
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRobust colorimetric detection based on the anti-aggregation of gold nanoparticles for bromide in rice samples†
Siwat Plaisenab,
Wilairat Cheewasedtham b and
Thitima Rujiralai*ab
b and
Thitima Rujiralai*ab
aDepartment of Chemistry, Center of Excellence for Innovation in Chemistry, Faculty of Science, Prince of Songkla University, Hat Yai, Songkhla, 90112 Thailand. E-mail: thitima.r@psu.ac.th
bAnalytical Chemistry and Environment Research Unit, Division of Chemistry, Department of Science, Faculty of Science and Technology, Prince of Songkla University, Pattani 94000, Thailand
First published on 13th June 2018
Abstract
Inorganic bromide (Br−) is an important contaminant ion as it can originate from the overuse of illegal methyl bromide as a fumigant in stored rice samples. Herein, we developed a simple and highly sensitive colorimetric sensor for bromide ion detection in rice samples. The sensor is based on the anti-aggregation of gold nanoparticles (AuNPs) by Br− in the presence of Cr3+, which made the method more selective than other typical aggregations of nanoparticles. The AuNPs underwent an aggregation process as a result of the coordination of Cr3+ and the carboxylate group of a citrate ion stabilized the AuNPs, resulting in a red-to-blue color change. When Br− was pre-mixed with the AuNPs and Cr3+ was added, the solution color changed from blue to red with an increase in the Br− concentration. The anti-aggregation process can be detected with the naked eye and monitored using UV-vis spectrophotometry. The linear calibration curve ranged between 0.31 and 3.75 μM Br− with a low LOD and LOQ of 0.04 and 0.13 μM. The recovery was excellent, ranging from 79.9–92.2% with an RSD of less than 4.0%. The good inter-day and intra-day precisions were 2.9–6.4% and 3.1–7.1%, respectively. The developed sensor has proved to provide a robust method for Br− detection in rice samples.
1. Introduction
Methyl bromide (CH3Br) is a fumigant that is often used for controlling a variety of pests such as insects, mites, moulds, nematodes, weeds, viruses and bacteria, as well as being used in soil disinfection and as a postharvest treatment for stored and dried foodstuffs including rice, tobacco, fresh fruits and vegetables.1–3 It is dominant over other fumigants due to the fact it is a colorless, odorless (at low concentrations) and non-flammable gas with a high rate of penetration.2–4 Its toxic effects on human health are (following short-term exposure) headache, dizziness, nausea, vomiting, blurred vision, slurred speech and convulsions. High concentrations of methyl bromide may cause unconsciousness and death. Prolonged exposure to methyl bromide may cause injury to the central nervous system.4 Furthermore, it can greatly destroy the ozone layer through emission from agricultural pesticide use, from the burning of biomass and leaded gasoline, and from the ocean.5,6 Thus, the use of methyl bromide is controlled by many organizations and it is already being phased out. From January 1 2005, the production and use of methyl bromide will be stopped in the U.S.A. with exemptions for emergency and critical use.5–7 The agricultural use of methyl bromide in China will also be terminated at the end of 2018.8 The application of methyl bromide in the European Union has been forbidden since 2010, but exceptions are made, especially with regard to the International Standards for Phytosanitary Measures 15.9After food commodities are fumigated, methylated products and inorganic bromide are formed as a result of chemical reactions with some of the constituents of the treated foodstuffs or the breakdown products of methyl bromide.1–3,10,11 Inorganic bromide has been detected in rice and glutinous rice in the ranges 0.13–11.93 mg kg−1 and 0.20–1.37 mg kg−1, respectively, and its concentration in all samples were within the Codex maximum residue limit value of 50 mg kg−1.2 Methyl bromide residues can persist in nuts and seeds for 10 weeks and in dried fruit for 4 weeks.1 Cova et al. reported that higher concentrations of bromide ions were detected in fumigated pastas with eggs, pastas with eggs and spinach, and rice compared to in those that were unfumigated.11
Thailand, as the world’s leading rice producer and also one of the largest rice consumers, imported an average of 357.69 and 103.85 tons per year of methyl bromide and a mixture between methyl bromide and chloropicrin, respectively, to eliminate undesired pests before commercializing their products between 2007 and 2012.2 Due to its toxicity, under the agreement of the Montreal Protocol, Thailand agreed to gradually reduce and to phase out the use of methyl bromide by 2015.12 The Thai Ministry of Public Health has defined the maximum residue limit (MRL) of methyl bromide in rice as 0.01 mg kg−1 and of bromide ions in rice as 50 mg kg−1.13 In 2013, methyl bromide was not detected in any rice samples (n = 13), however the bromide ion concentration in one sample was detected to be 77.2 mg kg−1 and after 4 days its concentration had decreased to 20.9 mg kg−1.14
Due to food globalization, rice consumption is also increasing around the world. The determination of methyl bromide and inorganic bromide contamination in rice is, therefore, required as a method for guaranteeing consumer safety. In most cases, bromide ion contamination in rice has been found when food commodities are fumigated with either low or high concentrations of methyl bromide, and are particularly over the limit when they have been fumigated with methyl bromide multiple times.1–3,11,14,15 Many studies have also focused on inorganic bromide in food in order to assess the daily intake of bromide ions from food and water into the human body.10,16,17 Therefore, bromide ions are the main target analyte for this work, whether from the accumulation of bromide ions from the environment or from the transformation of methyl bromide.
Prior to the detection step, bromide ions in foodstuffs have been treated using different procedures e.g., low energy microwave digestion,16 high temperature heating (∼850 °C),18 hydrolysis with alkaline and ashing at 550 °C,2 and suspension in 6 N sulfuric acid and propylene oxide followed by extraction with ethyl acetate.15 Subsequently, the pre-treated samples are determined using sophisticated instrumentation such as inductively coupled plasma-mass spectrometry,16 gas chromatography-electron capture detecting,2,15 and ion-exchange chromatography with visible light detection.19 However, these mentioned instrumentations require not only complex sample preparation and long analysis times but also complicated and expensive instruments.
Nowadays, gold nanoparticle (AuNP)-based colorimetric sensors have drawn increasing attention due to their low cost and their rapid, selective and reliable detection.20–23 In general, AuNPs change from the dispersion to the aggregation state and their color changes from ruby red to purple or blue.20–23 An absorption band in the visible range with a high extinction coefficient (e.g., 2.70 × 108 M−1 cm−1)23 appears when the incident photon frequency resonates with the excitation of the conduction electrons. This phenomenon is named surface plasmon resonance (SPR).20–23 Due to these advantages, AuNPs can be used as a powerful tool for real-time monitoring via naked-eye detection, rather than using any advanced instruments. AuNPs have also been applied for the detection of ions,20–22,24–28 gas29 and organic residues30–34 such as Hg2+, Cu2+, S2−, I−, SCN−, H2S, clenbuterol, aflatoxins, tetracycline, amoxicillin and pazufloxacinmesilate, in which the detection mostly depends on the aggregation. It was reported that AuNP aggregation is not selective and can provide false positive results due to the effects of many other external factors in real applications. Thus, the anti-aggregation or re-dispersion of AuNPs has become a good option to utilise in order to improve selectivity.21,22,25,27,29
So far, a colorimetric probe based on the AuNP anti-aggregation process has not been found for bromide ion determination in rice samples. In this work, we developed a AuNP colorimetric sensor for the facile, sensitive and selective detection of bromide ions in rice samples. The sensor is based on the fact that bromide ions prevent the aggregation of AuNPs. The addition of Cr3+ into a AuNP solution resulted in the aggregation of AuNPs with a red-to-blue color change. In the presence of Br−, the aggregation of AuNPs was interrupted and the corresponding color of solution changed from blue to red. The concentration of bromide ions in real samples can be easily observed with the naked eye, and also quantitatively measured using UV-visible spectroscopy.
2. Material and methods
2.1 Chemical and reagents
All chemicals were of analytical grade. Gold(III) chloride trihydrate (HAuCl4·3H2O) was purchased from Sigma-Aldrich (USA). Chromium(III) nitrate nanohydrate was purchased from Alfa-Aesar (USA). Trisodium citrate and disodium hydrogenphosphate dodecahydrate were purchased from Ajax Finechem (Australia). Hydrochloric acid, potassium hydroxide, ethanol and nitric acid were purchased from RCI Labscan (Thailand). Potassium bromide and sodium dihydrogen phosphate dehydrate were purchased from LobaChemi (India). All solutions were prepared using 18 MΩ cm−1 ultrapure water obtained from ELGA Maxima.Sodium phosphate buffer (pH 6.5 at 0.01 M) was prepared by mixing 34 mL of 0.02 M sodium dihydrogenphosphate and 16 mL of 0.02 M disodium hydrogenphosphate. Subsequently, the buffer was adjusted by either sodium dihydrogenphosphate or disodium hydrogenphosphate to obtain a pH of 6.5 and the volume was made up to 100 mL with ultrapure water. A stock solution of 12.52 mM bromide ion was prepared from potassium bromide. It was necessary that all the glassware was thoroughly cleaned in freshly prepared aqua regia solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v HCl–HNO3), washed with ultrapure water at least twice and then oven-dried at 150 °C before use to avoid aggregation of gold colloids due to any contaminants.
:1 v/v HCl–HNO3), washed with ultrapure water at least twice and then oven-dried at 150 °C before use to avoid aggregation of gold colloids due to any contaminants.
2.2 Synthesis of gold nanoparticles (AuNPs)
The gold nanoparticles (AuNPs) were synthesized via the trisodium citrate reduction of HAuCl4 according to the previous method with slight modification.20 A volume of 250 mL of 0.015% (m/v) HAuCl4 solution was added into a 500 mL Duran bottle and was heated to boiling with vigorous stirring. Subsequently, 8.75 mL of 1% (w/v) trisodium citrate solution was added rapidly into the boiling solution followed by stirring. The color of the solution changed from pale yellow to wine red in a few minutes as Au3+ was reduced to Au0, indicating the formation of citrate-capped gold nanoparticles (AuNPs). After that, the AuNP solution was continuously boiled for another 15 min to ensure the reaction was completed. It was then allowed to cool down to room temperature under stirring, and the volume was adjusted to 250 mL with ultrapure water in a volumetric flask. The spectrum of this synthesized AuNP solution was investigated. For further study, this AuNP solution was stored in an amber bottle at 4 °C. After synthesis, the maximum absorbance of the AuNP solution was 1.49 at a surface plasmon resonance (SPR) wavelength of 519 nm with a calculated particle concentration of 5.52 nM (Fig. S1 (ESI†)), according to Beer’s law using an extinction coefficient of 2.70 × 108 M−1 cm−1.23 We have proved that AuNPs synthesized via the above method can be kept in a fridge for up to 4 months without any loss of sensitivity together with an absorbance of 1.49 ± 0.01 at 519 nm (Fig. S2 (ESI†)).2.3 Characterization
The surface plasmon resonance (SPR) spectra of the synthesized AuNP solution were investigated in the range 400–800 nm using a UV-1800 spectrophotometer (Shimadzu, Japan) in a 1.5 mL disposable polystyrene cuvette. The size and shape of the AuNPs were measured using a transmission electron microscope (TEM, JEM-2010, JEOL, Japan) with a 200 kV acceleration voltage. Before TEM measurements, the samples were diluted 3 times with ultrapure water before deposition and 5 μL of the diluted sample was deposited on the carbon-coated copper grid and then evaporated at room temperature. Moreover, the zeta-potential value and size distribution of the synthesized AuNP solution were studied using a zeta potential analyzer (ZetaPALS, Brookhaven, USA).2.4 Colorimetric determination of bromide ions
To increase the selectivity of the developed method, the determination of the bromide ions in this study was based on the anti-aggregation of AuNPs. A volume of 100 μL of different concentrations of Br− was first added to 400 μL of 5.52 nM AuNP solution containing 500 μL of 10 mM phosphate buffer solution (pH 6.5), and the mixture was mixed well using a vortex mixer. After that, 100 μL of 57.70 μM Cr3+ was added to the mixture with vortex mixing and the solution was incubated for 10 min at room temperature (25 ± 2 °C) to allow for adequate reaction time and to ensure the color had completely developed. After interaction, the color of the solution was observed with the naked eye. Photographs of the resulting solution were taken and the UV-visible spectra of the mixtures from 400–800 nm were recorded immediately. The spiked samples for recovery in pre-treated samples were investigated using the same procedure.2.5 Sample preparation of rice
White rice samples (five commercial brands) were collected randomly from a supermarket in the Hat Yai district, Songkhla province, Thailand and stored in a refrigerator at 4 °C before the experiment. The rice sample was prepared according to the report of Sungwaranond et al. with modifications.2 The rice sample (100 g) was ground using a Ball mill (Retsch MM400, Germany) to obtain a powder (∼5 μm).Two grams of the rice powder was added into a 50 mL nickel crucible and digested with 5 mL of 1% potassium hydroxide in 50% ethanol. The sample was boiled on a hotplate (by gradually increasing the heating rate to avoid the sample bumping) until it was dry and no smoke was observed. Next, it was burned in a muffle furnace at 600 °C for 4 h to eliminate the organic residues in the rice samples. After the obtained ash was allowed to cool down, 15 mL ultrapure water was added and the solution was boiled for 15 min on a hotplate in order to dissolve the inorganic bromide, and was filtered with a 0.22 μm Nylon membrane through a 25 mL volumetric flask. The ash residue was placed in a nickel crucible and was again boiled using another 10 mL ultrapure water. This new solution was filtered through the previous volumetric flask and finally diluted with ultrapure water up to the mark. The sample solution was then diluted 1 fold with ultrapure water before the color reaction was performed.
2.6 Optimization of the colorimetric detection of bromide ions
To obtain the best sensitivity of the method, the analytical conditions for the detection of Br−, i.e., the concentration of Cr3+, the pH and concentration of the phosphate buffer, the concentration of the AuNPs and the reaction time, were optimized. Each parameter was studied using a one step at a time experiment. All experiments were repeated three times with the results reported as mean ± standard deviation (SD). The absorbances at 519 and 673 nm were found to be directly related to the dispersed, aggregated and anti-aggregated states of the AuNPs. Therefore, the absorbance intensity ratio of A519/A673 was used for the quantitative determination of Br−. In order to obtain a representative sample for method development and optimization, five different brands of rice were mixed together. All the statistical analyses were performed using Microsoft Office Excel 2007. The ANOVA at a 95% confidence level was used for data analysis.3. Results and discussion
3.1 Proposed sensing mechanism
Fig. 1 shows the proposed mechanism for the successful colorimetric sensor of bromide ions based on the anti-aggregation of AuNPs. The AuNP solution presented a maximum SPR absorption at 519 nm (Fig. 2a) with a red wine color (Fig. 1), which was in good agreement with previous reports.20–22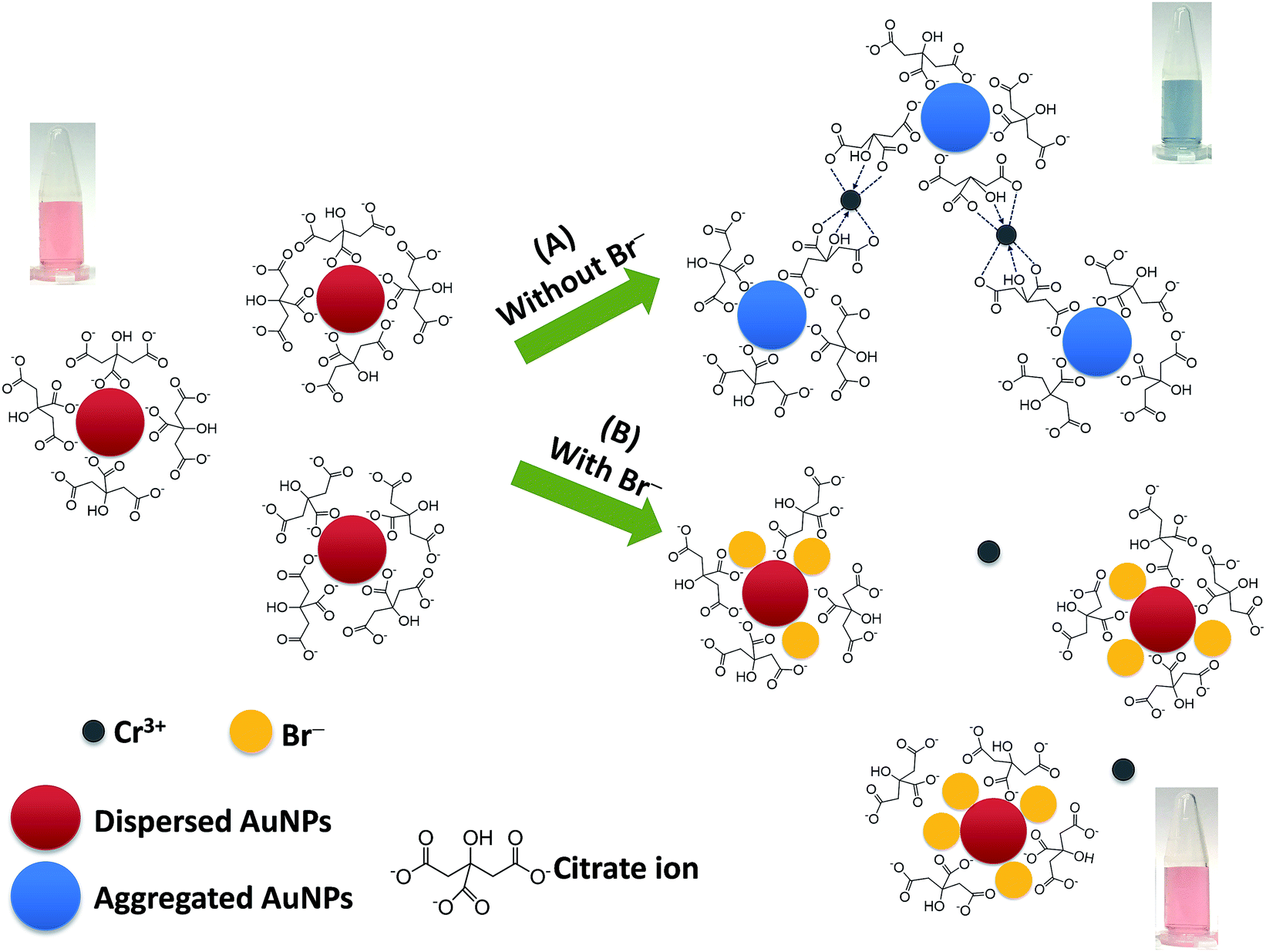 | ||
| Fig. 1 A proposed mechanism for the colorimetric sensor of bromide ions based on the anti-aggregation of AuNPs. | ||
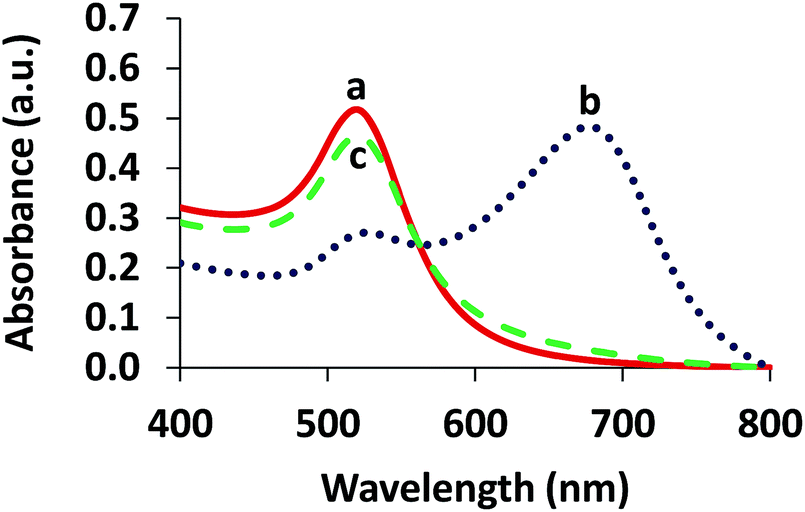 | ||
| Fig. 2 UV-vis absorption spectra of (a) AuNPs, (b) aggregation of the AuNPs in the presence of 4.80 μM Cr3+ and (c) anti-aggregation of the AuNPs in the presence of 3.13 μM Br− and 4.80 μM Cr3+. | ||
In this study, sodium citrate could act as a reductant and a stabilizer of colloidal AuNPs. Thus, the AuNPs were relatively stable due to the strong electrostatic repulsion between the negatively charged citrate ions on the surface of the AuNPs.35,36 It was reported that Cr3+ could coordinate with two citrate ions via the hydroxyl groups in each citrate ion with a high formation constant of 3.89 × 1029.37,38 Thus, citrate can also be used as the ligand for interaction with Cr3+, and Cr3+ can serve as a cross linking agent between the pairs of citrate-coated AuNPs.36 This ligand exchange is probably strong enough to overcome the electrostatic repulsion imposed by the citrate-AuNPs, thus inducing the aggregation of the AuNPs.
Upon addition of Cr3+ (Fig. 1A), the intensity of the SPR band of the AuNPs at 519 nm decreased, and a new absorption band appeared at 673 nm (Fig. 2b) due to the AuNP aggregation driven by Cr3+, demonstrating the successful conjugation of Cr3+ with the AuNPs. This caused a red-to-blue color change. The result of the aggregation was also confirmed via TEM measurement (Fig. 3A). We observed a spherical monodisperse colloid of individual AuNPs before aggregation (Fig. 3A(a)). The average particle size of the prepared AuNPs was 16.43 ± 0.81 nm. In the presence of Cr3+ (Fig. 3A(b)), heavy aggregation of the AuNPs was observed, confirming that Cr3+ induced large-scale aggregation of the AuNPs through chelating reactions, resulting in the particle size distribution of the aggregated AuNPs in the presence of Cr3+ being about 16 times higher than those without Cr3+ (calculated from the size ratio of 262.30/16.43 in Fig. 3B(d and e)). A similar phenomenon of aggregation was also reported by Liu and Wang.39
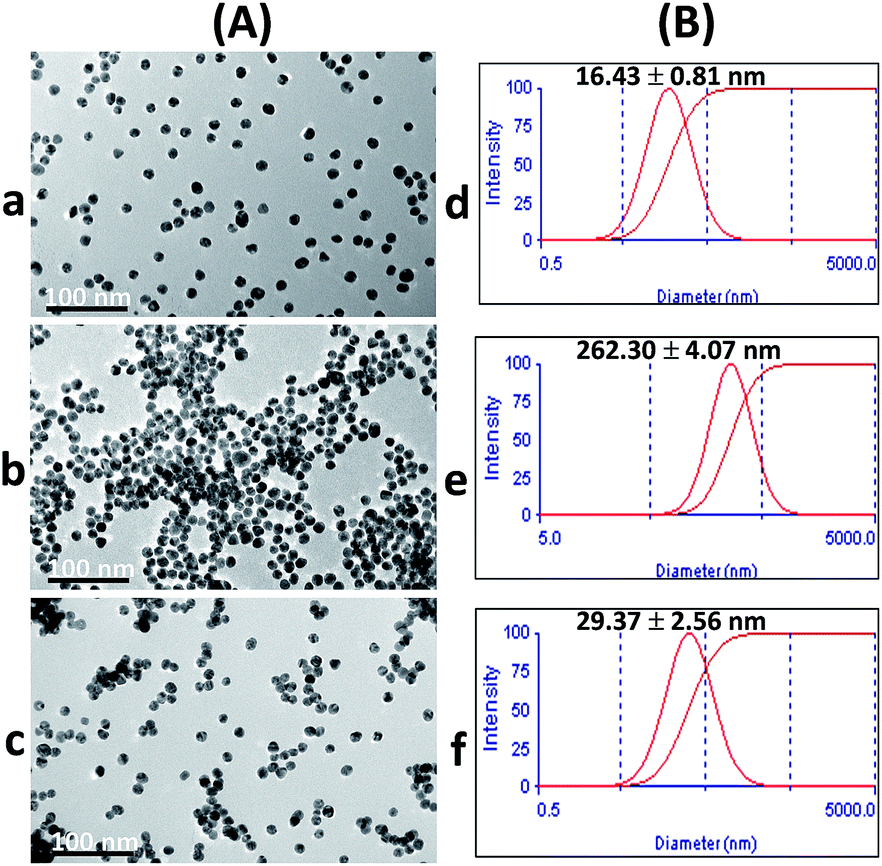 | ||
| Fig. 3 (A) TEM images and (B) particle size distributions of (a) and (d) AuNPs, (b) and (e) aggregation of the AuNPs in the presence of 4.80 μM Cr3+, and (c) and (f) anti-aggregation of the AuNPs in the presence of 3.13 μM Br− and 4.80 μM Cr3+. | ||
When the AuNPs were treated with Br− followed by mixing with Cr3+ (Fig. 1B), the color of the AuNPs was found to successfully change from blue to wine red, along with a decrease in the SPR absorption band at 673 nm and an increase in the band at 519 nm (Fig. 2c). This was called the anti-aggregation of the AuNPs, which was a result of the change of the AuNPs from the aggregation (blue color) to dispersion state (red color).
The anti-aggregation of the AuNPs could be caused by the adsorption of Br− onto the AuNP surface, leading to the prevention of aggregation driven by the Cr3+–citrate complex. The bromide ion is a chaotropic anion that can transfer its charge to the AuNP surface and thus, increase electrostatic stabilization.40 Moreover, the adsorption of Br− on the surface of the AuNPs may prevent aggregation due to a higher stability constant of the Au (1+ or 3+)–Br− complex, which is 12.3 and 32.8 for the Au+– and Au3+–Br− complex, respectively,41 compared to the lower stability constant of the Cr3+–Br− complex (−2.66).42
The zeta potential values of the AuNPs after addition of 0.63, 2.50 and 4.38 μM Br− were also investigated. As shown in Fig. S3 (ESI†), the zeta potential values increased in proportion to the concentration of Br−, indicating that colloidal AuNPs are more highly electrically stabilized. The addition of Br− could stabilize the AuNP colloid and enhance the repulsion force between the AuNPs. This increased the repulsion force of each AuNP, which could affect the chelation between Cr3+ and citrate. As a result, the color of the solution remained red as the Br− concentration increased, relating to the increased dispersion state. The TEM confirmed the observation that the AuNPs remained dispersed with the addition of Br− and Cr3+, as well as confirming the decrease in size of the AuNPs compared to their size in the aggregation state as shown in Fig. 3A(c) and (f), respectively. Thus, it suggested that the effective anti-aggregation is induced by Br−.
3.2 Optimization of colorimetric detection of bromide ions
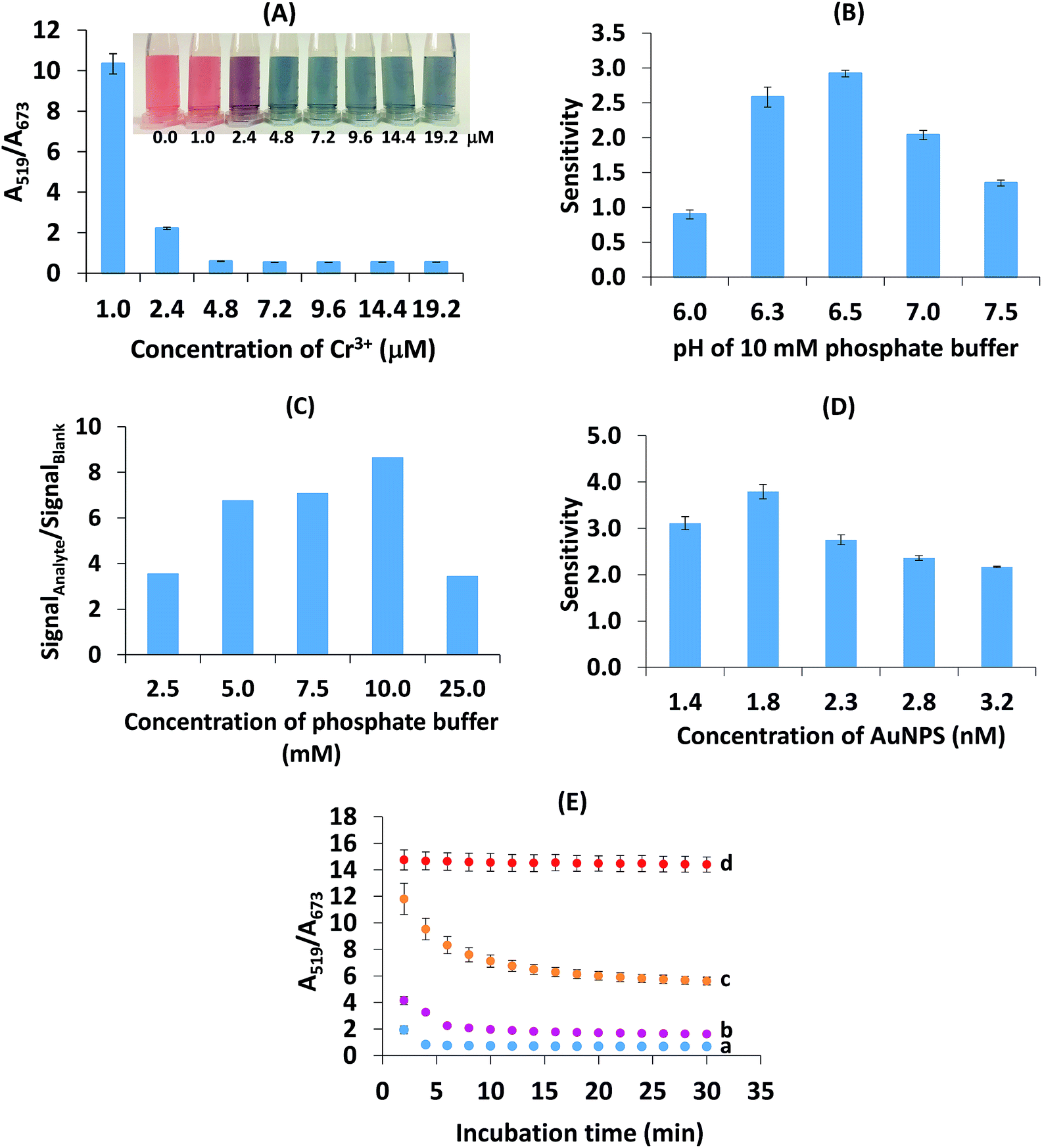 | ||
| Fig. 4 Optimization of the colorimetric detection of Br−. (A) Concentration of Cr3+. (B) pH of 10 mM phosphate buffer. The sensitivity was the slope of linearity plotted between the A519/A673 ratio (y axis) and the Br− concentration in the range 0.62–5.01 μM (x axis). (C) Concentration of the phosphate buffer (pH 6.5). The signalAnalyte and signalBlank were the anti-aggregation signal from the analyte and aggregation signal from the Cr3+, respectively. (D) Concentration of AuNPs. The sensitivity was the slope of linearity for the Br− concentration in the range 0.63–2.50 μM. (E) Reaction time: (a) blank (without Br−), (b) 1.25 μM Br−, (c) 2.50 μM Br− and (d) 5.01 μM Br−. | ||
These results reveal the sharp decrease in the absorbance ratio of A519/A673 of 0.96–4.81 μM Cr3+ (Fig. 4A). At a concentration of Cr3+ higher than 4.81 μM, the ratio became stable, indicating the maximum degree of aggregation. By considering the lowest concentration of Cr3+ that provided the maximum degree of aggregation, 4.81 μM Cr3+ was chosen as the optimal concentration.
Although Cr3+ is a toxic substance, it is considered as an essential trace element and is less toxic than Cr6+, since Cr6+ has high solubility and mobility in biological systems.43 The selected concentration of Cr3+ (equal to 4.81 μM or 5.76 nmol Cr3+) used in our test was very low.
Moreover, we have investigated other non-toxic substances such as Fe3+ and Al3+ for use as aggregation reagents as shown in Fig. S4 (ESI†). This showed that at the same concentration of ions, Cr3+ provided heavy aggregation of citrate-stabilized gold nanoparticles but Fe3+ and Al3+ did not aggregate AuNPs. The optimization of the pH of the phosphate buffer in the range 6.0–8.0 in the presence of Cr3+, Fe3+ and Al3+ was also carried out (Fig. S5 (ESI†)). Only Cr3+ gave the response of aggregation of the AuNPs. These two investigations confirmed that Cr3+ is more specific for Br− detection than other aggregation reagent ions (Fe3+ and Al3+).
It is known that the pKa1, pKa2 and pKa3 values of citric acid are 3.2, 4.8 and 6.4, respectively.38,44 When the pH value was lower than 6.4, the citric acid group can combine with H+, resulting in the reduction of the chelation interaction between the citrate ion and Cr3+. This negatively influenced the aggregation process, and as a result, a decrease in sensitivity was observed.
In the case of pH 6.5 (close to pKa3), citric acid exists in a trivalent anion and this has the highest ability to chelate Cr3+.37 This pH resulted in an increase in the affinity of chelation between the citrate ion and Cr3+. Therefore, high aggregation will lead to a high anti-aggregation process in the studied concentration range of Br− and then maximize sensitivity. When the pH was increased from 7.0–7.5, the sensitivity was reduced in this system because hydrolysis of Cr3+ probably occurred, resulting in the formation of colloidal Cr(OH)3 that could stop the chelating reaction of the citrate-capped-AuNPs with Cr3+.45,46 Another possible reason is that Br− was likely dispersed in the alkaline environment of the solution rather than adsorbed on the AuNP surface. To obtain the maximum response of detection, a phosphate buffer of pH 6.5 was chosen.
Liu and Wang mentioned that citrate-capped AuNPs would aggregate in aqueous media with high ionic strength.39 In this study, the maximum tolerable ionic strength was found to be 10.0 mM since this was when the maximum sensitivity was obtained. The results showed that a buffer concentration of 25.0 mM led to a large increase in the ionic strength, which could cause the anti-aggregation to decrease and thus cause the intensity ratio to decrease. By considering the maximal ratio between the signal of the analyte and the blank, 10.0 mM phosphate buffer (pH 6.5) was chosen for the next experiment.
3.3 Method validation
The concentration of bromide ions in a solution was confirmed by comparing its color and absorbance with a standard solution under the same conditions. The degree of anti-aggregation (seen by the color of solutions changing gradually from blue to red) depends on the concentration of Br−. Fig. 5 shows the absorption changes with the addition of Br−, which were further confirmed by looking at the color changes. The absorbance at 673 decreased with an increasing concentration of Br− and the peak at 519 nm increased due to the anti-aggregation of the AuNPs. The absorbance ratio of A519/A673 was used for the quantitative determination of Br− since it was found to be less sensitive to changes caused by different detection conditions. | ||
| Fig. 5 UV-vis absorption spectra changes of the AuNPs in the presence of 0.00–10.01 μM Br−. | ||
A good linearity of the standard calibration curve (y = 3.4803x − 0.6065, R2 = 0.9970) was found in the range 0.31–3.75 μM Br− (Fig. 6a). The limit of detection (LOD) and limit of quantification (LOQ) were the Br− concentrations that gave a signal equal to the blank signal (S/N) plus three and ten standard deviations of the blank, respectively (n = 20).47 The LOD and LOQ were as low as 0.04 and 0.13 μM, respectively. The recovery was investigated by spiking the Br− concentrations of 12.00 to 60.00 mg kg−1 into composite rice samples and then testing them using our developed system. As seen in Table 1, good recoveries ranging from 79.9–92.2% were obtained with a precision (RSD) of less than 4.0% (n = 3). The inter-day precision of the three concentration levels of Br− (0.63, 1.25, 3.75 μM) determined on each of 5 days fell between 3.07 and 7.12% (n = 45), whereas the intra-day precision obtained from the 5 measurements of each of the three concentration levels on the same day ranged from 2.87–6.35% (n = 15). These recoveries and precisions were acceptable within the range referred to in the AOAC standard method.48
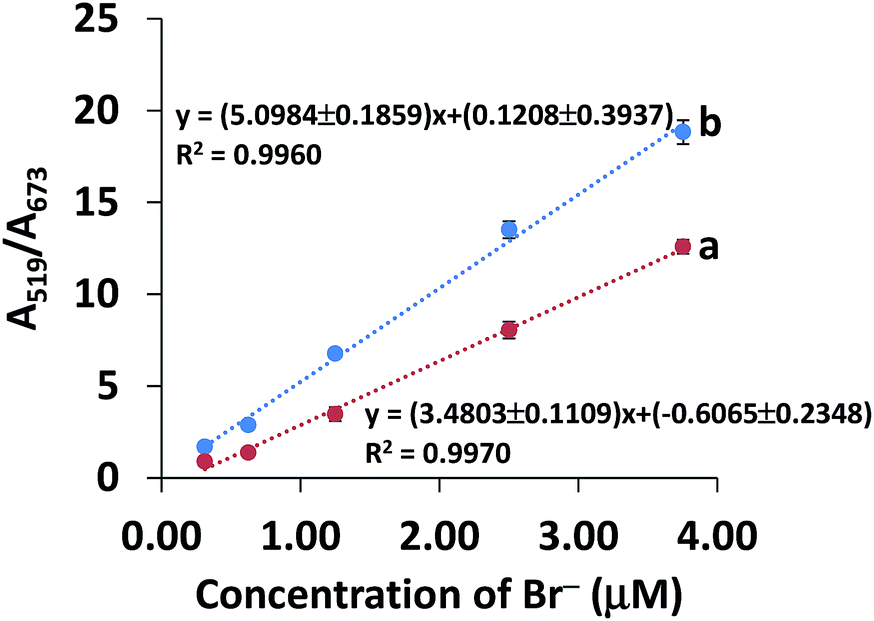 | ||
| Fig. 6 Curves of (a) the standard calibration curve and (b) the standard addition curve for Br− concentration in the range 0.31–3.75 μM. | ||
| Spiked (mg kg−1) | Detected (mg kg−1) | Recovery (%) | RSD (%) |
|---|---|---|---|
| 0.00 | 2.19 ± 0.57 | — | — |
| 12.00 | 11.78 ± 0.33 | 79.94 | 2.81 |
| 25.00 | 23.52 ± 0.94 | 85.35 | 3.98 |
| 50.00 | 48.28 ± 1.86 | 92.19 | 3.85 |
| 60.00 | 50.42 ± 1.18 | 80.40 | 2.35 |
In contrast to the aqueous standard solution, trace quantification of the analyte in real samples can suffer from the effects of the sample matrices, which can cause a positive or negative response by increasing background noise or suppressing the signal response.49 Hence, the matrix effect was investigated using a standard addition curve (Fig. 6b) in the range 0.31–3.75 μM Br− (spiked into rice water extracts), which was compared to a standard calibration curve (Fig. 6a). As shown in Fig. 6, the sensitivity of the standard addition curve (b) was significantly different from that of the standard curve (a) (P < 0.05), indicating that the matrix did have an effect on the analysis of Br− in rice samples. Therefore, we used a standard addition curve for accurate determination of Br− in rice samples.
3.4 Comparison of the developed sensor with other analytical methods
To further demonstrate the advantages of the developed sensor, the performance of our sensor was compared with other previously reported methods. Table 2 lists the comparison results, including sample amount, linear range, LOD and detection time, of our developed sensor with other methods. The LOD and linearity obtained by our sensor were clearly superior to those obtained in the other studies. In addition, our sensor has a short detection time compared with the expensive methods.3,52,53 Our sensor required a comparable amount of sample to be used for detecting Br− in real samples to the methods mentioned in Table 2. Overall, these results confirmed that our method for Br− determination based on the anti-aggregation of AuNPs was more efficient and sensitive.| Methoda | Sample | Sample amount | Linear range (μM) | LOD (μM) | Detection time (min) | Reference |
|---|---|---|---|---|---|---|
| a LLE-deriv.-GC-μECD: liquid–liquid extraction-derivatization-gas chromatography-micro electron capture detector; ICP-MS: inductive coupled plasma-mass spectrometry; LPIEC: low pressure ion-exchange chromatography. | ||||||
| LLE-deriv.-GC-μECD | Rice | 2 g | 62.58–938.63 | 25.03 | 20 | 3 |
| Microwave digestion-ICP-MS | Food | 1–2 g | 25.03–2503.00 | 8.51 | 1 | 16 |
| Phenol red spectrophotometry | Vegetables | 5 g | 6.26–56.32 | 3.13 | 3 | 18 |
| LPIEC-visible light detection | Foodstuffs | 25 g | 0.38–62.58 | 0.03 | 7.5 | 19 |
| Phenol red and chloramine-T colorimetry | Environmental samples | 20–300 μL | 0.00–375.45 | 1.38 | 5 | 50 |
| Aggregation and anti-aggregation of AgNPs | Water | — | 0.99–5.66 | 1.67 | — | 51 |
| Total reflection X-ray fluorescence spectrometry | Soil | 1 g | 15.64–156.44 | 3.13 | 25 | 52 |
| Fluorescent Ag nanocluster detection | Dried kelp | 1 g | 0.10–50.00 | — | 60 | 53 |
| Anti-aggregation of AuNPs | Rice | 2 g | 0.31–3.75 | 0.04 | <10 | This work |
3.5 Selectivity study
Other cations and anions in rice samples were investigated as they could probably interfere with the selectivity for Br−. It was reported that inorganic anions, i.e. F−, Cl−, NO2−, NO3− and SO42−, were found in rice samples from four provinces in China using ion chromatography, ranging from not detected to 20.90 mg kg−1.54 Some elements in rice extracts prepared using our alkaline digestion and ashing method were determined using inductively coupled plasma-optical emission spectrometry (ICP-OES) as presented in Table S1 (ESI†). It was shown that the samples had high concentrations of K, Mg, Ca and P in the range 6.66–13554.17 mg kg−1 whereas Al, Cr and Se were found in low concentrations (0.20–2.79 mg kg−1). The detection of K, Ca, Mg and Al agreed with the data of other reports.55,56 Thus, the 10 common ions, i.e. K+, Mg2+, Ca+, Al3+, NO2−, NO3−, PO43−, SO42−, F−, Cl−, were investigated alongside Br− under the same optimal conditions. We found that Cu, Fe, Mn, Ni and Zn were not detected, thus they were not included as interferences.
The selectivity was evaluated by testing the absorbance ratio (A519/A673) of 5 μM Br− to 5000 μM of other ions including K+, Mg2+, Ca2+, Al3+, NO2−, NO3−, PO43−, SO42−, F− and Cl−. As shown in Fig. 7, Br− (red bar) showed a distinct signal whereas in the absence of Br−, the signal of the other ions except for Cl− (blue bar) was almost identical to the blank even when their concentrations were 1000-fold higher than Br−, suggesting that no anti-aggregation of AuNPs was caused by the other ions. Furthermore, the response of Br− in the presence of a mixture of all the ions (violet bar) increased to a level similar to that of Br− alone (red bar). This can be explained by the fact that Br− is lightly hydrated and has higher polarizability which can be attractive when it is close to the AuNP surface compared to NO3−, SO42−, F− and Cl− while the other negative ions are repelled from the surface.57 Only the addition of 5000 μM Cl− can result in anti-aggregation. However, considering that a low abundance of Cl− (about 3.05 mg kg−1) was detected in the rice sample matrix, the signal caused by Cl− can be ignored.
 | ||
| Fig. 7 Selectivity of the Br− detection. The absorbance ratio response (A519/A673) of the proposed sensor for 5 μM Br− (red bar) was plotted against 5000 μM of ion interferences (K+, Mg2+, Ca2+, Al3+, NO2−, NO3−, PO43−, SO42−, F− and Cl− (blue bars)) and a mixture of Br− and the other ions (violet bar). All signals corresponded to the blank. The photograph is an example of the detection sensor for 5 μM Br−, the other ions and a mixture of Br− and all the other ions. | ||
Other anions such as I−, S2−, SO32−, and SCN− could affect the detection of Br−. Thus, the effect of I− and SCN− on the selectivity of the detection was investigated and the results are shown in Fig. S9 (ESI†), and the responses of these ions were similar to that of Br−. This means that they interfere with the anti-aggregation of AuNPs because I− and SCN− are classified as chaotropic anions like Br−.57 However, SCN−, S2− and SO32− have never been reported in rice samples, thus they are not of major concern for detection. It was reported that the concentration of I− is approximately 185 times lower than that of Br− in white rice samples,55 and therefore, the signal caused by the low abundance of I− in real samples can be ignored.
These results clearly revealed that the simultaneous presence of other competitive ions even at very high concentrations did not interfere with the determination of Br−. Thus, our colorimetric sensor exhibited a high selectivity for Br− over other ions in rice samples.
3.6 Application of the method to rice samples
To further evaluate the feasibility of the developed sensor, five different commercial brands of white rice were collected from supermarkets and examined for Br− residues. The first attempt to prepare the rice sample was made by soaking the rice in water as a simple method and the water extract of the rice was then passed through a solid phase sorbent cartridge (Oasis HLB) and analyzed using colorimetric determination. It appeared that no significant difference in color between the unspiked and spiked sample was observed (both samples presented a red solution), indicating that the interferences, especially organic substances as well as Br−, could dissolve in aqueous extracts.Srisawat et al. examined phenolic or flavonoid compounds in rice water extracts.58 These compounds could probably interfere with the Br− detection by: (i) adsorbing onto the surface of the AuNPs and hindering the ligand-exchange process in the Cr3+–citrate complex and (ii) forming a complex with Cr3+ instead of with the citrate. Therefore, the removal of the organic interferences is necessary.
Our sample digestion method using 1% potassium hydroxide in 50% ethanol and our ashing procedure to remove all the organic contents from the rice samples was studied and compared with an acidic digestion using hydrogen peroxide and hydrochloric acid. Metals in all the samples were analyzed using ICP-OES. As seen in Table S1 (ESI†), the concentrations of other cations such as Mg2+, Ca2+ and Al3+ obtained using our method were much lower than those obtained via acidic digestion except that a higher amount of K+ was detected. This was due to the addition of KOH for rice digestion; however, it has proved that K+ residue does not interfere with colorimetric detection using AuNPs as shown in Fig. 7.
In addition, high recoveries for the sample preparation and detection were obtained (Table 1). Since we used a high temperature (600 °C) for the ashing step, it was possible that Br− could evaporate, and so the remaining Br− in the samples was investigated using the X-ray fluorescence (XRF) technique. The composite rice samples were spiked with Br− concentrations of 50, 100, 200 and 400 M and prepared as mentioned in Section 2.5. After dilution, the solution was adjusted to pH 8.0 with 1 M nitric acid, and was subsequently analyzed using the XRF method. Fig. S10 (ESI†) shows the XRF spectra of the sample solutions spiked with Br− concentrations in the range 50–400 M. The peak signals of Br− increased with increasing Br− concentration. This confirmed that Br− will not evaporate at a temperature of 600 °C. Thus, these results confirmed that our alkaline digestion and ashing method can remove some metals and organic interferences without any loss of Br−.
Whereas Sungwaranond et al.2 performed the ashing step for 8 h, leading to a time consuming sample preparation, we further investigated the time for ashing (4 and 8 h). The results showed that statistically insignificant differences in the concentrations of cations with an ashing step of 4 and 8 h were observed in the samples (Table S1 (ESI†)). Thus, in this work, the rice samples were treated using the alkaline digestion and ashing method for 4 h.
Table 3 shows the determination of Br− in rice samples using our sensor, together with the corresponding colorimetric results. The color chart, made by adding a Br− standard ranging from 6.25 to 50.00 mg kg−1 into the samples, was made for detection with the naked eye. Br− residues were detected in all the studied samples in the range 3.12–5.21 mg kg−1, which all fell in the range 0.00–6.25 mg kg−1 shown in the color chart and were higher than those detected by Parengam et al. (0.40–1.4 mg kg−1).55 However, the detectable concentrations of Br− did not exceed the MRL limit set by The Thai Ministry of Public Health (50 mg kg−1).13 Moreover, it showed that no interferences due to co-extracted compounds were found as seen in the colorimetric results of the samples (Table 3(A)).
| Sample | Found (mg kg−1) | |
|---|---|---|
| Developed sensor | Ion chromatography | |
| White rice 1 | 3.12 ± 0.83 | 3.41 ± 1.61 |
| White rice 2 | 4.23 ± 1.10 | 6.25 ± 0.80 |
| White rice 3 | 4.07 ± 1.70 | 3.41 ± 2.41 |
| White rice 4 | 4.16 ± 0.48 | 4.00 ± 2.41 |
| White rice 5 | 5.21 ± 0.91 | 6.82 ± 4.82 |
 |
||
At present, there is no established standard method for the detection of Br− in rice samples. We have validated our developed sensor with the ion chromatographic (IC) technique (Table 3). It was found that the concentrations of Br− detected using the developed sensor were not significantly different from those detected using IC (P > 0.05).
These results confirmed that the developed sensor method can be used for the analysis of Br− in real complex samples.
4. Conclusions
We have developed a robust colorimetric sensor for bromide ions using citrate-capped AuNPs in the presence of Cr3+. The method was simple, highly sensitive and selective, and was based on the anti-aggregation induced by Br−, which results in a color change from blue to red upon the addition of different concentrations of Br−. This system presents a low LOD and LOQ without any noticeable interfering effects from the other components in rice samples. Satisfying recovery and precision were achieved with our developed colorimetric sensor.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Research Assistantship (Contract No. 1-2558-02-003), Faculty of Science, Prince of Songkla University, the Department of Chemistry, the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Office of the Higher Education Commission, Ministry of Education (OHEC), and the Graduate School, Prince of Songkla University.References
- K. N. T. Norman, Pest Manage. Sci., 2000, 56, 154–158 CrossRef.
- B. Sungwaranond, P. Jongmeevasana and G. Thoophom, Bulletin of the Department of Medical Sciences, 1998, 40(2), 171–177 Search PubMed.
- W. Wittayanan and W. Jongmeevasana, Bulletin of the Department of Medical Sciences, 2015, 57(3), 219–232 Search PubMed.
- U.S Department of Health and Human Services, Occupational health guideline for methyl bromide, 1978, pp. 1–5 Search PubMed.
- United States Environmental Protection Agency (U.S. EPA), Protection of Stratospheric Ozone: Incorporation of Montreal Protocol Adjustment for a 1999 Interim Reduction in Class I, Group VI Controlled Substances. Fed. Regist., 1999, 64(104), 29240–29245 Search PubMed.
- W. B. Thomas, J. Nematol., 1996, 28(4S), 586–589 Search PubMed.
- FAO (1999) Food Agriculture Organization-World Health Organization, Codex maximum limits for pesticide residues, Codex Alimentarius Commission, FAO plant production and protection paper.
- CCM, China reveals a list of newly banned insecticides, http://www.cnchemicals.com/Press/89543-China%20reveals%20a%20list%20of%20newly%20banned%20insecticides.html, accessed 18. 11. 2017.
- X. Baur, L. T. Budnik, Z. Zhao, M. Bratveit, R. Djurhuus, L. Verschoor, F. M. Rubino, C. Colosio and J. R. Jepsen, J. Occup. Med. Toxicol., 2015, 10(19), 1–18 Search PubMed.
- M. Miyahara and Y. Saito, J. Agric. Food Chem., 1994, 42, 1126–1131 CrossRef.
- D. Cova, G. F. Molinari and L. Rossini, Food Addit. Contam., 1986, 3(3), 235–240 CrossRef PubMed.
- The Montreal Protocol on Substances that Deplete the Ozone Layer/Article 2H: Methyl bromide, http://www.ozone.unep.org/en/handbook-montreal-protocol-substances-deplete-ozone-layer/16, accessed 9. 04. 2018.
- Notification of the Ministry of Public Health (No. 361) B.E. 2556, 2013, page 5.
- P. Pimpun, Methyl bromide and bromide ion, http://www.thaipan.org/sites/default/files/file/4%20MB-Br%20.Prapassara.pdf, accessed 4. 11. 2017.
- A. A. Mahmoud, R. Gouda and L. Ryad, Journal of Plant Protection and Pathology, 2014, 5(11), 1015–1023 Search PubMed.
- T. K. D. Nguyen and R. Ludwig, Anal. Sci., 2014, 30, 1089–1092 CrossRef PubMed.
- G. Chiu and R. D. Eubanks, Microchim. Acta, 1989, 2, 145–148 CrossRef.
- E. Baso-Cejas, G. Brito, C. Díaz and E. M. Peña-Méndez, Bull. Environ. Contam. Toxicol., 2007, 78, 417–420 CrossRef PubMed.
- L. Yu, X. Zhang, J. Jin, S. Che and L. Yu, Czech J. Food Sci., 2011, 29(6), 634–640 CrossRef.
- Y. Zhou, H. Dong, L. Liu, M. Li, K. Xiao and M. Xu, Sens. Actuators, B, 2014, 196, 106–111 CrossRef.
- Y. Li, P. Wu, H. Xu, Z. Zhang and X. Zhong, Talanta, 2011, 84, 508–512 CrossRef PubMed.
- M. R. H. Nezhad and S. A. Moayed, Talanta, 2014, 129, 227–232 CrossRef PubMed.
- D. Liu, Z. Wang and X. Jiang, Nanoscale, 2011, 3, 1421–1433 RSC.
- M. Zhang, Y. Q. Liu and B. C. Ye, Analyst, 2011, 136, 4558–4562 RSC.
- T. Lou, L. Chen, C. Zhang, Q. Kang, H. You, D. Shen and L. Chen, Anal. Methods, 2012, 4(2), 488–491 RSC.
- G. Wang, Z. Chen, W. Wang, B. Yan and L. Chen, Analyst, 2011, 136(1), 174–178 RSC.
- L. Chen, W. Lu, X. Wang and L. Chen, Sens. Actuators, B, 2013, 182, 482–488 CrossRef.
- Z. Zhang, J. Zhang, C. Qu, D. Pan, Z. Chen and L. Chen, Analyst, 2012, 137, 2682–2686 RSC.
- Z. Zhang, Z. Chen, S. Wang, C. Qu and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6(9), 6300–6307 Search PubMed.
- J. Kang, Y. Zhang, X. Li, L. Miao and A. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 1–5 Search PubMed.
- A. Sharma, Z. Matharu, G. Sumana, P. R. Solanki, C. G. Kim and B. D. Malhotra, Thin Solid Films, 2010, 519, 1213–1218 CrossRef.
- Y. Luo, J. Xu, Y. Li, H. Gao, J. Guo, F. Shen and C. Sun, Food Control, 2015, 54, 7–15 CrossRef.
- M. Akhond, G. Absalan and H. Ershadifar, Spectrochim. Acta, Part A, 2015, 143, 223–229 CrossRef PubMed.
- S. Kong, M. Liao, Y. Gu, N. Li, P. Wu, T. Zhang and H. He, Spectrochim. Acta, Part A, 2016, 157, 244–250 CrossRef PubMed.
- J. Song, F. Wu, Y. Wan and L. Ma, Food Control, 2015, 50, 356–361 CrossRef.
- M. Elavarasi, A. Rajeshwari, N. Chandrasekaran and A. Mukherjee, Anal. Methods, 2013, 5, 6211–6218 RSC.
- A. Heller, A. Barkleit, H. Foerstendorf, S. Tsushima, K. Heim and G. Bernhard, Dalton Trans., 2012, 41, 13969–13983 RSC.
- V. I. Kornev and G. A. Mikryukova, Russ. J. Coord. Chem., 2004, 30(12), 946–950 CrossRef.
- Y. Liu and X. Wang, Anal. Methods, 2013, 5, 1442–1448 RSC.
- C. Pfeiffer, C. Rehbock, D. Hühn, C. Carrillo-Carrion, D. J. de Aberasturi, V. Merk, S. Barcikowski and W. J. Parak, J. R. Soc., Interface, 2014, 11(20130931), 1–13 Search PubMed.
- G. Senanayake, Miner. Eng., 2004, 17, 785–801 CrossRef.
- E. G. Vinokurov and V. V. Bondar, Russ. J. Coord. Chem., 2003, 29(1), 66–72 CrossRef.
- K. Shrivas, S. Sahu, G. K. Patra, N. K. Jaiswal and R. Shankar, Anal. Methods, 2016, 8, 2088–2096 RSC.
- M. Chen, H.-H. Cai, F. Yang, D. Lin, P.-H. Yang and J. Cai, Spectrochim. Acta, Part A, 2014, 118, 776–781 CrossRef PubMed.
- X. Wang, Y. Wei, S. Wang and L. Chen, Colloids Surf., A, 2015, 472, 57–62 CrossRef.
- L. Spiccia and W. Marty, Inorg. Chem., 1986, 25, 266–271 CrossRef.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson Education Limited, England, UK, 5th edn, 2005, Essex Search PubMed.
- AOAC, Guidelines for Standard Method Performance Requirements, Appendix F, 2016, pp. 2–18 Search PubMed.
- T. Rujiralai, N. Raekasin, W. Cheewasedtham and C. Cheewasedtham, Chem. Pap., 2014, 68(8), 1041–1048 Search PubMed.
- B. J. Lepore and P. Barak, Soil Sci. Soc. Am. J., 2009, 73, 1130–1136 CrossRef.
- S. Bothra, R. Kumar, R. K. Pati, A. Kuwar, H.-J. Choi and S. K. Sahoo, Spectrochim. Acta, Part A, 2015, 149, 122–126 CrossRef PubMed.
- H. Gallardo, I. Queralt, J. Tapias, L. Candela and E. Margui, Chemosphere, 2016, 156, 294–301 CrossRef PubMed.
- L. Fu, C. Li, Y. Li, S. Chen, Y. Long and R. Zeng, Sens. Actuators, B, 2017, 240, 315–321 CrossRef.
- H. Sun, L. Xia, S. Liang and S. Shen, Food Analytical Methods, 2014, 7, 1791–1797 CrossRef.
- M. Parengam, K. Judprasong, S. Srianujata, S. Jittinandana, S. Laoharojanaphand and A. Busamongko, J. Food Compos. Anal., 2010, 23, 340–345 CrossRef.
- L. Lu, S. Tian, H. Liao, J. Zhang, X. Yang, J. M. Labavitch and W. Chen, PLoS One, 2013, 8(2), e57360 Search PubMed.
- V. Merk, C. Rehbock, F. Becker, U. Hagemann, H. Nienhaus and S. Barcikowski, Langmuir, 2014, 30, 4213–4222 CrossRef PubMed.
- U. Srisawat, W. Panunto, N. Kaendee, S. Tanuchit, A. Itharat, N. Lerdvuthisopon and P. Hansakul, J. Med. Assoc. Thailand, 2010, 93(7), S83–S91 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03497d |
| This journal is © The Royal Society of Chemistry 2018 |