
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDoubling the photocatalytic performance of SnO2 by carbon coating mixed-phase particles†
Qingbo Li *a,
Hongkai Zhaoa,
Honggang Sunb,
Xian Zhao*a and
Weiliu Fan*c
*a,
Hongkai Zhaoa,
Honggang Sunb,
Xian Zhao*a and
Weiliu Fan*c
aState Key Laboratory of Crystal Materials, Shandong University, Jinan, 250100, P. R. China. E-mail: liqingbo2016@sdu.edu.cn
bSchool of Machanical, Electrical & Information Engineering, Shandong University, Weihai, 264209, P. R. China
cSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, P. R. China
First published on 28th August 2018
Abstract
In this work, carbon-coated mixed-phase (tetragonal/orthorhombic) SnO2 photocatalysts were successfully fabricated with a chemical precipitation method, followed by a facile hydrothermal process. This material exhibits the highest activity during photocatalytic methyl orange and phenol degradation, and the degradation rate constant is almost 3 times than that of pure tetragonal SnO2. The enhancement of photocatalytic activity could be mainly attributed to the synergistic effect among tetragonal SnO2, orthorhombic SnO2 and carbon (serves as a charge transfer mediator), which was found to lead to more efficient separation of photogenerated electron–hole pairs. Phase junctions in mixed-phase SnO2 were beneficial to the separation of photogenerated electrons and holes and carbon further facilitated the charge transfer between tetragonal and orthorhombic SnO2 nanoparticles. This study may provide a method for other mixed-phase semiconductors with promising performance for potential applications in environmental protection.
1. Introduction
Water contamination, resulting from organic pollutants in industrial waste and domestic sewage, is becoming an overwhelming problem all over the world.1–4 Heterogeneous photocatalysts show great potential for the decomposition of organic pollutants under light irradiation.5–10 Mixed-phase materials have received a great deal of attention recently since their unique features of identical chemical composition but different crystal structure that can significantly improve the charge separation efficiency in photocatalysis and photochemistry. Li's group put forward the phase junction concept for the first time and found that the phase junction formed between the surface anatase TiO2 nanoparticles and rutile TiO2 particles could greatly enhance the photocatalytic activity for H2 production.11 Then, they reported that α-β Ga2O3 showed enhanced photocatalytic performance for overall water splitting because of the formation of efficient mixed-phase junctions.12 In addition, more mixed-phase semiconductor photocatalysts with high photocatalytic performance have been successfully prepared, such as BiFeO3/Bi25FeO40, α-β AgAl0.4Ga0.6O2, α-β Fe2O3 and zincblende–wurtzite mixed-phase ZnO.13–16 As polymorphic semiconductors are quite common in nature, atomically well-matched phase junctions can be conveniently fabricated by fine-tuning the phase transformation conditions. Hence, it is reasonably speculated that the design of phase junctions is an efficient approach to promote photocatalytic performance.SnO2 is an important n-type semiconductor material with a wide direct band gap of 3.6 eV, which has been widely investigated as gas sensors, electrodes, and photocatalysts.17–19 Generally speaking, there are two main phases of SnO2 in common.20–23 Tetragonal SnO2 is regarded as a stable phase under ambient conditions. Orthorhombic phase is metastable at ambient pressure and is hard to be prepared through traditional methods under low pressure and temperature. To the best of our knowledge, there seems to be no investigation conducted yet on the photocatalytic activity of mixed-phase SnO2. Recently, mixed-phase SnO2 nanorods were successfully synthesized via chemical precipitation, but only their gas-sensing performance were investigated.24 This approach opens up a promising possibility to prepare mixed-phase SnO2 under ambient conditions. However, due to the special preparation method, the as-prepared nanorods are polycrystalline and consist of uniform nanocrystals. Pores can also be observed in the SnO2 nanorods, indicating a loose connection and a poor interfacial contact between particles, which limits the effective interparticle charge transfer. The same conclusions were reported in the studies of mesoporous TiO2.25,26 Thus, it is obliged to resolve the particle contact and interfacial charge transfer problems in the preparation process of mixed-phase SnO2.
Generally, carbon materials, like carbon nanotubes or graphene, are ideal electron transport materials for heterostructured photocatalyst design because they have high conductivity and superior electron mobility.27–32 In carbon-based heterostructures, carbon materials could serve as excellent electron acceptors and transfer media to decrease the recombination rate of charge carriers, thus enhancing photocatalytic activity. Among all carbon materials, the thin carbon shell is noted for its great promise of enhancing the photocatalytic behavior from its favorable electron transfer ability.33–39 We believe that it is a feasible scheme to use thin carbon shell as electron transfer media to resolve the particle contact and interfacial charge transfer problems in efficient mixed-phase SnO2 design.
Herein, we designed a novel carbon-coated mixed-phase SnO2 nanorods composed of a large number of nanoparticles by a chemical precipitation method, followed by a facile hydrothermal process. Moreover, the photocatalytic ability of the as-prepared carbon-coated mixed-phase SnO2 was assessed by degradating MO and phenol under UV light irradiation. Finally, the possible photocatalytic mechanism of the carbon-coated mixed-phase SnO2 photocatalyst was proposed and elaborated.
2. Experimental
2.1 Synthesis of SnO2 photocatalysts
SnO2 nanorods were synthesized by a chemical precipitation method.24 In a typical preparation procedure, 1.801 g of H2C2O4·2H2O was dissolved into a mixed solution of 10 mL of PEG (Mw = 400) and 20 mL of ethanol under continuous magnetic stirring. Then 4.513 g of SnCl2·2H2O was then added to the solution. Subsequently, 20 mL of deionized water was added dropwise. After stirring for 30 min, the as-prepared precipitation was separated by centrifugation, washed several times with distilled water and absolute ethanol. The dried products were heat treated at 550 °C or 650 °C for 2 h, which is noted as to-SnO2 (tetragonal–orthonormal mixed-phase SnO2) or t-SnO2 (tetragonal SnO2), respectively.Carbon-coated SnO2 photocatalysts were obtained from glucose solution under hydrothermal conditions. Briefly, a certain amount of t-SnO2 was dispersed for 10 minutes in 30 mL of deionized water. Then a certain amount of glucose was added and stirred for an additional 30 minutes. The solution was then sealed in a 50 mL Teflon-lined autoclave and was kept at 180 °C for 6 h. The final carbon-coated t-SnO2 was obtained after calcination of the hydrothermal products at 550 °C for 4 h under a nitrogen atmosphere, denoted as t-SnO2–C. Carbon-coated to-SnO2 was prepared under similar conditions, denoted as to-SnO2–C.
2.2 Characterization
The morphology and structure of the as-prepared samples were analyzed by scanning electron microscopy (SEM, Hitachi, S-4800), transmission electron microscopy (TEM, JEM-2100). X-ray diffraction data (XRD) for as-prepared samples were measured via X-ray diffraction analysis (XRD, Bruker D8 Advance diffractometer) with Cu-Kα radiation operated in a 2θ range of 10–80° with a step width of 0.02°. X-ray photoelectron spectroscopy (XPS) analysis of the samples was performed on an ESCALAB 250 spectrometer equipped with an Al Kα source, and the binding energies of the composing elements were referenced to the C 1s peak at 284.6 eV. Ultraviolet-visible (UV-Vis) diffuse reflectance spectra was carried out using a SHIMADZU UV-2550 spectrometer and BaSO4 as the reference. A Builder 4200 instrument was used to measure the Brunauer–Emmett–Teller (BET) specific surface areas of the samples through nitrogen adsorption–desorption.2.3 Photocatalytic and photoelectrochemical performance tests of SnO2 photocatalysts
The photocatalytic activities of the as-prepared samples were detected through the degradation of methyl orange (MO) and phenol operated by a 300 W Xe arc lamp (PLS-SXE300CUV, Beijing Perfect Light Co., Ltd., China). The optical power density was measured to be 84.30 mV cm−1 by UV radiometer (UV-A 141108). For each experiment, 30 mg of photocatalyst was added into 50 mL of MO solution (20 mg L−1) or phenol (30 mg L−1) in a 100 mL beaker with a diameter of ∼53 mm. The lamp is about 10 cm above from the liquid surface. Before illumination, the suspensions were stirred in dark for 30 minutes to reach the adsorption–desorption equilibrium. During the degradation, at given time intervals, 5.0 mL of suspension was withdrawn and centrifuged to remove the photocatalysts and then was tested by a Hitachi U-3500 UV-Vis spectrometer to determine the concentration of MO or phenol through observing the 463 nm or 270 nm absorption peak. The degradation ratio was defined as (C0 − C)/C0, where C is the pollutant's concentration after photocatalysis, and C0 is the pollutant's concentration after dark adsorption.The photoelectrochemical analysis was conducted by transient photocurrent response and electrochemical impedance spectroscopy (EIS) measurements in a three-electrode system on a LAN-LIKE electrochemical workstation, using the t-SnO2, to-SnO2, t-SnO2–C and to-SnO2–C powders deposited onto tin oxide conducting glass with a working area of 0.25 cm2 as the working electrode, Ag/AgCl (saturated KCl) as the reference electrode, Pt foil as the counter electrode and Na2SO4 (0.5 M) as electrolyte. A 300 W Xe lamp was used for excitation and the photocurrents with light on and off were measured at 0.5 V. EIS was conducted over a frequency range of 10 mHz to 100 kHz.
2.4 Computational details
DFT with a GGA-PBE0 functional,40 as implemented in the CASTEP program,41 was used for all the calculations. Norm-conserving pseudopotential was used for the treatment of core electrons.42 An energy cutoff of 600 eV was employed throughout our calculations. The self-consistent convergence accuracy was set at 1 × 10−5 eV per atom, the convergence criterion for the force between atoms was 0.03 eV Å−1, and the maximum displacement was 0.001 Å. Monkhorst–Pack grids of 3 × 3 × 4 and 3 × 2 × 2 k-points were used for tetragonal and orthorhombic SnO2, respectively.3. Results and discussion
3.1 Structure and morphology
Fig. 1 shows the XRD patterns of all the as-prepared samples. It can be observed that t-SnO2 shows peaks corresponding to pure tetragonal phase of SnO2 (JCPDS no. 41-1445). Obviously, to-SnO2 shows an extra peak at 2θ = 30°, which correspond to the (113) planes of orthorhombic phase of SnO2 (JCPDS no. 229-1484), confirming to-SnO2 sample is mixed-phase crystalline.24,43 The XRD patterns of t-SnO2–C and to-SnO2–C are similar to that of t-SnO2 and to-SnO2, respectively, indicating that the introduction of carbon does not change the crystal structure of SnO2. The peaks of carbon are not visible in the carbon-coated SnO2 samples, which could be attributed to the low content of carbon. It is noticed that the (113) diffraction peaks of to-SnO2–C still appear and the intensity do not change significantly, suggesting that orthorhombic SnO2 did not transform into tetragonal SnO2 during the hydrothermal and calcination process, which could be attributed to the protection of nitrogen atmosphere and carbon modification.28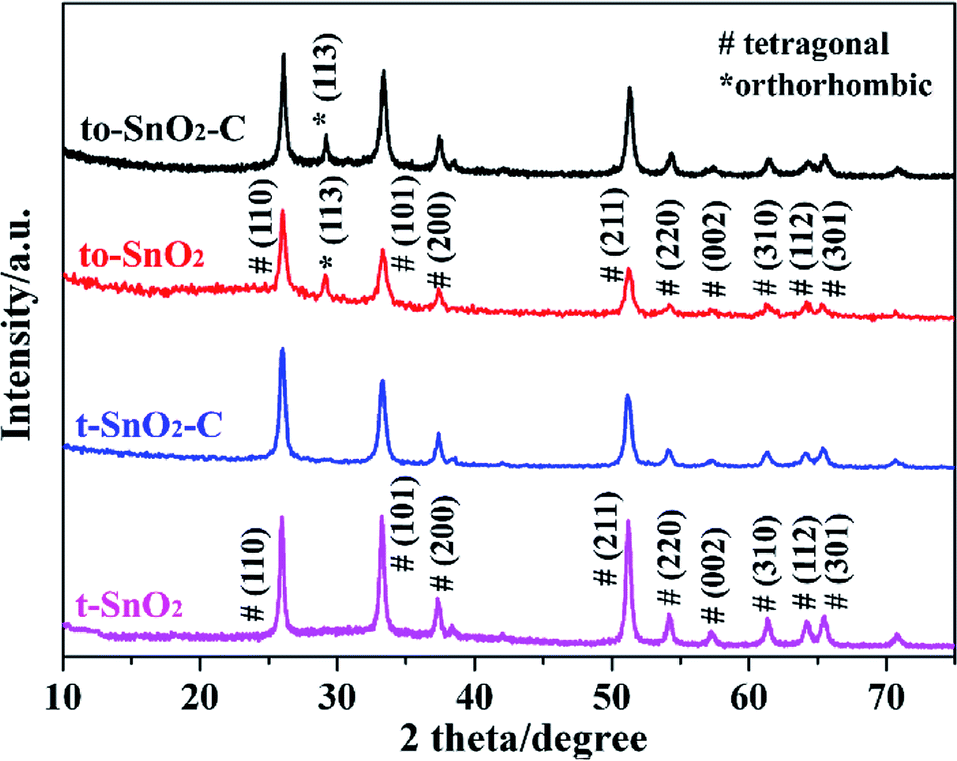 | ||
| Fig. 1 XRD patterns of t-SnO2, to-SnO2, t-SnO2–C and to-SnO2–C samples. | ||
The surface morphologies and microstructure of to-SnO2, t-SnO2, to-SnO2–C and t-SnO2–C samples were studied by SEM and TEM techniques and the results are shown in Fig. 2 and 3. Fig. 2a and b display the SEM images of to-SnO2 and t-SnO2, revealing their uniform nanorod shapes, with a diameter of around 300 nm and length around 2 μm. Further observation indicates that the nanorods surfaces are rough, and it is obvious that the nanorods are composed of many small nanoparticles. Moreover, to-SnO2–C and t-SnO2–C samples morphologies display no obvious changes in comparison with to-SnO2 and t-SnO2.
 | ||
| Fig. 2 SEM of sample to-SnO2 (a), t-SnO2 (b), to-SnO2–C (c) and t-SnO2–C (d). | ||
 | ||
| Fig. 3 TEM and HRTEM of sample to-SnO2 (a1 and a2), t-SnO2 (b1 and b2), to-SnO2–C (c1 and c2) and t-SnO2–C (d1 and d2). | ||
More insight into the crystal structure of as-prepared samples was obtained from TEM and HRTEM shown in Fig. 3. To-SnO2 and t-SnO2 are polycrystalline and composed of many small nanoparticles with a mean size of 10–20 nm (Fig. 3a1 and b1). Obviously, pores can also be observed in to-SnO2 and t-SnO2, indicating a loose connection between the nanoparticles. Fig. 3a2 displays well-defined lattice fringes with planar distance of 0.331 and 0.285 nm corresponding to the (110) plane of tetragonal SnO2 and (020) plane of orthorhombic SnO2, respectively, indicating the coexistence of tetragonal and orthorhombic SnO2 nanoparticles in to-SnO2. Fig. 3b2 reveals that only tetragonal SnO2 nanoparticles exist in t-SnO2. After carbon coating, both to-SnO2–C and t-SnO2–C can maintain nanorod morphologies and consist of uniform nanoparticles with a mean size of 10–20 nm (Fig. 3c1 and d1). This indicates that the morphologies and structure of SnO2 samples could not be destroyed by hydrothermal and calcination treatments, which is in agreement with the XRD results. HRTEM images clearly present that only tetragonal SnO2 exist in t-SnO2–C (Fig. 3c2) and both tetragonal and orthorhombic SnO2 nanoparticles remain in to-SnO2–C (Fig. 3d2). This demonstrates that the transformation from orthorhombic SnO2 to tetragonal SnO2 did not happen during carbon coating process, which is also consistent with the XRD results. Notably, the nanoparticles surfaces in to-SnO2–C were coated by a thin carbon layer (marked with blue arrows), in other words, carbon was filled into the interparticle space in to-SnO2–C. Similarly, a thin carbon layer can be observed in t-SnO2–C sample.
The composition and chemical status of the elements in the as-prepared samples were investigated by XPS spectra. Fig. 4a shows the surveys of the four samples. As expected, there are only Sn, O and C related peaks, indicating that the four samples possess a very high chemical purity. Fig. 4b displays the Sn 3d spectra of to-SnO2, t-SnO2, to-SnO2–C and t-SnO2–C samples. Two peaks centered at 486.6 and 495.0 eV that correspond to the Sn 3d5/2 and Sn 3d3/2 peaks of Sn4+ in SnO2 are observed for to-SnO2 and t-SnO2 samples, which is in good agreement with the energy splitting reported for SnO2.43 No distinct difference is observed between the spectra of to-SnO2 and t-SnO2, indicating that the surface chemical states of to-SnO2 and t-SnO2 are similar. The same phenomenon can be seen in mixed-phase TiO2.44 The Sn 3d peaks in the to-SnO2–C (t-SnO2–C) sample display slightly shift as compared to the Sn 3d peaks in the to-SnO2 (t-SnO2), which should be attributed to the interaction between SnO2 and carbon. With respect to the XPS spectra of O 1s in Fig. 4c, for both to-SnO2 and t-SnO2 samples, two peaks of 530.4 and 531.35 eV have been fitted, which should be ascribed to lattice oxygen and OH groups in t-SnO2.44 Again, after carbon coating, slight shift of O 1s peaks in to-SnO2–C (t-SnO2–C) sample can be observed in comparison with to-SnO2 (t-SnO2), also confirming the interaction between SnO2 and carbon. Fig. 4d shows the high-resolution spectra of C 1s. In the spectra of to-SnO2 and t-SnO2, C 1s peaks center at 284.6 eV (C1) and no other peaks appear. In contrast, the C 1s XPS spectra of to-SnO2–C and t-SnO2–C can be deconvolved into four peaks. The main C 1s peak at binding energy of 284.6 eV is attributed to adventitious carbon and sp2-hybridized carbon. The sharp peak located at 285.3 eV is ascribed to defect-containing sp2-hybridized carbon. Besides, there are two other weak peaks positioned at 286.1 and 288.0 eV, corresponding to hydroxyl carbon (C–O) and carboxyl carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), respectively, indicating the presence of carbon in to-SnO2–C and t-SnO2–C samples. No other obvious peaks, such as peaks corresponding to C–Sn bond, were detected, suggesting that the main form of carbon element here should be mostly coated on the surface of SnO2 nanoparticles, rather than in the form of doping, as reported in other works.45 The intimate contact between the carbon and SnO2 favors the charge separation, thus, carbon can act as a media to promote the charge carriers' separation in the to-SnO2–C sample.46
O), respectively, indicating the presence of carbon in to-SnO2–C and t-SnO2–C samples. No other obvious peaks, such as peaks corresponding to C–Sn bond, were detected, suggesting that the main form of carbon element here should be mostly coated on the surface of SnO2 nanoparticles, rather than in the form of doping, as reported in other works.45 The intimate contact between the carbon and SnO2 favors the charge separation, thus, carbon can act as a media to promote the charge carriers' separation in the to-SnO2–C sample.46
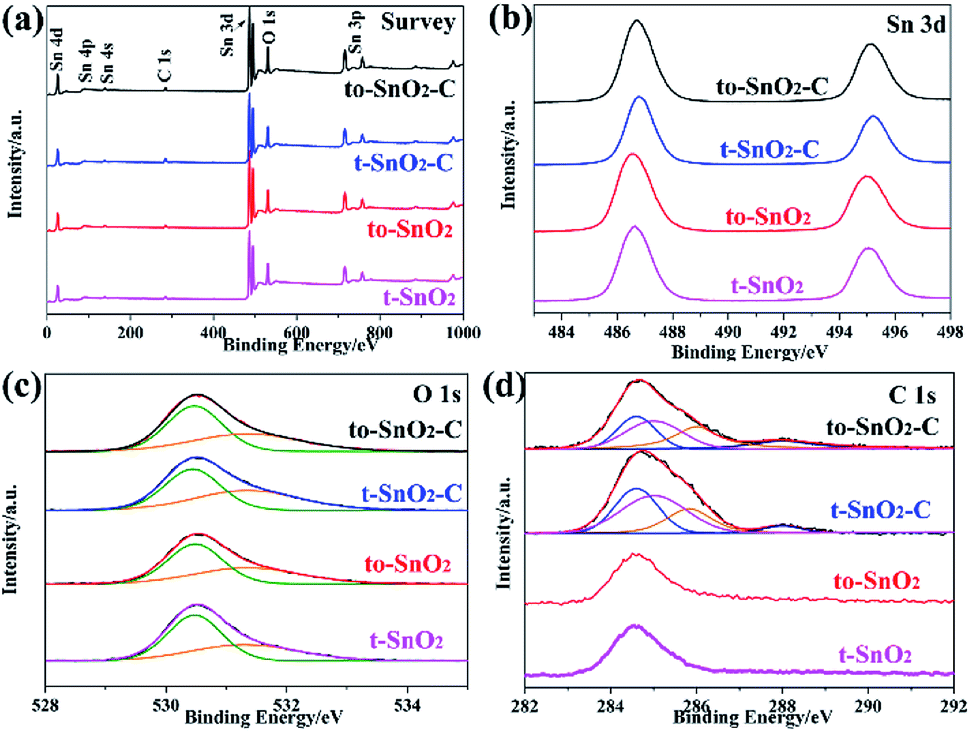 | ||
| Fig. 4 XPS of to-SnO2, t-SnO2, to-SnO2–C and t-SnO2–C samples. (a) Survey of the samples; (b) Sn 3d; (c) O 1s; (d) C 1s. | ||
3.2 Photocatalytic activity
The photocatalytic activities and stability of the as-prepared samples were evaluated by the photodegradation of the typical water pollutants, MO and phenol, under UV irradiation. Before visible-light irradiation, the establishment of adsorption/desorption equilibrium between the photocatalyst and the degrading pollutants is used to evaluate the adsorption ability of photocatalysts. As is shown in Table S1,† MO or phenol adsorption rate of as-prepared photocatalysts is slight, indicating that is not a critical factor to determine the different photocatalytic performance of as-prepared photocatalysts. Moreover, to exclude the influence of adsorption factor, the degradation curve in our work is reported as (C0 − C)/C0, where C is the pollutant's concentration after photocatalysis, and C0 is the pollutant's concentration after dark adsorption. During the degradation, the self-degradation of MO or phenol in the absence of a photocatalyst is negligible, indicating that the MO or phenol is stable under UV light irradiation. It is observed that to-SnO2 and t-SnO2 show limited photocatalytic activity i.e. 39% and 52%, respectively for MO degradation reaction under UV light irradiation (Fig. 5a). The mixed-phase to-SnO2 shows better degradation efficiency than tetragonal t-SnO2, indicating that the formation of phase junctions exhibits a significant influence on the photocatalytic activity of SnO2 samples. After carbon coating, the photocatalytic activities of t-SnO2–C and to-SnO2–C are enhanced. Remarkably, the MO is almost decomposed completely in the test of the to-SnO2–C after 60 min of light irradiation, which indicates carbon coating can notably improve the photocatalytic activity for degrading MO. Fig. 5b shows that to-SnO2–C also exhibits more efficiency than t-SnO2, to-SnO2, and t-SnO2–C in the degradation of phenol.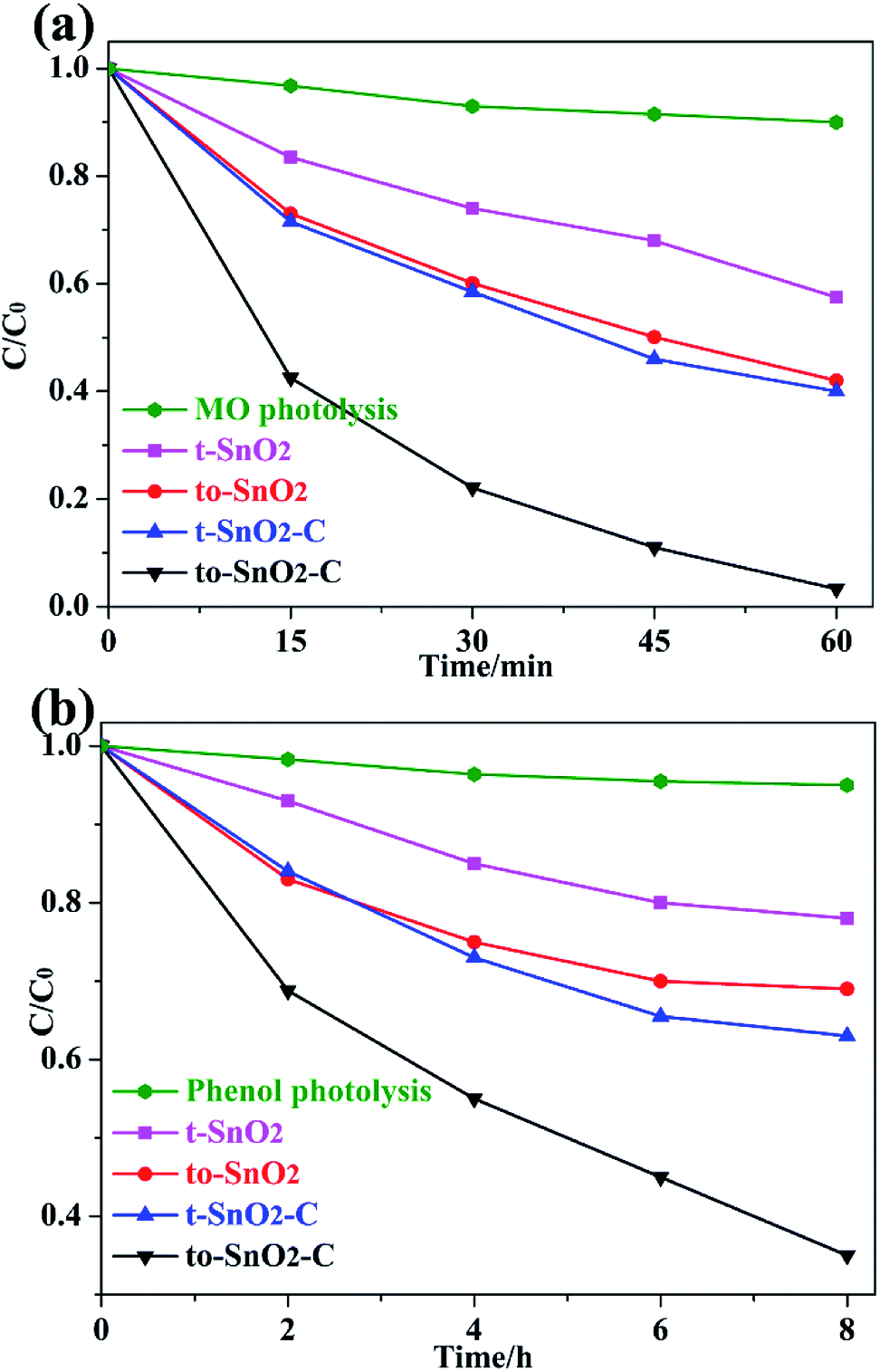 | ||
| Fig. 5 Photocatalytic degradation performance of MO (a) and phenol (b) under irradiation of 300 W Xe arc lamp. | ||
To gain a better understanding of the reaction kinetics, the experimental data are fitted by a pseudo-first-order model, and the corresponding apparent reaction constants (k) are summarized in Table 1. Taking the effects of surface area into consideration, we normalized the reaction rate constants of the samples by surface area. Based on the N2 adsorption–desorption isotherms, the BET surface area (ABET) of the samples were calculated, and are also summarized in Table 1. To-SnO2 and t-SnO2 have similarly BET surface area, which is in response to the TEM analysis and results. The normalized rate constant of to-SnO2 are higher than that of t-SnO2, indicating that the formation of phase junctions has an important effect on the enhancement of photocatalytic activity. Notably, to-SnO2–C shows the highest normalized rate constant, which is almost 3.0 and 3.4 times than those of to-SnO2 and t-SnO2–C, respectively, suggesting that surface area is not a major factor influencing the photocatalytic efficiency of the SnO2 samples.
More importantly, Fig. 6 shows that very high stability was obtained over to-SnO2–C. After three repeat cycles, to-SnO2–C retained relatively consistent activity without apparent deactivation of its photocatalytic effects, indicating its high stability and great promise in practical applications.
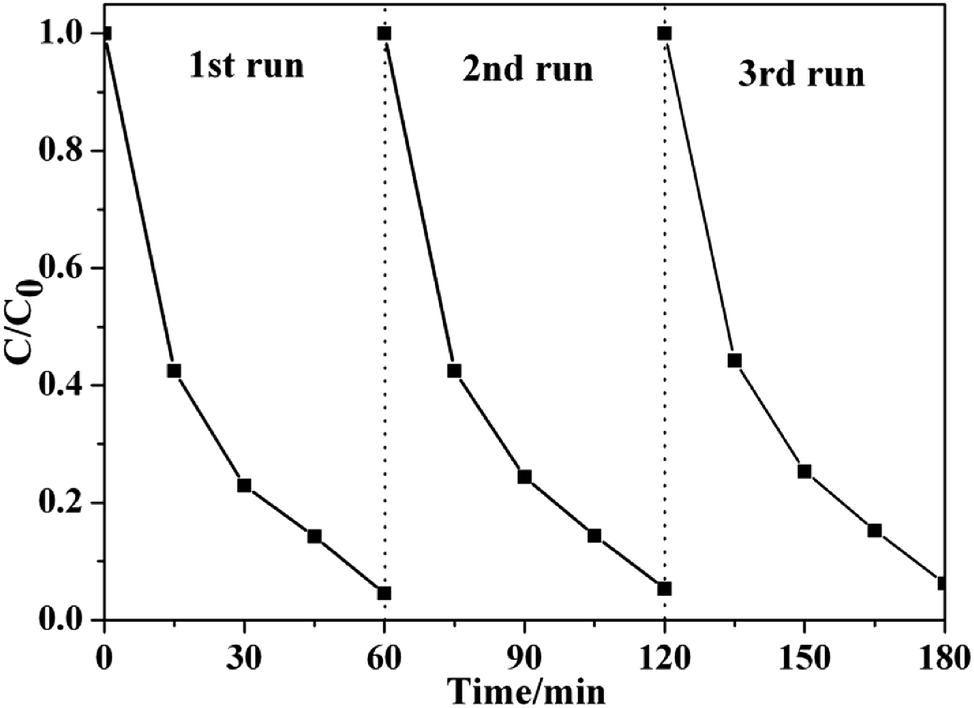 | ||
| Fig. 6 Cyclic photocatalytic degradation experiments of MO by to-SnO2–C sample. | ||
3.3 Photocatalytic mechanism
The optical properties of the as-prepared samples were investigated by UV-Vis diffuse reflectance spectra (DRS). As shown in Fig. 7, the absorption edges of pure t-SnO2 is located at ∼360 nm. In comparison with t-SnO2, the absorption edge of to-SnO2 (∼350 nm) shows a slight blue shift, which can be ascribed to the larger energy band gap of orthorhombic SnO2. After carbon coating, no obvious migration of the absorption edge of to-SnO2–C (t-SnO2–C) can be observed in comparison with to-SnO2 (t-SnO2), possibly ruling out the lattice doping of carbon into SnO2. This is due to the fact that the amount of carbon is low and, also, the carbon are mostly coated on the surface of SnO2 nanoparticles, which is consistent with previous experimental reports.45,47 The bandgap energies (Eg) calculated on the basis of the corresponding absorption edges are ∼3.45 eV (t-SnO2 and, t-SnO2–C) and ∼3.54 eV (to-SnO2 and to-SnO2–C). Moreover, to-SnO2–C and t-SnO2–C samples exhibit absorption tails at the entire visible range, which is due to the background absorption of the carbon. Several literatures have reported the rise of the visible-light absorption tails in carbon-covered titania systems.45,47 Obviously, to-SnO2–C samples have better optical absorption than other samples. Photocatalytic tests of all samples were conducted under 320 nm UV light irradiation by 300 W Xe lamp with a band-pass filter (Fig. S1†). The results show that to-SnO2–C still exhibits superior photocatalytic performance than other samples in the condition of excluding visible light. Therefore, it can be concluded that the excellent photocatalytic activity of to-SnO2–C is not mainly attribute to narrower band gap or visible light absorption tail.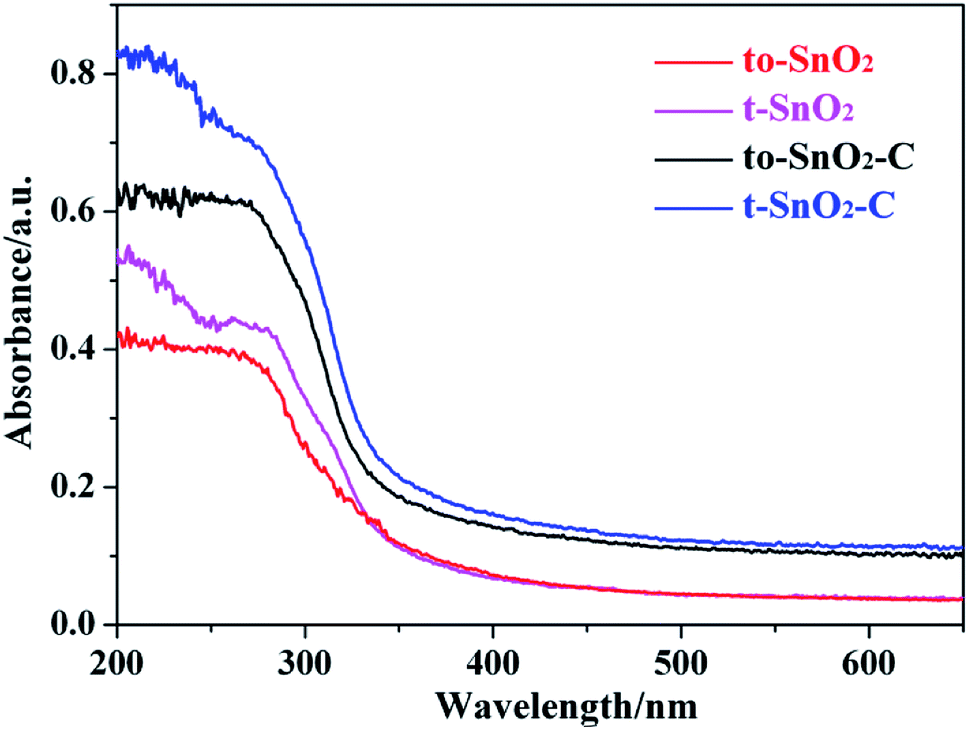 | ||
| Fig. 7 UV-Vis diffuse reflectance spectra of to-SnO2, t-SnO2, to-SnO2–C and t-SnO2–C samples. | ||
Photoelectrochemical measurements were performed to understand the electron transfer and separation of the as-prepared samples. The transient photocurrent responses of to-SnO2, t-SnO2, to-SnO2–C and t-SnO2–C are shown in Fig. 8a. Obviously, mixed-phase to-SnO2 exhibits a higher photocurrent than pure tetragonal t-SnO2, indicating that the separation efficiency of the photogenerated electron–hole pairs is more effective in to-SnO2. In comparison with to-SnO2 sample, to-SnO2–C exhibits a drastically increased larger current density. This demonstrates that the transfer and separation of the photogenerated electron–hole pairs in to-SnO2–C is significantly improved by the introduction of carbon into the interparticle space in to-SnO2. The result is consistent with the proposed mechanism. EIS spectra are obtained to confirm the transfer and migration processes of the photogenerated electron–hole pairs. In general, the radius of the arc reflects the charge transfer resistance and photogenerated electron–hole separation efficiency of samples. A smaller arc radius indicates higher efficiency in charge transfer and separation. It can be clearly seen from Fig. 8b that to-SnO2 possesses a smaller arc compared to t-SnO2 sample. We know that the formation of phase junctions in mixed-phase photocatalysts is one of the most efficient methods to diminish the electron–hole recombination. This is not surprising efficient charge separation and transfer across the phase junctions in the mixed-phase to-SnO2. It should be noted that to-SnO2–C shows an obvious smaller arc than to-SnO2, indicating that the introduction of carbon can further facilitate the transfer and separation of photogenerated electron–hole pairs in the phase junction interface. The result is in consistent with photocurrent responses results. Therefore, the results of the transient photocurrent responses and EIS analysis demonstrate that the construction of phase junctions is beneficial for improving the transfer and separation efficiency of photogenerated electron–hole pairs, and the introduction of carbon as a charge transfer mediator can further enhanced the charge separation efficiency.
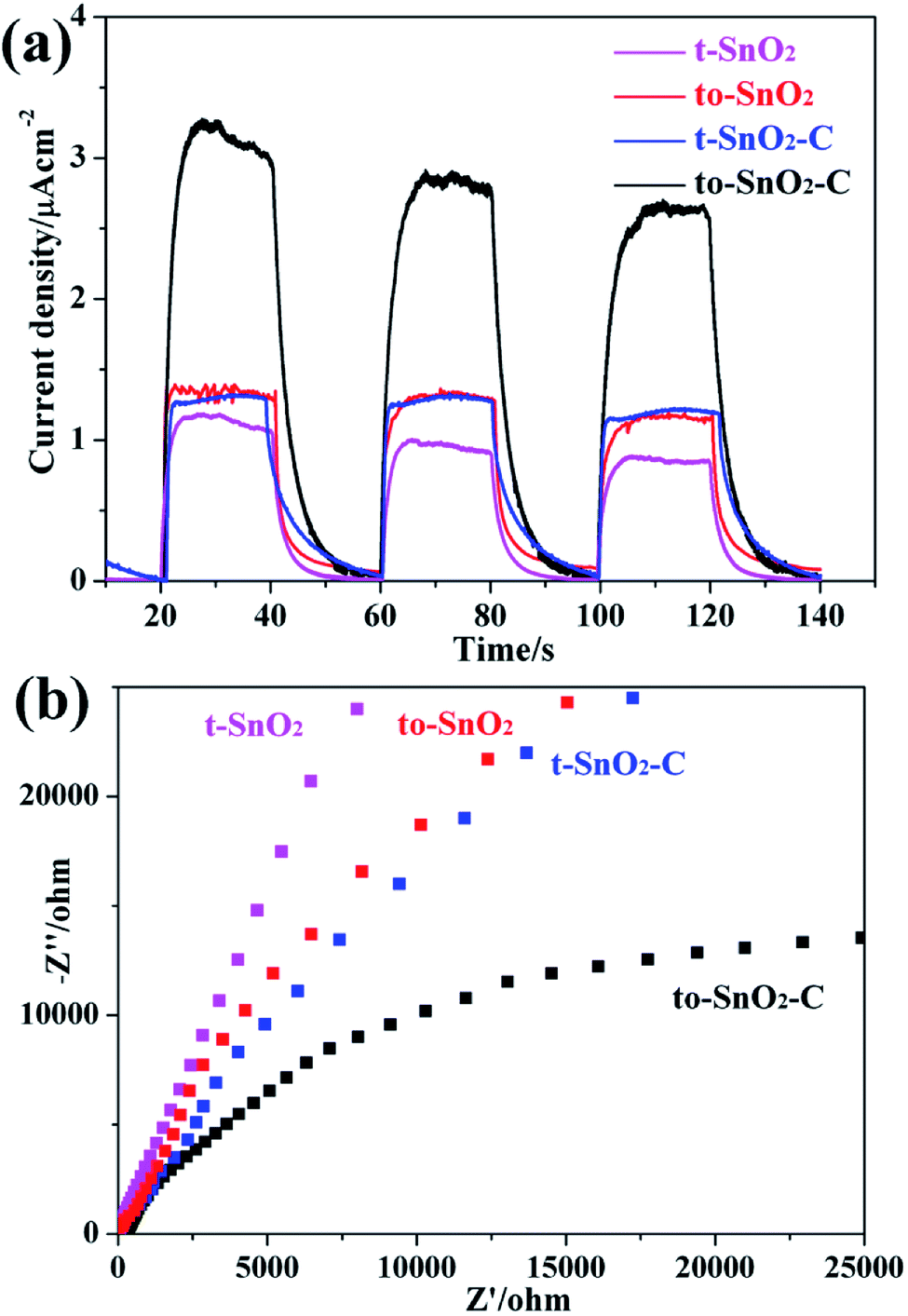 | ||
| Fig. 8 Photocurrent response under visible light irradiation (a) and electrochemical impedance spectroscopy plots (b) for as-prepared samples. | ||
To understand the photocatalytic mechanism, a series of control experiments with the addition of different scavengers for quenching various radicals was performed. Three scavengers, triethanolamine (TEOA), tert-butylalcohol (TBA) and benzoquinone (BQ), are used to detect functions of holes (h+), hydroxyl radicals (˙OH), and superoxide radicals (˙O2−), respectively (Fig. 9). The results show that all the three species play role in the degradation but the major species is ˙O2− because the degradation efficiency falls from 98.0% to 16.2% by the addition of BQ. It is also observed that the efficiency of MO removal is obviously inhibited by TEOA or TBA, implying that both ˙OH and h+ played important roles in the MO degradation by to-SnO2–C. Based on this, it can be deduced that the degradation of MO over to-SnO2–C sample is dominated by ˙O2− radicals, and involves the ˙OH and h+ oxidation process to some extent.
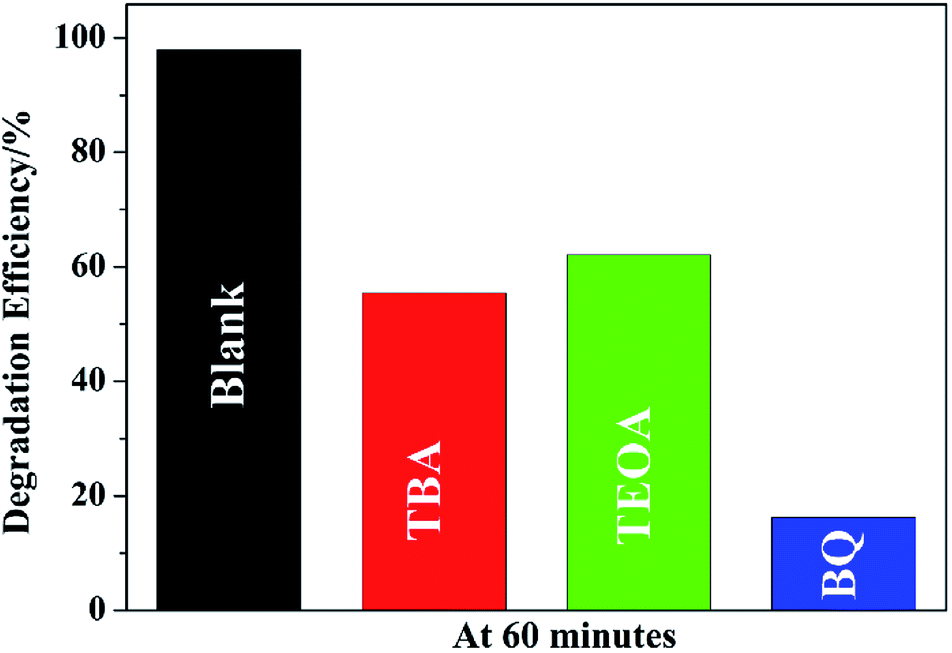 | ||
| Fig. 9 Trapping of active species during the photodegradation of MO over to-SnO2–C under irradiation of 300 W Xe arc lamp for 60 min. | ||
Because pure orthorhombic SnO2 (o-SnO2) is hard to be prepared by using this chemical precipitation method, we perform a first-principles study to investigate the band structure of the tetragonal SnO2 and orthorhombic SnO2, and further reveal that the energy band structure matches between the two phase SnO2 (Fig. 10). Based on the calculated results, the band gap of orthorhombic SnO2 is smaller than that of tetragonal SnO2 (Fig. 10a), which is consistent with the reported results.48 Fig. 10b displays the band structure of the band alignment in to-SnO2 sample. As the schematic diagram suggests, the phase junction could be built between the tetragonal SnO2 and orthorhombic SnO2. Under UV light irradiation, both tetragonal SnO2 and orthorhombic SnO2 would be excited, producing electrons and holes. The photogenerated electrons in CB of tetragonal SnO2 tend to transfer to that of orthorhombic SnO2 because the energy of CB of tetragonal SnO2 is much higher than that of orthorhombic SnO2. Besides, the holes excited from orthorhombic SnO2 flow into the tetragonal SnO2. In this way, photogenerated electron–hole pairs could be separated effectively due to the formation of phase junctions. Notably, although the band schemes of the tetragonal SnO2 and orthorhombic SnO2 benefit the charge separation, the loose connection between the mixed-phase SnO2 nanoparticles obstructs the charge transfer. As can be shown in Fig. 11, in to-SnO2, partial photogenerated electrons and holes cannot transfer across phase junctions in time and combine again, which is disadvantageous for photocatalytic activity improvement. When to-SnO2 was coated by carbon (conducting material), the recombination of photogenerated electron–hole pairs was decreased, due to the fact that the carbon could act as electronic transfer media and further facilitate the charge transfer between tetragonal SnO2 and orthorhombic SnO2 nanoparticles. Thus, more charge carriers could participate in the photodegradation process in to-SnO2–C sample, which are propitious to the enhancement of photocatalytic performance. Hence, the formation of phase junctions and the promotion of interfacial transfer and separation efficiency of photogenerated carriers induced by carbon coating were responsible for the enhancement of photocatalytic activity of to-SnO2–C.
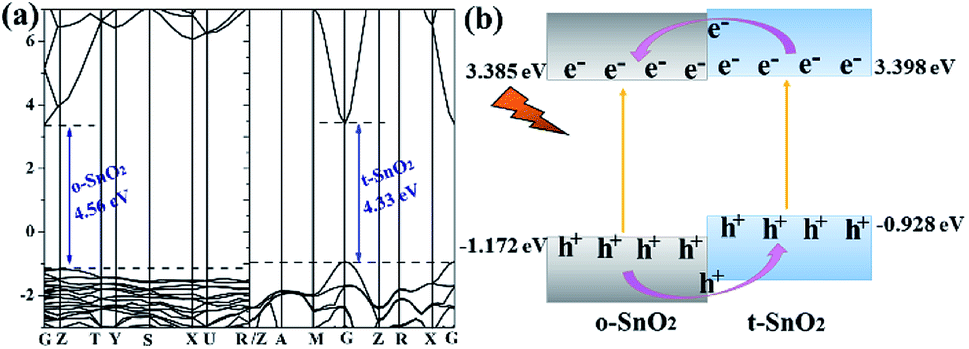 | ||
| Fig. 10 (a) Band structures of o-SnO2 and t-SnO2; (b) band alignments in mixed-phase to-SnO2. | ||
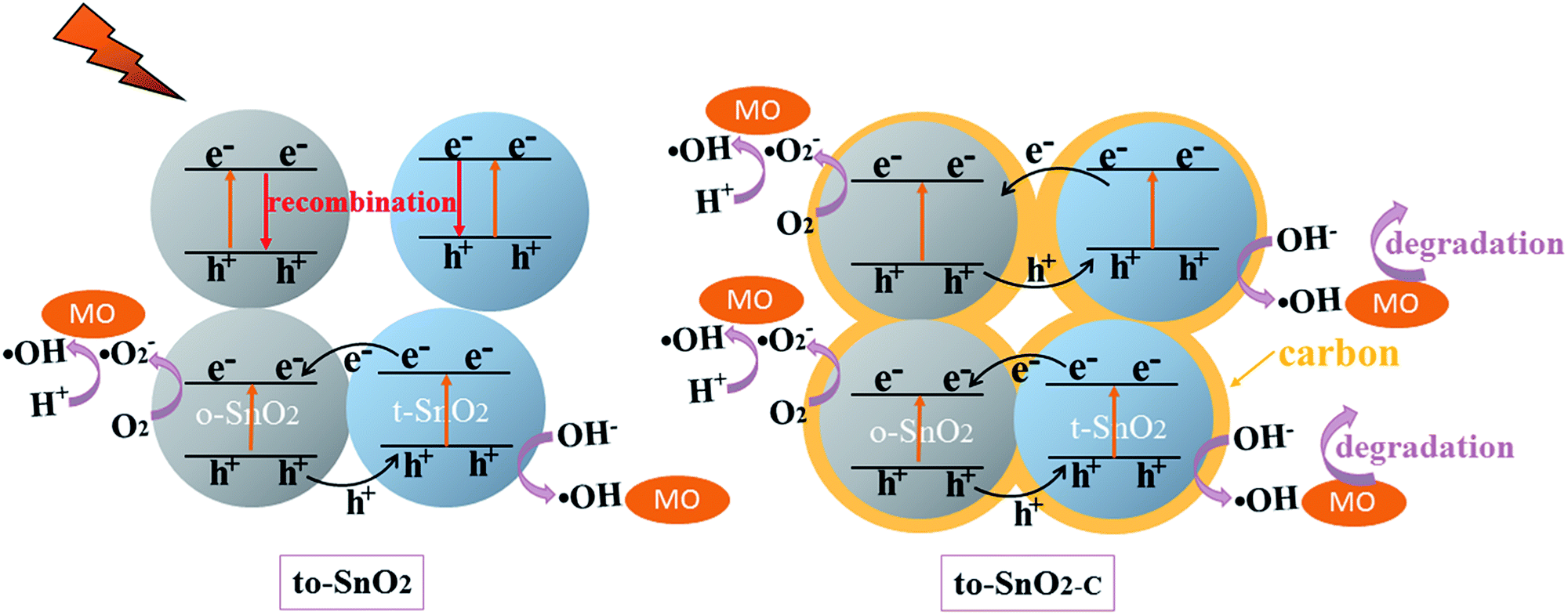 | ||
| Fig. 11 Schematic illustration of the proposed mechanism for photogenerated charge carrier transfer in the to-SnO2 and to-SnO2–C under UV light irradiation. | ||
4. Conclusions
In this study, we have developed a novel carbon-coated mixed-phase SnO2 nanorods composed of a large number of nanoparticles with a chemical precipitation method, followed by a facile hydrothermal process. The as-prepared carbon-coated mixed-phase SnO2 nanorods show a higher photocatalytic activities for degrading MO and phenol under UV light irradiation, compared with the pure tetragonal SnO2. According to the results of photoelectrochemical measurements and first-principles calculations, the photogenerated electron–hole pairs can be separated effectively due to the formation of phase junctions, and the introduction of carbon can further facilitate the charge transfer process between tetragonal and orthorhombic SnO2 nanoparticles. The synergistic effect of the band alignment between tetragonal and orthorhombic SnO2 and the conductivity of the carbon media in the interparticle space is believed to contribute to the improvement of photocatalytic activity of to-SnO2–C photocatalyst. This study provides a facile and convenient method to synthesize mixed-phase semiconductor photocatalysts with promising performance for the potential application in environmental protection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Speical Funds for Postdoctoral of Shangdong Province (Grant No. 201703010) and the Natural Science Foundation of China (Grant No. 21771119).References
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef PubMed.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef PubMed.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef.
- C. S. Turchi and D. F. Ollis, J. Catal., 1990, 122, 178–192 CrossRef.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef.
- Q. Li, X. Zhao, J. Yang, C.-J. Jia, Z. Jin and W. Fan, Nanoscale, 2015, 7, 18971–18983 RSC.
- C. Yu, G. Li, S. Kumar, K. Yang and R. Jin, Adv. Mater., 2014, 26, 892–898 CrossRef PubMed.
- Y. Yang, C. Sun, L. Wang, Z. Liu, G. Liu, X. Ma and H. M. Cheng, Adv. Energy Mater., 2014, 4, 1400057 CrossRef.
- J. Tian, Y. Sang, Z. Zhao, W. Zhou, D. Wang, X. Kang, H. Liu, J. Wang, S. Chen and H. Cai, Small, 2013, 9, 3864–3872 CrossRef PubMed.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef PubMed.
- X. Wang, Q. Xu, M. Li, S. Shen, X. Wang, Y. Wang, Z. Feng, J. Shi, H. Han and C. Li, Angew. Chem., Int. Ed., 2012, 51, 13089–13092 CrossRef PubMed.
- S. Kalikeri and V. S. Kodialbail, Environ. Sci. Pollut. Res., 2018, 1–13 Search PubMed.
- M. Zhang, L. Dang, C. Tian, S. Zhao and Q. Lu, Superlattices Microstruct., 2017, 111, 423–437 CrossRef.
- F.-T. Li, Y. Liu, Z.-M. Sun, Y. Zhao, R.-H. Liu, L.-J. Chen and D.-S. Zhao, Catal. Sci. Technol., 2012, 2, 1455–1462 RSC.
- B. M. Rajbongshi and S. Samdarshi, Appl. Catal., B, 2014, 144, 435–441 CrossRef.
- K. Vinodgopal and P. V. Kamat, Environ. Sci. Technol., 1995, 29, 841–845 CrossRef PubMed.
- W. W. Wang, Y. J. Zhu and L. X. Yang, Adv. Funct. Mater., 2007, 17, 59–64 CrossRef.
- Z. Liu, D. D. Sun, P. Guo and J. O. Leckie, Nano Lett., 2007, 7, 1081–1085 CrossRef PubMed.
- A. Kolmakov, D. Klenov, Y. Lilach, S. Stemmer and M. Moskovits, Nano Lett., 2005, 5, 667–673 CrossRef PubMed.
- K. Suito, N. Kawai and Y. Masuda, Mater. Res. Bull., 1975, 10, 677–680 CrossRef.
- Z. Dai, J. Gole, J. Stout and Z. Wang, J. Phys. Chem. B, 2002, 106, 1274–1279 CrossRef.
- N. Zhao, G. Wang, Y. Huang, B. Wang, B. Yao and Y. Wu, Chem. Mater., 2008, 20, 2612–2614 CrossRef.
- D. Hu, B. Han, S. Deng, Z. Feng, Y. Wang, J. Popovic, M. Nuskol, Y. Wang and I. Djerdj, J. Phys. Chem. C, 2014, 118, 9832–9840 CrossRef.
- P. Hartmann, D.-K. Lee, B. M. Smarsly and J. Janek, ACS Nano, 2010, 4, 3147–3154 CrossRef PubMed.
- N. Lakshminarasimhan, E. Bae and W. Choi, J. Mater. Chem. C, 2007, 111, 15244–15250 Search PubMed.
- Y. Yu, C. Y. Jimmy, J.-G. Yu, Y.-C. Kwok, Y.-K. Che, J.-C. Zhao, L. Ding, W.-K. Ge and P.-K. Wong, Appl. Catal., A, 2005, 289, 186–196 CrossRef.
- J. Yu, T. Ma and S. Liu, Phys. Chem. Chem. Phys., 2011, 13, 3491–3501 RSC.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef PubMed.
- N. Li, G. Liu, C. Zhen, F. Li, L. Zhang and H. M. Cheng, Adv. Funct. Mater., 2011, 21, 1717–1722 CrossRef.
- K. Zhou, Y. Zhu, X. Yang, X. Jiang and C. Li, New J. Chem., 2011, 35, 353–359 RSC.
- L. W. Zhang, H. B. Fu and Y. F. Zhu, Adv. Funct. Mater., 2008, 18, 2180–2189 CrossRef.
- L. Zhang, H. Cheng, R. Zong and Y. Zhu, J. Phys. Chem. B, 2009, 113, 2368–2374 Search PubMed.
- P. Zhang, C. Shao, Z. Zhang, M. Zhang, J. Mu, Z. Guo, Y. Sun and Y. Liu, J. Mater. Chem., 2011, 21, 17746–17753 RSC.
- K. R. Reddy, V. G. Gomes and M. Hassan, Mater. Res. Express, 2014, 1, 015012 CrossRef.
- Z. Li, B. Gao, G. Z. Chen, R. Mokaya, S. Sotiropoulos and G. L. Puma, Appl. Catal., B, 2011, 110, 50–57 CrossRef.
- B. Jiang, Y. Tang, Y. Qu, J.-Q. Wang, Y. Xie, C. Tian, W. Zhou and H. Fu, Nanoscale, 2015, 7, 5035–5045 RSC.
- Y. Guo, H. Wang, C. He, L. Qiu and X. Cao, Langmuir, 2009, 25, 4678–4684 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. Probert, K. Refson and M. C. Payne, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 Search PubMed.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1227 CrossRef.
- H. J. Song, L. C. Zhang, C. L. He, Y. Qu, Y. F. Tian and Y. Lv, J. Mater. Chem., 2011, 21, 5972–5977 RSC.
- X. Liu, Y. X. Li, D. Y. Deng, N. Chen, X. X. Xing and Y. D. Wang, CrystEngComm, 2016, 18, 1964–1975 RSC.
- J. G. Yu, T. T. Ma and S. W. Liu, Phys. Chem. Chem. Phys., 2011, 13, 3491–3501 RSC.
- G. An, W. Ma, Z. Sun, Z. Liu, B. Han, S. Miao, Z. Miao and K. Ding, Carbon, 2007, 45, 1795–1801 CrossRef.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef.
- J. Bae, J. Park, H. Kim and J.-S. Park, Appl. Catal., B, 2015, 7, 12074–12079 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03794a |
| This journal is © The Royal Society of Chemistry 2018 |