DOI:
10.1039/C8RA04356F
(Paper)
RSC Adv., 2018,
8, 25719-25724
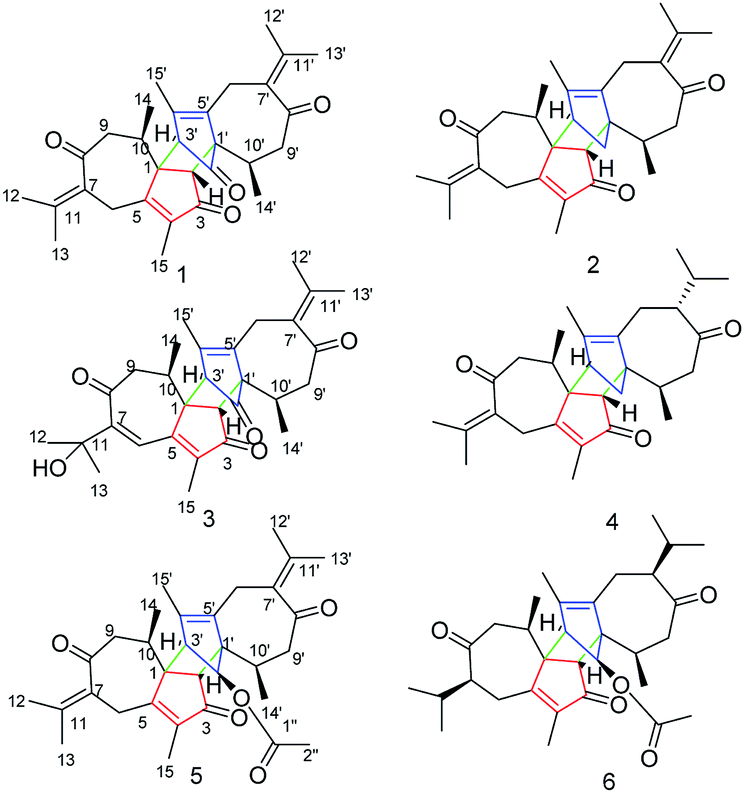
Xyloplains A–F, six new guaiane-type sesquiterpenoid dimers from Xylopia vielana†
Received
22nd May 2018
, Accepted 22nd June 2018
First published on 18th July 2018
Abstract
Six new guaiane dimers, xyloplains A–F (1–6), with connecting patterns through two direct C–C bonds (C-1 to C-3′, C-2 to C-1′), were isolated from the roots of Xylopia vielana. Their structures were elucidated clearly using extensive analysis of 1D NMR and 2D NMR, combined with Cu-Kα X-ray diffraction and circular dichroism (CD) experiments. In additon, all of the isolates were tested for anti-inflammatory activity by measuring the amount of nitric oxide produced. To our delight, compounds 2 and 6 exhibited moderate inhibitory activity against the production of nitric oxide with IC50 value of 34.5 and 31.1 μM, respectively, in RAW264.7 cells stimulated by LPS.
Introduction
The genus Xylopia, which is a member of the family Annonaceae, provides bark and leaves that are used for the treatment of irregular menstruation, rheumatism and pain.1,2 Studies examining the genus Xylopia have demonstrated its antimicrobial,3 antitumor,4 antioxidant5 and antiinflammatory6 activities. Recently, several novel dimeric guaianes or dimeric diterpenes have been isolated from the leaves of X. vielana by researchers.7–9 Thus far, only four guaiane-type sesquiterpenoid dimers (C-1 to C-3′, C-2 to C-1′) have been reported, and their absolute configurations were determined by X-ray diffraction and CD experiments. As an important part of our research into searching for novel and bioactive sesquiterpenoid dimers from natural products, we first investigated the systematic isolation of dimers from the roots of X. vielana. As a result, six new guaiane-type sesquiterpenoid dimers xylopains A–F (Fig. 1) were isolated successfully. In addition, their inhibitory activity against the generation of NO was tested. Notably, the absolute configurations of 1 and 5 were elucidated by Cu Kα X-ray diffraction.
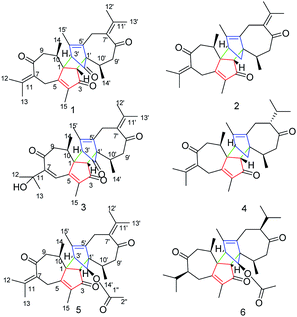 |
| | Fig. 1 Chemical structures of compounds 1–6. | |
Results and discussion
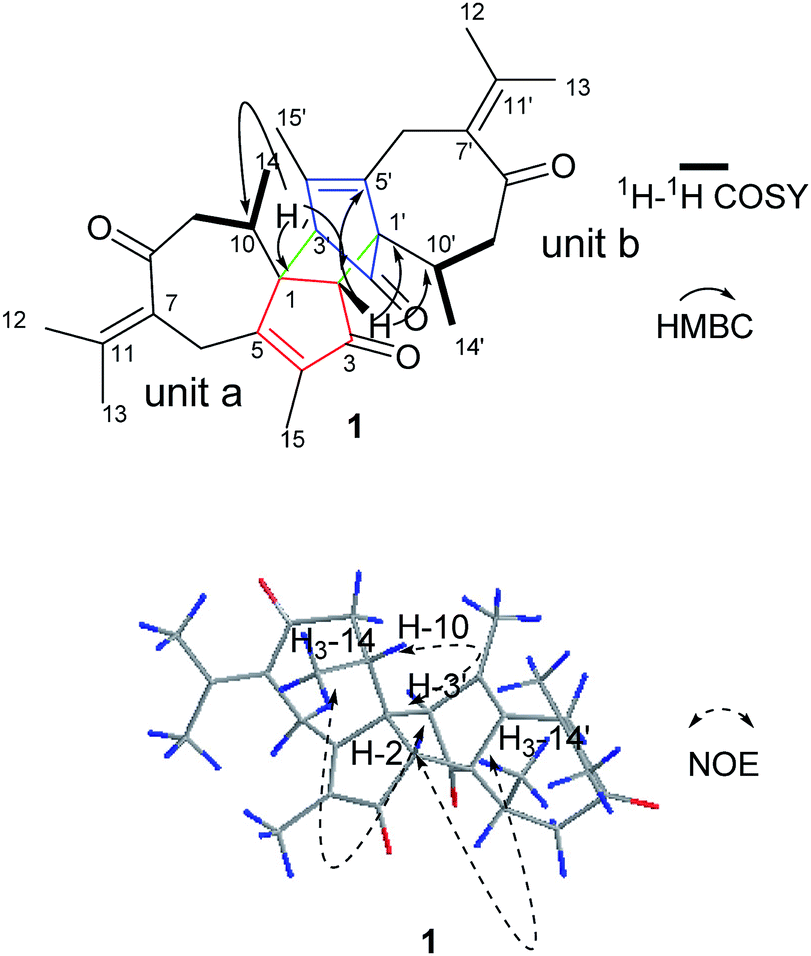
Compound 1 was obtained as colorless needle crystals. Its molecular formula C30H36O4 was established by HR-ESI: m/z 483.2517 [M + Na]+ (calcd for C30H36O4Na+, 483.2506), indicating 13 degrees of unsaturation. All of 30 carbon signals in the 13C NMR spectrum were classified by DEPT and HMQC experiments as eight methyl, four methylene, four methine and 14 quarternary carbons (Table 1). In addition, detailed analyses of 1D and 2D NMR data implies the presence of two guaiane units (a and b). In unit (a), the 1H–1H COSY spectrum of 1 gave the following correlations: H3-14/H-10 and H-10/H2-9. Furthermore, the HMBC cross-peaks from H-2 to C-1/C-5 and from H3-15 to C-3/C-4/C-5 suggests the appearance of a five-membered ring (I). In addition, the five-membered ring (I) is connected to a seven membered ring (II) at C-1 and C-5, according to the analyses of the HMBC cross-peaks from H2-6 to C-1/C-7/C-5, from H-10 to C-1/C-8/C-9 and from H2-9 to C-1/C-8/C-10 (Fig. 2). Furthermore, two other methyl groups (H3-14 and H3-15) were linked to C-4 and C-10, respectively, by observing the HMBC cross-peaks from H3-14 to C-10 and from H3-15 to C-4 (Fig. 2)
Table 1 1H and 13C NMR spectroscopic data of 1–4
| No. |
1a |
2b |
3b |
4a |
| δC |
δH |
δC |
δH |
δC |
δH |
δC |
δH |
| δ in ppm; J in Hz within parentheses; measured at 125 MHz for 13C NMR and 500 MHz for 1H NMR in chloroform-d. δ in ppm; J in Hz within parentheses; measured at 125 MHz for 13C NMR and 500 MHz for 1H NMR in CD3OD. |
| 1 |
58.4 s |
|
65.1 s |
|
56.8 s |
|
64.7 s |
|
| 2 |
48.5 d |
2.64 s |
55.1 d |
2.65 s |
51.4 d |
2.81 s |
54.9 d |
2.52 s |
| 3 |
204.9 s |
|
209.6 s |
|
205.2 s |
|
208.4 s |
|
| 4 |
143.6 s |
|
138.1 s |
|
147.1 s |
|
138.1 s |
|
| 5 |
167.5 s |
|
171.9 s |
|
160.1 s |
|
169.1 s |
|
| 6 |
29.6 t |
3.58 d (14.5) |
30.1 t |
3.79 d (14.1) |
124.1 d |
7.31 s |
30.8 t |
3.64 d (14.1) |
| |
|
2.93 d (14.5) |
|
3.15 d (14.1) |
|
|
|
2.91 d (14.1) |
| 7 |
129.8 s |
|
129.6 s |
|
157.4 s |
|
129.6 s |
|
| 8 |
203.3 s |
|
204.5 s |
|
204.7 s |
|
203.3 s |
|
| 9 |
47.8 t |
2.88 m |
46.8 t |
3.45 dd (15.0, 2.0) |
50.7 t |
3.20 m |
47.3 t |
3.23 dd (15.2, 2.1) |
| |
|
2.40 m |
|
2.40 m |
|
2.73 m |
|
2.54 m |
| 10 |
32.9 d |
2.25 m |
34.6 d |
2.38 m |
32.7 d |
2.13 m |
34.8 d |
2.28 m |
| 11 |
143.3 s |
|
146.9 s |
|
72.1 s |
|
146.3 s |
|
| 12 |
22.9 q |
1.90 s |
22.9 q |
2.03 s |
28.7 q |
1.51 s |
23.7 q |
1.95 s |
| 13 |
23.3 q |
1.93 s |
22.8 q |
2.04 s |
28.3 q |
1.43 s |
23.6 q |
2.03 s |
| 14 |
16.1 q |
0.85 d (6.9) |
15.1 q |
0.85 d (6.9) |
16.8 q |
0.98 d |
16.1 q |
0.87 d (7.1) |
| 15 |
8.6 q |
1.66 s |
7.4 q |
1.65 s |
7.2 q |
1.82 s |
8.6 q |
1.61 s |
| 1′ |
63.1 s |
|
63.6 s |
|
62.2 s |
|
64.4 s |
|
| 2′ |
199.6 s |
|
47.1 t |
1.78 dd (8.6, 1.4) |
199.4 s |
|
47.1 t |
1.66 dd (8.6, 1.3) |
| |
|
|
|
1.52 dd (8.6, 1.4) |
|
|
|
1.59 m |
| 3′ |
60.3 d |
2.98 s |
52.1 d |
2.99 S |
60.1 d |
2.98 s |
52.6 d |
2.82 d (1.3) |
| 4′ |
134.9 s |
|
140.1 s |
|
135.7 s |
|
139.8 s |
|
| 5′ |
134.3 s |
|
135.3 s |
|
133.6 s |
|
136.4 s |
|
| 6′ |
25.9 t |
3.10 d (15.2) |
25.5 t |
3.04 d (15.1) |
25.5 t |
3.19 d (15.1) |
26.0 t |
2.17 m |
| |
|
2.67 d (15.2) |
|
2.52 d (15.1) |
|
2.74 d (15.1) |
|
1.90 m |
| 7′ |
132.2 s |
|
132.1 s |
|
132.1 s |
|
56.0 d |
2.47 m |
| 8′ |
208.8 s |
|
211.2 s |
|
210.1 s |
|
214.2 s |
|
| 9′ |
49.2 t |
2.85 m |
50.4 t |
2.59 dd (16.8, 4.0) |
49.0 t |
2.76 m |
48.7 t |
2.31 m |
| |
|
2.41 m |
|
2.29 dd (16.8, 12.2) |
|
2.55 m |
|
2.18 m |
| 10′ |
27.1 d |
2.84 m |
28.3 d |
2.86 m |
26.7 d |
2.88 m |
28.9 d |
2.87 m |
| 11′ |
132.2 s |
|
134.1 s |
|
133.2 s |
|
28.3 d |
1.80 m |
| 12′ |
21.1 q |
1.63 s |
18.7 q |
1.69 s |
20.4 q |
1.64 s |
21.2 q |
0.78 s |
| 13′ |
20.1 q |
1.64 s |
20.5 q |
1.64 s |
18.4 q |
1.71 s |
20.0 q |
0.89 s |
| 14′ |
15.9 q |
1.22 d (6.9) |
14.9 q |
1.07 d (6.9) |
14.8 q |
1.27 d |
17.3 q |
1.05 d (6.8) |
| 15′ |
14.1 q |
1.61 s |
12.5 q |
1.61 s |
12.5 q |
1.57 s |
14.1 q |
1.55 s |
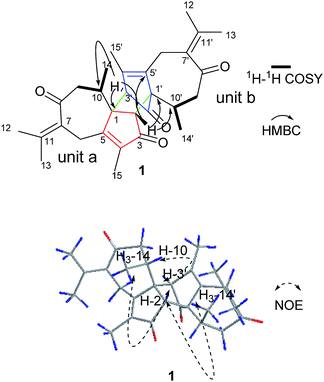 |
| | Fig. 2 Key HMBC, 1H–1H COSY and NOESY correlations of 1. | |
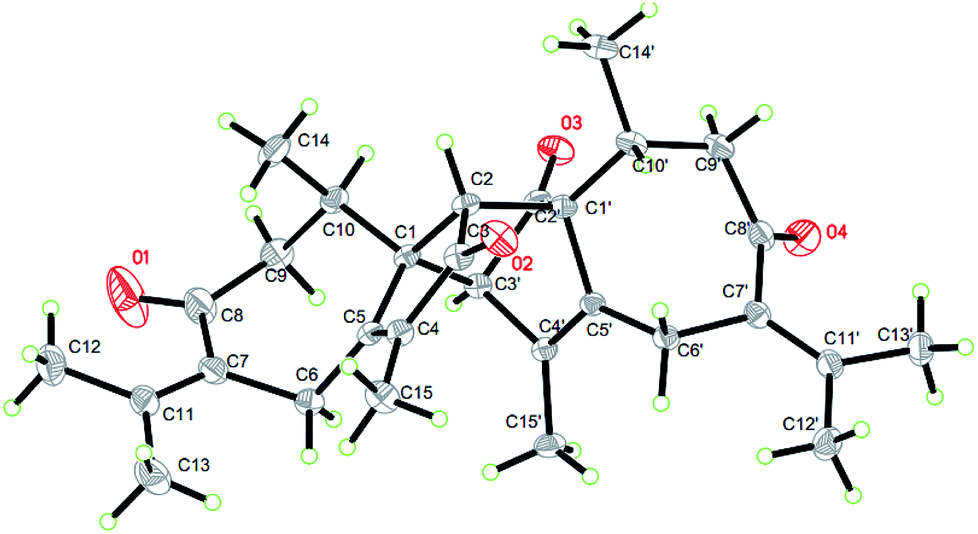
On the basis of the above spectral analysis, unit (a) was established as a guaiane sesquiterpenoid. Similarly, unit (b) was also assigned as a guaiane sesquiterpenoid, supported by key HMBC cross-peaks from H2-6′ to C-1’/C-7’/C-5′, H-10′ to C-1′/C-8′/C-9′ and H2-9′ to C-1′/C-8′/C-10′. The linkage of units (a) and (b) via two direct C–C bonds (C-1 to C-2′, C-2 to C-1′) was deduced from the key HMBC cross-peaks from H-2 to C-1′/C-2’/C-10’/C-5′, and H-3′ to C-1/C-2/C-10. Thus, the planar structure of 1 was confirmed, as shown in Fig. 2. The key NOE correlations of H3-14/H-2 and H3-14′/H-2 indicate that these hydrogens are in the same face and were assigned as β-oriented, while the correlations of H-3′/H-10 place them on the opposite side and were assigned as α-oriented (Fig. 2). The absolute configuration was further determined by a Cu Kα X-ray diffraction study. All relevant chiral centers in 1 were unambiguously assigned as 1S, 2R, 10R, 1′S, 3′R, 10′R and named xyloplain A (Fig. 3).
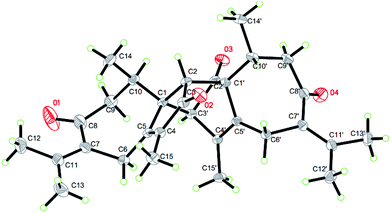 |
| | Fig. 3 X-ray structure of 1. | |
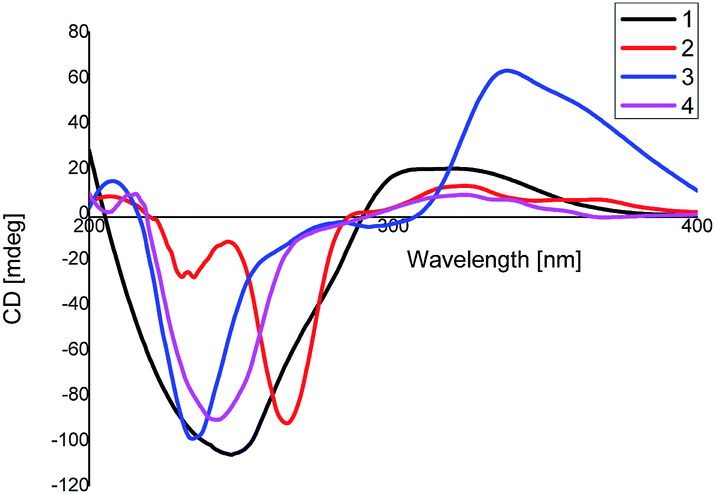
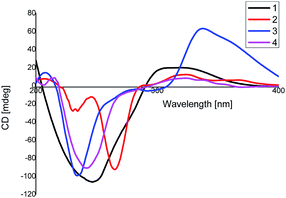
Compound 2 was assigned a molecular formula of C30H38O3, established from the HR-ESI MS positive peak at m/z 469.2717 [M + Na]+ (calcd for C30H38O3Na+, 469.2713), which represents 12 degrees of unsaturation. The 1D NMR spectrum exhibits 30 carbon resonance peaks, involving eight CH3 groups, five CH2 groups, four CH groups, and 13 quaternary carbons (Table 1). Further analysis of the 2D NMR spectra of 2 was in good agreement with 1 except for the absence of the carbonyl group at C-3. This was further supported by 1H–1H COSY correlations H2-2′/H-3′. The relative configuration of 2 was identical to that of 1 via the same key NOE correlations. The absolute configurations of 2 were finally determined by a similar CD experiment (Fig. 4) and given the name xyloplain B.
 |
| | Fig. 4 CD spectrum of compounds 1–4. | |
Compound 3, obtained as yellow oil, shows a positive ion HR-ESIMS peak at m/z 499.2462 [M + Na]+, which is consistent with a molecular formula of C30H36O5 by (calcd for C30H36O5Na+, 499.2455), indicating 13 degrees of unsaturation. The 13C NMR, DEPT and HSQC spectra exhibit 30 carbon resonance peaks, of which 16 carbons were assigned as eight methylic, three methylenic, and five methinic carbons, while the other carbons were assigned as four ketonic, eight olefinic, and two quaternary carbons (Table 1). A detailed comparison of the 2D NMR data of 3 with those of 1 indicate that they have the same basic skeleton. In an HMBC experiment, the correlations between the olefinic proton H-6 to C-7/C-11, H3-12 to C-7/C-11 and H3-13 to C-7/C-11 are observed, revealing 2-oxygenisopropyl connected to the seven-membered ring at C-9. The relative configuration of 3 is in accordance with 1 via the same key NOE correlations. The absolute configurations of 3 were finally determined to be 1S, 2R, 10R, 1′S, 3′R, 10′R by a similar CD experiment (Fig. 4).
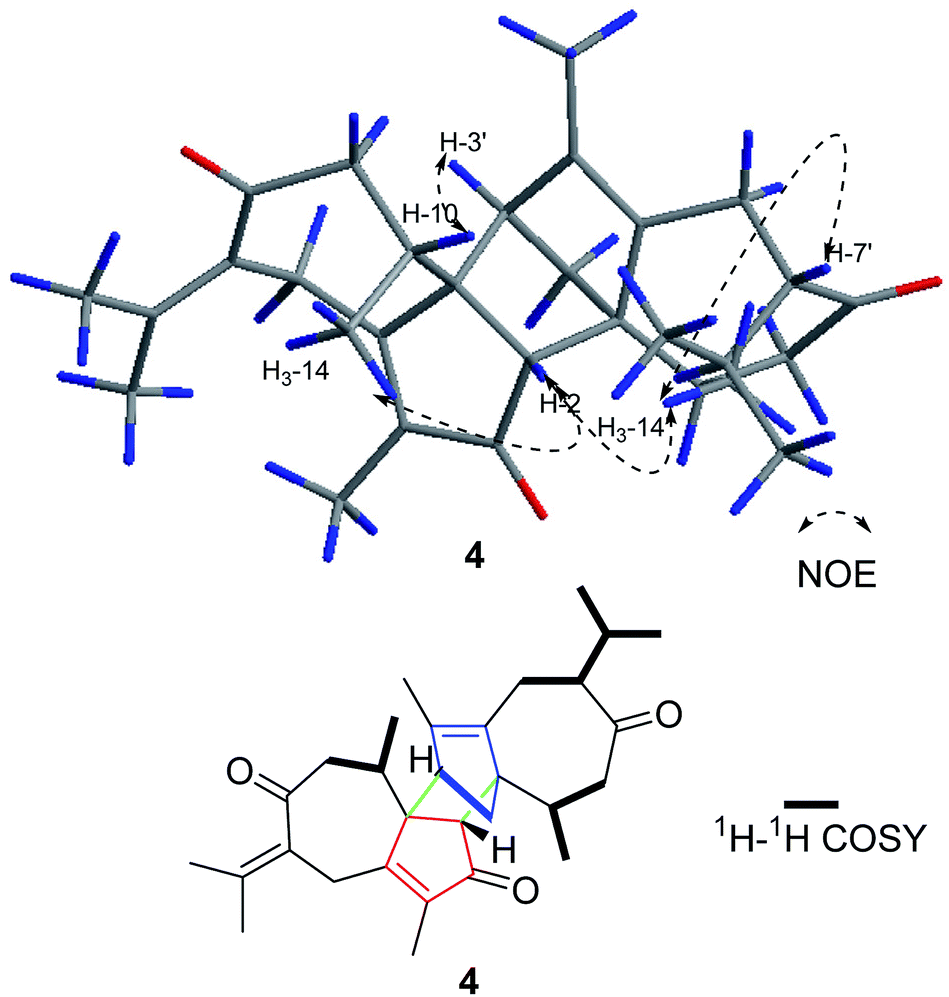
Compound 4 was obtained as a yellow oil with a molecular formula of C30H40O3, which was determined by positive ion HR-ESI-MS at m/z 471.2883 (calcd for C30H40O3Na+, 471.2870), accounting for 11 degrees of unsaturation. The 1H-NMR data of 4 displays eight methyl, five methylene, and four methine carbons (Table 1). Furthermore, the 2D NMR data suggests that the structure of 4 is closely related to that of 2, except for the disappearance the double bond at C-7′. This was confirmed by the 1H–1H COSY correlations, namely, H3-12′/H-11′, H3-13′/H-11′, and H-7′/H-11′. Moreover, in the NOESY spectrum, H3-14′, H-7′, H-2 and H3-14 were determined to be β-oriented by the key correlation of H3-14′/H-7′, H3-14/H-2, and H3-14′/H-2, while the α-orientations of H-3′ are determined by the key correlations of H-3′/H-10 (Fig. 5). Furthermore, The CD spectrum of 4 is in accordance with that of 1. Therefore, the absolute configurations of 4 was established as 1S, 2R, 10R, 1′S, 3′R, 7′S, 10′R and named xyloplain D (Fig. 4).
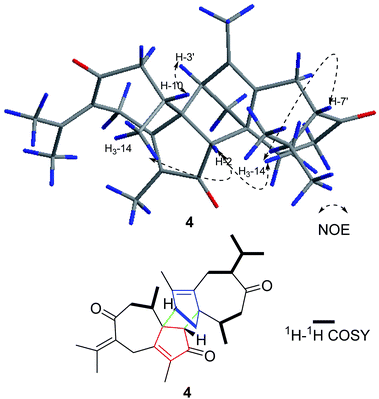 |
| | Fig. 5 Key NOESY and 1H–1H COSY correlations of 4. | |
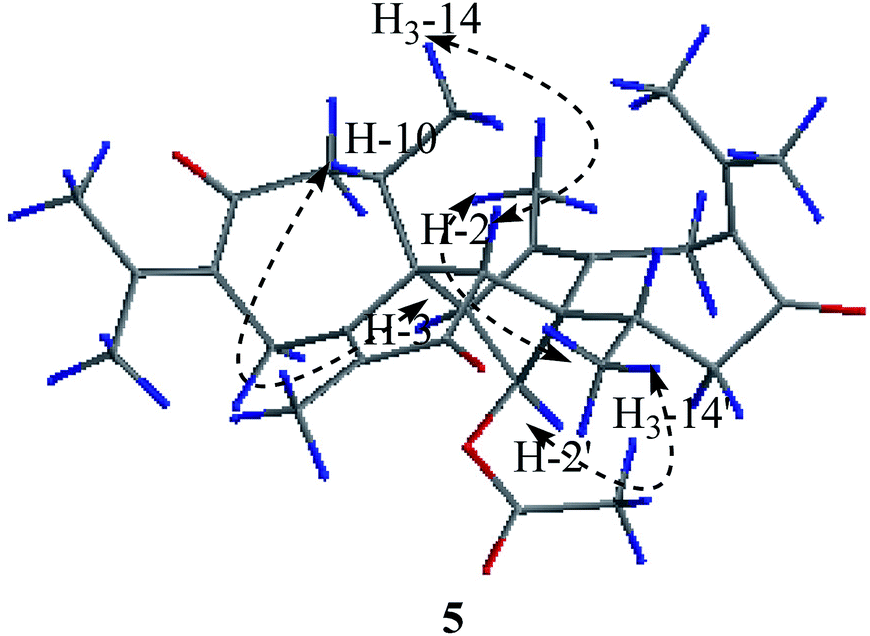
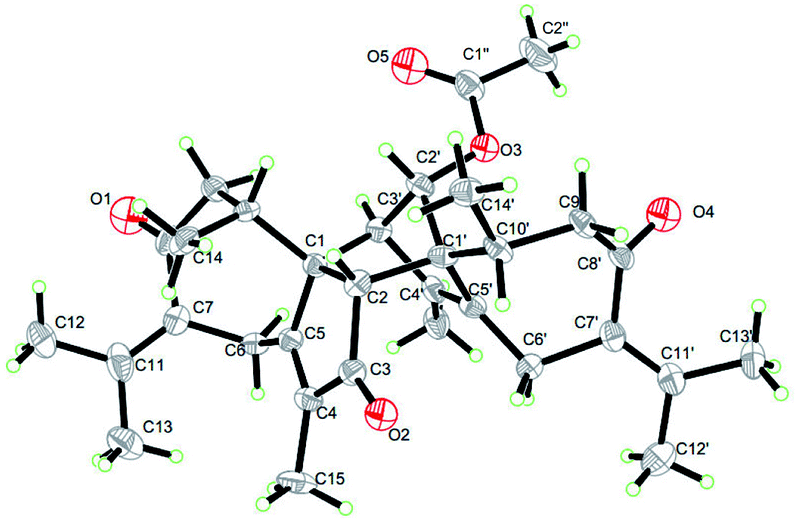
Compound 5 was obtained as colorless needle crystals. Its molecular formula C32H40O5 was established from the HR-ESI MS peak at m/z 527.2772 [M + Na]+ (calcd for C32H40O5, 527.2768), accounting for 14 degrees of unsaturation. The 1D NMR and 2D NMR data of 5 show great similarity to those of 1 except that the group at C-2′ is an acetoxy group instead of ketonic (Table 2). This is supported by the presence of the chemical shifts of the acetate group at δC 171.5 (C), 20.8 (CH3), and δH 2.00 (CH3). The relative configurations of H3-14, H3-14′, H-2 and H-2′ were assigned as β-orientations by the NOESY correlation of H3-14/H-2, H3-14′/H-2, and H3-14′/H-2′, while the α-orientations of H-3′ were deduced by the key correlations of H-3′/H-10′ (Fig. 6). The absolute configurations of 5 were finally determined by single-crystal X-ray diffraction was thus assigned as 1R, 2R, 10R, 1′S, 2′S, 3′R, 10′R (Fig. 7) and named xyloplain E.
Table 2 1H and 13C NMR spectroscopic data of 5 and 6
| No. |
5a |
6b |
| δC |
δH |
δC |
δH |
δ in ppm; J in Hz within parentheses; measured at 125 MHz for 13C NMR and 500 MHz for 1H NMR in chloroform-d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD3OD 1:2. δ in ppm; J in Hz within parentheses; measured at 125 MHz for 13C NMR and 500 MHz for 1H NMR in chloroform-d. :CD3OD 1:2. δ in ppm; J in Hz within parentheses; measured at 125 MHz for 13C NMR and 500 MHz for 1H NMR in chloroform-d. |
| 1 |
60.1 s |
|
64.4 s |
|
| 2 |
51.5 d |
2.58 s |
54.7 d |
2.39 s |
| 3 |
207.5 s |
|
205.1 s |
|
| 4 |
147.4 s |
|
142.8 s |
|
| 5 |
169.7 s |
|
168.1 s |
|
| 6 |
30.6 t |
3.65 d (14.2) |
31.2 t |
2.66 m |
| |
|
2.94 d (14.2) |
|
2.11 m |
| 7 |
129.1 s |
|
54.9 d |
2.65 m |
| 8 |
203.4 s |
|
214.0 s |
|
| 9 |
47.2 t |
3.23 dd (14.6, 1.9) |
48.3 t |
2.72 m |
| |
|
2.50 m |
|
2.17 m |
| 10 |
34.7 d |
2.44 m |
28.2 d |
2.93 m |
| 11 |
147.4 s |
|
28.7 d |
2.05 m |
| 12 |
23.8 q |
2.01 s |
19.6 q |
0.96 d (6.8) |
| 13 |
23.8 q |
2.01 s |
21.2 q |
0.91 d (6.8) |
| 14 |
15.9 q |
0.87 d (7.0) |
17.0 q |
1.08 d (6.9) |
| 15 |
8.3 q |
1.64 s |
8.3 q |
1.54 s |
| 1′ |
64.1 s |
|
59.4 s |
|
| 2′ |
86.2 d |
4.88 s |
87.5 d |
4.89 d (1.8) |
| 3′ |
56.4 d |
3.06 s |
56.2 d |
3.40 d (1.8) |
| 4′ |
135.4 s |
|
135.1 s |
|
| 5′ |
131.7 s |
|
132.2 s |
|
| 6′ |
25.4 t |
3.05 m |
25.8 t |
2.31 d (15.4) |
| |
|
2.56 m |
|
1.87 d (15.4) |
| 7′ |
133.3 s |
|
56.9 d |
2.43 m |
| 8′ |
209.9 s |
|
211.8 s |
|
| 9′ |
50.9 t |
2.43 m |
49.6 t |
3.19 dd (11.5, 4.5) |
| |
|
|
|
2.42 m |
| 10′ |
27.3 d |
2.98 m |
34.5 d |
2.45 m |
| 11′ |
140.8 s |
|
27.9 d |
1.97m |
| 12′ |
19.7 s |
1.66 s |
20.1 q |
0.81 d (6.8) |
| 13′ |
21.2 q |
1.65 s |
21.2 q |
0.91 d (6.8) |
| 14′ |
15.8 q |
1.09 d (6.8) |
17.1 q |
0.73 d (6.9) |
| 15′ |
13.7 q |
1.56 s |
13.8 q |
1.60 s |
| 1′′ |
171.5 s |
|
170.8 s |
|
| 2′′ |
20.8 q |
2.00 s |
21.1 q |
2.03 s |
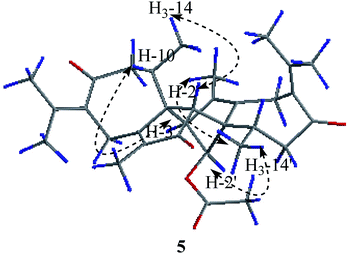 |
| | Fig. 6 Key NOESY correlations of 5. | |
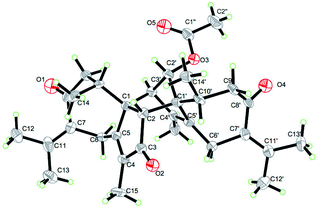 |
| | Fig. 7 X-ray structure of 5. | |
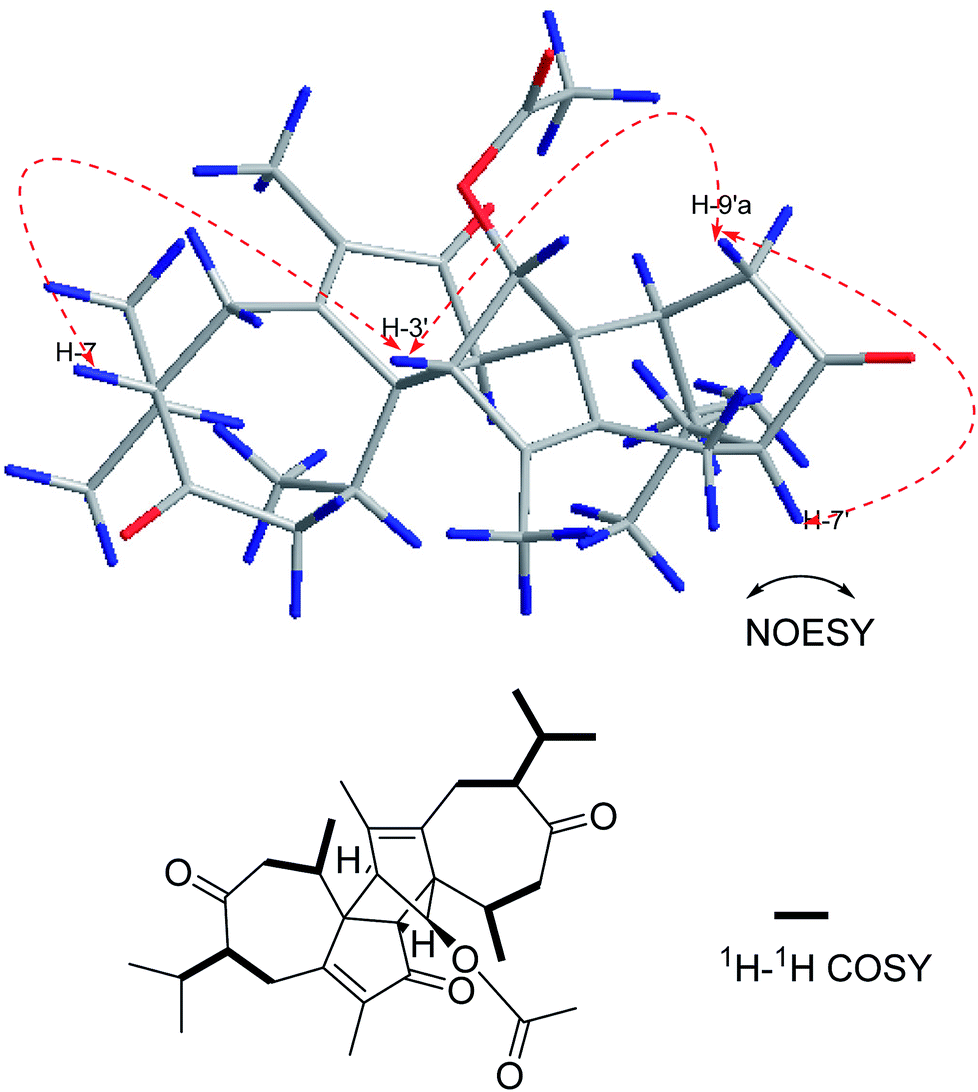
Compound 6 was isolated as a yellow oil. Its positive ion HR-ESI-MS revealed a peak for a molecular ion at m/z 531.3092 [M + Na]+, corresponding to a molecular formula of C32H44O5. The difference of 4 Da between the molecular weights of 5 and 6 suggests that 6 is a dihydro product of 5, which was confirmed by the change of the chemical shift for C-7, C-11 and C-7′, C-11′ (Table 2) and by the 1H–1H COSY correlations shown in Fig. 8. The relative configurations of 6 at H-7 and H-7′ were determined to be α-oriented based on the NOESY correlations H-7/H-3′, H-3′/H-9′a and H-7′/H-9′a (Fig. 8). The absolute configuration of 6 was assumed to be the same as that of 5 due to the similar CD and named xyloplain F (Fig. S49†).
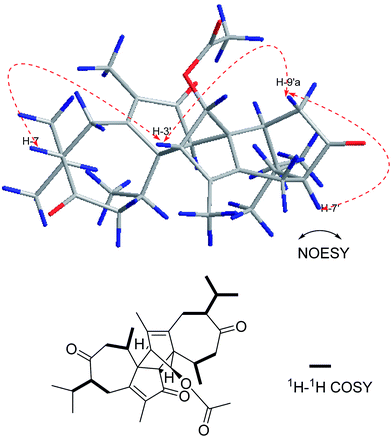 |
| | Fig. 8 Key NOESY and 1H–1H COSY correlations of 6. | |
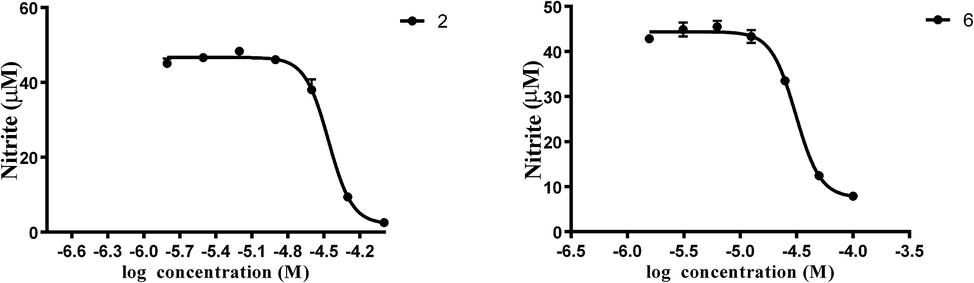
Additionally, we measured the inhibitory effects of these compounds on nitric oxide production in LPS stimulated RAW 264.7 macrophages to elucidate their anti-inflammatory potentials. All of the compounds showed no cytotoxic effects. Interestingly, compounds 2 and 6 displayed moderate inhibition effects against nitric oxide production, while the others showed a weak inhibition at concentration of 50 μM (Fig. S50†).
Crystallographic data for compounds 1 and 5 have been deposited at the Cambridge Crystallographic Data Centre with the deposition numbers CCDC 1834140 and 1834138, respectively.
Conclusions
In summary, we reported, for the first time, the isolation and structural characterization of six new guaiane-type sesquiterpenoid dimers (1–6) from the roots of X. vielana. Their structures were elucidated by extensive analyses of NMR spectroscopic data and CD spectrum. The absolute configurations of 1 and 5 were first determined by single-crystal X-ray diffraction, while the others were established by similar CD experiments. More importantly, we provide a method to confirm the absolute configuration of guaiane-type sesquiterpenoid dimers (C-1 to C-3′, C-2 to C-1′). All of the isolated compounds were screened for inhibitory activity against LPS induced nitric oxide production in RAW 264.7 macrophages. Interestingly, compounds 2 and 6 displayed moderate inhibition effects against nitric oxide production with IC50 values of 34.5 and 31.1 μM, respectively, while the others showed weak inhibition at concentration of 50 μM (Fig. 9).
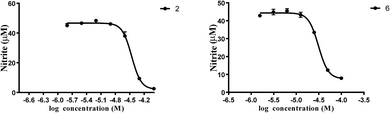 |
| | Fig. 9 The dose inhibition curves of nitric oxide produced by compounds 2 and 6. The data were obtained from three independent experiments and expressed as the means ± SEM. | |
Experimental
General experimental procedures
TLC analysis was conducted on HSGF254 silica gel plates (10–40 μm, Yantai, China). Column chromatography was performed using silica gel (100–200 mesh, Yantai, China) and silica gel H (10–40 μm, Qingdao, China). Preparative HPLC (Shimadzu LC-6AD) was performed on a preparative column (Shimadzu PRC-ODS EV0233). 1D and 2D NMR spectra were recorded on a Bruker Avance 500 NMR spectrometer, and chemical shifts are shown in δ (ppm) with TMS as an internal reference, and coupling constants were in Hz. ESI MS spectra were recorded on Agilent LC/MSD Trap XCT spectrometer (Waters, USA) and HR-ESI MS on Q-Tof micro YA019 mass spectrometer (Waters, USA).
Plant material
The roots of X. vielana were collected from Fangchenggang, Guangxi province, P. R. China, in December 2016. The identification of the plant material was authenticated by Prof. Zhong Liu, School of Pharmacy, Shanghai Jiao Tong University. A voucher specimen (No. 201611MBS) has been deposited in the School of Pharmacy, Shanghai Jiao Tong University.
Extraction and isolation
The dried roots of X. vielana (22.3 kg) were powdered and extracted with 95% ethanol (50 L) three times at room temperature and then evaporated in vacuum to obtain a crude extract (764.1 g). The crude extract was further partitioned with petroleum ether (PE 3 L × 3) and EtOAc (EA 3 L × 3) to obtain two fractions. The PE fraction (32.4 g) was subjected to silica gel column chromatography and eluted with a step gradient of PE–EA (0:100–0:100, v/v) to yield ten fractions (A–J). Fraction D (4.2 g) was further isolated and eluted with PE–acetone (100:1–10:1, v/v), resulting in six fractions (D-1–D-6). Fraction D-3 was purified by preparative HPLC (MeOH–H2O, 65:35) to yield 1 (18.2 mg), 2 (10.6 mg) and 6 (9.0 mg). Fraction D-5 was subjected to preparative HPLC (MeOH–H2O, 70:30) to yield 3 (9.8 mg), 4 (8.8 mg) and 5 (11.2 mg).
Xyloplain A (1). Colorless crystals; [α]20D + 63.2 (c 0.16 CH2Cl2); mp 189–192 °C; IR (KBr) vmax 3446, 2920, 1769, 1694, 1457, 1380, 1221, 1036, 755 cm−1; CD (c 6 × 10−5 M, MeOH), Δε (nm): −103.9 (248), + 19.1 (311); for 1H-NMR and 13C-NMR spectroscopic data, see Table 1; HR-ESI MS (positive): m/z 483.2517 [M + Na]+ (calcd as 483.2506).
Xyloplain B (2). Yellow oil; [α]20D +29.2 (c 0.10 CH2Cl2); IR (KBr) vmax 3438, 2923, 1777, 1678, 1639, 1459, 1378, 1026, 730 cm−1; CD (c 6 × 10−5 M, MeOH), Δε (nm): −92.1 (235), + 12.1 (323); for 1H-NMR and 13C-NMR spectroscopic data, see Table 1; HR-ESI MS (positive): m/z 469.2717 [M + Na]+ (calcd as 469.2713).
Xyloplain C (3). Yellow oil; [α]20D +44.3 (c 0.11 CH2Cl2); IR (KBr) vmax 3433, 2961, 1696, 1643, 1402, 1261, 998, 836, 610 cm−1; CD (c 6.5 × 10−5 M, MeOH), Δε (nm): −97.9 (234), + 59.1 (332); for 1H-NMR and 13C-NMR spectroscopic data, see Table 1; HR-ESI MS (positive): [M + Na]+ m/z 499.2462 (calcd as 499.2455).
Xyloplain D (4). Yellow oil; [α]20D + 44.3 (c 0.11 CH2Cl2); IR (KBr) vmax 3379, 2838, 1742, 1669, 1396, 1247, 1131, 996, 596 cm−1; CD (c 6 × 10−5 M, MeOH), Δε (nm): −91.9 (241), + 8.1 (321); for 1H-NMR and 13C-NMR spectroscopic data, see Table 1; HR-ESI MS (positive): [M + Na]+ m/z 471.2883 (calcd as 471.2870).
Xyloplain E (5). Colorless crystals; [α]20D −62.3 (c 0.12 CH2Cl2); mp 176–179 °C; IR (KBr) vmax 3410, 2911, 1736, 1675, 1411, 1269, 1031, 654 cm−1; CD (c 4 × 10−5 M, MeOH), Δε (nm): +10.9 (224), −5.1 (263); for 1H-NMR and 13C-NMR spectroscopic data, see Table 2; HR-ESI MS (positive): m/z 527.2772 [M + Na]+ (calcd as 527.2768).
Xyloplain F (6). Yellow oil; [α]20D −54.3 (c 0.10 CH2Cl2); IR (KBr) vmax 3423, 2901, 1739, 1677, 1396, 1253, 1121, 998, 606 cm−1; CD (c 4.5 × 10−5 M, MeOH), Δε (nm): + 11.2 (220), −8.1 (260); for 1H-NMR and 13C-NMR spectroscopic data, see Table 2; HR-ESI MS (positive): m/z 531.3092 (calcd as 531.3081).
Cell culture
The RAW 264.7 mouse macrophage cell line was obtained from China Cell Line Bank (Beijing, China). The cells were grown in RPMI-1640 supplemented with 10% heat-inactivated FBS, penicillin (100 U mL−1) and streptomycin (100 U mL−1) at 37 °C under a humidified atmosphere of 5% CO2.
Bioassay for nitric oxide production
Cells were plated onto 96-well plates (4 × 104 cells per well) and pretreated with 100 μL of different concentrations of compounds with two-fold concentration dilution for 1 h before stimulation with 100 ng mL−1 (for RAW 264.7) of LPS. The compounds were initially subjected to maximum dilution of 100 μM. After incubation for 24 h, an equal volume (50 μL) of the supernatant was mixed with Griess Reagent (Beyotime Institute of Biotechnology, China) in a 96-well plate. The mixture was allowed to react for 15 min, and the release of nitric oxide was measured at 540 nm on a Flexstation 3.10
MTT assay for cell viability
Cells were seeded in 96-well plates at 1 × 104 cells per well. After 24 h, the cells were treated with 100 μL of different concentrations of compounds (10 μM or 50 μM) for 24 h. Subsequently, 10 μL of 5 mg mL−1 MTT in DD water was added to each well, and the cells were incubated for 4 h. The medium was removed and resolved with 100 μL per well DMSO. The optical density was measured at 490 nm on a Flexstation 3 (Molecular Devices, Silicon Valley, CA, USA).11
Statistical analysis
The data were obtained from three independent experiments and expressed as the mean ± SEM. All statistical analyses were calculated by the GraphPad Prime 7 Software (GraphPad, Avenida, CA, USA).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by Professor of Chang Jiang Scholars Program, NSFCs (81520108030, 21472238), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (16DZ2280200), the Scientific Foundation of Shanghai China (13401900103, 13401900101), the National Key Research and Development Program of China (2017YFC1700200). We thank Prof. Xiaoli Bao and Lingling Li from the Instrumental Analysis Center of Shanghai Jiao Tong University for X-ray crystallographic analysis.
Notes and references
- D. Martins, E. Osshiro, N. F. Roque, V. Marks and H. E. Gottlieb, Phytochemistry, 1998, 48, 677–680 CrossRef.
- C. Kamperdick, N. M. Phuong, G. Adam and T. Van Sung, Phytochemistry, 2001, 56, 335–340 CrossRef PubMed.
- O. T. Asekun and B. A. Adeniyi, Fitoterapia, 2004, 75, 368–370 CrossRef PubMed.
- R. P. C. Ferraz, G. M. B. Cardoso, T. B. da Silva, J. E. d. N. Fontes, A. P. d. N. Prata, A. A. Carvalho, M. O. Moraes, C. Pessoa, E. V. Costa and D. P. Bezerra, Food Chem., 2013, 141, 196–200 CrossRef PubMed.
- A. Mohammed and M. S. Islam, Biomed. Pharmacother., 2017, 96, 30–36 CrossRef PubMed.
- V. B. Oliveira, A. V. M. Ferreira, M. C. Oliveira, M. M. Teixeira and M. G. L. Brandão, Food Res. Int., 2014, 62, 541–550 CrossRef.
- C. Kamperdick, N. M. Phuong, G. Adam and T. Van Sung, Phytochemistry, 2003, 64, 811–816 CrossRef PubMed.
- W. Vilegas, J. D'Arc Felicio, N. F. Roque and H. E. Gottlieb, Phytochemistry, 1991, 30, 1869–1872 CrossRef.
- Y. L. Zhang, X. W. Zhou, X. B. Wang, L. Wu, M. H. Yang, J. Luo, Y. Yin, J. G. Luo and L. Y. Kong, Org. Lett., 2017, 19, 3013–3016 CrossRef PubMed.
- J. Ren, C. Fan, Y.-G. Guo, S.-K. Yan, R. D. Ye, Y. Zhang, H.-Z. Jin and W.-D. Zhang, Tetrahedron, 2017, 73, 1611–1617 CrossRef.
- L. Wu, Y. Fan, C. Fan, Y. Yu, L. Sun, Y. Jin, Y. Zhang and R. D. Ye, Eur. J. Pharmacol., 2017, 801, 46–53 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1834140 and 1834138. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra04356f |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Wei-dong Zhang*ab
*a and
Wei-dong Zhang*ab