
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Synthesis of non-toxic, biocompatible, and colloidal stable silver nanoparticle using egg-white protein as capping and reducing agents for sustainable antibacterial application†
Kalaiyarasan Thiyagarajan a,
Vijay K. Bharti*a,
Shruti Tyagib,
Pankaj K. Tyagib,
Anami Ahujab,
Krishna Kumara,
Tilak Raja and
Bhuvnesh Kumarc
a,
Vijay K. Bharti*a,
Shruti Tyagib,
Pankaj K. Tyagib,
Anami Ahujab,
Krishna Kumara,
Tilak Raja and
Bhuvnesh Kumarc
aDefence Institute of High Altitude Research (DIHAR), Defence Research and Development Organization (DRDO), C/o 56 APO, Leh-Ladakh-194101, India. E-mail: vijaykbharti@rediffmail.com; Fax: +0172-2638900; Tel: +0172-2642900
bDepartment of Biotechnology, Meerut Institute of Engineering & Technology, Meerut, Uttar Pradesh-250005, India
cDefence Institute of Physiology and Allied Sciences, Defence Research and Development Organization (DRDO), Timarpur, Delhi-110 054, India
First published on 25th June 2018
Abstract
For nearly a decade, silver nanoparticles (AgNPs) have been the most prevalent commercial nanomaterials products widely used in different biomedical applications due to their broad-spectrum antimicrobial activity. However, their poor long-term stability in different environments, namely, pH, ionic strength, and temperature, and cytotoxicity toward mammalian cells has restricted their more extensive applications. Hence, there is urgent need to develop highly biocompatible, non-toxic, and stable silver nanoparticles for wide-ranging environments and applications. In the present study, a simple, sustainable, cost-effective and green method has been developed to prepare highly stable aqueous colloidal silver nanoparticles (AgNPs-EW) using the ovalbumin, ovotransferrin, and ovomucoid of egg-white as reducing and capping agents accomplished under the irradiation of direct sunlight. Then, we evaluated the effects of freezing–drying (lyophilization) and freeze–thaw cycles on the stability of AgNPs-EW in aqueous solution under visual inspection, transmission electron microscopy, and absorbance spectroscopy. In addition, we studied the antibacterial activity against Salmonella typhimurium and Escherichia coli, carried out biocompatibility studies on chicken blood, and tested acute, chronic toxicity in Drosophila melanogaster. The results suggest that AgNPs-EW did not aggregate upon freeze-thawing and lyophilization, thus exhibiting remarkable stability. The antibacterial activity results showed that the AgNPs-EW had the highest antibacterial activity, and the minimum inhibitory concentration (MIC) of AgNPs-EW for E. Coli and S. typhimurium were 4 and 6 μg ml−1, respectively. The biocompatibility study revealed that the AgNPs-EW did not induce any hemolytic effect or structural damage to the cell membranes of chicken erythrocytes up to a concentration of 12 μg ml−1. Similarly, no acute and chronic toxicity was observed on melanization, fecundity, hatchability, viability, and the duration of development in the 1st generation of Drosophila melanogaster at the concentration range of 10 mg L−1 to 100 mg L−1 of AgNPs-EW, and all the flies completed their full developmental cycle. Therefore, the present study successfully demonstrated the green and sustainable preparation of non-toxic AgNPs-EW having good biocompatibility, enhanced colloidal stability, and antibacterial activity. Hence, the synthesized AgNPs-EW could be used for the development of an antimicrobial formulation for controlling microbial infection.
1. Introduction
Infectious diseases are mostly responsible for one-third of mortality in human beings and represent a constant threat to human healthcare. Moreover, several antibiotics are being used indiscreetly in controlling these disease-causing microbes, which could lead to the emergence of resistant pathogenic bacterial strains toward one or several conventional antibiotics (penicillin, streptomycin, tetracycline, vancomycin, etc.).1–5 Recently, the World Health Organization (WHO) released a list of drug-resistant bacteria that possess the greatest threat to human health, namely Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacteriaceae, Enterococcus faecium, Staphylococcus aureus, Helicobacter pylori, Campylobacter spp. Salmonellae, Neisseria gonorrhoeae, Streptococcus pneumonia, Haemophilus influenza, and Shigella spp.6 This report clearly suggested that given the seriousness of drug-resistant bacteria, the development of new alternatives antibiotics/antimicrobials is urgently needed. The use of alternative antimicrobial agents could reduce the application of conventional antibiotics and the further development of resistant to these antibiotics. Recent advances in nanotechnology have attracted increasing attention owing to their unique electrical, optical, and physicochemical properties, which effectively influence their biomedical application, including antimicrobial prescriptions.7 Several kinds of nanomaterials have been developed and evaluated for their antibacterial properties, such as silver nanoparticles (AgNPs), graphene oxide nanosheets, carbon nanotubes (CNTs), gold nanoparticles (AuNPs), zinc oxide nanoparticles (ZnO), titanium dioxide nanoparticles (TiO2), chitosan, and cationic peptides.4,8 Among these, silver nanoparticles (AgNPs) have received much attention in medical research, owing to their effective antimicrobial activity toward a broad spectrum of bacteria, including viruses and fungi, and therefore they hold great promise for a wide range of biomedical and environmental applications. Recently, Zhou et al. and Panacek et al. reported a high antimicrobial and bactericidal activity of AgNPs against Gram-positive and Gram-negative bacteria.9,10 In addition, Shahverdi et al. and Fayaz et al. demonstrated the combined use of AgNPs with antibiotics (e.g., penicillin G, amoxicillin, erythromycin, and vancomycin), and the results revealed enhanced and synergistic antimicrobial effects against E. coli and S. aureus.11,12 These studies indicated that AgNPs have potentially high-quality antibacterial activity; however, in general, they have poor stability and are prone to be aggregated in the aqueous phase within three days of storage, leading to a loss of antibacterial activity, restricting their practical applications as nanomaterials.11,12 Furthermore, Tomankova et al. reported that the cytotoxicity of Ag+ and AgNPs to mammalian cells significantly narrowed the application of AgNPs as a sustainable antimicrobial agent.13 So, there is an urgent need to develop a methodology for the synthesis of non-toxic silver nanoparticles. In fact, metal nanoparticles can be synthesized by using various methods, such as chemical, electrochemical, chemical vapor deposition, photochemical, and molecular beam epitaxy.14–16 Noteworthy, the experimental factors, the interaction of metal precursor ions with reducing agents, and the interface of capping agents with metal nanoparticles significantly influence the size, shape, stability, and physicochemical properties of the metal nanoparticles. Therefore, the selection of an appropriate capping agent is often a prerequisite for stabilizing the nanoparticles, their size, shape, and surface charges, and their interactions with the surrounding solvent. Apart from these, capping agents also play a vital role in cytotoxicity and the biocompatibility of nanomaterials.17,18 To address these issues, various biomolecules, viruses, microorganisms, and plant extracts have been employed in the synthesis and reducing aggregation of AgNPs during long-term storage. In comparison with all the capping agents, the use of proteins and peptides in the synthesis of nanomaterials is drawing more interest due to their unique tertiary and quaternary structures, which offer interesting opportunities for the template growth and organization of nanoparticles.19,20 Huang et al.19 reported novel BSA-based Ag microspheres exhibiting good biocompatibility, while similarly Mauro et al.21 also demonstrated that the glyco-polypeptide architecture has excellent antimicrobial properties against S. aureus and P. aeruginosa. Moreover, to improve the stability and antibacterial efficacy of AgNPs, researchers tend to do immobilization and surface modification. However, these processes require complex and tedious preparation procedures with a high cost. Thus, great efforts are still necessary to develop novel kinds of facile methods for the synthesis of stable materials with good biocompatibility and non-toxic antibacterial properties to meet the increasing demand for broad-spectrum antibacterial applications and to reduce dependence on conventional antibiotics, so as to overcome the antimicrobial resistance to those antibiotics. In this context, we present a new method for the synthesis of ovalbumin, ovotransferrin, and ovomucoid of egg-white-capped silver nanoparticles (AgNPs-EW), featuring high colloidal stability during freezing–drying (lyophilization) and freeze–thaw cycles without using any immobilization, surface modification or cryoprotectants. The AgNPs-EW were also evaluated for antibacterial activity toward Salmonella typhimurium and Escherichia coli with good biocompatibility and non-toxicity to Drosophila. As far we know, this is the first report extensively studied about which protein among all the egg-white proteins plays the key role in the reduction of silver ions (Ag+) to silver nanoparticles (Ag0), and in the antibacterial activity and toxicity studies on Drosophila for egg-white-capped silver nanoparticles.2. Experimental section
2.1 Reagents
The chemicals used for this study were purchased from Sigma-Aldrich (USA) and HiMedia Laboratories (Mumbai, India), and unless otherwise mentioned, all the reagents and solvents were of analytical grade. Throughout all the experiments, Millipore water (18.2 Ω) was used (Millipore Applied Systems, USA), and before the experiments, all the glassware used was cleaned in aqua regia (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 HNO3, HCl).
:3 HNO3, HCl).
2.2 Preparation of silver nanoparticles (AgNPs-EW)
The synthesis of AgNPs-EW was carried out by a green chemistry approach, whereby in brief, chicken eggs were collected from the local market and the egg-white was collected by breaking the eggs in a sterile Petri dish (Borosil Pvt Ltd, Gujarat, India). Thereafter, 2 ml of egg-white extract was fully dissolved in 92 ml of Milli-Q water with a strong magnetic stirrer (120 rpm) for 30 min. The cloudy white solution was filtered through filter paper (Whatman no 1, USA) and a clear solution was obtained. The resulting solution was mixed with 6 ml of silver nitrate solution (1000 ppm) drop by drop with strong stirring (120 rpm) and the reaction mixture pH was set at 7.2 and thereafter kept under sunlight and at a temperature of 38 °C to 40 °C. The reaction mixture showed an instant color change from white to pale yellow with the sharp surface plasmon resonance (SPR: λmax 430 nm) band within 2 min of sunlight exposer, which was an indication of the nanoparticles formation, which was further confirmed by UV-Vis spectroscopy examination (the detailed experimental setup is presented in Table 1). In order to optimize the maximum the nanoparticles formation, aliquots of the reaction mixture was collected at different time intervals (i.e., 5, 10, 15, 30, 45, 60, and 95 min) and examined by UV-Vis spectroscopy. The synthesized nanoparticles were dialyzed to remove the un-bounded proteins from solution through dialysis tubing (flat width 25 mm, MWCO 12000 Da, Sigma, USA), the water was changed two times during 24 h dialysis period. The dialyzed silver nanoparticles were examined under UV-Vis spectroscopy before and after dialysis (ESI Fig. 1†). The dialyzed nanoparticles were dried at 50 °C in a hot-air oven and the dried powder was used for characterization of the nanomaterials, and in the in vivo acute and chronic toxicity evaluation, biocompatibility studies, and antibacterial activity.
| Exp. setup | Egg-white (ml) | Water (ml) | Silver nitrate (1000 ppm) solution (ml) | Homogenisation speed | Reaction mixture exposed to sunlight | Observations |
|---|---|---|---|---|---|---|
| E1 | 1 | 90 | 9 | 120 rpm for 30 minutes | Reaction mixture exposed to sunlight | (1) The colour of resulting solution was turned from white to pale yellow after 30 minutes of exposure. |
| (2) The solution was precipitated/aggregated after 45 minutes of exposure to sunlight. | ||||||
| E2 | 1 | 92 | 7 | No colour change was seen. | ||
| E3 | 2 | 90 | 8 | (1) The colour of resulting solution was turned from white to pale yellow after 10 minutes of exposure. At every 30 minutes, 2 ml of sample was collected and examined under UV-Vis Spectroscopy. However, no significant change in absorbance (OD) was observed beyond 60 minutes of exposure. | ||
| (2) The synthesised nanoparticles were stored at room temperature (away from direct light) and precipitation/aggregation was observed within 24 hours of storage. | ||||||
| E4 | 2 | 92 | 6 | (1) The colour of resulting solution was turned from white to pale yellow after 10 minutes of exposure. At every 30 minutes, 2 ml of sample was collected and examined under UV-Vis Spectroscopy. However, no significant change in absorbance (OD) was observed beyond 60 minutes of exposure. | ||
| (2) The synthesised nanoparticles were stored at room temperature (away from direct light) and no precipitation/aggregation was observed till 30 days of storage. | ||||||
| E5 | 2 | 88 | 10 | (1) The colour of resulting solution was turned from white to pale yellow after 12 minutes of exposure. | ||
| (2) The solution was precipitated/aggregated after 2 days of storage. |
2.3 Characterization of the nanoparticles
The AgNPs-EW were primarily characterized by UV-Vis absorption spectroscopy (Shimadzu UV spectrophotometer UV-2600). Fourier transform infrared spectroscopy (FTIR) was employed to characterize the interaction between the silver nanoparticles and protein present in the egg-white, and FTIR spectra were recorded in the range from 4000 to 400 cm−1 using a Spectrum RX-IFTIR (Perkin Elmer Massachusetts, USA). Lyophilized egg-white served as a control. Dynamic light scattering (DLS) analysis was carried out using a zeta sizer (Malvern Instrument Co. Ltd., UK) for evaluation of the AgNPs-EW stability. This analysis was performed using duplicate measurements and the size distribution on correlator software version Zetasizer 7.11. All the samples were diluted 100× with double distilled water before analysis.Zeta potential measurements were carried out using a zeta sizer (Malvern Instrument Co. Ltd., UK) in a disposable cell at 25 °C. DLS and zeta potential measurements were performed to study the stability of AgNPs-EW. These measurements were carried out between 1 to 3 h after the preparation of the AgNPs-EW. The surface morphology and particles sizes of the AgNPs-EW were observed using high-resolution transmission electron microscopy (Hitachi H-7500).
2.4 Effect of freezing–thawing on the AgNPs-EW
To perform this study, a conical tube containing the AgNPs-EW (10 ml) solution was stored at −20 °C in a refrigerator (Samsung Pvt Ltd). The sample was frozen for 1 h and then thawed at room temperature (26 ± 1.5 °C); thereafter freeze–thaw cycles were repeated three times. The thawed solutions were analyzed using a UV-Vis spectrophotometer to know the change in the SPR band (wavelength peak shift due to agglomeration), while DLS, and surface charge analysis (zeta potential) were also performed to know the freezing–thawing effect on the AgNPs-EW stability.2.5 Effect of lyophilization (freeze-drying) on the AgNPs-EW
The effects of lyophilization (freeze-drying) on the stability of the AgNPs-EW were assessed without using any cryoprotectants. Glass vials containing 2.0 ml of AgNPs-EW solutions were frozen, keeping at −80 °C for 30 min and then immediately shifted into a freeze-dried device (FreeZone Plus, US) with a vacuum (0.050 mBar) for 48 h, with the condenser surface temperature maintained at −80 °C throughout the experiments.000 rpm for 15 min and AgNPs-EW pellets were collected, and the pellets washed with Milli-Q water and dried at 50 °C in a hot-air oven. From this, 5 μg of dried AgNPs-EW was dissolved in 2.0 ml of 100 mM of phosphate, bicarbonate, and Tris–HCl, buffers at pH 6.5 and 7.4 and incubated for 24 h at 25 °C ± 1.5 °C. After the incubation, the solutions were examined by UV-Vis spectroscopy and the control experiment was performed by dissolving AgNPs-EW pellets in Milli-Q water while keeping the other parameters the same.2.6 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis
Egg-white has various proteins, therefore SDS-PAGE analysis was performed according to Desert et al.22 with modification in order to understand the type of proteins that play the vital role in the reduction of silver ions (Ag+) into silver nanoparticles (Ag0) and protein bonding on the surface of AgNPs-EW. In brief, 25 ml of freshly synthesized AgNPs-EW was centrifuged at 12000 rpm for 20 min to remove the unbound proteins. Thereafter, the supernatant was collected in a 15 ml sterile tube to estimate the unbound proteins. The pellets (AgNPs-EW) were washed three times with Milli-Q water and resuspended in 5 ml Milli-Q water. Then, the bound proteins with AgNPs-EW were digested by adding 10 mM dithiothreitol, and incubated at 95 °C for 6 min in a running water bath. Thereafter, this solution was cooled down to room temperature, and then centrifuged at 12000 rpm for 15 min and the supernatants were collected in a 1.5 ml sterile tube and stored at 4 °C until future analysis. SDS-PAGE was performed using 4% acrylamide in stacking gels and 12% acrylamide in separating gels. Then, in a 200 μl vial, 50 μl of sample buffer and 50 μl of protein samples (i.e., an egg-white protein used for nanoparticles synthesis, unbound proteins, and bounded proteins digested with dithiothreitol) were added and incubated at 95 °C for 5 min in a running water bath, and after that, the vials were brought to room temperature. Then, the samples were loaded over the gel. The migration buffer comprised 25 mM Tris, 192 mM glycine, and 0.1% SDS, and electrophoresis (Bio-Rad Electrophoresis Equipment) was carried out at 75 V in the stacking gel and 120 V in the separating gel. After that, the gel was stained with Coomassie brilliant blue, and further a photograph was captured using a gel documentation system (Bio-Rad-Gel Doc™ XR+) and the thickness (quantified) of the protein bands were analyzed using ImageJ software tools.
2.7 Evaluation of antibacterial activity
The bacterial strains used in this study, namely Salmonella typhimurium (MTCC-3224) and Escherichia coli (MTCC-062), were procured from Microbial Type Culture Collection (MTCC), Institute of Microbial Technology, Chandigarh, India. The brain–heart infusion (BHI) (5% peptone, and 1.5% agar) and nutrient agar (5% peptone, and 1.5% agar) slants were used as the culture media. In brief, freshly grown bacterial strains (Salmonella typhimurium, Escherichia coli) species from the brain–heart infusion agar slants and nutrient agar slants were inoculated in a brain–heart infusion broth and nutrient broth and then incubated at 37 °C at 120 rpm. Thereafter, the cells were harvested from the early stationary phase, i.e., 12 to 16 h, by centrifugation at 6000 rpm for 10 min and washed three times with phosphate buffer (10 mM, pH 7.2). The cell pellets were finally resuspended in the same phosphate buffer to obtain a cell density of 0.1 OD (optical density), approximately 105 CFU ml−1.To quantify the antibacterial activity of the AgNPs-EW, serial dilution of the bacterial suspension was done, and from this, 100 μl of bacterial suspension was speared on the brain–heart infusion and nutrient agar plates. Then, all the plates were incubated at 37 °C for 24 h. Photographs of Salmonella typhimurium and E. coli grown on the plates were obtained and a number of colonies were manually counted and the antibacterial activity of AgNPs-EW was calculated. The lowermost concentration was taken for bacteriological progress on the brain–heart infusion plates and nutrient agar plates selected for the MBC (minimum bactericidal concentrations).
2.8 Antibacterial mechanisms
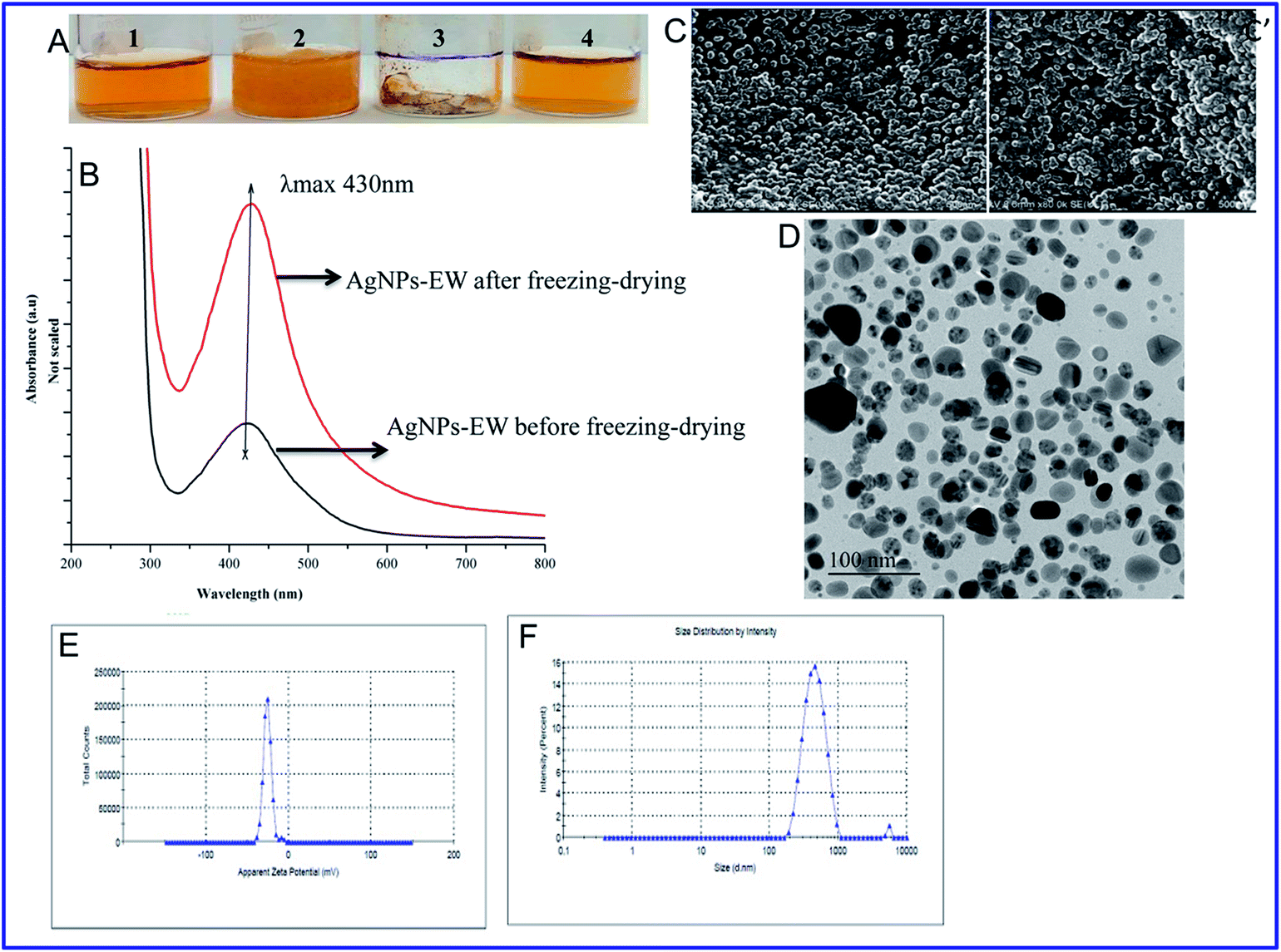
000 rpm for 10 min, and the supernatants were measured for absorbance at 260 and 280 nm using a plate reader (Spectramax i3x, Molecular Devices, CA, USA).| % Cell death = A570 of treated cells/A570 of control cells × 100 |
2.9 Evaluation of the hemocompatibility
The hemolysis assay was employed with chicken whole blood as described by Kutwin27 to know the biocompatibility of the synthesized AgNPs-EW. In brief, heparinized blood collected from the slaughterhouse was incubated at 37 °C at 120 rpm with 1, 2, 4, 6, 8, 10, and 12 μg ml−1 AgNPs-EW. Thereafter, the blood was centrifuged at 1200 rpm for 10 min and the supernatant collected. The supernatant was analyzed for the concentration of hemoglobin at 540 nm using a plate reader (Spectramax i3x, Molecular Devices, CA, USA). Samples incubated with Triton X-100 were considered as a positive control (100% hemolysis) and the sample with PBS buffer as a negative control (0% hemolysis). The percentage of hemolysis was calculated using the formula as per the Shiny et al. method.28 The structural damage of erythrocytes (RBCs) was also analyzed by using upright light microscopes at 40× (Leica Microsystems, Germany).2.10 Evaluation of the acute and chronic toxicity effects of AgNPs-EW on Drosophila melanogaster
The in vivo toxicity of AgNPs-EW was investigated using the Drosophila melanogaster animal model. Fecundity, hatchability, viability, larva to adult development, and pigmentation were studied upon exposure to AgNPs-EW at 10, 20, 30, 40, 50, and 100 mg L−1 concentrations through maize-agar food. In brief, before starting, the newly enclosed virgin females and males were fed separately on different concentrations of AgNPs-EW-mixed food medium. The AgNPs-EW-fed flies were mated in the early morning between 6 and 8 am at 21 °C for two days prior to monitoring their egg-laying capacity. 10 ml of proper AgNPs-EW-mixed cooled media with and without AgNPs-EW food media was poured into the culture vial (25 × 100 mm, Tarson, Kolkata) and 10 no. of mated female were subjected to culture and allowed to lay eggs. Five replicates were rum simultaneously for all the treatments and control. The number of eggs laid by the mated females within 24 h were counted and divided by 10 fly in order to determine the rate of fecundity per fly.For hatchability, the 50 no. of eggs (10 eggs/replicates) were transferred into culture vials containing the food medium of normal feed and mixed with a different concentrations of AgNPs-EW. The hatchability was determined by counting the rupture chorion, i.e., 1st instar larvae after 24 h at 25 °C. Development of the 1st instar larvae into adult enclosing was determined by scoring the number of adult's flies that emerged from the culture vials. All the replicates used in the hatchability were considered for scoring the newly emerged flies, with this phenomenon called the viability. Moreover, after emergence of the 1st instar larvae, the larvae follow the developmental phases of the life cycle and finally eclosion in the adults. The viability was monitored by calculating the percentage of larvae that could reach the pre-pupal, pupal, and adult stages. The duration of eggs development into adults (hatchability + viability) was also monitored in hours for calculating the time it took the flies to develop from eggs to adults.
For analyzing the change in pigmentation of D. melanogaster, the females were observed for the most prominent abdominal tergites (A2–A6), indicated by a “stripe” of dark melanins at the posterior edge of the segment as well as a peak of this dark color along the dorsal midline. These variations in pigmentation were compared among different concentrations of AgNPs-EW containing food media with the normal food medium (control).
2.11 Examination of the antibacterial activity of AgNPs-EW after freezing–drying
In order to investigate the impact of freezing–drying on the antibacterial activity of AgNPs-EW, the freshly grown bacterial strains of Salmonella typhimurium and Escherichia coli in brain–heart infusion agar slants and nutrient agar slants were inoculated in brain–heart infusion broth and nutrient broth and incubated at 37 °C at 120 rpm. Thereafter, cells were harvested from the early stationary phase, i.e., 12 to 16 h, by centrifugation at 6000 rpm for 10 min and washed three times with phosphate buffer (10 mM, pH 7.2). The cell pellets were finally resuspended in the same phosphate buffer to obtain the cell density of 0.1 OD (optical density), approximately 105 CFU ml−1. The antibacterial activity was studied as per the protocol described earlier.3. Results and discussion
3.1 Rapid biosynthesis of AgNPs-EW
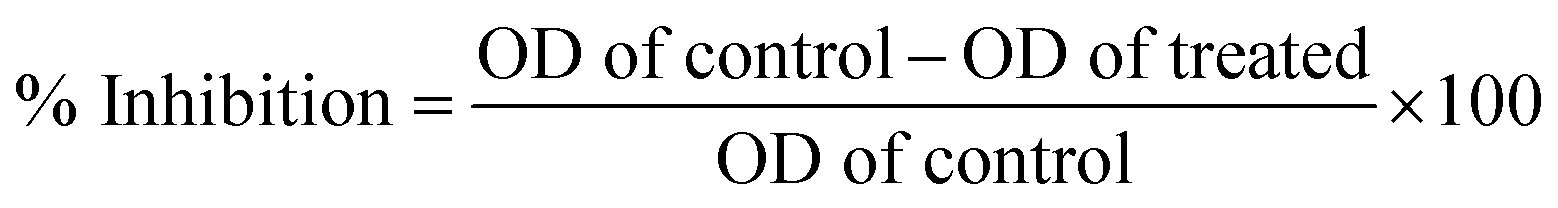
At present, the green and sustainable synthesis of metal nanomaterials using proteins, biopolymers, and polysaccharides has drawn great interest because it has more advantages compared with chemically prepared nanomaterials.20 Here, we demonstrated a simple photochemical (i.e., direct sunlight) reduction method to prepare silver nanoparticles (AgNPs-EW) using egg-white as reductant and capping agents. The mixing of silver nitrate and egg-white solution resulted in a gradual color change of the solution from white to pale yellow and deep yellow, which indicated the formation of AgNPs-EW (Fig. 1A). Further, the formation was confirmed by UV-Vis spectroscopy. In order to acquire high-quality stable nanoparticles, we performed different experimental condition setups (Table 1) and observed that E1, E2, E3, E5 agglomerated within 24 h to 48 h after synthesis of the nanoparticles, whereas E4 was highly stable and the maximum absorption at 430 nm, which was due to the surface plasmon resonance (SPR) of AgNPs (Fig. 1B).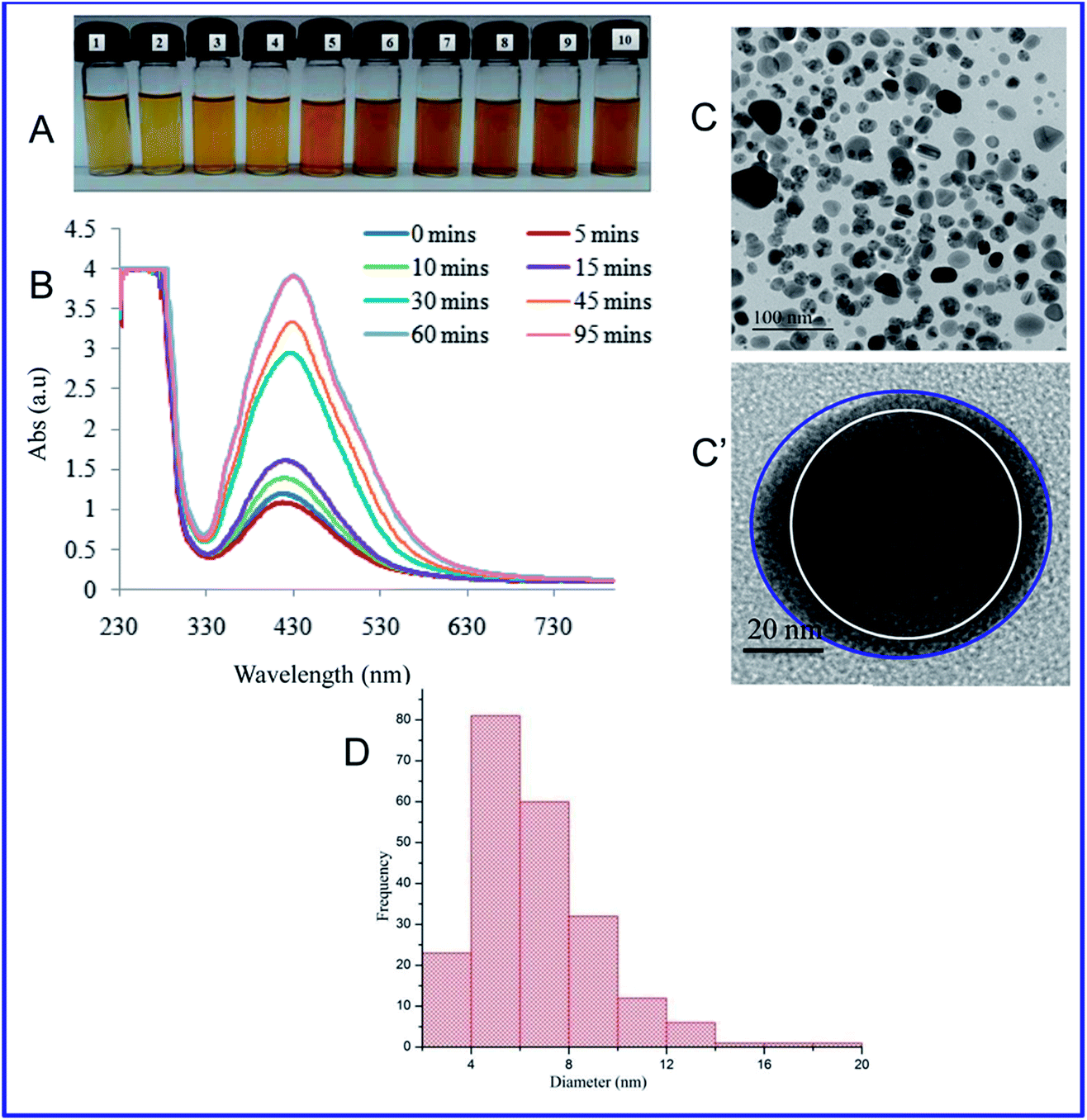 | ||
| Fig. 1 (A) Vials showing showing nanoparticles formation at different time intervals (1: 5 min, 2: 10 min, 3: 15 min, 4: 30 min, 5: 45 min, 6: 60 min, 7: 90 min, 8: 120 min, 9: 160 min, 10: 190 min). (B) UV-Vis spectra for nanoparticles formation (reaction kinetics); (C and C′) TEM image of AgNPs-EW, rounded signify protein bounded on the surface of nanoparticles. (D) Size distribution of AgNPs-EW estimated by image J software. | ||
Then, we performed the reaction kinetics (duration of reaction) of E4, which indicated a continuous increase in the absorbance with regard to the time, an indication of nanoparticles formation, reaching its maximum (Abs) at 60 min of sunlight exposure. However, with an extension of duration up to 95 min, there was no significant change in the spectra (λmax) as compared to 60 min. These findings indicated that AgNPs-EW formation was accomplished within 60 min, which indicates the completion of the reaction. Furthermore, nanoparticles formation was investigated in the absence of sunlight at room temperature (26 ± 1.5 °C) and no change in color of the reaction mixture was observed until 12 h (ESI, Fig. 2†). The reaction mixture only started to change from white to pale yellow after 12 h and gradually turned into yellowish brown. This is an indication of nanoparticles formation, which was further confirmed by using UV-Vis spectrophotometry at different time intervals, namely 12, 24, 36, 72, 84 h, which revealed changes in the spectra (λmax) from 0.35 Abs to 0.96 Abs at 72 h (ESI, Fig. 2†). Though, an extension of duration up to 84 h indicated that most of the AgNPs-EW formation was completed within 72 h, and therefore no significant change in the spectra (λmax) after 72 h was observed. These results suggested that sunlight-mediated nanoparticles synthesis is faster and a greater quantity of nanoparticles formation occurs, as indicated by the high absorbance (a.u.), than in the process without sunlight-exposed mediated synthesis (Fig. 1A and B). This is because the sunlight provides a higher electron transfer reaction than at room temperature and enhances the reduction of Ag+ to Ag0 in a short time interval.29,30
Moreover, we compared our results with previously reported results,31,32 and observed that the method explained in the present study produced nanoparticles in a higher quantity, i.e., ∼3 Abs units, and in a shorter duration, i.e., 60 min. Therefore, sunlight-mediated egg-white-capped nanoparticles synthesis is faster and able to produce a high quantity of nanoparticles.
Then, the size and morphology of the AgNPs-EW were measured by HRTEM analysis, which clearly revealed the capping of egg-white protein over silver nanoparticles (Fig. 1C and C′). The TEM picture estimated by image J software tools revealed ∼2 to ∼20 nm sized nanoparticles (Fig. 1D). Zeta potential analysis of the synthesized AgNPs-EW showed a ξ −24.2 ± 3.39 negative surface charge, which was due to protein coating on the surface of AgNPs-EW. The bioconjugation of egg-white protein with AgNPs-EW was further confirmed by FTIR spectroscopy. The FTIR spectra of the control egg-white protein and AgNPs-EW are shown in Fig. 2. The IR spectrum of control egg-white protein showed sharp absorption bands at 3280, 3295 for C–H, and 2993 and 3001 cm−1 for O–H stretching vibrations of the proteins. The result indicated the presence of characteristic peaks of carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (1646, 1631, 1587 cm−1) and amide-I (N–H) (1594 cm−1). Further, the FTIR spectrum of AgNPs-EW showed peaks at 3280, 3295, 3001, 1647, 1631, 1587, 1594 cm−1 due to the symmetric stretching vibration of surface-bound protein molecules. Therefore, these results clearly demonstrated that egg-white protein acted both as a reducing and capping agent in AgNPs-EW formation.
O) (1646, 1631, 1587 cm−1) and amide-I (N–H) (1594 cm−1). Further, the FTIR spectrum of AgNPs-EW showed peaks at 3280, 3295, 3001, 1647, 1631, 1587, 1594 cm−1 due to the symmetric stretching vibration of surface-bound protein molecules. Therefore, these results clearly demonstrated that egg-white protein acted both as a reducing and capping agent in AgNPs-EW formation.
 | ||
| Fig. 2 FTIR spectra showing egg-white in red colour and AgNPs-EW in black colour spectral lines. | ||
3.2 Evaluation of stability under freeze–thaw cycles
The nanoparticles were exposed to various changes during freezing–thawing and drying, such as “solute exclusion” phenomena and the formation of liquid-rich phases within the ice mass with a high concentration of nanoparticles, which resulted in changes in the pH, ionic strength, and the aggregation of nanoparticles. The stability of nanoparticles is an important criterion during pharmaceutical formulations, which generally changes during to freeze-thawing and freeze-drying (lyophilization process).33–36 Hence, these variations in chemical–physical properties limit their biomedical application. Therefore, the effect of freezing–thawing and freeze-drying was investigated regarding the stability of the synthesized AgNPs-EW.The synthesized AgNPs-EW on repeated freezing at −20 °C and subsequent thawing at room temperature indicated no significant change in color of the solution or in plasmon resonance λmax (ESI, Fig. 3A and B†). These observations indicated the stability of the nanoparticles after the freezing–thawing cycle, while the dynamic light scattering and zeta potential measurements were further evidence that no aggregation took place during the freeze-thawing cycle (ESI, Fig. 3C and D†).
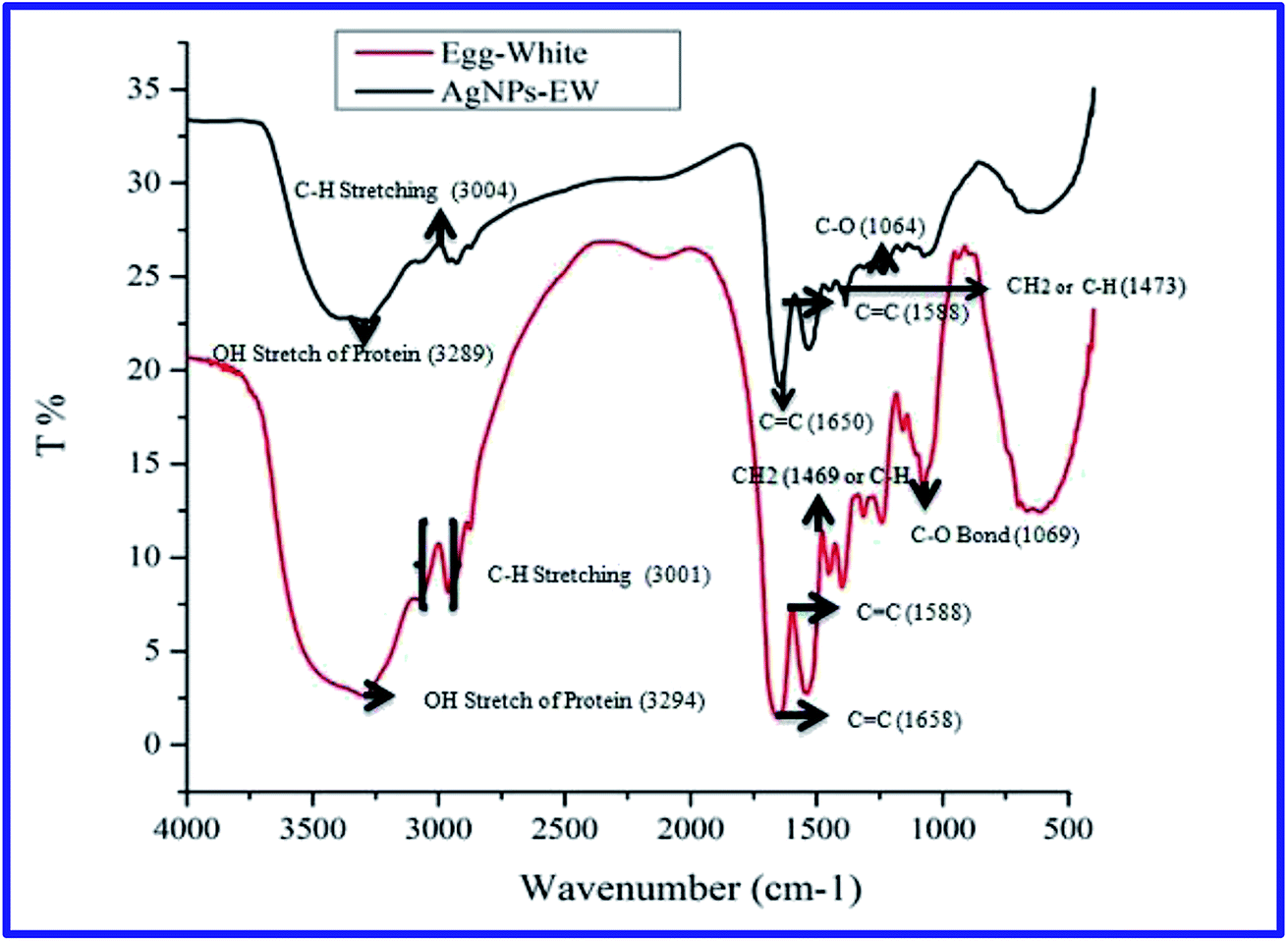
Furthermore, we analyzed the stability of AgNPs-EW during freeze-drying (lyophilization) without using any cryoprotectants. The lyophilized nanoparticles were fully dissolved in Milli-Q water and no color change was observed by visual observation (Fig. 3A and B). This was further analyzed through UV-Vis spectroscopy, dynamic light scattering, and zeta potential measurements, while the morphology changes were investigated using FESEM and TEM. The FESEM and TEM images offered that the AgNPs-EW were well-separated from each other and had an insignificance change in the morphology (Fig. 3C, C′ and D–F), which also revealed that the nanoparticles were stable during freeze-drying without any aggregation.
 | ||
| Fig. 3 (A) Photographs of nanoparticles containing vials: 1: prior to lyophilization, 2: during ice phase 3: after lyophilization, 4: reconstituted AgNPs-EW; (B) UV-Vis spectra of reconstituted AgNPs-EW solution after freeze-drying (UV-Vis spectrum for AgNPs-EW prior to lyophilization is plotted as a reference spectrum); (C and C′) FE-SEM image of AgNPs-EW before and after freezing–thawing; (D) TEM image of AgNPs-EW after freeze-drying (E and F) zeta potential and DLS analysis for reconstituted AgNPs-EW. | ||
Cryoprotectants are usually added at various concentrations into colloidal solutions to prevent the aggregation of nanoparticles during freezing–thawing and freezing–drying. However, the presence of cryoprotectants influences the size, charge, morphology, and adsorption surface of nanoparticles, which limits their biomedical application.37–39 Hence, the present study indicated that the egg-white-mediated green synthesis of silver nanoparticles had good stability during freezing–thawing and freezing–drying without using any cryoprotectants. On the basis of the obtained results, during freezing–thawing/freezing–drying, the conjugation between silver ions and egg-white protein plays a crucial role in the stability of the nanoparticles. The egg-white contains different proteins and other biomolecules, which might have acted as natural cryoprotectants.40 Egg-white is a cocktail of proteins, mainly ovalbumin (54%), ovotransferrin (12%), ovomucoid (11%), ovomucin (3.5%), and lysozyme (3.5%).41 Therefore, we hypothesized that during synthesis of the nanoparticles, ovalbumin, ovotransferrin, ovomucoid bind on the surface of AgNPs-EW producing a resistance to freezing denaturation. These proteins are unique in structure containing carbohydrate or glycan chains, which makes them highly resistant to physicochemical changes during freezing–thawing and freezing–drying.42–47 In order to confirm this hypothesis, we digested bound proteins on the surface of AgNPs-EW and analyzed with SDS-PAGE.
Further, our findings suggest that, due to electric repulsions and sterical hinderance between the negatively charged proteins (ovalbumin, ovotransferrin, ovomucoid) at the surface of nanoparticles, electrosterical stabilization of the conjugates occurs since the bioconjugates remain stable during freezing–thawing and freezing–drying cycles without using any cryoprotectants. Hence, our studies bring new information on the understanding of nanoparticles stability during freezing–thawing and freezing–drying without using any cryoprotectants, immobilization, or surface modification of nanoparticles.
3.3 SDS-PAGE analysis
In order to analyse the bound and unbound protein on the surface of AgNPs-EW, the nanoparticles were digested and SDS-page was performed. The results are presented in Fig. 4, and we observed that the proteins were separated according to their size placed on 12% acrylamide gels. In total, three protein bands were unambiguously identified by comparison with standard proteins marker and the findings revealed the presence of three major proteins on the surface AgNPs-EW. These findings were compared with the molecular weight of the concerned proteins in the database, which revealed ovotransferrin (MW ∼76 kDa), ovomucoid (MW ∼44 kDa), and ovalbumin (MW-37 kDa).48–50 In comparison to two other major proteins of egg-white, i.e., ovalbumin and ovomucoid, we observed significant unbound proteins in the total percentage of egg-white (Fig. 4A). Moreover, semi-quantitative densitometry analysis of the bands indicated that ovotransferrin (i.e., 12% of total egg-white) was fully bound on the surface of AgNPs-EW (Fig. 4B). On the basis of the obtained results, ovalbumin and ovotransferrin were the highest percentage of proteins bound on the surface of AgNPs-EW, followed by ovomucoid (Fig. 4B). This could be due to total egg-white proteins consisting of 54% ovalbumin, 12%, ovotransferrin, and 11% ovomucoid.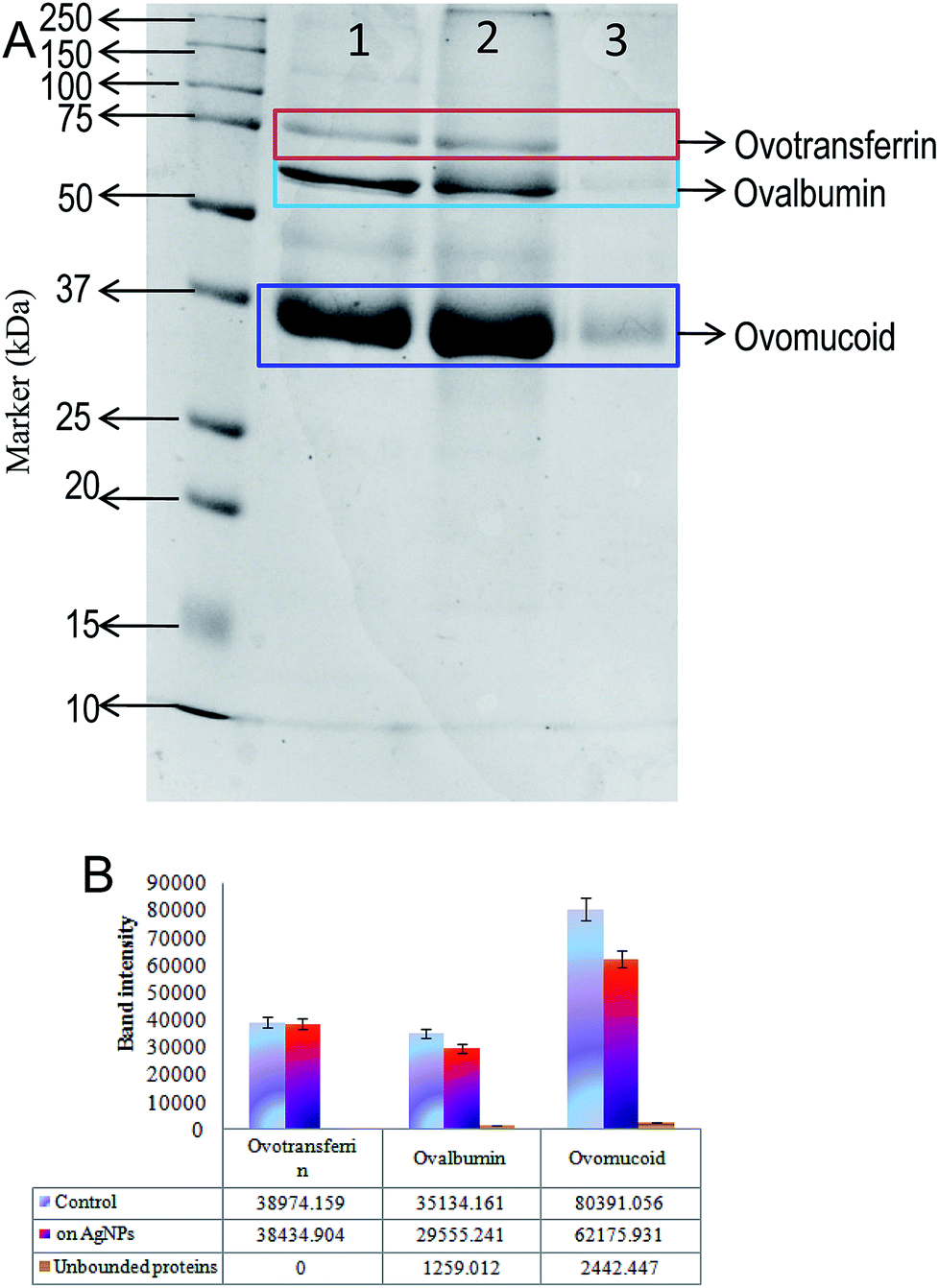 | ||
| Fig. 4 (A) SDS-PAGE gel of egg-white proteins showing bounding of various proteins on the surface of AgNPs-EW: Lane 1: egg-white proteins alone, Lane 2: bound egg-white protein present over synthesized AgNPs, and Lane 3: unbound egg-white proteins present in the reaction mixture. (B) Histogram representing the total band intensity of bound and unbound egg-white proteins over synthesized AgNPs-EW. | ||
Hence, the present study very clearly advocates that the three major proteins, i.e., ovomucoid, ovotransferrin, and ovalbumin, play vital roles in the reduction of silver ions (Ag+) to silver nanoparticles (Ag0) and anchor on the surface of the nanoparticle. Therefore, we concluded that these proteins are highly resistant to the chemical and physical conditions. This could be a key factor facilitating the colloidal stability of AgNPs-EW during freezing–thawing/freezing–drying without any surface modifications, cryoprotectants, or immobilization of nanoparticles.
3.4 Evaluation of AgNPs-EW stability in physiological buffers
ESI Fig. 4† presents the UV-Vis absorption spectra of AgNPs-EW stability in physiological buffers at different concentrations. The colloidal stability of AgNPs-EW was studied in different physiological buffer solutions at pH 6.5 and 7.4 in the concentration range from 25 to 200 mM by re-dispersing the 5 μg ml−1 of dried AgNPs-EW, while UV-Vis spectroscopy examination was carried after incubation for 24 h at room temperature. The results indicated no notable changes in the absorption spectra (i.e., λmax retained at 430 nm). Therefore, our results demonstrated that the egg-white-capped nanoparticles (AgNPs-EW) had excellent colloidal stability in physiological pH (i.e., 6.5 and 7.4). This high stability of AgNPs-EW could be due to the egg-white proteins used for the nanoparticles synthesis, which have high stability at different ionic strengths.47 Further, the egg-white proteins bound on the surface of nanoparticles gave a constant stabilizing potential to the hard protein corona.51 This result suggested that AgNPs-EW did not aggregate and retained their optical and physical properties at different ionic strengths.3.5 Antibacterial activity studies
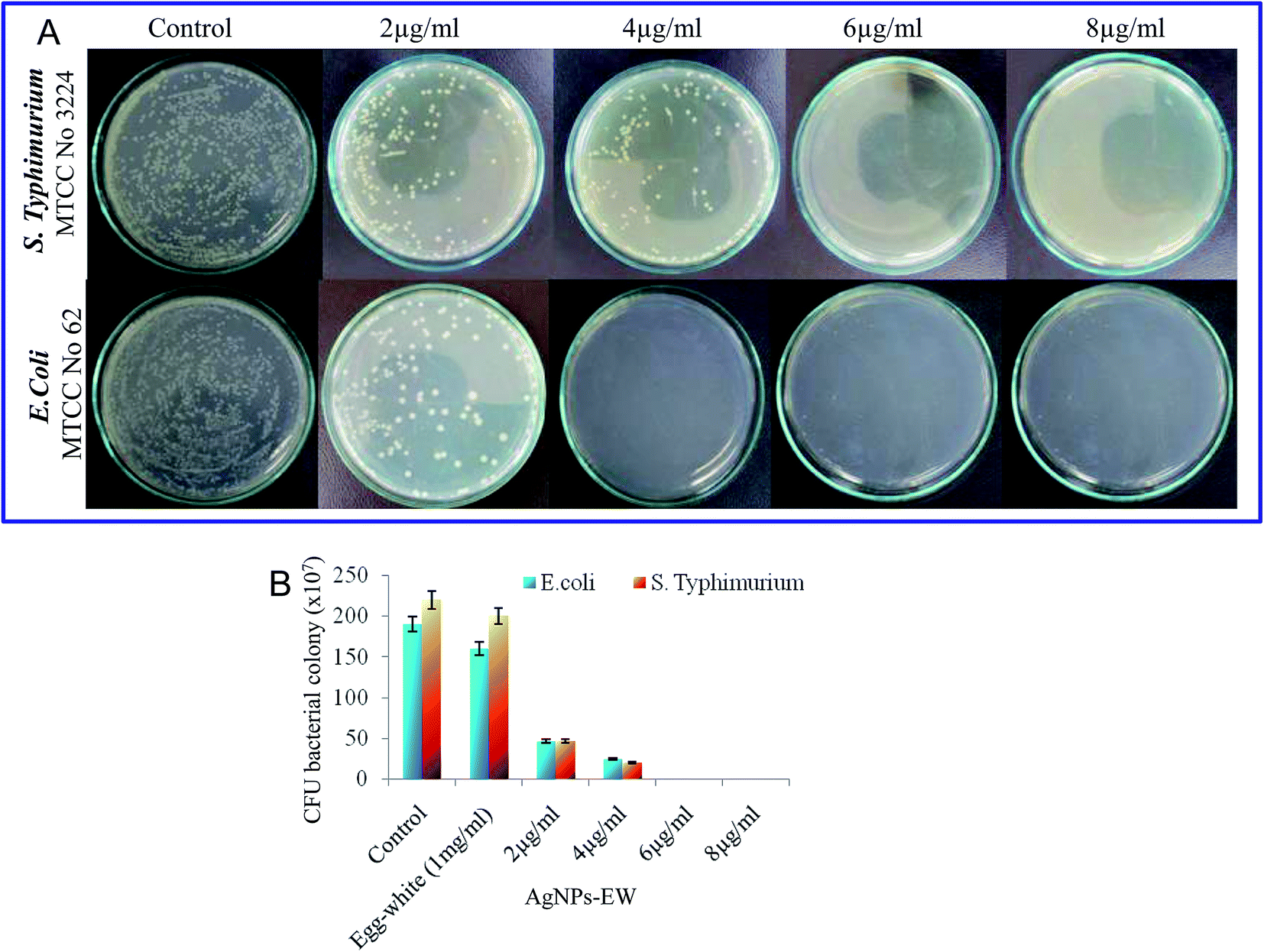 | ||
| Fig. 5 (A) Photograph showing the dose-dependent inhibition of CFU count (colony forming unit), antimicrobial efficiency of AgNPs-EW against Salmonella Typhimurium on BHI, E. coli on LB plates. (B) Quantitative evaluation of the antibacterial activity of AgNPs-EW by counting the colonies grown on agar and BHI plates. Values are expressed as the mean ± SD (n = 3). | ||
3.6 Antibacterial mechanisms
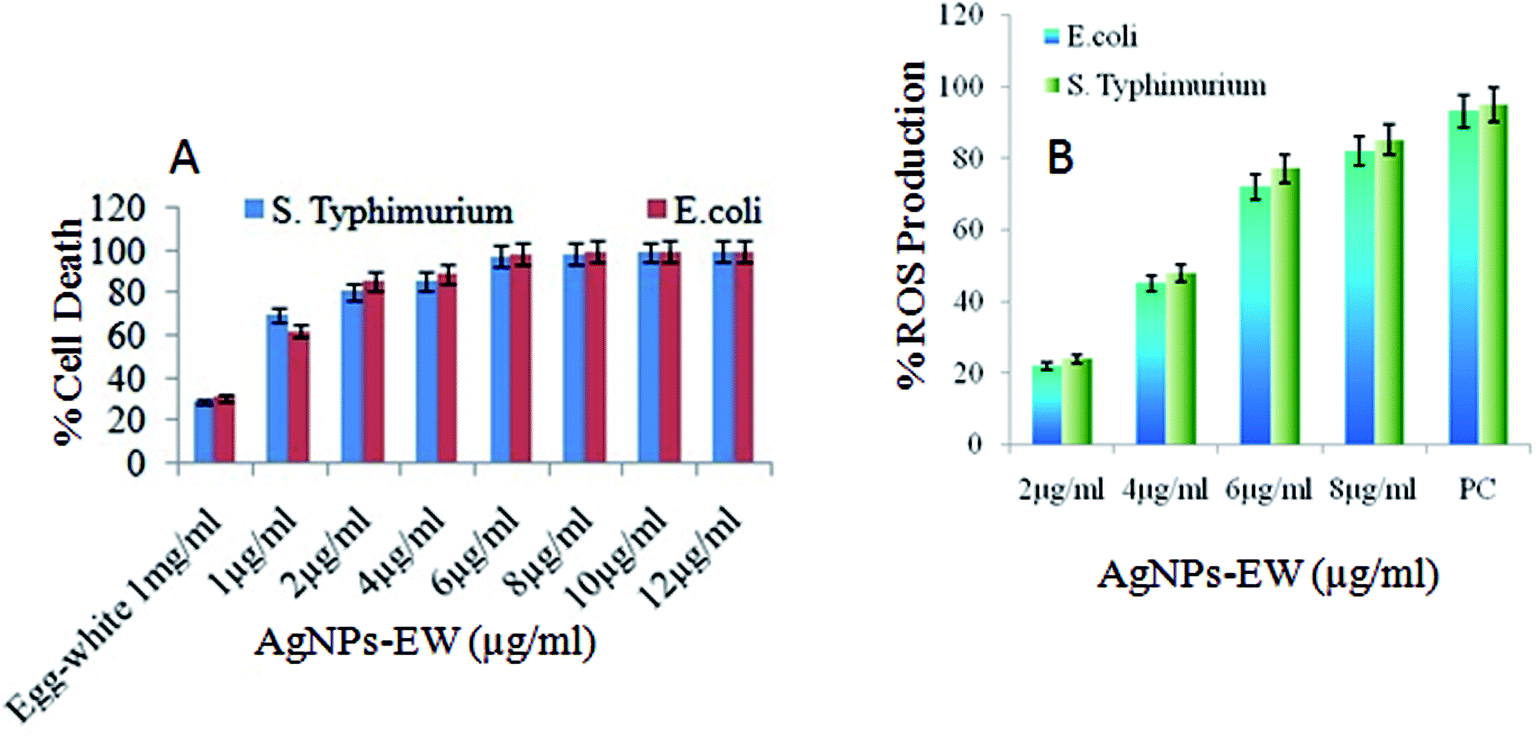 | ||
| Fig. 6 (A) MTT assay for the viability of E. coli and Salmonella typhimurium after treatment with different concentrations of AgNPs-EW (1, 2, 4, 6, 8, 10, 12 μg ml−1). (B) Intracellular ROS production in the bacterial cell suspensions treated with AgNPs-EW. Values are expressed as the mean ± SD (n = 3). | ||
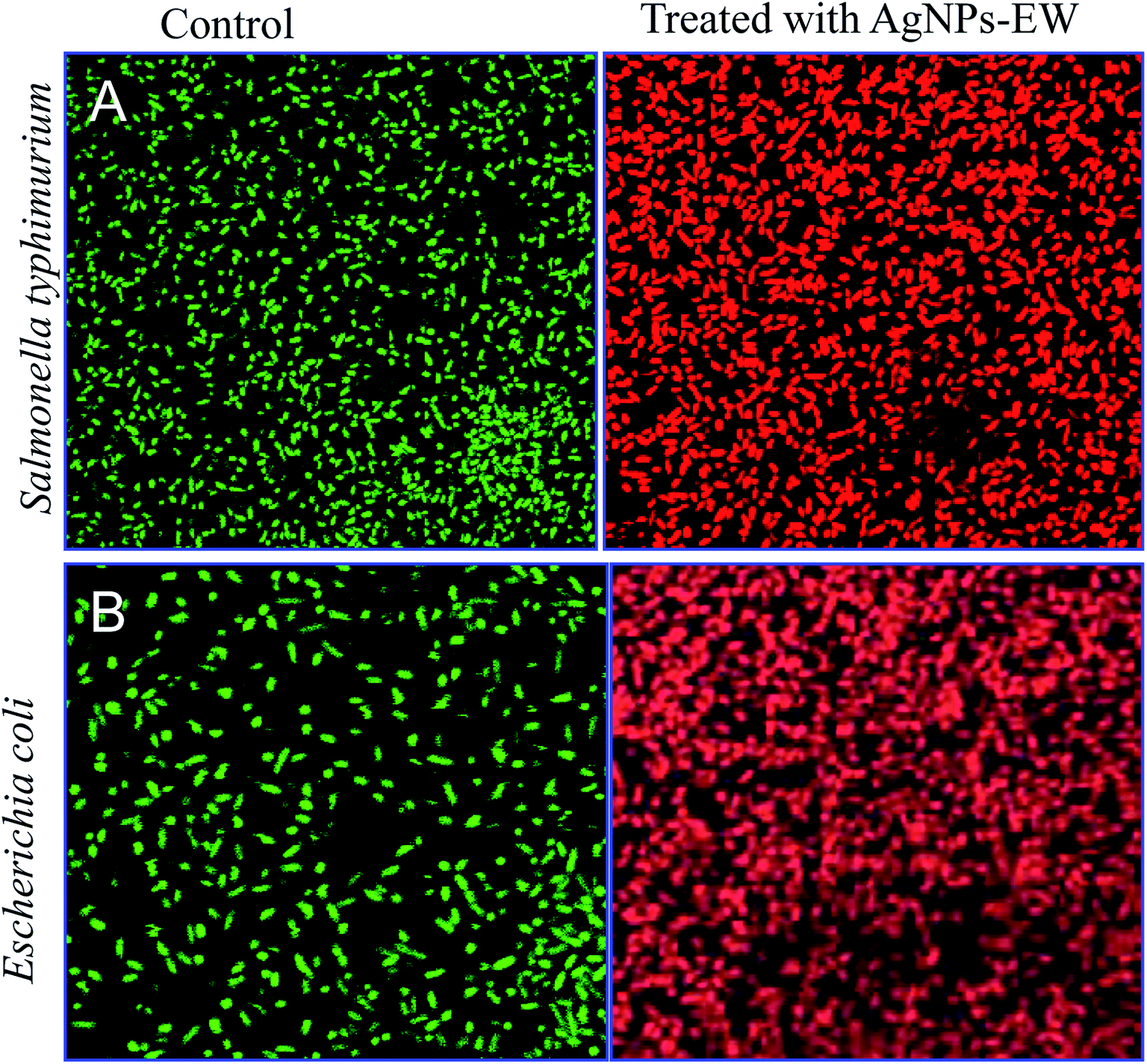 | ||
| Fig. 7 Confocal images of Salmonella Typhimurium and E. coli after treatment with AgNPs-EW (4 μg ml−1) for 4 h and stained with acridine orange (green, indicator of intact membrane, live cells) and propidium iodide (red, indicator of membrane damage, dead cells) (scale bar 20 μm). | ||
3.7 Evaluation of hemocompatibility
The hemolytic behavior and structural damage of chicken erythrocytes were investigated under upright light microscopy to evaluate the hemocompatability (biocompatibility) behavior of the AgNPs-EW. The amount of hemolysis and the change in erythrocytes morphology caused by AgNPs-EW exposed to chicken whole blood for 1 h incubation are shown in Fig. 8. The percent hemolysis increased with the increasing mass concentration of AgNPs-EW in the blood. In our study, nanoparticles caused 0.1% hemolysis at 12 μg ml−1 concentration. Interestingly, no morphological shape (normal biconcave shape) distortion was observed even at 12 μg ml−1 concentration. Exposure of the AgNPs-EW to RBCs showed spikes over a few cell surfaces, which was non-significant. Both these findings indicated very good hemocompatibility of the AgNPs-EW. In comparison with reported data,57 23% hemolysis was obtained in regard to human RBC, whereby, according to the criterion in the ASTM E2524-08 standard, a percent hemolysis >5% indicates that the test material causes damage to RBC.57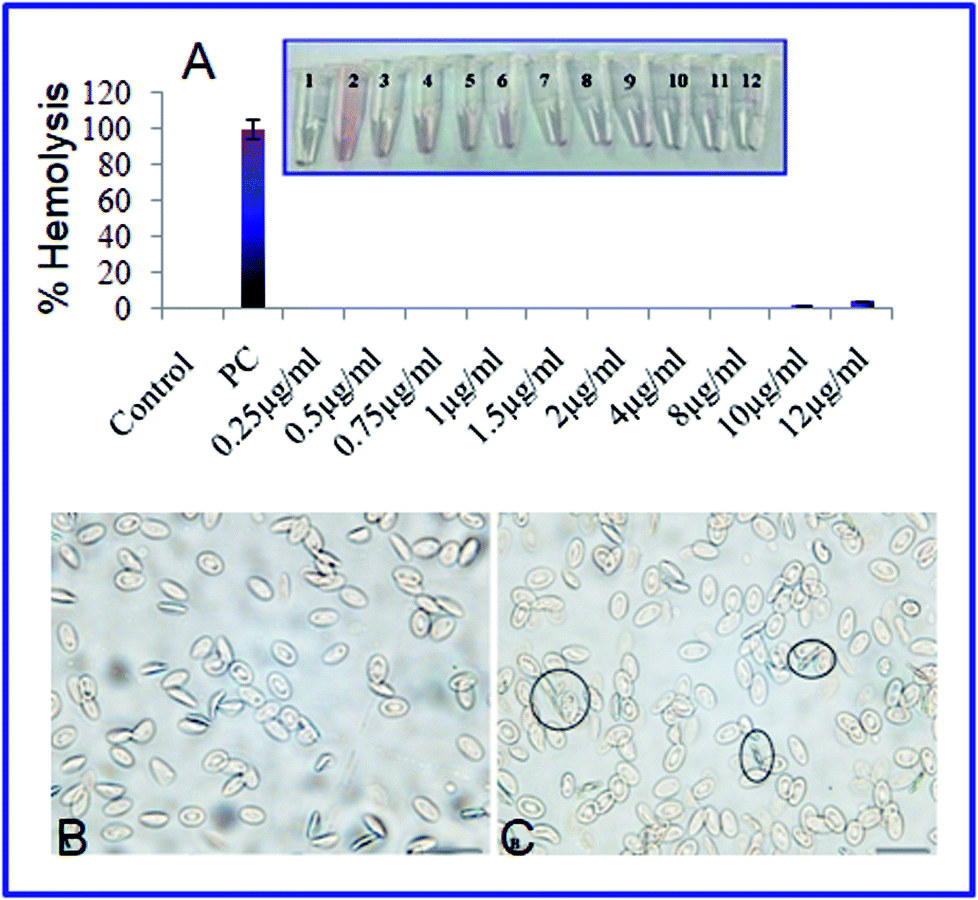 | ||
| Fig. 8 Morphology of chicken erythrocytes under upright light microscopy (40× magnification), (A) image of chicken erythrocytes before and (B) after the treatment with 12 μg ml−1 of AgNPs-EW ((C) rounded circle indicates the damaged erythrocytes). | ||
3.8 Evaluation of acute and chronic toxicity effects of AgNPs-EW on Drosophila melanogaster
The acute and chronic toxic effects of AgNPs-EW on the developmental stages of Drosophila melanogaster were investigated at six different concentrations of AgNPs-EW ranging from 10 mg L−1, 20 mg L−1, 30 mg L−1, 40 mg L−1, 50 mg L−1, and 100 mg L−1. The histogram of fecundity/fly clearly indicates that the AgNPs-EW did not cause any significant difference in the egg-laying capability as compared with the control (Fig. 9A). The control fly laid on average 55 eggs per fly per day, which is near about to that in all the replicates fed different concentrations of AgNPs-EW. Therefore, the results suggested that the AgNPs-EW did not induce any toxicity on the fecundity of Drosophila. Further, the present study finding indicated 98% hatchability in the control and 96% to 98% in the AgNPs-EW supplemented with different concentrations (Fig. 9B). The results propose that AgNPs-EW did not cause any significant difference in hatchability of Drosophila as compared with untreated flies (control). The viability was observed at 98–99% in the control as well as in the AgNPs-EW-treated group (Fig. 9C). Furthermore, the duration of development of the control flies was 251 h, whereas, the treated with AgNPs-EW flies took 252 h (Fig. 9D). Moreover, pigmentation, which is primarily determined by the amount, the type, and the distribution of melanin, showed a remarkable diversity in populations. Therefore, we selected this trait for analyzing the acute and chronic toxicity effects of AgNPs-EW in the pigmentation. The observed results indicated that the control female flies had a 6.55 ± 0.12 abdominal pigmentation sum score (sum of the segments 2, 3, 4, 5 and 6), whereas a score pf 6.47 ± 0.35 was reported for the AgNPs-EW-treated female flies (Fig. 9D). The results of both sums values have minor differences but statistically insignificant. Therefore, the present study revealed no toxicity of AgNPs-EW on Drosophila pigmentation in female flies. On the basis of these findings, it was demonstrated that the concentration range from 10 to 100 mg L−1 of AgNPs-EW did not cause any significant acute and chronic toxic effects on the reproductive traits, namely the fecundity, hatchability, viability, duration of development, and pigmentation of Drosophila. We compared these findings with previously reported AgNPs toxicity studies on Drosophila and summarize the results in Table 2,58–62 which clearly shows that AgNPs toxicity is based on the capping agents used for nanoparticles synthesis, size, and dose. Based on our literature study, we understand that this is the first report of AgNPs-EW having no acute and chronic toxicity effects on Drosophila at the maximum concentration, which is 100 mg L−1. This may be due to the capping of ovalbumin, ovotransferrin, and ovomucoid over the nanoparticles, making these particles non-toxic. Moreover, this is a higher concentration than the minimum inhibitory concentration (MIC) of test bacterial strains used in this study. In fact, our results suggested that nanoparticles synthesis via green chemistry approach and using protein/peptide as reducing and capping can reduce the toxicity of nanomaterials and improve their practical applications of nanomaterials. | ||
| Fig. 9 Acute and chronic toxicity effects of AgNPs-EW on Drosophila melanogaster, (A) rate of fecundity (%), (B) hatchability% (C) viability% (D) toxic effects of AgNPs-EW on duration of developmental stage of Drosophila melanogaster (E) abdominal pigmentation. Values are expressed as the mean ± SD (n = 5). | ||
| S. no. | Nanoparticles | Capping agents | Toxicity range | Particles size (nm) | Reference |
|---|---|---|---|---|---|
| a Indicating better biocompatibility and non-toxic nature even at higher concentration exposure, which is important for wider biomedical application. | |||||
| 1 | AgNPs | Citrate | 30 mg L−1 | 18.2 ± 8.2 | 58 |
| 2 | AgNPs | D-Maltose | Above 5 mg L−1 | 29 ± 4 | 59 |
| 3 | AgNPs | Citrate | Above 5 mg L−1 | ∼20 nm | 60 |
| 4 | AgNPs | Commercially available | Above 50 mg L−1 | 20 ± 100 | 61 |
| 5 | AgNPs | Polysaccharide | Above 30 mg L−1 | 10 | 62 |
| 6 | AgNPs | Egg-white proteins | Above 100 mg L−1 | 2 to 20 | aPresent study |
3.9 Examination of the antibacterial activity of AgNPs-EW after freezing–drying
The study on the impact of freezing–drying on the antibacterial activity of AgNPs-EW indicated no significant difference in the antibacterial activity upon freezing–drying as compared with before freezing–drying (Fig. 10). Therefore, these findings revealed that the freezing–drying of AgNPs-EW did not cause aggregation and the physico-chemical properties were retained the same as the freshly synthesized. This could be the cause of the unaltered antibacterial properties of AgNPs-EW even after freezing–drying. Interestingly, the sunlight-mediated egg-white-capped silver nanoparticles featured high stability and antibacterial potential. Hence, the synthesized AgNPs-EW investigated have immense potential for controlling bacterial infections.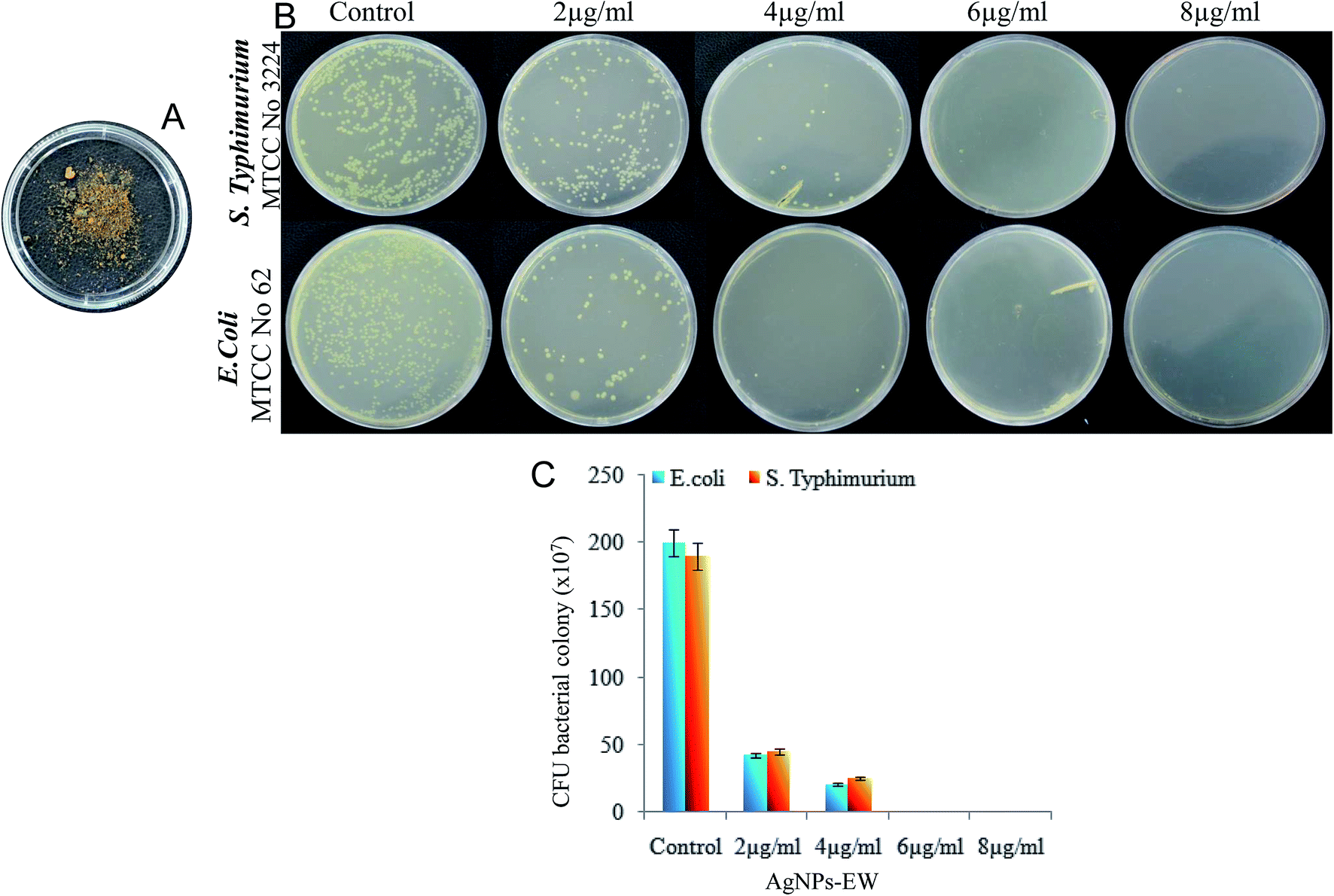 | ||
| Fig. 10 Impact of freezing–drying on the antibacterial activity of AgNPs-EW: (A) freeze-dried AgNPs-EW used for the antibacterial study (B) photograph of plates showing impact of freezing–drying on the antibacterial efficiency of AgNPs-EW against Salmonella Typhimurium on BHI and E. coli on LB plates. (C) Quantitative evaluation of the antibacterial activity of AgNPs-EW by counting the colonies grown on agar and BHI plates. Values are expressed as the mean ± SD (n = 3). | ||
4. Conclusion
In conclusion, we herein presented a novel method for the rapid, cost-effective, eco-friendly synthesis of non-toxic AgNPs-EW using egg-white protein as capping agents under the irradiation of daily sunlight. The synthesized nanoparticles employing this method had good biocompatibility, enhanced colloidal stability, and antibacterial activity. It is significant to point out that the AgNPs-EW were readily accomplished via a one-step reaction, i.e., the photocatalytic synthesis of AgNPs, which was simple, cheap, and environmentally friendly. The egg-white protein was tightly anchored on the surface of AgNPS-EW, facilitating long-term stability under the ambient environmental condition of more than one year and further, the AgNPS-EW were resistant to repeated freezing–thawing and freezing–drying processes without using any cryoprotectants. Furthermore, the antibacterial activity preserved >99% antibacterial activity against Salmonella typhimurium and Escherichia coli. In addition, the AgNPs-EW possessed adaptable biocompatibility at the lowest concentration on chicken erythrocytes and yielded feeble cytotoxicity on Drosophila. These results will enable the exploration of novel applications in the field of nano-biomaterials and antimicrobial therapy.Conflicts of interest
All the authors declare that there are no conflicts of interest.Acknowledgements
The authors are thankful to Defence Research and Developmental Organization (DRDO), New Delhi, India, for financial support. We are also thankful to The Director, Institute of Nanoscience and Technology, Mohali, Punjab, India for providing the TEM, FTIR, and zeta facilities and help of Mr Sandeep Vashishth, Research fellows, INST, Mohali is duly acknowledged. All the authors are thankful to Sophisticated Analytical Instrumentation Facility, Panjab University, Chandigarh, India for providing FESEM facilities and Institute of microbial technology (IMTECH, Chandigarh, India) for providing confocal microscopy facility and Deepak Butt, Technical Officer for their help during image capturing.References
- J. S. Kim, E. Kuk, K. N. Yu, J. H. Kim, S. J. Park, H. J. Lee, S. H. Kim, Y. K. Park, Y. H. Park, C. Y. Hwang, Y. K. Kim, Y. S. Lee, D. H. Jeong and M. H. Cho, Nanomedicine, 2007, 3, 95–101 CrossRef PubMed.
- D. I. Andersson and D. Hughes, Nat. Rev. Microbiol., 2010, 8, 260–271 CrossRef PubMed.
- B. Le Ouay and F. Stellacci, Nano Today, 2015, 10, 339–354 CrossRef.
- Y. Wang, J. Wan, R. J. Miron, Y. Zhao and Y. Zhang, Nanoscale, 2016, 8, 11143–11152 RSC.
- M. Talukdar, M. Bordoloi, P. P. Dutta, S. Saikia, B. Kolita, S. Talukdar, S. Nath, A. Yadav, R. Saikia, D. K. Jha and T. C. Bora, J. Appl. Microbiol., 2016, 121, 973–987 CrossRef PubMed.
- C. Willyard, Nature, 2017, 543, 15 CrossRef PubMed.
- A. Ravindran, P. Chandran and S. S. Khan, Colloids Surf., B, 2013, 105, 342–352 CrossRef PubMed.
- T. Kalaiyarasan, V. K. Bharti and O. P. Chaurasia, RSC Adv., 2017, 7, 51130–51141 RSC.
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová, V. K. Sharma, T. Nevěčná and R. Zbořil, J. Phys. Chem. B, 2006, 110, 16248–16253 CrossRef PubMed.
- Y. Zhou, X. Jiang, J. Tang, Y. Su, F. Peng, Y. Lu, R. Peng and Y. He, J. Mater. Chem. B, 2014, 2, 691–697 RSC.
- A. R. Shahverdi, A. Fakhimi, H. R. Shahverdi and S. Minaian, Nanomedicine Nanotechnology, Biol. Med., 2007, 3, 168–171 CrossRef PubMed.
- A. M. Fayaz, K. Balaji, M. Girilal, R. Yadav, P. T. Kalaichelvan and R. Venketesan, Nanomedicine, 2010, 6, 103–109 CrossRef PubMed.
- K. Tomankova, J. Horakova, M. Harvanova, L. Malina, J. Soukupova, S. Hradilova, K. Kejlova, J. Malohlava, L. Licman, M. Dvorakova, D. Jirova and H. Kolarova, Food Chem. Toxicol., 2015, 85, 20–30 CrossRef PubMed.
- H. Wang, X. Qiao, J. Chen and S. Ding, Colloids Surf., A, 2005, 256, 111–115 CrossRef.
- L. Mahmudin, E. Suharyadi, A. B. S. Utomo and K. Abraha, AIP Conf. Proc., 2016, 1725, 020041, DOI:10.1063/1.4945495.
- B. Ajitha, Y. Ashok Kumar Reddy and P. Sreedhara Reddy, Powder Technol., 2015, 269, 110–117 CrossRef.
- W. Chunyan and S. Valiyaveettil, RSC Adv., 2013, 3, 14329–14338 RSC.
- B. Ajitha, Y. A. Kumar Reddy, P. S. Reddy, H.-J. Jeon and C. W. Ahn, RSC Adv., 2016, 6, 36171–36179 RSC.
- P. Huang, D.-P. Yang, C. Zhang, J. Lin, M. He, L. Bao and D. Cui, Nanoscale, 2011, 3, 3623–3626 RSC.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935–497821 CrossRef PubMed.
- N. Mauro, D. Schillaci, P. Varvarà, M. G. Cusimano, D. M. Geraci, M. Giuffrè, G. Cavallaro, C. M. Maida and G. Giammona, ACS Appl. Mater. Interfaces, 2018, 10(1), 318–331 CrossRef PubMed.
- C. Desert, C. Guérin-Dubiard, F. Nau, G. Jan, F. Val and J. Mallard, J. Agric. Food Chem., 2001, 49, 4553–4561 CrossRef PubMed.
- N. G. Heatley and W. Dunn, Biochem. J., 1944, 38, 61–65 CrossRef PubMed.
- G. A. Martınez-Castanon, N. Nino-Martınez and F. Martınez-Gutierrez, et al., J. Nanopart. Res., 2008, 10, 1343–134825 CrossRef.
- H. Wang, H. Cheng, F. Wang, D. Wei and X. Wang, J. Microbiol. Methods, 2010, 82, 330–333 CrossRef PubMed.
- R. Y. Pelgrift and A. J. Friedman, Adv. Drug Delivery Rev., 2013, 65, 1803–1815 CrossRef PubMed.
- M. Kutwin, E. Sawosz, S. Jaworski, N. Kurantowicz, B. Strojny and A. Chwalibog, Nanoscale Res. Lett., 2014, 9, 1–6 CrossRef PubMed.
- P. J. Shiny, A. Mukherjee and N. Chandrasekaran, Bioprocess Biosyst. Eng., 2014, 37, 991–997 CrossRef PubMed.
- K. Behlol, A. Ahmed, R. Senthilnathan, S. Megarajan and V. Anbazhagan, J. Photochem. Photobiol., B, 2015, 151, 39–45 CrossRef PubMed.
- K. Behlol, A. Ahmed, S. B. Subramaniyan, S. F. Banu, P. Nithyanand and V. Anbazhagan, Colloids Surf., B, 2018, 163, 209–217 CrossRef PubMed.
- D. Cui, Z. Wang and L. Guo, Int. J. Nanomed., 2012, 7, 2101–2107 Search PubMed.
- A. Tirado-Guizar, G. Rodriguez-Gattorno, F. Paraguay-Delgado, G. Oskam and G. E. Pina-Luis, MRS Commun., 2017, 7, 695–700 CrossRef.
- F. Alihosseini, S. Ghaffari, A. R. Dabirsiaghi and S. Haghighat, Braz. J. Pharm. Sci., 2015, 51, 798–802 Search PubMed.
- M. A. Moretton, D. A. Chiappetta and A. Sosnik, J. R. Soc., Interface, 2012, 487–502 CrossRef PubMed.
- P. V Date, A. Samad and P. V Devarajan, AAPS PharmSciTech, 2010, 11, 304–313 CrossRef PubMed.
- R. I. Maccuspie, J. Nanopart. Res., 2011, 13, 2893–2908 CrossRef.
- P. Fonte, S. Soares, A. Costa, J. C. Andrade, V. Seabra, S. Reis and B. Sarmento, Biomatter, 2012, 2(4), 329–339 CrossRef PubMed.
- P. Fonte, S. Soares, F. Sousa, A. Costa, V. Seabra, S. Reis and B. Sarmento, Biomacromolecules, 2014, 15(10), 3753–3765 CrossRef PubMed.
- E. Cao, Y. Chen, Z. Cui and P. R. Foster, Biotechnol. Bioeng., 2003, 82, 684–690, DOI:10.1002/bit.10612.
- Y. Yeh and R. E. Feeney, Chem. Rev., 1996, 96, 601–617 CrossRef PubMed.
- E. D. N. S. Abeyrathne, H. Y. Lee and D. U. Ahn, Poult. Sci., 2013, 92, 3292–3299 CrossRef PubMed.
- S. Yu, J. Hu, X. Pan, P. Yao and M. Jiang, Langmuir, 2006, 22(6), 2754–2759 CrossRef PubMed.
- T. H. I. Hong and L. A. N. P. Thi, J. Food Sci., 1981, 46, 1336–1338 CrossRef.
- H. F. Deutsch, J. Biol. Chem., 1949, 181(2), 499–510 Search PubMed.
- H. Wang, H. Cheng, F. Wang, D. Wei and X. Wang, J. Microbiol. Methods, 2010, 82, 330–333 CrossRef PubMed.
- J. N. Medical, Immunology, 1973, 25, 377–383 Search PubMed.
- T. Koseki, N. Kitabatake and E. Doi, J. Biochem., 1990, 107, 389–394 Search PubMed.
- O. Wellman-Labadie, J. Picman and M. T. Hincke, Br. Poult. Sci., 2008, 49, 125–132 CrossRef PubMed.
- E. D. N. S. Abeyrathne, H. Lee, J. S. Ham and D. U. Ahn, Poult. Sci., 2017, 92, 1091–1097 CrossRef PubMed.
- B. Prajanban, L. Shawsuan, S. Daduang and J. Kommanee, J. Proteomics, 2012, 75, 1940–1959 CrossRef PubMed.
- J. S. Gebauer, M. Malissek, S. Simon, S. K. Knauer, M. Maskos, R. H. Stauber, W. Peukert and L. Treuel, Langmuir, 2012, 28, 9673–9679 CrossRef PubMed.
- W. Du, S. Niu, Y. Xu, Z. Xu and C. Fan, Carbohydr. Polym., 2009, 75, 385–389 CrossRef.
- A. J. Kora and R. B. Sashidhar, J. Antibiot., 2015, 68, 88–97 CrossRef PubMed.
- D. Wu and P. Yotnda, J. Visualized Exp., 2011, 2, 1–5 Search PubMed.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef PubMed.
- C. Carlson, S. M. Hussain, A. M. Schrand, K. L. Hess, R. L. Jones and J. J. Schlager, J. Phys. Chem. B, 2008, 112, 13608–13619 CrossRef PubMed.
- A. K. Bhunia, P. K. Samanta, D. Aich and S. Saha, J. Phys. D: Appl. Phys., 2015, 48, 235305–235315 CrossRef.
- B. Mao, Z. Chen, Y. Wang and S. Yan, Sci. Rep., 2018, 8, 1–16 CrossRef PubMed.
- A. Panacek, R. Prucek, D. Safarova, M. Dittrich, J. Richtrova, K. Benickova, R. Zboril and L. Kvitek, Environ. Sci. Technol., 2011, 45, 4974–4979 CrossRef PubMed.
- C. Ong, Q. Y. Lee, Y. Cai, X. Liu, J. Ding, L. L. Yung, B. Bay and G. Baeg, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- A. Raj, P. Shah and N. Agrawal, PLoS One, 2017, 1–14 Search PubMed.
- R. Posgai, C. B. Cipolla-mcculloch, K. R. Murphy, S. M. Hussain, J. J. Rowe and M. G. Nielsen, Chemosphere, 2011, 85, 34–42 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03649g |
| This journal is © The Royal Society of Chemistry 2018 |