DOI:
10.1039/C8RA03384F
(Paper)
RSC Adv., 2018,
8, 28533-28537
Photophysical behaviour of BODIPY-phenylacetylene macrocyclic dyads for light-harvesting applications†
Received
20th April 2018
, Accepted 1st August 2018
First published on 10th August 2018
Abstract
The synthesis and study of a family of BODIPY-phenylacetylene macrocycles where donor groups have been added to the macrocycle in order to tune the physicochemical properties and absorption profile is reported. Energy transfer is observed between this phenylacetylene antennae and BODIPY core and fluorescence emission from the BODIPY, at any excitation wavelength, is consistent with energy transfer from the macrocycle.
Introduction
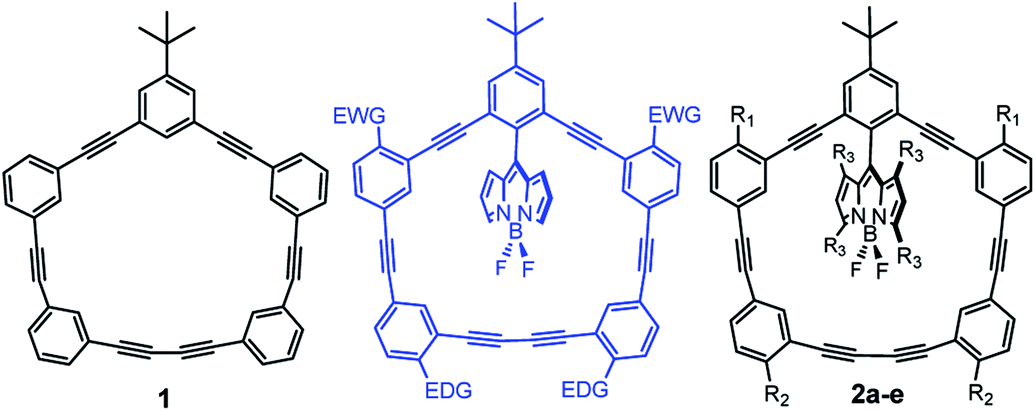
Owing to their rigidity and minimal degrees of conformational freedom, shape-persistent phenylacetylene macrocycles (1; Fig. 1) have attracted much attention in supramolecular chemistry and materials science.1–4 Their extended π-structures are responsible for intense light absorption, and have been explored in a number of optoelectronic and light-harvesting applications.5 The molecular architecture of large macrocycles with well-defined geometries possesses similarity to natural light harvesting complexes which can absorb light and transfer energy through excitation energy transfer (EET) processes.6,7 Increasing interest in π-conjugated macrocycles is attributed to the their promising application in material science,8 host–guest systems,9 storage devices,10 digital computation,11 and molecular rotors.12–14
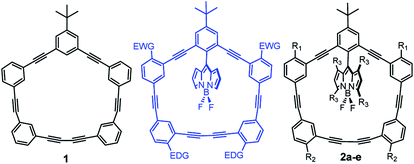 |
| | Fig. 1 BODIPY-phenylacetylene macrocycles in this study. 2a (R1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) R2R3H), 2b (R1R3H; R2OCH3), 2c (R1OCH3; R2R3H), 2d (R1R2OCH3; R3H), 2e (R1R2–H; R3–CH3). R2R3H), 2b (R1R3H; R2OCH3), 2c (R1OCH3; R2R3H), 2d (R1R2OCH3; R3H), 2e (R1R2–H; R3–CH3). | |
We previously reported the synthesis and physicochemical characterization of benchmark macrocycle 1 and BODIPY-phenylacetylene macrocycle 2a (Fig. 1).15 By combining the well-studied BODIPY dye systems16–20 with phenylacetylene architectures we had hoped to develop supramolecular-inspired organic dyes for dye-sensitised solar cell applications.21–24 Particularly, we were interested in designing novel π-spacers that could be exploited for panchromatic absorption within the DSSC manifold. Curiously, we thought that if we could use the phenylacetylene macrocycle as an antenna, then we could effectively harvest more light. In our previous report we observed energy transfer from the macrocycle (λex = 292 nm) to the BODIPY core (quantum yield of 0.89) as the fluorescence emanated from the BODIPY.15 However, the absorption profile of the macrocycle lied predominantly in the UV region, ultimately preventing the use of this molecule architecture for terrestrial photovoltaic applications. To address this limitation, we sought to red-shift the absorption profile of the macrocycle and attempted to further increase the efficiency of the energy transfer process by altering the rigidity and orthogonality of the BODIPY core. Initially we tried to modify the phenylacetylene-macrocycle by putting electron donating group (EDG, e.g., –OMe) and electron withdrawing group (EWG; e.g., –NO2) as shown in Fig. 1. However, because of synthetic challenges we were unable to add EWGs on the macrocycle and decided to explore EDGs only. Herein, we report the synthesis and physicochemical characterization of di- and tetra-methoxy substituted phenylacetylene-BODIPY macrocyclic dyads 2b–d (Scheme 1), as well as the more rigid 2e dyad containing a tetra-methylated BODIPY core.
 |
| | Scheme 1 Synthesis of BODIPY-phenylacetylene macrocycles 2a–2e. | |
Synthesis and characterization
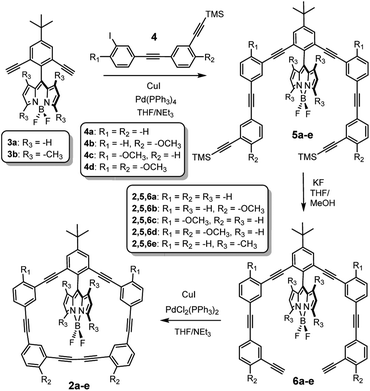
The synthesis of the control macrocycle 1 and BODIPY-macrocycle 2a have been previously reported,15 and the general synthetic route leading to 2b–e is depicted in Scheme 1. Sonogashira cross-coupling reaction between BODIPY 3a–b and TMS-protected acetylenes 4a–d yielded 5b–e, which were subsequently deprotected with KF to afford 6b–e. BODIPY macrocycles 2b–e were obtained via oxidative, Glazer-like cross-coupling using PdCl2(PPh3)2 and CuI.
Complete experimental details and characterization can be found in the ESI, but as expected, the 1H, 13C, 11B and 19F NMR spectra reveal numerous similarities between the five BODIPY-phenylacetylene macrocycles, as well as supporting their proposed structures. The structural identity of 2b and 2c was further confirmed using X-ray crystallography (Fig. 2) as needles were grown via slow evaporation of saturated dichloromethane/hexanes (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Despite the presence of the donating methoxy groups at different locations in the molecule, little structural differences are observed when considering key bond lengths and angles (tabulated in Table 1). In both compounds, the phenylacetylene macrocycle is planar and the BODIPY is canted relative to this phenylacetylene macrocyclic plane (see dihedral angle). It was hoped that the donating group would attenuate the bond lengths more, but both structures possess similar bond lengths and angles, and the –OMe group does little to alter the solid state structure. Unfortunately, the crystal structure of 2d was never obtained because the tiny needle-like crystals were prone to solvent loss and cracking.
:1). Despite the presence of the donating methoxy groups at different locations in the molecule, little structural differences are observed when considering key bond lengths and angles (tabulated in Table 1). In both compounds, the phenylacetylene macrocycle is planar and the BODIPY is canted relative to this phenylacetylene macrocyclic plane (see dihedral angle). It was hoped that the donating group would attenuate the bond lengths more, but both structures possess similar bond lengths and angles, and the –OMe group does little to alter the solid state structure. Unfortunately, the crystal structure of 2d was never obtained because the tiny needle-like crystals were prone to solvent loss and cracking.
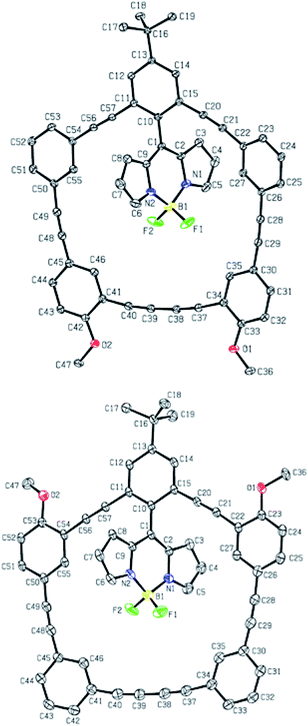 |
| | Fig. 2 Crystal structures of 2b (top) and 2c (bottom); ORTEP, 30% thermal ellipsoids. Hydrogen atoms and solvent molecules were removed for clarity. | |
Table 1 Select bond angles and distances from Fig. 2a
| Dyad |
Distance (C45–C50) |
Distance (C34–C41) |
Distance (C26–C30) |
Dihedral angle C15–C10–C1–C9 |
| Bond lengths/distance have been reported above in Angstroms. |
| 2b |
4.048 |
6.572 |
4.050 |
117.0 |
| 2c |
4.067 |
6.552 |
4.043 |
116.5 |
Results and discussion
To better appreciate the effect of adding electron donating groups around the periphery of the macrocycles, optical measurements (absorption/fluorescence) were performed and correlated to theoretical calculations. The spectra of compounds 2b–e (in DCM) have been presented in Fig. 3–6 respectively, and they have been contrasted to benchmark 2a. In addition, fluorescence emission is depicted for λex, and in all cases emission emanates from the BODIPY. The presence of –OMe groups on the macrocycle (2b–d) only modestly effects the absorption profile (between 330–380 nm) of these dyads, and specific comparisons relating to this lower energy absorption shoulder (belonging to the phenylacetylene macrocycle) are collated in Table 2.
Table 2 Photophysical data for low energy shoulder (330–380 nm) of 2a–ea
| Dye |
Absorptionb |
TD-DFT assignmentc |
Fluorescence |
ETEe (%) |
| λmax (nm) |
(ε × 104 LM−1 cm−1) |
λem (nm) |
Φd |
| More detailed theoretical assignments are found in the ESI. Low energy phenylacetylene absorption shoulder for 2a, 2b, 2c, 2d and 2e in DCM. Assigned FMO transitions based on TDDFT (B3LYP/6-31G) calculations. Measurements were made in DCM (dichloromethane). Quantum yield calculated at 22 °C relative to Rhodamine 6G (QY = 0.95). Energy transfer efficiency (ETE) calculated based on integrating the emission profile and comparing the high and low energy emission (from excitation at ∼300 nm). |
| 2a |
338 |
3.60 |
HOMO − 1 to LUMO + 3 |
528 |
0.89 |
>95 |
| 2b |
364 |
2.65 |
HOMO to LUMO + 1 |
527 |
0.84 |
∼70 |
| 2c |
337 |
4.70 |
HOMO − 3 to LUMO + 1 |
530 |
0.85 |
>95 |
| 2d |
376 |
2.64 |
HOMO to LUMO + 1 |
533 |
0.81 |
∼70 |
| 2e |
335 |
3.12 |
HOMO − 5 to LUMO |
521 |
0.91 |
∼90 |
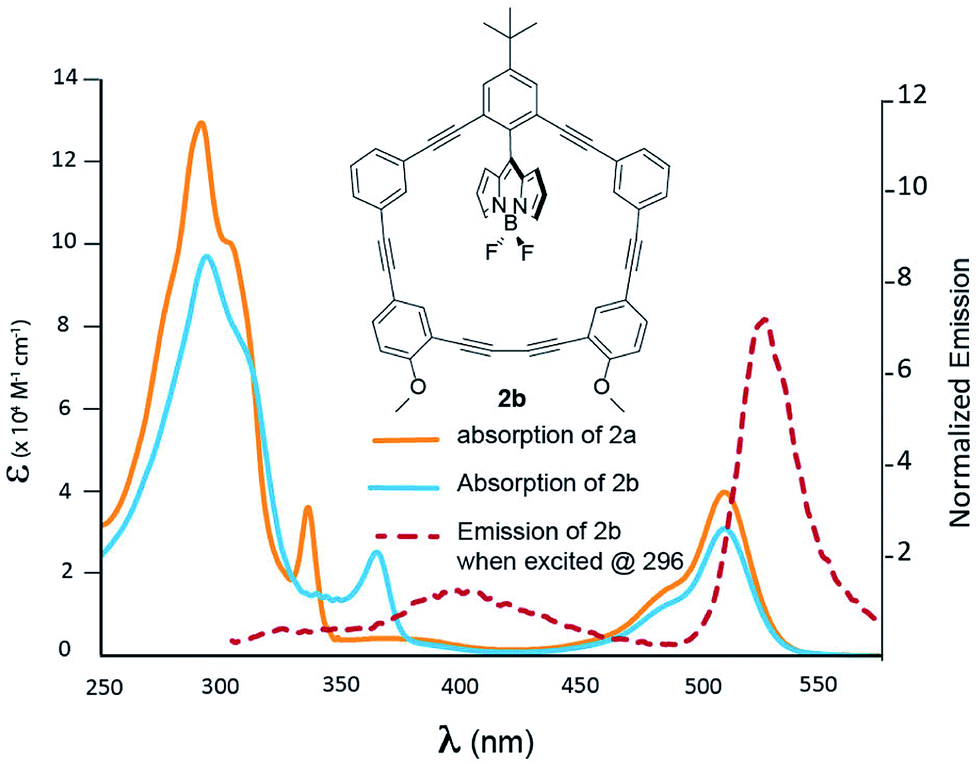
Fig. 3 shows the photophysical behaviour for compound 2b; a more detailed TD-DFT assignment for 2b can be found in Table S2 (ESI†). A closer examination of Fig. 3 reveals that when contrasted to 2a, the phenylacetylene shoulder is red-shifted to 364 nm; otherwise the two spectra share many of the same features. This absorption has been assigned to the HOMO to LUMO + 1 transition, which is heavily centred on the π-system proximal to the –OMe groups. While, excitation at 364 nm resulted in BODIPY emission with a Stokes shift of 163 nm, the energy transfer is not quantitative, as there is a solvent-dependent fluorescence (see ESI Fig. 74†) centred at 400 nm.
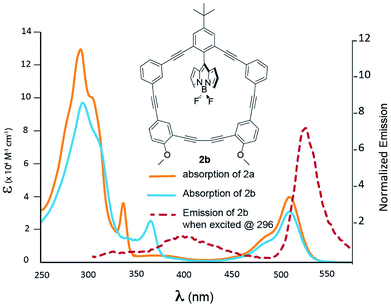 |
| | Fig. 3 Absorption spectra of 2a (blue) and 2b (orange) recorded in DCM. Emission of 2b is presented by the dashed red line. | |
Fig. 4 shows the optical properties for compound 2c; a more detailed TD-DFT assignment for 2c can be found in Table S3 (ESI†). When contrasted to 2a, the macrocycle profile is nearly identical, with the presence of an inflection point at 315 nm. This absorption shoulder has been assigned to the HOMO − 3 to LUMO + 1 transition, where electron density of the HOMO − 3 is heavily centred on the π-system distal to the –OMe groups. As a result, the methoxy groups do little to perturb the energetics of the involved orbitals, and the energy transfer efficiency resembles that of 2a.15
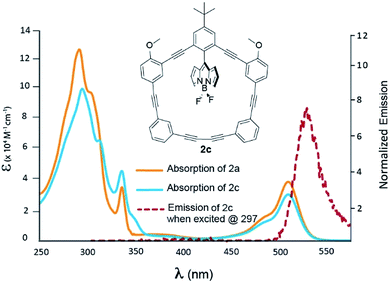 |
| | Fig. 4 Absorption spectra of 2a (blue) and 2c (orange) recorded in DCM. Emission of 2c is presented by the dashed red line. | |
Fig. 5 depicts the absorption and emission behaviour for compound 2d; a more detailed TD-DFT assignment for 2d can be found in Table S4 (ESI†). When contrasted with 2a, it is now possible to see more substantive differences in the absorption profile of 2d. Although modest, the addition of distal and proximal methoxy groups red-shifts the high energy region of the absorption profile by 8 nm (292 nm for 2a to 300 nm for 2d) resembling the superposition of what is observed in 2b and 2c. The lowest energy macrocycle transition has again been assigned to the HOMO to LUMO + 1 transition, where the HOMO now has a greater degree of delocalization throughout the macrocycle π-system. In this instance, the –OMe groups raise the HOMO energy which ultimately red-shifts the absorption spectrum. Again, excitation at 376 nm resulted in BODIPY emission with a Stokes shift of 157 nm. Another observation to note (from Table 2) is that as –OMe groups are added to the macrocycle, there is a decrease in the quantum yield, and as with 2b, a solvent dependent fluorescence is observed. This is consistent with the notion that increased degrees of freedom, should lead to more non-radiative pathways that would ultimately decrease fluorescence intensity.
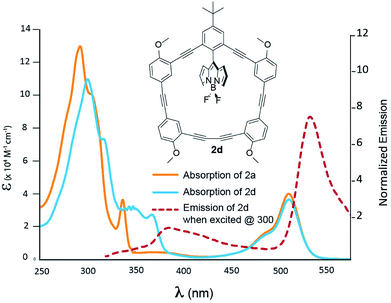 |
| | Fig. 5 Absorption spectra of 2a (blue) and 2d (orange) recorded in DCM. Emission of 2d is presented by the dashed red line. | |
Fig. 6 shows the photophysical behaviour for compound 2e; a more detailed TD-DFT assignments for 2e can be found in Table S5 (ESI†). This particular dyad does not have –OMe groups attached, and as such it is an outlier when considering macrocycle modification, but it was designed to determine if increased rigidity of the BODIPY within the macrocycle would result in altered absorption, energy transfer or emissive behaviour. At first glance, the high energy absorption is nearly identical to 2a (the non-methylated BODIPY) with a slight difference in relative extinction coefficients. This would be expected as tuning the orthogonal BODIPY should have little impact on the macrocycle absorption. Curiously, the extinction coefficient owing to the BODIPY transition (at 530 nm) increases nearly 40% compared to 2a. The efficiency of the energy transfer is difficult to gauge (and theoretical calculations can be found in the ESI†), but having methyl groups attached to the BODIPY was designed to ‘stuff it’ in the macrocycle and minimize canting/rotational degrees of freedom – which may mitigate through-bond exchange owing to increase orthogonality between the macrocycle and BODIPY. The low energy macrocycle shoulder was assigned as a HOMO − 5 to LUMO transition, and excitation at 335 nm leads to a BODIPY emission with an increased quantum yield (Table 2), and a Stokes shift of 188 nm.
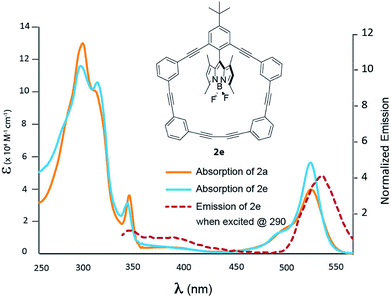 |
| | Fig. 6 Absorption spectra of 2a (blue) and 2e (orange) recorded in DCM. Emission of 2e is presented by the dashed red line. | |
Conclusions
Four novel BODIPY-phenylacetylene macrocycles (2b–e) were synthesized and their photophysical behaviour contrasted with the previously reported benchmark 2a. With the intent of red-shifting the absorption profile of the macrocycle using strong resonance donors (i.e., –OMe groups), it was revealed that modification of the BODIPY-phenylacetylene framework does lead to bathochromically shifted absorption spectra. While the effect was modest, this strategy still exhibited the desired energy transfer (from the macrocycle to the BODIPY core), with only a slight decrease in fluorescence quantum yield when –OMe groups were added distal to the BODIPY (2b, 2d). This work also suggests that modifying the macrocycle and simultaneously rigidifying the BODIPY within the macrocyclic cavity (as seen with 2e) could be a strategy employed to mitigate the losses in quantum yield while maintaining the desired bathochromic shift. Still many questions remain about the nature (rates, Dexter vs. Forester, etc.) of the energy transfer. However, outfitted with this family of macrocyclic dyads, further structure–property insights regarding these questions could be obtained with the appropriate collaboration. Furthermore, the design of a [2]-rotaxane analogue where only Forester energy transfer is possible, would also garner unique energy transfer insights, and this will be articulated in future work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
BDK would like to acknowledge NSERC & Ryerson University for their financial support. ESV and XZB would like to acknowledge, Fondap/serc chile/15110019 and Fondecyt 1161416 and 1180565, respectively.
Notes and references
- N. Aratani, D. Kim and A. Osuka, Acc. Chem. Res., 2009, 42, 1922–1934 CrossRef PubMed.
- J. Yang, M.-C. Yoon, H. Yoo, P. Kim and D. Kim, Chem. Soc. Rev., 2012, 41, 4808–4826 RSC.
- K. Nakao, M. Nishimura, T. Tamachi, Y. Kuwatani, H. Miyasaka, T. Nishinaga and M. Iyoda, J. Am. Chem. Soc., 2006, 128, 16740–16747 CrossRef PubMed.
- M. Iyoda, J. Yamakawa and M. J. Rahman, Angew. Chem., Int. Ed., 2011, 50, 10522–10553 CrossRef PubMed.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef.
- T. Pullerits and V. Sundstrom, Acc. Chem. Res., 1996, 29, 381–389 CrossRef.
- A. W. Roszak, T. D. Howard, J. Southall, A. T. Gardiner, C. J. Law, N. W. Isaacs and R. J. Cogdell, Science, 2003, 302, 1969–1972 CrossRef PubMed.
- D. Venkataraman, S. Lee, J. S. Zhang and J. S. Moore, Nature, 1994, 371, 591–593 CrossRef.
- Y. Hosokawa, T. Kawase and M. Oda, Chem. Commun., 2001, 1948–1949 RSC.
- M. A. Reed, M. J. Chen, A. M. Rawlett, D. W. Price and J. M. Tour, Appl. Phys. Lett., 2001, 78, 3735–3737 CrossRef.
- J. M. Tour, M. Kozaki and J. M. Seminario, J. Am. Chem. Soc., 1998, 120, 8486–8493 CrossRef.
- T. R. Kelly, Acc. Chem. Res., 2001, 34, 514–522 CrossRef PubMed.
- D. Moessinger, J. Hornung, S. Lei, S. De Feyter and S. Hoeger, Angew. Chem., Int. Ed., 2007, 46, 6802–6806 CrossRef PubMed.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281–1376 CrossRef PubMed.
- M. Yousaf, A. J. Lough, E. Schott and B. D. Koivisto, RSC Adv., 2015, 5, 57490–57492 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1209 CrossRef PubMed.
- R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611–631 RSC.
- E. Heyer and R. Ziessel, J. Org. Chem., 2015, 80, 6737–6753 CrossRef PubMed.
- J. Banuelos, Chem. Rec., 2016, 16, 335–348 CrossRef PubMed.
- C. Bonnier, D. D. Machin, O. Abdi and B. D. Koivisto, Org. Biomol. Chem., 2013, 11, 3756–3760 RSC.
- S. Erten-Ela, M. D. Yilmaz, B. Icli, Y. Dede, S. Icli and E. U. Akkaya, Org. Lett., 2008, 10, 3299–3302 CrossRef PubMed.
- S. Kolemen, O. A. Bozdemir, Y. Cakmak, G. Barin, S. Erten-Ela, M. Marszalek, J. H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Gratzel and E. U. Akkaya, Chem. Sci., 2011, 2, 949–954 RSC.
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1836414 and 1836415. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03384f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
Alan J. Loughe and
Bryan D. Koivisto
d,
Alan J. Loughe and
Bryan D. Koivisto