
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral ionic liquids supported on natural sporopollenin microcapsules†
Ivan Palazzoa,
Andrea Mezzettaa,
Lorenzo Guazzelli *a,
Stefania Sartinia,
Christian Silvio Pomellia,
Wallace O. Parker Jrb and
Cinzia Chiappe*a
*a,
Stefania Sartinia,
Christian Silvio Pomellia,
Wallace O. Parker Jrb and
Cinzia Chiappe*a
aDipartimento di Farmacia, Università di Pisa, Via Bonanno 33, 56126, Pisa, Italy. E-mail: cinzia.chiappe@unipi.it; lorenzo.guazzelli@unipi.it
bEni, Research & Technical Innovation, Physical Chemistry Department, Via Maritano 26, San Donato Milanese, 20097, Italy
First published on 8th June 2018
Abstract
Supported chiral ionic liquids (SILs) were prepared choosing the starting material for the ionic liquid part from the enantiopure stock of the chiral pool (monoterpenoids and an amino acid) and the sporopollenin as an environmentally friendly support. Sporopollenins are microcapsules with naturally well-defined sizes and shapes that can be obtained from pollen grains after removal of the internal cytoplasm and the second shell layer. As thermally stable organic biocompatible structures, sporopollenins have attracted increasing interest in recent years for several applications. Herein, bio-based ILs were anchored onto the surface of sporopollenins obtained from the pollen of Populus deltoides, selected as a model pollen grain. These new structures, which present an external positively charged shell, were characterized by physico-chemical techniques (ATR-FTIR, TGA, SEM, EDX, and solid-state 13C NMR). A metathesis reaction was also performed on selected bio-based IL modified sporopollenins, demonstrating the possibility to switch the surface properties by exploiting well-known IL chemistry.
Introduction
In the last two decades, ionic liquids (ILs) have received increasing interest in the area of green chemistry,1,2 where they can play an important role as alternative environmentally friendlier media with unique solvent properties, including the peculiar ability to dissolve biopolymers,3,4 and/or as catalysts for several important reactions.5,6 However, in contrast to what was generally thought only 10 years ago, it is well-known that ILs also present some drawbacks that need to be addressed: ILs can be more toxic that molecular solvents, their cost is often higher than that of molecular solvents and they are generally based on petrol derived raw materials.7,8 Thus, recently, considerable attention has been directed toward the replacement of the most common IL constituent ions with bio-based materials9,10 which have higher biodegradability and unlimited availability from renewable resources. On the other hand, immobilization of ILs on suitable supports represent another strategy to reduce costs and increase sustainability. In organic catalytic reactions, supported ionic liquids (SILs) offer several advantages over free ILs, reducing the amount of used IL, avoiding its leaching, and improving the products recoverability and reusability of IL and catalysts.11 Nonetheless, as novel functional materials in surface science and material chemistry, SILs can modify and improve the properties of solids, modifying wettability, lubricating properties, separation efficiency and electrochemical response.12 To date, zeolite-, silica-, carbon nanotube-, polystyrene- and cross-linked polymers represent the most used materials to obtain SILs. However, many of these IL supports possess some disadvantages: complicated preparation technologies and a high production costs are required, in particular, when empty size homogeneous systems with peculiar microscale and nanoscale architectures are mandatory. Thus, the possibility to have highly effective and cheap polymeric supports for the heterogeneous catalysts is high desirable. Nature can provide a myriad of unique, widely available biological materials with surprising microscale and nanoscale architectures. One interesting natural alternative to synthetic organic polymers is represented by sporopollenin, a highly cross-linked polymer, which is the main component of outer shell of pollen grains and plant spores (exine).13 It is characterized by a remarkable chemical, physical and biological stability even at high temperatures, that arise from its high cross-linked structure involving tertiary carbons and ethers. Although it is still a matter of discussion,14–21 sporopollenin possesses aliphatic chains bound to aromatic moieties, conjugated phenols, aliphatic alcohols, lactones, and carboxylic acids. Morphologically, sporopollenin exine capsules (SEC) are microcapsules with a hydrophobic cavity and a variable number of pores, species specific in shape and size, with elaborated 3D surface architectures. SEC have been used to sequester edible oils from emulsions efficiently, encapsulate various polar and nonpolar materials, and act as drug delivery systems with excellent enhanced bioavailability.22–27 On the other hand, the presence of functional groups on the outer surface allows for their further modification. Chemically modified SEC have been employed in water remediation studies,28 as stationary phases chromatography,29 and as solid supports for peptide synthesis,30 enzyme immobilization,31,32 and chemical reactions.33,34 In the context of drug delivery, efficient drug encapsulation systems have also been prepared by coating SEC with natural biopolymer composites.35,36 Recently, sporopollenin has also been used in the area of biomass valorization. SEC surfaces have been modified by sulfonation, and the obtained material tested as a heterogeneous catalyst in the dehydration of D-xylose and xylan to produce furfural.37Despite its potentiality also the chemistry of SEC presents some drawbacks in term of sustainability. Extraction of SEC from the whole pollen grains and plant spores traditionally entails the use of harsh working protocols/conditions (e.g. acetic anhydride/sulfuric acid and heating,38 HF,39 4-methylmorpholine N-oxide,40 KOH/H3PO4 (ref. 41)). Moreover, these procedures, which are necessary to remove the internal cytoplasm and the second shell layer (intine), comprised mainly of cellulose, hemicellulose, and pectin, can determine significant modifications on the polymer composition and capsule shape, negatively affecting their use in a variety of the above mentioned applications.
However, an improvement to SEC extraction, in terms of time and environmental impact, has been proposed by Mundargi et al.42 and, more recently, the possibility to use ILs as extraction media has been demonstrated by Chiappe et al., although the product was not fully characterized or explored as polymeric support.43
Herein we show that sporopollenin microcapsules, purified using an IL, can be used to prepare, through grafting procedures, charged natural polymeric capsules. Despite the increasing number of studies aimed at using SEC as solid supports, surprisingly, these microcapsules have never been employed for the immobilization of ILs. The excellent stability and durability of the employed materials and the tunability of anchored ILs, which are also chiral, give the possibility to obtain new environmentally friendly functional materials with high potentialities for application in catalytic reactions, separation technologies and electrochemistry.
Results and discussion
Sporopollenin exine capsules (SEC) were isolated from defatted pollen of Populus deltoides using the recently reported extraction protocol based on acidic ILs.43 Briefly, pollen grains were suspended in 1-(4-sulfobutyl)-3-methylimidazolium hydrogen sulfate, [HSO3C4mim][HSO4], and heated to 160 °C for 90 minutes. The ATR-FTIR spectrum of the isolated SEC (Fig. 1) is in agreement with those reported previously. Comparison with the spectrum of the pristine pollen clearly shows the removal of the polysaccharide portion (absence of the strong C–O–C and C–OH stretching at 1047 cm−1), of the protein component (absence of the amide II band at 1544 cm−1), and of the lipid fraction (absence of signals at 1738 cm−1 and 1408 cm−1). These data confirm that the treatment with [HSO3C4mim][HSO4] can be considered an adequate chemical protocol to eliminate all the undesired components, including also proteins that may cause potential allergic reactions. However, as previously observed,43 the main peculiarity of the FTIR spectrum of these SEC was the presence of two peaks at 1183 cm−1 and 1036 cm−1, which are usually not detected. Such signals can be assigned to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO group stretching modes. In agreement, elemental analysis showed the presence of sulfur in isolated SEC which was confirmed also by the EDX analysis. Although, in principle, these bands could be related to the presence of residual IL, having the SO3H group on the cation and hydrogen sulphate as the anion, their persistence also after extensive washing with ethanol, methanol, toluene, and water strongly suggested the presence of a covalent linkage. Previously, we hypothesized43 the transformation of the hydroxyl groups on sporopollenin into the corresponding sulphate monoesters, through a process probably catalyzed by the acidic IL. This supposition finds a further support in the fact that the FTIR spectrum of a sulfonated sporopollenin, recently prepared by treating sporopollenin with chlorosulfonic acid,37 turns out to be very similar to the spectrum presented in this work, the two aforementioned peaks being present in both spectra.
SO group stretching modes. In agreement, elemental analysis showed the presence of sulfur in isolated SEC which was confirmed also by the EDX analysis. Although, in principle, these bands could be related to the presence of residual IL, having the SO3H group on the cation and hydrogen sulphate as the anion, their persistence also after extensive washing with ethanol, methanol, toluene, and water strongly suggested the presence of a covalent linkage. Previously, we hypothesized43 the transformation of the hydroxyl groups on sporopollenin into the corresponding sulphate monoesters, through a process probably catalyzed by the acidic IL. This supposition finds a further support in the fact that the FTIR spectrum of a sulfonated sporopollenin, recently prepared by treating sporopollenin with chlorosulfonic acid,37 turns out to be very similar to the spectrum presented in this work, the two aforementioned peaks being present in both spectra.
 | ||
| Fig. 1 ATR-FTIR spectra of sporopollenin (SEC), 3-chloropropyl-functionalized sporopollenin CPS-SEC and IL-functionalized sporopollenins: 5b and 6b. | ||
Although this functionalization could negatively affect the subsequent functionalization reactions, fortunately, the treatment of the isolated SEC with a solution of (3-chloropropyl)triethoxysilane (CPTES), following a procedure similar to that recently reported in the literature,44 gave the desired Si-functionalized sporopollenin, CPS-SEC. CPS-SEC were further transformed into IL-modified sporopollenin capsules (5a–d) by reaction with four different 1-alkylimidazoles (4a–d), Scheme 1.
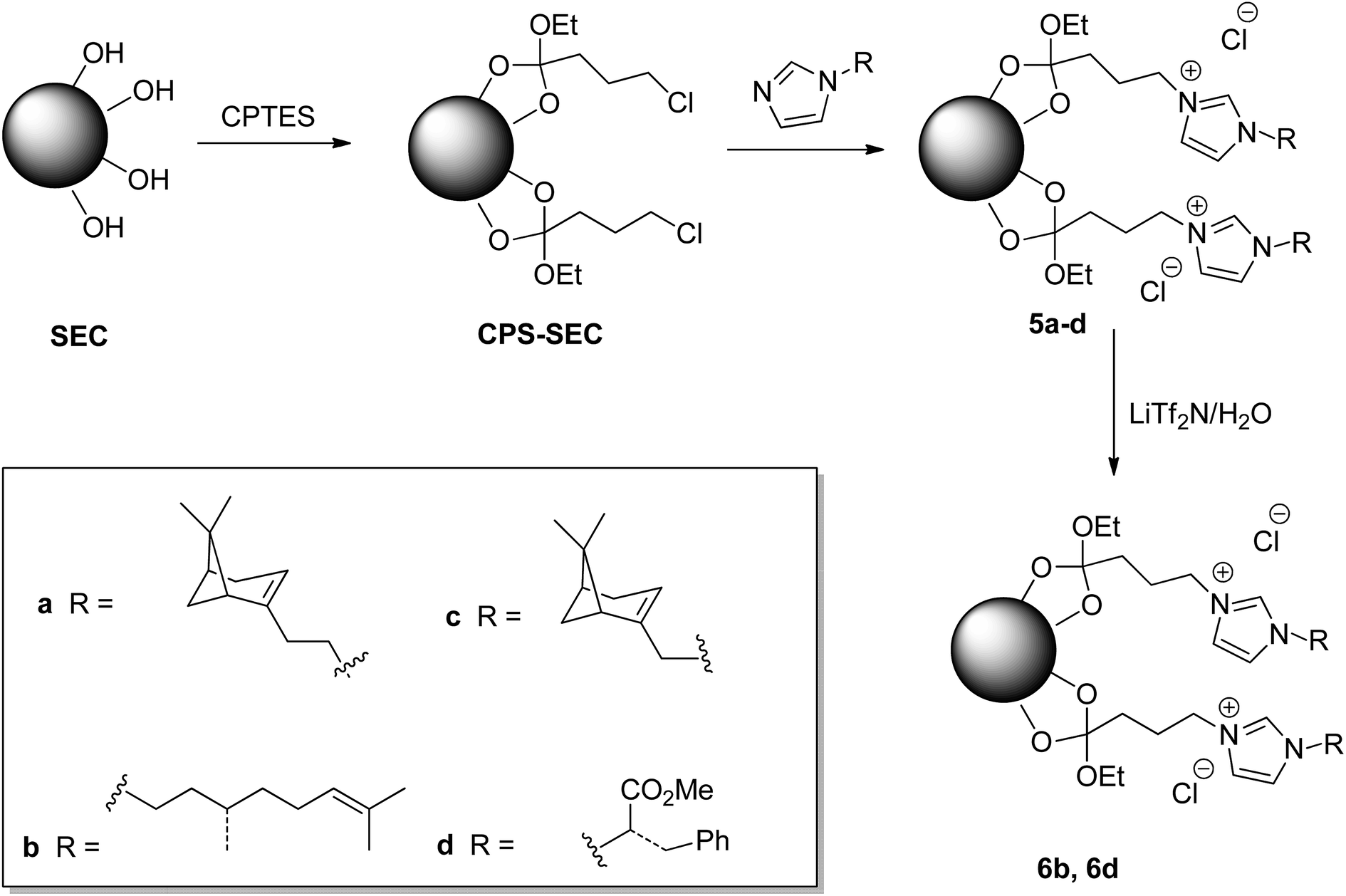 | ||
| Scheme 1 Preparation of ILs functionalized SEC. | ||
In particular, three natural alcohols, namely (−)-nopol (1a), (S)-(−)-β-citronellol (1b) and (−)-myrtenol (1c), which are also chiral, were used as starting materials to synthesize the respective 1-alkylimidazoles in a two step process. (−)-Nopol and (S)-(−)-β-citronellol were transformed, in very high yields, into the corresponding mesylates (2a–b) by reaction with methanesulfonyl chloride, in the presence of Et3N, and the isolated compounds 2a–b were reacted with an equimolar amount of sodium imidazolate. Interestingly, when the same approach was performed on (−)-myrtenol, only the corresponding allyl chloride (3c) was isolated in good yield. This probably arises from the displacement by the chloride of the first formed mesylate. Reaction of 3c with sodium imidazolate gave the expected product (4c) in good yield (Scheme 2).
 | ||
| Scheme 2 Preparation of alkylimidazoles. | ||
Chiral imidazole 4d was obtained instead starting from L-phenylalanine, glyoxal, formaldehyde, and ammonia through the Debus–Radziszewski reaction.45 The desired product 4d was isolated in 48% yield, in agreement with the literature.45
To provide new properties to the IL-modified SEC, an anion metathesis aimed at substituting the chloride anion with the bis(trifluoromethylsulfonyl)imide anion was performed on compounds 5a and 5d. This kind of anion metathesis is usually performed to prepare highly hydrophobic ionic liquids. Likewise, the presence of this heavily fluorinated anion could determine the hydrophilic–hydrophobic switch of the sporopollenin surface, contemporaneously, increasing the thermal stability of the resulting material. The well-known tunability of ILs can become an efficient method to modify the whole sporopollenin surface. The reaction was carried out by suspending the IL-functionalized sporopollenin in an aqueous solution of LiTf2N and stirring the mixture at room temperature for 48 hours, to give derivatives 6b and 6d (Scheme 1).
IL modified sporopollenins 5a–d, the related Tf2N derivatives, and their precursors were characterized by means of ATR-FTIR, SEM, EDX, solid-state 13C NMR analyses and TGA.
ATR-FTIR analysis and focal plane array imaging of the IL-functionalized SEC
Pollen grains and their modified forms were initially characterized by ATR-FTIR. The spectrum of CPTES immobilized sporopollenin CPS-SEC (Fig. 1) shows the characteristic adsorption band of the Si–O stretching at 1036 cm−1, demonstrating that the modification was successful. In Fig. 1 are also reported the IR spectra of the sporopollenin derived from the treatment with [C4mimSO3H][HSO4] (SEC) and the (S)-(−)-β-citronellol IL-functionalized sporopollenin both with Cl− and Tf2N− counterions, as examples of bio-based supported derivatives. These latter, although very similar to the CPS-SEC spectrum, show the distinctive absorption bands of the imidazolium moiety (around 1600 cm−1 and between 1109 cm−1 and 1132 cm−1). On the other hand, after anion exchange the typical strong absorption of CF3 and SO2 can be evidenced (asymmetric stretching of CF3, around 1200 −1, asymmetric and symmetric stretching of SO2, around 1350 and 1190–1130 cm−1, respectively). It is noteworthy that, although these bands in the case of free ILs often show the most pronounced dependence on the state of salt (liquid–solid) and CF3 and SO2 are the principal moieties involved in the cation and anion interactions, in this case moderate (if any) changes have been observed with respect to analogous not supported ILs, based on 1-alkyl-3-methylimidazolium cations. This suggest that, as desired, the grafting of the IL on the SEC surface is not accompanied by strong interactions between IL and support: thus, the IL should largely maintain their peculiar physico-chemical features.For selected sporopollenin samples, false-colour images were obtained using the Focal Plane Array (FPA) imaging technique, as shown in Fig. 2. For each sample, a real-colour optical image was first collected to select the area for imaging. Subsequently, after the FTIR spectra of the sample were recorded for each pixel, a wavenumber was selected in order to obtain information about the spatial distribution of specific chemical components on the micro-capsules. The colours of the pixels span from red to blue, passing through orange, yellow, green, and light blue, indicating an increasing absorbance on that pixel at the specified wavenumber. Fig. 2 illustrates visible and FPA images of 3-chloropropyl-functionalized sporopollenin CPS-SEC, and IL-functionalized sporopollenin, respectively. Data referred to pollen grains treated with [C4mimSO3H][HSO4] are not shown as they are in good agreement with results obtained in our previous work.43 The blue colour distribution in the FPA images of Fig. 2a and b (2D and 3D images, respectively), related to CPS-SEC sample, confirms not only that Si-derivatization has been carried with success but also that Si is well-distributed on the SEC surface. In this case, the wavenumber selected is 1035 cm−1, which is the characteristic band of Si–O bond stretching mode. In the lower row of Fig. 3, the optical image of CPS-SEC functionalized with (S)-(−)-β-citronellol Cl ionic liquid, sporopollenin 5b, is shown on the left. On the right, is the false-colour image at two selected wavenumbers. In Fig. 2c, the wavenumber is 1035 cm−1 corresponding to silanyl moieties, corroborating the fact that Si–O groups are always well distributed, while in Fig. 3d the referring band is 1563 cm−1, typical of imidazolium-based compounds. As seen from the false colour image, blue-green is also in this case quite well distributed on the surface of the analyzed sample, revealing an efficient functionalization of sporopollenin with chiral bio-based ionic liquid.
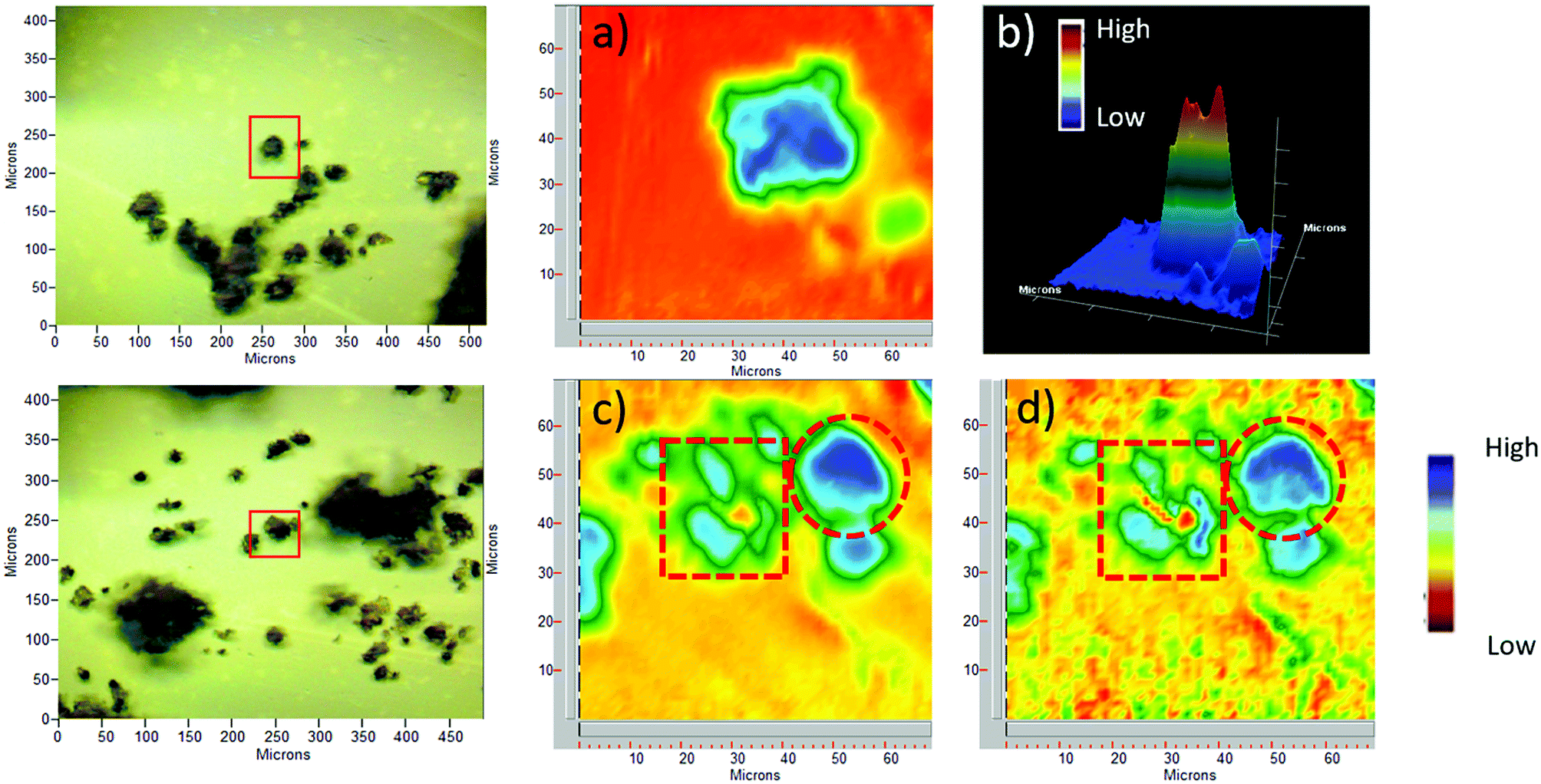 | ||
| Fig. 2 Optical and FPA images: (a) 2D image (blue is high) of 3-chloropropyl-functionalized sporopollenin CPS-SEC, selected wavenumber 1035 cm−1; (b) 3D image (red is high) of CPS-SEC; (c) 2D image of IL-functionalized sporopollenin 5b at 1035 cm−1; (d) 2D Image of 5b at 1563 cm−1. Red circles indicate intact pollen grains, whereas red squares show pollen fragments. | ||
 | ||
| Fig. 3 SEM images of (a) sporopollenin extracted with the IL [HSO3C4mim][HSO4], (b) 3-chloropropyl-functionalized sporopollenin CPS-SEC and (c) IL-functionalized sporopollenin 5b. | ||
SEM imaging and energy-dispersive X-ray (EDX) spectroscopy of the IL-functionalized SEC
In order to observe their morphology, SEM images were collected for the sporopollenin samples (Fig. 3). The sporopollenins capsule obtained after treatment with the acidic ionic liquid [HSO3C4mim][HSO4] is shown in panel a. The characteristic microstructure of the exine surface is partially lost after the treatment; moreover, the diameter of the capsule decreases slightly due to the removal of the intine layer and the interior protoplast. However, the circular shape is still well recognizable. Panel b illustrates a SEM image of the 3-chloropropyl-modified sporopollenin CPS-SEC, in which the presence on the sporopollenin capsule of an additional external layer can be observed, as well as the presence of some minor fragments on the capsule surface. Finally, panel c represents IL-functionalized sporopollenin 5b. As observed for all the other bio-based IL-functionalized sporopollenin synthesized (ESI†), the SEM image is similar to that of CPS-SEC, showing that the last reaction, IL grafting, has only a very few (if any) effect on capsule size and morphology.Finally, to detect elements not revealed by elemental analysis EDX microanalyses were performed on some of the sporopollenin samples (Fig. S3†). As expected, the spectrum of the pristine pollen shows a large variety of elements apart from carbon, nitrogen, oxygen, and sulphur. In particular, sodium, magnesium, phosphorus, potassium, iron, and lead were also detected. All these elements disappear in the EDX spectrum of the 3-chloropropyl-functionalized sporopollenin CPS-SEC, whereas silicon and chlorine are clearly visible. According to this analysis, the silicon concentration turns out to be around 0.58 mmol per gram of CPS-SEC. Among the IL-modified sporopollenin materials, 5a and 5b were chosen to perform the EDX microanalysis. The spectra show the presence of carbon, nitrogen, oxygen, sulphur, silicon, and chlorine as the most abundant elements. In particular, the ratio between Si/N changes in these latter samples in accordance with the IL functionalization.
Solid-state 13C NMR analyses
IL-functionalized sporopollenin 5a was examined also by 13C CP-MAS NMR spectroscopy. The spectrum obtained with 12 kHz sample rotation (Fig. 4) shows no appreciable spinning side-bands (95 ppm intervals), as verified by spinning at 8 kHz (see ESI Fig. S1†), indicating that the organic species giving the most intense peaks are not rigid solids. Several resolved peaks (10, 38, 41, 46, 122 ppm) correspond to those expected for 5a (Fig. S2†) can be indeed easily evidenced. However, there are also some extra species giving signals near 70 and 100 ppm (C–O), 150 ppm (oxygenated aromatics), 170 ppm (carboxylate) and 200 ppm (ketones), attributable to the SEC support.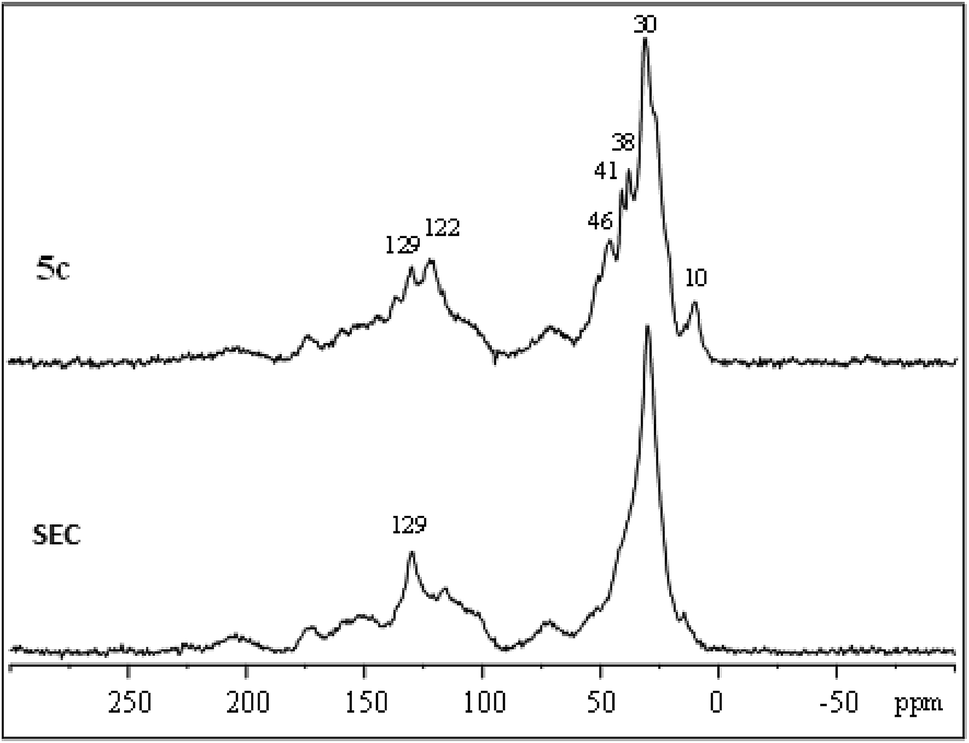 | ||
| Fig. 4 13C CP-MAS NMR spectrum of sporopollenin (SEC) and IL-functionalized sporopollenin 5a. | ||
The largest signal at 30 ppm, due to aliphatic carbons, was also observed for SEC, along with the sharp signal at 129 ppm. Sporopollenins contain aromatic (129 ppm) and aliphatic (30 ppm) carbons thought to originate from the phenylpropanoid pathway and from waxes of the exine.46 These two 13C signals have surprisingly short longitudinal relaxation times, being well evidenced even with re-cycle delays of only 0.3 s (Fig. S1†). Clearly, the carbon nuclei giving these signals are near paramagnetic sites (unpaired electrons). The paramagnetic properties of these materials, which will be more in detail examined in the future, could be of interest for specific applications, for instance for the development of bio-inspired paramagnetic particle labels to be used in magneto immunoassay, as well as for a more generic antioxidant activity.
Thermogravimetric analyses of IL-functionalized SEC
Finally, functionalized and non-functionalized sporopollenin microcapsules were also characterized by thermal gravimetric analysis, under nitrogen. Since it is well-known that temperature ramp in TGA measurements causes mass loss resulting from either thermal decomposition or evaporation of ILs and the temperature profile depends on some operating factors, such as heating rate, instrumentation, type of carrier gas, and sample amount, measurements were carried out under identical conditions. The maximum thermal degradation temperature of sporopollenin arising from IL treatment was recorded at 431 °C (Fig. S4†), in agreement with the high thermal stability of this biopolymer. It is worth noting the presence of a small shoulder at around 250 °C on the DTG plot that can be attributed to a first partial degradation, due to oxygen release. On the other hand, TGA of lightly washed sporopollenin, containing residual acidic IL, gave a well-distinguishable additional peak at about 200 °C, which disappeared after refluxing the sporopollenin in methanol (data not shown). The CPTES-modified sporopollenin showed a similar trend to the washed sporopollenin, with the principal peaks shifted toward higher temperatures (451 °C). Furthermore, the total weight loss was about 5% less for the modified sporopollenin than the unmodified one (Fig. 5a). This is probably due to the presence of silica atoms in the former. The thermogram of the IL-modified sporopollenin 5b presents two new peaks at 208 °C and 291 °C, which can be attributed to the degradation of the grafted IL moiety (Fig. 5b), whereas the maximum thermal degradation occurs at 448 °C, i.e. at a temperature which is close to the value of the unmodified sporopollenin. Interestingly, the degradation profile of the IL-modified sporopollenin 6b (Fig. 5b) shows instead only a small degradation peak at 204 °C (<5%), it principally decomposes at 437 °C. As expected, the thermal stability of the sporopollenin supported ionic liquids depends on counter anion: analogously to the situation characterizing free ILs, a significant thermal stability increase has been observed changing the anion from chloride to Tf2N. Still, this thermal stability is very high for an IL supported on capsules with biological origin and it is a desirable property for a carrier to be used in wide range of temperatures. | ||
| Fig. 5 a) Temperature-ramped TGA thermographs of CPS-SEC (dash dot line), IL functionalized sporopollenin 5b (solid line) and IL functionalized sporopollenin 6b (short dash line) with a heating rate of 10 °C min−1. (b) Temperature-ramped TGA thermographs of CPS-SEC (dash dot line), IL functionalized sporopollenin 5b (solid line) and IL functionalized sporopollenin 6b (short dash line). | ||
Conclusions
We believe that our approach, based on simple reactions, can provide new smart 3D-microcapsules exploiting the unique properties of pollen grains and tailor-made ionic liquids.Treatment with [C4mimSO3H][HSO4] allows to obtain easily sporopollenin capsules through an efficient removal of the other components, mainly cellulose and proteins.
SEM images show that, both after the first treatment and the following ones, the sporopollenin retained its distinctive spherical shape and collapse phenomena having detrimental effects on absorption/release mechanisms, generally observed when harsh purification procedures are used, are largely avoided.
ATR-FTIR/focal plane array imaging and SEM/EDX analyses show that Si and ILs are well distributed on the capsule surface.
Solid-state 13C NMR suggests interesting paramagnetic properties of these materials.
Finally, thermal analyses of IL decorated sporopollenin capsules evidence a high stability of these materials, which depend largely on IL anion. The degradation profile of the IL-modified sporopollenin 6b shows indeed only a small degradation peak at 204 °C (<5%), it principally decomposes at 437 °C.
To the best of our knowledge, this is the first example of sporopollenin capsules decorated with positively charged moieties. Normally, sporopollenin possess ionisable groups on their surface that become increasingly negatively charged with an increase in pH.47 The presence of positive charges could instead facilitate the adhesion of these materials to negatively-charged surfaces, such as negatively charged synthetic materials or the cell membranes of a range of bacteria.
Nonetheless, the obtained thermal stable bio-based IL-SEC hybrid structures may lead to improved systems for many of the fields mentioned in the introduction and may clear the way for the development of new applications. For instance, surface modification of sporopollenin has recently allowed for the development of micromotors able to transport bovine serum albumin loaded into the microcavity.48 The proposed modification, which also permit the tuning of the hydrophobic/hydrophilic surface properties by means of the metathesis reaction, could further boost research into this direction, with an extra option of target oriented micromotors exploiting the chiral centers present on the bio-based ILs moiety.
Experimental section
Materials
(S)-(−)-β-Citronellol, (−)-myrtenol, and (−)-nopol were purchased from Sigma Aldrich. 1-Methylimidazole was obtained by Alfa Aesar. Phenylalanine and aqueous ammonia (33%) were obtained from Fluka, whereas aqueous glyoxal (40%) and aqueous formaldehyde (37%) were purchased from Sigma Aldrich and Acros Organics, respectively. (3-Chloropropyl)triethoxysilane was obtained from Sigma Aldrich. Defatted pollen from Populus deltoides was purchased by Sigma Aldrich. All substances were used as received, without further purification, unless specified otherwise. All organic solvents were purchased from Sigma Aldrich and used without further purification.Methods
TLC analysis were performed on aluminium-supported silica sheets (Merck TLC Silica gel 60 F254). The identification of the spots was carried out with an UV lamp (CAMAG, λ = 254 nm) and/or by wetting the TLC with a 100g L−1 ethanol solution of phosphomolybdic acid (MoO3·H3PO4) and heating it. Solution state NMR spectra were recorded on a Bruker Avance II 250 spectrometer at 250.13 MHz (1H) and 52.90 MHz (13C) using tetramethylsilane as internal standard. The following abbreviations are used: s = singlet, d = doublet, dd = double doublet, t = triplet, tt = triplet of triplets, td = triplet of doublets, qa = quartet, qi = quintuplet, m = multiplet. Solid state 13C NMR spectra were collected at 126 MHz, using a Varian 500 spectrometer and high power 1H “spinal” decoupling, for samples contained in 4 mm rotors spinning at 14 kHz (CP) or at 8 kHz (one pulse). A contact time of 2 ms was used for 1H–13C cross-polarization (“tancpx” program). One pulse 13C spectra were collected using a composite (90°–180°–180°) excitation pulse, “onepuldpth” with 16-phase cycling to suppress the Kel-F spacer signal adapted from ref. 49, 4.2 μs = 90° pulse, variable relaxation delay and shifts referenced to adamantane (38.5 and 29.4 ppm). ATR-FTIR spectra were recorded with an IR Cary 660 FTIR spectrometer (Agilent Technologies) using a macro-ATR accessory with a Zn/Se crystal. The spectra were measured in a range from 4000 to 500 cm−1, with 32 scans both for background and samples. Chemical imaging data was collected by Agilent's ATR-imaging technique using a FTIR Cary 620 imaging system equipped with a 64 × 64 Focal Plane Array (FPA) detector cooled by liquid nitrogen. Spectra were measured, from 3300 to 900 cm−1, with 256 scans for both background and pollen samples. Thermal gravimetric analysis was performed on a TGA Q 500 (TA instruments), under nitrogen. For the sporopollenin-based materials: ramp 10.00 °C min−1 to 900.00 °C, isothermal for 2.00 min. Elemental analyses of the pristine and the IL-treated pollen were carried out on a Thermo Finnigan model Flash 1112 CHNS-O elemental analyzer at the RIAIDT of the University of Santiago de Compostela (USC, Spain). SEM images were acquired on sporopollenin and IL-modified sporopollenin using a Quanta FEG 450 SEM from FEI. Typical imaging conditions were: pressure 90 Pa (low-vacuum mode), accelerating voltage 15 kV, back scattered electrons detector. EDX analysis were performed with an X-Flash 6|10 instrument from Bruker.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH 95:5) demonstrated total consumption of alcohol. If the alcohol was still present after 24 hours, 0.2 equivalents of Et3N and methansulfonyl chloride were added to the mixture. The reaction was quenched by adding 50 mL of a saturate solution of NaHCO3, and the aqueous phase was extracted three times with 20 mL of CH2Cl2. The collected organic phases were washed with 50 mL of milliQ water, and dried over MgSO4. After filtration, the solvent was removed under reduced pressure obtaining 12.11 g (49.6 mmol) of (1R,5S)-2-(6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)ethyl methanesulfonate (2a). Yield 99%. TLC: Rf = 0.88 (SiO2, CH2Cl2:MeOH 95:5).
:MeOH 95:5) demonstrated total consumption of alcohol. If the alcohol was still present after 24 hours, 0.2 equivalents of Et3N and methansulfonyl chloride were added to the mixture. The reaction was quenched by adding 50 mL of a saturate solution of NaHCO3, and the aqueous phase was extracted three times with 20 mL of CH2Cl2. The collected organic phases were washed with 50 mL of milliQ water, and dried over MgSO4. After filtration, the solvent was removed under reduced pressure obtaining 12.11 g (49.6 mmol) of (1R,5S)-2-(6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)ethyl methanesulfonate (2a). Yield 99%. TLC: Rf = 0.88 (SiO2, CH2Cl2:MeOH 95:5).1H NMR (250.13 MHz, CDCl3) δ(ppm): 5.35 (s, 1H), 4.18 (t, 2H), 2.97 (s, 3H), 2.37 (m, 3H), 2.21 (s, 2H), 2.03 (m, 2H), 1.25 (s, 3H), 1.12 (d, 1H), 0.81 (s, 3H).
13C NMR (52.90 MHz, CDCl3) δ(ppm): 142.4, 119.8, 67.9, 45.4, 40.4, 37.9, 37.3, 36.2, 34.6, 31.2, 26.1, 21.0.
The synthesis of (3S)-3,7-dimethyloct-6-en-1-yl-methanesulfonate (2b) is analogous to that described for compound 2a. Yield 99%.
TLC: Rf = 0.88 (SiO2, CH2Cl2:MeOH 95:5).
1H NMR (250.13 MHz, CDCl3) δ(ppm): 5.03 (tt, 1H), 4.21 (td, 2H), 2.95 (s, 3H), 1.94 (m, 2H), 1.64 (s, 3H), 1.56 (s, 3H), 1.83–1.08 (m, 5H), 089 (d, 3H).
13C NMR (52.90 MHz, CDCl3) δ(ppm): 131.6, 124.5, 68.8, 37.4, 36.9, 29.1, 25.9, 25.4, 19.3, 18.8.
The synthesis of (1R,5S)-2-(chloromethyl)-6,6-dimethylbicyclo[3.1.1]hept-2-ene (3c) is analogous to that described for compound 2a. Yield 96%.
TLC: Rf = 0.89 (SiO2, CH2Cl2:MeOH 95:5).
1H NMR (250.13 MHz, CDCl3) δ(ppm): 5.59 (s, 1H), 3.97 (s, 2H), 2.41 (m, 1H), 2.26 (m, 3H), 2.08 (m, 1H), 1.29 (s, 3H), 1.17 (d, 1H), 082 (s, 3H).
13C NMR (52.90 MHz, CDCl3) δ(ppm): 144.4, 122.7, 48.9, 44.5, 40.7, 38.3, 31.9, 31.6, 26.3, 21.4.
:MeOH 95:5), 3.42 g (15.8 mmol) of a light yellow liquid was obtained. Yield 87%. [α]20D −21.6 (c 1.1, CHCl3).TLC: Rf = 0.35 (SiO2, CH2Cl2:MeOH 95:5).
1H NMR (250.13 MHz, CDCl3) δ(ppm): 7.38 (s, 1H), 6.96 (s, 1H), 6.84 (s, 1H), 5.20 (m, 1H), 3.88 (t, 2H), 2.34 (m, 3H), 2.14 (m, 2H), 1.98 (m, 2H), 1.22 (s, 3H), 1.06 (d, 1H), 0.75 (s, 3H).
13C NMR (52.90 MHz, CDCl3) δ(ppm): 143.7, 137.0, 129.2, 119.3, 118.7, 45.5, 45.2, 40.6, 38.4, 38.0, 31.6, 31.2, 26.2, 21.1.
The synthesis of 1-[(3S)-3,7-dimethyloct-6-en-1-yl]-imidazole (4b) is analogous to that described for compound 4a. Yield 94%. [α]20D −2.7 (c 0.9, CHCl3).
TLC: Rf = 0.43 (SiO2, CH2Cl2:MeOH 95:5).
1H NMR (250.13 MHz, CDCl3) δ(ppm): 7.40 (s, 1H), 6.99 (s, 1H), 6.85 (s, 1H), 5.01 (t, 1H), 3.90 (m, 2H), 1.63 (s, 3H), 1.54 (s, 3H), 2.00–1.00 (m, 7H), 0.89 (s, 3H).
13C NMR (52.90 MHz, CDCl3) δ(ppm): 137.2, 131.8, 129.5, 124.4, 118.9, 45.2, 38.3, 36.9, 29.9, 25.9, 25.5, 19.4, 17.8.
The synthesis of 1-{[(1R,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl]methyl}-imidazole (4c) is analogous to that described for compound 4a. Yield 76%. [α]20D −29.0 (c 1.4, CHCl3).
TLC: Rf = 0.43 (SiO2, CH2Cl2:MeOH 95:5).
1H NMR (250.13 MHz, CDCl3) δ(ppm): 7.40 (s, 1H), 6.99 (s, 1H), 6.83 (s, 1H), 5.43 (s, 1H), 4.36 (s, 2H), 2.50–1.80 (m, 5H), 1.19 (s, 3H), 1.09 (d, 1H), 0.72 (s, 3H).
13C NMR (52.90 MHz, CDCl3) δ(ppm): 143.2, 137.2, 139.1, 121.3, 119.2, 51.9, 43.4, 40.4, 37.9, 31.6, 31.1, 25.9, 20.9.
TLC: Rf = 0.31 (SiO2, EtOAc).
1H NMR (250.13 MHz, CDCl3) δ(ppm): 7.35 (s, 1H), 7.17 (m, 2H), 7.00 (s, 2H), 6.95 (m, 2H), 4.85 (dd, 1H), 3.70 (s, 3H), 3.41 (dd, 1H), 3.16 (dd, 1H), 1.22 (s, 3H), 1.06 (d, 1H), 0.75 (s, 3H).
13C NMR (52.90 MHz, CDCl3) δ(ppm): 169.41, 136.75, 135.09, 129.29, 128.59, 128.50, 127.22, 117.86, 61.21, 57.20, 39.39.
1H NMR (250.13 MHz, CD3OD) δ(ppm): 9.01 (s, 1H), 7.70 (t, 1H), 7.61 (t, 1H), 4.30 (t, 2H), 3.79 (s, 3H), 2.88 (t, 2H), 2.09 (qi, 2H), 1.81 (qi, 2H).
13C NMR (52.90 MHz, CD3OD) δ(ppm): 138.8, 125.7, 124.5, 52.3, 51.0, 37.3, 30.7, 23.6.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank UNIPI for the financial support.References
- P. J. Hallet and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef PubMed.
- C. Chiappe and D. Pieraccini, J. Phys. Org. Chem., 2005, 18, 275 CrossRef.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef PubMed.
- A. Mezzetta, L. Guazzelli and C. Chiappe, Green Chem., 2017, 19, 1235 RSC.
- C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929 CrossRef PubMed.
- C. Chiappe and C. S. Pomelli, Eur. J. Org. Chem., 2014, 28, 6120 CrossRef.
- C. Chiappe and C. S. Pomelli, Ionic liquids and green Chemistry, in Analytical application of ionic liquids, ed. M. Koehl, World Scientific, New Jersey, 2016 Search PubMed.
- S. P. F. Costa, A. M. O. Azevedo, P. C. A. G. Pinto and M. L. M. F. S. Saraiva, ChemSusChem, 2017, 10, 2321 CrossRef PubMed.
- J. Hulsbosch, D. E. De Vos, K. Binnemans and R. Ameloot, ACS Sustainable Chem. Eng., 2016, 4, 2917 CrossRef.
- A. Mezzetta, L. Guazzelli, M. Seggiani, C. S. Pomelli, M. Puccini and C. Chiappe, Green Chem., 2017, 19, 3103 RSC.
- Supported Ionic Liquids: Fundamentals and Applications, ed. R. Fehrmann, A. Riisager and M. Haumann, Wiley-VCH Verlag GmbH & Co. KGaA, 2014 Search PubMed.
- B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43, 7171 RSC.
- S. Wilmesmeier and R. Wiermann, J. Plant Physiol., 1995, 146, 22 CrossRef.
- G. Shaw, in Sporopollenin, Academic Press, London, New York, 1971, pp. 305–348 Search PubMed.
- E. Domínguez, J. A. Mercado, M. A. Quesada and A. Heredia, Sex. Plant Reprod., 1999, 12, 171 CrossRef.
- H. Bubert, J. Lambert, S. Steuernagel, F. Ahlers and R. Wiermann, Z. Naturforsch. C Bio. Sci., 2002, 57, 1035 CrossRef PubMed.
- K. Wehling, C. Niester, J. J. Boon, M. T. M. Willemse and R. Wiermann, Planta, 1989, 179, 376 CrossRef PubMed.
- J. Brooks and G. Shaw, Chem. Geol., 1972, 10, 69 CrossRef.
- S. Barrier, A. Loebbert, A. J. Boasman, A. N. Boa, M. Lorch, S. L. Atkin and G. MacKenzie, Green Chem., 2010, 12, 234 RSC.
- F. A. Loewus, B. G. Baldi, V. R. Franceschi, L. D. Meinert and J. J. McCollum, Plant Physiol., 1985, 78, 652 CrossRef PubMed.
- E. Domínguez, A. Heredia, J. A. Mercado and M. A. Quesada, Grana, 1998, 37, 93 CrossRef.
- S. Barrier, A. S. Rigby, A. Diego-Taboada, M. J. Thomasson, G. Mackenzie and S. L. Atkin, LWT--Food Sci. Technol., 2010, 43, 73 CrossRef.
- A. Berthod, M. J. Ruiz-Angel and S. Huguet, Anal. Chem., 2005, 77, 4071 CrossRef PubMed.
- S. Barrier, A. Diego-Taboada, M. J. Thomasson, L. Madden, J. C. Pointon, J. D. Wadhawan, S. T. Beckett, S. L. Atkin and G. Mackenzie, J. Mater. Chem., 2011, 21, 975 RSC.
- S. U. Atwe, Y. Ma and H. S. Gill, J. Controlled Release, 2014, 194, 45 CrossRef PubMed.
- M. G. Potroz, R. C. Mundargi, J. J. Gillissen, E. L. Tan, S. Meker, J. H. Park, H. Jung, S. Park, D. Cho, S. I. Bang and N. J. Cho, Adv. Funct. Mater., 2017, 27, 1 Search PubMed.
- E. Maltas, I. H. Gubbuk and S. Yildiz, Biochem Biophys Rep, 2017, 7, 201 Search PubMed.
- A. Cimen, A. Bilgic, A. N. Kursunlu, I. H. Gubbuk and H. I. Ucan, Desalin. Water Treat., 2014, 52, 4837 CrossRef.
- E. Pehlivan, U. S. Vural, A. Ayar and S. Yildiz, Sep. Sci. Technol., 1996, 31, 1643 CrossRef.
- G. Mackenzie and G. Shaw, Int. J. Pept. Protein Res., 1980, 15, 298 CrossRef PubMed.
- E. Yilmaz, M. Sezgin and M. Yilmaz, J. Mol. Catal. B: Enzym., 2011, 69, 35 CrossRef.
- H. Tutar, E. Yilmaz, E. Pehlivan and M. Yilmaz, Int. J. Biol. Macromol., 2009, 45, 315 CrossRef PubMed.
- M. Keles, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 575 CrossRef.
- T. Baran, I. Sargin, M. Kaya, A. Menteş and T. Ceter, J. Colloid Interface Sci., 2017, 486, 194 CrossRef PubMed.
- S. M. Alshehri, H. A. Al-Lohedan, E. Al-Farraj, N. Alhokbany, A. A. Chaudhary and T. Ahamad, Int. J. Pharm., 2016, 504, 39 CrossRef PubMed.
- S. M. Alshehri, H. A. Al-Lohedan, A. A. Chaudhary, E. Al-Farraj, N. Alhokbany, Z. Issa, S. Alhousine and T. Ahamad, Eur. J. Pharm. Sci., 2016, 88, 158 CrossRef PubMed.
- Y. Wang, T. Len, Y. Huang, A. Diego Taboada, A. N. Boa, C. Ceballos, F. Delbecq, G. Mackenzie and C. Len, ACS Sustainable Chem. Eng., 2017, 5, 392 CrossRef.
- G. Erdtman, Sven. Bot. Tidskr., 1960, 54, 561 Search PubMed.
- F. A. Loewus, B. G. Baldi, V. R. Franceschi, L. D. Meinert and J. J. McCollum, Plant Physiol., 1985, 78, 652 CrossRef PubMed.
- E. Domínguez, A. Heredia, J. A. Mercado and M. A. Quesada, Grana, 1998, 37, 93 CrossRef.
- F. Zetzsche and K. Huggler, Liebigs Ann., 1928, 461, 89 CrossRef.
- R. C. Mundargi, M. G. Potroz, J. H. Park, J. Seo, E.-L. Tan, J. H. Lee and N.-J. Cho, Sci. Rep., 2016, 6, 19960 CrossRef PubMed.
- C. Chiappe, G. C. Demontis, V. Di Bussolo, M. J. Rodriguez Douton, F. Rossella, C. S. Pomelli, S. Sartini and S. Caporali, Green Chem., 2017, 19, 1028 RSC.
- T. Baran, I. Sargin, M. Kaya, A. Menteş and T. Ceter, J. Colloid Interface Sci., 2017, 486, 194 CrossRef PubMed.
- W. Bao, Z. Wang and Y. Li, J. Org. Chem., 2003, 68, 591 CrossRef PubMed.
- C. Descolas-Gros and C. Scholzel, New Phytol., 2007, 176, 390 CrossRef PubMed.
- B. P. Binks, A. N. Boa, M. A. Kibble, G. Mackenzie and A. Rochera, Soft Matter, 2011, 7, 4017 RSC.
- H. Wong, M. G. Potroz, J. A. Jackman, B. Khezri, T. Maric, N.-J. Cho and M. Pumera, Adv. Funct. Mater., 2017, 27, 1702338 CrossRef.
- M. R. Bendall and R. E. Gordon, Magn. Reson. Med., 1985, 2, 91 CrossRef PubMed.
- A. Meyer, M. A. Taige, T. Strassner and E. Gordon, J. Organomet. Chem., 2009, 694, 1861 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03455a |
| This journal is © The Royal Society of Chemistry 2018 |