ 886.1222; found 886.1222.Synthesis of [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}Mo(CO)4] (5)
A solution of 2 (0.03 g, 0.051 mmol) and [Mo(CO)4(piperidine)2] (0.019 g, 0.051 mmol) in dichloromethane (8 mL) was stirred for 4 h at room temperature. The solvent was evaporated under vacuum, and the residue was washed with petroleum ether (3 × 5 mL) and dried under vacuo to give analytically pure product of 5 as a colorless solid. Yield: 79% (0.034 g). Mp: 202–205 °C. IR (KBr disk): νCO = 2023, 1925, 1901 cm−1. Anal. calcd for C42H29MoN3O4P2·0.5CH2Cl2: C, 60.76; H, 3.59; N, 5.00. Found C, 60.98; H, 3.67; N, 5.25%. 1H NMR (500 MHz, CDCl3): δ 7.68–7.62 (m, 2H), 7.53–7.47 (m, 2H), 7.47–7.42 (m, 3H), 7.39–7.32 (m, 5H), 7.31–7.27 (m, 4H), 7.24–7.15 (m, 5H), 7.00 (t, J = 7.4 Hz, 1H), 6.95 (dd, J = 7.7, 6.2 Hz, 1H), 6.90 (t, J = 7.7 Hz, 2H), 6.86–6.79 (m, 4H). 31P{1H} NMR (202 MHz, CDCl3): δ 35.58 (d, 2JPP = 19.0 Hz), 23.67 (d, 2JPP = 19.0 Hz). HRMS (ES) m/z calcd for C42H30N3O4P2Mo ([M + H])+ 800.0770; found 800.0770.Synthesis of [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}W(CO)4] (6)
Mp: 217–220 °C. IR (KBr disk): νCO = 2018, 1918, 1892 cm−1 Anal. calcd for C42H29O4N3P2W: C, 56.97; H, 3.3; N, 4.75. Found C, 56.63; H, 3.35; N, 4.57%. 1H NMR (400 MHz, CDCl3): δ 7.63 (ddd, J = 11.7, 7.5, 1.8 Hz, 2H), 7.51–7.41 (m, 5H), 7.39–7.27 (m, 8H), 7.27–7.20 (m, 5H), 7.16 (dd, J = 13.1, 5.4 Hz, 2H), 7.01 (t, J = 7.4 Hz, 1H), 6.97–6.86 (m, 3H), 6.86–6.79 (m, 3H). 31P NMR (162 MHz, CDCl3): δ 18.9 (1JWP = 238.3 Hz, 2JPP = 12.8 Hz), 6.9 (1JWP = 247.3 Hz, 2JPP = 12.8 Hz). HRMS (ES) m/z calcd for C42H30N3O4P2W ([M + H])+ 886.1222; found 886.1222.
Synthesis of [μ-PN,P{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2AuCl)}-κ2-P,N}W(CO)4] (7)
To a solution of complex 4 (0.051 g, 0.0576 mmol) in dichloromethane (5 mL) was added dropwise a solution of [AuCl(SMe2)] (0.017 g, 0.0576 mmol) in the same solvent (5 mL) and stirred for 4 h at room temperature. The solvent was afterward evaporated under vacuum; the residue obtained was washed with petroleum ether (2 × 5 mL) and dried under vacuo to give analytically pure product of 7 as an orange red solid. Yield: 84% (0.057 g). Mp: 222–225 °C. IR (KBr disk): νCO = 2010, 1901, 1888, 1857 cm−1 Anal. calcd for C42H29AuClO4N3P2W: C, 45.12; H, 2.61; N, 3.76. Found C, 45.09; H, 2.35; N, 3.94%. 1H NMR (500 MHz, CDCl3): δ 8.10 (ddd, J = 10.8, 7.4, 2.2 Hz, 2H), 7.84–7.71 (m, 5H), 7.69–7.61 (m, 4H), 7.57 (dt, J = 6.2, 3.1 Hz, 5H), 7.23 (dd, J = 11.7, 4.4 Hz, 2H), 7.07 (ddd, J = 6.6, 5.8, 4.2 Hz, 2H), 6.92 (t, J = 7.8 Hz, 2H), 6.82 (ddd, J = 9.6, 7.3, 3.2 Hz, 3H), 6.74 (dd, J = 8.1, 1.1 Hz, 2H), 6.27 (dd, J = 14.6, 7.3 Hz, 2H). 31P NMR (202 MHz, CDCl3): δ 22.89 (1JWP = 237.5 Hz), 12.75.Synthesis of [(η5-C5H5)RuCl{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-κ2-P,P] (8)
A solution of [(η5-C5H5)Ru(PPh3)2Cl] (0.0369 g, 0.051 mmol) in toluene (8 mL) was added slowly to a solution of 2 (0.030 g, 0.051 mmol) in toluene (8 mL) at room temperature. The reaction mixture was refluxed at 100 °C for 12 h to give a clear yellow solution. The solvent was removed under vacuum and the residue obtained was washed with hot petroleum ether (3 × 8 mL) and dissolved in chloroform and layered with petroleum ether, which on slow diffusion gave red colour crystals of 8. Yield 74% (0.03 g). Mp: 270–273 °C. Anal. calcd for C43H34ClN3P2Ru: C, 65.27; H, 4.33; N, 5.31. Found C, 65.41; H, 3.97; N, 5.05%. 1H NMR (500 MHz, CDCl3): δ 7.70 (s, 2H), 7.60 (dd, J = 18.2, 10.2 Hz, 3H), 7.50 (t, J = 6.5 Hz, 4H), 7.43 (s, 5H), 7.30 (t, J = 7.5 Hz, 1H), 7.26–7.20 (m, 2H), 7.19–7.10 (m, 2H), 6.93 (t, J = 7.4 Hz, 1H), 6.85 (t, J = 7.1 Hz, 1H), 6.76 (t, J = 7.1 Hz, 2H), 6.70 (t, J = 6.5 Hz, 2H), 6.66–6.60 (m, 2H), 6.45 (d, J = 7.0 Hz, 2H), 4.25 (s, 5H). 31P{1H} NMR (202 MHz, CDCl3): δ 54.36 (d, J = 51.9 Hz), 36.34 (d, 2JPP = 51.9 Hz). HRMS (ES): m/z calcd for C43H35ClN3P2Ru ([M + H]+): 792.1041; found: 792.1042.Synthesis of [(η6-p-cymene)RuCl{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-κ2-P,N]Cl (9)
A solution of [Ru(η6-p-cymene)Cl2]2 (0.0174 g, 0.0285 mmol) in dichloromethane (3 mL) was added drop wise to a solution of 2 (0.0338 g, 0.057 mmol) also in dichloromethane (4 mL) and stirred for 4 h at room temperature. The solvent was removed under reduced pressure and the residue obtained was washed with petroleum ether (3 × 5 mL) and dried under vacuo to give compound 9 as a red solid. Yield 93% (0.048 g) Mp: 257–260 °C. Anal. calcd for C48H43Cl2N3P2Ru: C, 64.36; H, 4.84; N, 4.69. Found C, 64.19; H, 4.53; N, 4.36%. 1H NMR (500 MHz, CDCl3): δ 8.15 (dd, J = 12.7, 7.7 Hz, 1H), 7.88–7.62 (m, 7H), 7.57 (s, 4H), 7.48–7.40 (m, 4H), 7.34 (t, J = 7.6 Hz, 1H), 7.18–7.16 (m, 2H), 6.97 (dt, J = 40.8, 7.1 Hz, 6H), 6.73 (t, J = 7.0 Hz, 2H), 6.54 (d, J = 3.7 Hz, 1H), 6.43 (d, J = 4.7 Hz, 1H), 6.03 (t, J = 8.0 Hz, 2H), 5.93 (d, J = 6.0 Hz, 1H), 5.25 (d, J = 6.0 Hz, 1H), 3.18–3.03 (m, 1H), 1.72 (s, 3H), 1.24 (d, J = 6.5 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H). 31P{1H} NMR (202 MHz, CDCl3): δ 35.52 (s), −21.34 (s). HRMS (ES): m/z Calcd. for C48H43ClN3P2Ru ([M − Cl]+): 860.1669; found: 860.1667.Synthesis of [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}PdCl2] (10)
To a stirred solution of [Pd(COD)Cl2] (0.015 g, 0.051 mmol) in dichloromethane (4 mL) was added drop wise a solution of 2 (0.03 g, 0.051 mmol) also in dichloromethane (4 mL) and the reaction mixture was stirred for 4 h at room temperature. The solvent was removed under reduced pressure and the residue obtained was washed with petroleum ether (2 × 5 mL) and dried under vacuo to give compound 10 as a yellow solid. Yield: 85% (0.0331 g). Mp: 271–274 °C. Anal. calcd for C38H29 Cl2N3P2Pd: C, 59.51; H, 3.81; N, 5.48. Found C, 59.57; H, 3.44; N, 5.12%. 1H NMR (500 MHz, CDCl3): δ 8.02 (s, 2H), 7.86 (dd, J = 13.0, 7.4 Hz, 2H), 7.63 (dd, J = 11.3, 8.2 Hz, 3H), 7.57–7.49 (m, 3H), 7.48–7.41 (m, 3H), 7.33 (dd, J = 12.1, 7.8 Hz, 5H), 7.23 (d, J = 7.6 Hz, 1H), 7.18–7.10 (m, 2H), 7.09–7.02 (m, 2H), 6.99 (t, J = 7.6 Hz, 2H), 6.85 (td, J = 7.8, 3.2 Hz, 2H), 6.62 (d, J = 7.2 Hz, 2H). 31P{1H} NMR (202 MHz, CDCl3): δ 25.9 (s), 12.9 (s). HRMS (ES) m/z calcd for C38H29N3P2PdCl ([M − Cl])+ 730.0565; found 730.0565.Synthesis of [{o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}-κ2-P,P}PtCl2] (11)
A solution of Pt(COD)Cl2 (0.019 g, 0.051 mmol) in dichloromethane (4 mL) was added drop wise to a solution of 2 (0.03 g, 0.051 mmol) also in dichloromethane (4 mL) and the solution was stirred for 4 h at room temperature. The solvent was removed under reduced pressure and the residue obtained was washed with petroleum ether (2 × 5 mL) and concentrated to a small bulk and 2 mL of chloroform was added and stored at 25 °C to give 11 as a colorless crystalline solid. Yield: 79% (0.0362 g). Mp: 260–263 °C. Anal. calcd for C38H29Cl2N3P2Pt·0.5CH2Cl2: C, 51.49; H, 3.37; N, 4.68. Found C, 51.70; H, 3.40; N, 4.59%. 1H NMR (400 MHz, CDCl3): δ 7.96 (s, 2H), 7.79 (dd, J = 13.1, 7.5 Hz, 2H), 7.66–7.48 (m, 7H), 7.48–7.39 (m, 3H), 7.35–7.27 (m, 4H), 7.22 (d, J = 7.5 Hz, 1H), 7.18–7.10 (m, 2H), 7.09–7.03 (m, 2H), 6.99 (t, J = 7.7 Hz, 2H), 6.83 (td, J = 7.8, 3.2 Hz, 2H), 6.61 (d, J = 7.2 Hz, 2H). 31P{1H} NMR (202 MHz, CDCl3): δ 8.04 (d, 1JPt–P = 3581.7 Hz, 2JPP = 17.7 Hz), −4.06 (d, 1JPt–P = 3720.7 Hz, 2JPP = 17.7 Hz). HRMS (ES) m/z calcd for C38H29N3P2PtCl ([M − Cl])+ 819.1171; found 820.1162.Synthesis of [Au2Cl2{ o-Ph2P(C6H4){1,2,3-N3C(Ph)C(PPh2)}}-μ-P,P] (12)
A dichloromethane (5 mL) solution of AuCl(SMe2) (0.030 g, 0.102 mmol) was added drop wise to a solution of 2 (0.030 g, 0.051 mmol) in the same solvent (6 mL) and stirred for 4 h at room temperature. The solvent was removed under reduced pressure and the residue obtained was washed dissolved in a mixture of petroleum ether (5 mL) and chloroform (1 mL) and stored at room temperature for 24 h to give 12 as a colourless crystalline solid. Yield: 68% (0.041 g). Mp: >275 °C. Anal. calcd for C38H29Au2Cl2N3P2·CHCl3: C, 39.91; H, 2.58; N, 3.58. Found C, 39.37; H, 2.31; N, 3.65%. 1H NMR: (400 MHz, CDCl3): δ 7.80–7.63 (m, 5H), 7.61–7.45 (m, 13H), 7.25 (d, J = 6.2 Hz, 3H), 7.23–7.16 (m, 2H), 7.10 (t, J = 7.4 Hz, 1H), 7.00 (ddd, J = 19.3, 11.2, 6.2 Hz, 4H), 6.58 (dd, J = 7.6, 4.7 Hz, 1H). 31P{1H} NMR (202 MHz, CDCl3): δ 24.49 (s), 12.18 (s). HRMS (ES): m/z Calcd. for C38H29Au2ClN3P2 ([M − Cl]+): 1018.0851; found: 1018.0853.General procedure for tandem Cu-free Sonagashira alkynylation/cyclization reaction
The reactions were performed in a closed vessel containing a mixture of alkyne (1.2 equiv.), substituted 2-iodophenol (1.0 equiv), Cs2CO3 (1.5 equiv) and catalyst 10 (1 mol%) in DMSO (3 mL). The reaction mixture was heated at 80 °C for 6 h. The solution was diluted with H2O (10 mL) extracted twice with ethyl acetate (10 mL). The combined organic fractions were dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude product obtained was purified by silica gel column chromatography using petroleum ether/ethyl acetate.X-ray crystallography
Single crystal X-ray data collection was performed at 100–150 K using a Rigaku Saturn724 diffractometer (Enhance Mo-Kα X-ray source, λ = 0.71073 Å) with a graphite monochromater for compounds 2 and 4–12 with the ω-scan technique. Crystal data and the details of the structure determination of 2 and 4–12 are given in Tables 4 and 5, whereas the selected bond parameters are listed in Tables 6 and 7. The data were reduced using CrysalisPro Red 171.38.43 software. The structures were solved by Olex2 (ref. 29) with the ShelXT30 structure solution program using intrinsic phasing and refined with the ShelXL31 refinement package using least squares minimization. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and included as riding contributions with isotropic displacement parameters tied to those of the attached non-hydrogen atoms. In compounds 4, 8 and 9 the disordered solvent molecules could not be identified or modelled to a known solvent hence it was SQUEEZED using PLATON.32 Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1832214 (compound 2) and CCDC 1832216–1832224 (compounds 4–12).| a R = Σ||Fo| − |Fc||/Σ|Fo|.b wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2 w = 1/[σ2(Fo2) + (xP)2] where P = (Fo2 + 2Fc2)/3. | |||||
|---|---|---|---|---|---|
| 2 | 4 | 5 | 6 | 7 | |
| Formula | C38H29N3P2 | C42H29N3O4P2W | C43H30Cl3MoN3O4P2 | C43H30Cl3N3O4P2W | C42H29AuClN3O4P2W |
| Formula weight | 589.58 | 885.47 | 916.93 | 1004.84 | 1117.89 |
| T (K) | 150 | 150 | 150 | 100 | 150 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/c | P21/n |
| a, Å | 15.5170(11) | 12.6150(6) | 14.1841(3) | 14.1865(3) | 12.5915(3) |
| b, Å | 11.9307(7) | 13.3420(9) | 11.2966(2) | 11.2881(3) | 20.0214(6) |
| c, Å | 18.3226(15) | 14.0151(8) | 25.2699(6) | 25.1974(9) | 15.3286(4) |
| α, deg | 90 | 71.437(5) | 90 | 90 | 90 |
| β, deg | 111.801(9) | 69.756(5) | 97.008(2) | 97.169(2) | 92.147(2) |
| γ, deg | 90 | 65.940(5) | 90 | 90 | 90 |
| V, Å3 | 3149.4(4) | 1977.1(2) | 4018.80(15) | 4003.53(19) | 3861.62(18) |
| Z | 4 | 2 | 4 | 4 | 4 |
| ρcalcg/cm3 | 1.243 | 1.487 | 1.515 | 1.667 | 1.923 |
| M (MoKα)/mm−1 | 0.169 | 3.046 | 0.653 | 3.213 | 6.97 |
| F(000) | 1232 | 876 | 1856 | 1984 | 2136 |
| Crystal size (mm3) | 0.151 × 0.14 × 0.065 | 0.171 × 0.146 × 0.108 | 0.314 × 0.154 × 0.095 | 0.224 × 0.119 × 0.097 | 0.168 × 0.053 × 0.039 |
| Theta range (deg) | 4.33 to 49.994 | 4.082 to 50 | 4.624 to 49.998 | 3.958 to 49.996 | 4.112 to 49.998 |
| Reflections collected | 21113 |
22276 |
42569 |
21398 |
18829 |
| Independent reflections | 5544 | 6935 | 7077 | 6857 | 6764 |
| S | 1.059 | 1.054 | 1.033 | 1.04 | 1.034 |
| R1a | 0.0555 | 0.0336 | 0.0472 | 0.0379 | 0.0323 |
| wR2b | 0.1586 | 0.0861 | 0.1198 | 0.0949 | 0.0781 |
| a R = Σ||Fo| − |Fc||/Σ|Fo|.b wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2 w = 1/[σ2(Fo2) + (xP)2] where P = (Fo2 + 2Fc2)/3. | |||||
|---|---|---|---|---|---|
| 8 | 9 | 10 | 11 | 12 | |
| Formula | C87H69Cl5N6P4Ru2 | C48H43Cl2N3P2Ru | C39H30Cl5N3P2Pd | C39H30Cl5N3P2Pt | C39H30Au2Cl5N3P2 |
| Formula weight | 1701.75 | 895.76 | 887.25 | 974.94 | 1173.78 |
| T (K) | 150 | 150 | 150 | 150 | 150 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P |
P |
P21/c | P212121 | P21/c |
| a, Å | 13.7636(3) | 12.7908(7) | 13.3510(4) | 10.2544(3) | 12.7791(5) |
| b, Å | 14.7629(4) | 15.2107(10) | 11.0729(2) | 12.3816(4) | 14.3916(6) |
| c, Å | 22.1839(5) | 15.2959(10) | 25.9296(6) | 28.9853(11) | 20.6657(8) |
| α, deg | 76.1417(19) | 114.525(6) | 90 | 90 | 90 |
| β, deg | 85.9054(18) | 103.851(5) | 95.690(2) | 90 | 95.247(4) |
| γ, deg | 85.3961(19) | 102.567(5) | 90 | 90 | 90 |
| V, Å3 | 4355.90(17) | 2453.6(3) | 3814.38(17) | 3680.1(2) | 3784.7(3) |
| Z | 2 | 2 | 4 | 4 | 4 |
| ρcalcg/cm3 | 1.297 | 1.212 | 1.545 | 1.76 | 2.06 |
| M (MoKα)/mm−1 | 0.619 | 0.526 | 0.954 | 4.297 | 8.216 |
| F(000) | 1732 | 920 | 1784 | 1912 | 2232 |
| Crystal size (mm3) | 0.123 × 0.102 × 0.065 | 0.176 × 0.153 × 0.098 | 0.222 × 0.091 × 0.05 | 0.175 × 0.157 × 0.072 | 0.17 × 0.07 × 0.05 |
| Theta range (deg) | 4.17 to 50 | 3.174 to 49.994 | 4.178 to 49.994 | 3.29 to 49.994 | 4.272 to 50 |
| Reflections collected | 83601 |
47691 |
36825 |
24542 |
30848 |
| Independent reflections | 15315 |
8624 | 6721 | 6484 | 6649 |
| S | 1.054 | 1.037 | 1.053 | 1.109 | 1.025 |
| R1a | 0.0582 | 0.084 | 0.0406 | 0.0562 | 0.0269 |
| wR2b | 0.1493 | 0.2392 | 0.0835 | 0.134 | 0.065 |
| 4 (M = W) | 5 (M = Mo) | 6 (M = W) | 7 (M = W) | |
|---|---|---|---|---|
| Bond distances | ||||
| P1–C2 | 1.840(4) | 1.846(3) | 1.844(5) | 1.854(6) |
| P2–C7 | 1.824(4) | 1.834(3) | 1.828(5) | 1.827(6) |
| P1–M | 2.5156(11) | 2.5335(9) | 2.5263(13) | 2.5080(15) |
| P2–M | — | 2.5245(9) | 2.5131(13) | — |
| M–CAvg | 2.002(5) | 2.017(4) | 2.018(6) | 2.009(6) |
| N1–N2 | 1.369(5) | 1.353(4) | 1.354(5) | 1.360(6) |
| N2–N3 | 1.313(5) | 1.303(4) | 1.311(6) | 1.312(6) |
| C39–O1 | 1.161(6) | 1.149(5) | 1.157(7) | 1.141(7) |
| C40–O2 | 1.156(6) | 1.147(4) | 1.146(6) | 1.157(7) |
| M–N2 | 2.242(3) | — | — | 2.244(5) |
| P2–Au | — | — | — | 2.2134(15) |
| Au–Cl | — | — | — | 2.2893(15) |
|
||||
| Bond angles | ||||
| P1–M–P2 | — | 87.76(3) | 87.81(4) | — |
| P1–M–N2 | 75.10(9) | — | — | 76.14(9) |
| P1–M–C40 | 97.74(15) | 92.21(10) | 92.27(16) | 97.74(15) |
| P2–M–C39 | — | 92.06(10) | 92.27(16) | — |
| N2–M–C39 | 96.93(18) | — | — | 96.93(18) |
| P1–M–C39 | 171.68(15) | 174.34(11) | 173.87(15) | 166.05(17) |
| N2–M–C40 | 172.32(16) | — | — | 176.10(19) |
| P2–M–C40 | — | 178.17(10) | 178.09(14) | — |
| C41–M–C42 | 173.32(17) | 174.19(14) | 174.8(2) | 168.8(2) |
| N1–N2–N3 | 107.3(3) | 107.4(3) | 107.2(4) | 108.6(4) |
| P2–Au–Cl | — | — | — | 175.86(5) |
| 8 (M = Ru) | 9 (M = Ru) | 10 (M = Pd) | 11 (M = Pt) | |
|---|---|---|---|---|
| Bond distances | ||||
| P1–M | 2.2851(13) | 2.3060(19) | 2.2606(9) | 2.249(4) |
| P2–M | 2.2902(13) | — | 2.2549(9) | 2.241(4) |
| N2–M | — | 2.136(6) | — | — |
| M–Cl1 | 2.4245(12) | 2.4074(19) | 2.3243(9) | 2.336(4) |
| M–Cl2 | 2.3507(9) | 2.352(4) | ||
| M–Cavg | 2.2066 | 2.237 | — | — |
| P1–C2 | 1.835(5) | 1.825(8) | 1.835(3) | 1.834(17) |
| P2–C7 | 1.827(4) | 1.838(8) | 1.818(3) | 1.853(19) |
| N3⋯H34 | 2.505 | — | — | — |
|
||||
| Bond angels | ||||
| P1–M–P2 | 91.86(5) | — | 94.72(3) | 94.11(16) |
| P1–M–N2 | — | 80.42(16) | — | — |
| P1–M–Cl1 | 88.72(4) | 89.85(7) | 93.56(3) | 89.09(16) |
| P1–M–Cl2 | — | — | 168.66(3) | 174.15(16) |
| P2–M–Cl1 | 93.59(4) | — | 167.96(4) | 171.60(16) |
| P2–M–Cl2 | — | — | 88.64(3) | 90.92(16) |
| P1–M–P2 | 91.86(5) | — | 94.72(3) | 94.11(16) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Science & Engineering Research Board, New Delhi, India, for financial support of this work through grant No. SB/S1/IC-08/2014. LR and MKP thank UGC, New Delhi for the fellowship. We also thank the Department of Chemistry Instrumentation Facilities, IIT Bombay, for spectral and analytical data. We thank Dr Debasish Sengupta for verifying some catalytic data.References
- (a) P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680–699 CrossRef; (b) R. J. Long, V. C. Gibson, A. J. P. White and D. J. Williams, Inorg. Chem., 2006, 45, 511–513 CrossRef PubMed; (c) N. Dwadnia, J. Roger, N. Pirio, H. Cattey and J.-C. Hierso, Coord. Chem. Rev., 2018, 355, 74–100 CrossRef.
- E. M. Schuster, M. Botoshansky and M. Gandelman, Organometallics, 2009, 28, 7001–7005 CrossRef.
- M. Hirotsu, K. Santo, Y. Tanaka and I. Kinoshita, Polyhedron, 2018, 143, 201–208 CrossRef.
- (a) E. Poverenov, M. Gandelman, L. J. W. Shimon, H. Rozenberg, Y. Ben-David and D. Milstein, Organometallics, 2005, 24, 1082–1090 CrossRef; (b) R. Lindner, B. van den Bosch, M. Lutz, J. N. H. Reek and J. I. van der Vlugt, Organometallics, 2011, 30, 499–510 CrossRef; (c) G. Mancano, M. J. Page, M. Bhadbhade and B. A. Messerle, Inorg. Chem., 2014, 53, 10159–10170 CrossRef PubMed; (d) A. Ghisolfi, A. Waldvogel, L. Routaboul and P. Braunstein, Inorg. Chem., 2014, 53, 5515–5526 CrossRef PubMed.
- (a) V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines, Wiley-VCH, New York, 2nd edn, 2008 CrossRef; (b) D. Kalny, M. Elhabiri, T. Moav, A. Vaskevich, I. Rubinstein, A. Shanzer and A.-M. Albrecht-Gary, Chem. Commun., 2002, 1426–1427 RSC.
- (a) J. R. Dilworth and N. Wheatley, Coord. Chem. Rev., 2000, 199, 89–158 CrossRef; (b) S. Kumar, G. Mani, S. Mondal and P. K. Chattaraj, Inorg. Chem., 2012, 51, 12527–12539 CrossRef PubMed.
- (a) P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193, 499–556 CrossRef; (b) C. Zeng, N. Wang, T. Peng and S. Wang, Inorg. Chem., 2017, 56, 1616–1625 CrossRef PubMed.
- (a) Y. Canac, N. Debono, L. Vendier and R. Chauvin, Inorg. Chem., 2009, 48, 5562–5568 CrossRef PubMed; (b) I. Abdellah, Y. Canac, C. D. Mboyi, C. Duhayon and R. Chauvin, J. Organomet. Chem., 2015, 776, 149–152 CrossRef; (c) S. A. Bhat, M. K. Pandey, J. T. Mague and M. S. Balakrishna, Dalton Trans., 2017, 46, 227–241 RSC.
- (a) A. Otero, F. Carrillo-Hermosilla, P. Terreros, T. Expósito, S. Rojas, J. Fernández-Baeza, A. Antiñolo and I. López-Solera, Eur. J. Inorg. Chem., 2003, 2003, 3233–3241 CrossRef; (b) S. A. Bhat, J. T. Mague and M. S. Balakrishna, Dalton Trans., 2015, 44, 17696–17703 RSC; (c) S. A. Bhat, J. T. Mague and M. S. Balakrishna, Inorg. Chim. Acta, 2016, 443, 243–250 CrossRef.
- (a) T. Benincori, E. Brenna, F. Sannicolò, L. Trimarco, P. Antognazza, E. Cesarotti, F. Demartin and T. Pilati, J. Org. Chem., 1996, 61, 6244–6251 CrossRef PubMed; (b) N. G. Andersen, M. Parvez and B. A. Keay, Org. Lett., 2000, 2, 2817–2820 CrossRef PubMed.
- (a) D. M. Zink, T. Grab, T. Baumann, M. Nieger, E. C. Barnes, W. Klopper and S. Bräse, Organometallics, 2011, 30, 3275–3283 CrossRef; (b) D. M. Zink, T. Baumann, J. Friedrichs, M. Nieger and S. Bräse, Inorg. Chem., 2013, 52, 13509–13520 CrossRef PubMed; (c) D. M. Zink, D. Volz, T. Baumann, M. Mydlak, H. Flügge, J. Friedrichs, M. Nieger and S. Bräse, Chem. Mater., 2013, 25, 4471–4486 CrossRef.
- (a) D. Liu, W. Gao, Q. Dai and X. Zhang, Org. Lett., 2005, 7, 4907–4910 CrossRef PubMed; (b) Q. Dai, W. Gao, D. Liu, L. M. Kapes and X. Zhang, J. Org. Chem., 2006, 71, 3928–3934 CrossRef PubMed; (c) R. J. Detz, S. A. Heras, R. de Gelder, P. W. N. M. van Leeuwen, H. Hiemstra, J. N. H. Reek and J. H. van Maarseveen, Org. Lett., 2006, 8, 3227–3230 CrossRef PubMed; (d) S.-i. Fukuzawa, H. Oki, M. Hosaka, J. Sugasawa and S. Kikuchi, Org. Lett., 2007, 9, 5557–5560 CrossRef PubMed.
- B. Choubey, L. Radhakrishna, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2016, 55, 8514–8526 CrossRef PubMed.
- L. Bonnafoux, L. Ernst, F. R. Leroux and F. Colobert, Eur. J. Inorg. Chem., 2011, 2011, 3387–3397 CrossRef.
- (a) M. Ardon, G. Hogarth and D. T. W. Oscroft, J. Organomet. Chem., 2004, 689, 2429–2435 CrossRef; (b) D. F. Brayton and D. M. Heinekey, Organometallics, 2008, 27, 3901–3906 CrossRef.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef PubMed.
- (a) P. Fuentealba, H. Preuss, H. Stoll and L. Von Szentpály, Chem. Phys. Lett., 1982, 89, 418–422 CrossRef; (b) D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. L. Caricato, X. H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- D. A. Zhurko and G. A. Zhurko, ChemCraft 1.5; Plimus, San Diego, CA, Available at http://www.chemcraftprog.com Search PubMed.
- S. Leonid, Chemissian 1.7, 2005–2010, Available at http://www.chemissian.com Search PubMed.
- I. Fleming, Frontier Orbital and OrganicChemical Reactions, John Wiley & Sons, New York, 1976 Search PubMed.
- (a) Q.-S. Li, C.-Q. Wan, R.-Y. Zou, F.-B. Xu, H.-B. Song, X.-J. Wan and Z.-Z. Zhang, Inorg. Chem., 2006, 45, 1888–1890 CrossRef PubMed; (b) Q.-L. Ni, X.-F. Jiang, T.-H. Huang, X.-J. Wang, L.-C. Gui and K.-G. Yang, Organometallics, 2012, 31, 2343–2348 CrossRef.
- R. J. Lundgren, M. A. Rankin, R. McDonald, G. Schatte and M. Stradiotto, Angew. Chem., Int. Ed., 2007, 46, 4732–4735 CrossRef PubMed.
- C. K. Seubert, Y. Sun and W. R. Thiel, Dalton Trans., 2009, 4971–4977 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- (a) J. Zhang, L. Li, Y. Wang, W. Wang, J. Xue and Y. Li, Org. Lett., 2012, 14, 4528–4530 CrossRef PubMed; (b) M. Heravi, V. Zadsirjan, H. Hamidi and P. H. Tabar Amiri, RSC Adv., 2017, 7, 24470–24521 RSC.
- (a) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef PubMed; (b) R. Wang, S. Mo, Y. Lu and Z. Shen, Adv. Synth. Catal., 2011, 353, 713–718 CrossRef; (c) M. Yamaguchi, T. Akiyama, H. Sasou, H. Katsumata and K. Manabe, J. Org. Chem., 2016, 81, 5450–5463 CrossRef PubMed.
- R. Zhou, W. Wang, Z.-j. Jiang, K. Wang, X.-l. Zheng, H.-y. Fu, H. Chen and R.-x. Li, Chem. Commun., 2014, 50, 6023–6026 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef.
- D. J. Darensbourg and R. L. Kump, Inorg. Chem., 1978, 17, 2680–2682 CrossRef.
- M. I. Bruce, C. Hamiester, A. G. Swincer and R. C. Wallis, Inorg. Synth., 1982, 21, 78–82 Search PubMed.
- M. A. Bennett, T. N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74–76 Search PubMed.
- D. Drew and J. R. Doyle, Inorg. Synth., 1990, 28, 346–349 Search PubMed.
- M.-C. Brandys, M. C. Jennings and R. J. Puddephatt, J. Chem. Soc., Dalton Trans., 2000, 4601–4606 RSC.
- M. J. Bosiak, ACS Catal., 2016, 6, 2429–2434 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1832214, 1832216–1832224. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra04086a |
| This journal is © The Royal Society of Chemistry 2018 |