
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhospholipid-mediated exfoliation as a facile preparation method for graphene suspensions†
Aled T. Williamsa,
Roberto Donno *b,
Nicola Tirellibc and
Robert A. W. Dryfea
*b,
Nicola Tirellibc and
Robert A. W. Dryfea
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
bLaboratory of Polymers and Biomaterials, Fondazione Istituto Italiano di Tecnologia, Via Morego 30, Genoa, Italy. E-mail: roberto.donno@iit.it
cNorthWest Centre for Advanced Drug Delivery, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 24th May 2018
Abstract
This paper deals with simple, inexpensive and ‘green’ methods of production for graphene in colloidal dispersion. Herein, we report on such a method by preparing aqueous graphene dispersions via ultrasonic exfoliation in the presence of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). The product predominantly consists of few-layer graphene flakes coated by DOPC with a lateral size of a few tens to hundreds of nm, as confirmed by Raman and X-ray photoelectron spectroscopies, thermogravimetric analysis (TGA), dynamic light scattering (DLS) and atomic force microscopy (AFM). The novelty of this method lies in its dependence on a typical soft matter property: the fluidity of the hydrophobic chains. Stiffer phospholipids such as 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC, which possesses two palmitoyl chains) or 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC, one palmitoyl, one oleyl chain) are ineffective at dispersing graphene; however, in the presence of cholesterol these phospholipids also become effective mediators. The phospholipid coating renders the flakes compatible with biological environments.
Introduction
Stable graphene colloidal dispersions are versatile materials. They can find application in their own morphology, e.g. as imaging agents or drug carriers,1 or may be used as precursors to other graphene-based materials, e.g. nano-structured films that can be exploited for various applications.2,3 Much attention has been devoted to their preparative processes:4 currently liquid-phase exfoliation5–7 is probably the most attractive and scalable process and, under optimal conditions yields materials ranging from monolayer to few-layer (n ≤ 5) graphene (FLG).5,8–10 Exfoliation requires both the provision of energy, e.g. as ultrasound, and molecules that minimize the surface energy of the dispersed colloids.9 Hernandez et al. reported that solvents provide optimal exfoliation if their surface tension = 40–50 mJ m−2,9 as is the case for N-methyl-2-pyrrolidone (NMP) and other polar aprotic solvents; in particular, NMP allows for graphene concentrations of up to 1.2 mg mL−12. Both for ease of removal (lower boiling point) and environmental considerations, water would be a better exfoliation medium, provided that its high surface tension (72.75 mJ m−2 at 20 °C)11 is reduced by surfactants2,5,12 or polymers.13,14Herein we demonstrate that phospholipids are effective agents for producing graphene by aqueous exfoliation; we note that phospholipids have been previously used in a chloroform-based exfoliation method15 and a multi-step aqueous exfoliation method,16 but we are unaware of phospholipids being used in direct single-step waterborne exfoliation of graphite. In terms of molecular variables, it is known that an appropriate phospholipid curvature (arising from the interfacial mismatch between the hydrophilic and hydrophobic portions of the molecule) is critical for their adaptation to a curved carbon surface (nanotubes).17 Here, we show that another molecular parameter, specifically the fluidity of the hydrophobic environment, is critical for graphene exfoliation.
Experimental methods
Materials
All starting materials and solvents were used as received. The primary graphite powder (grade 2369) used was obtained from Graphexel Ltd (UK). 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) (≥99%), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) (≥99%), 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC) (≥95.5%) and cholesterol (≥99%) were purchased from Sigma-Aldrich and kept refrigerated. Sodium phosphate dibasic (Na2HPO4) (BioXtra, ≥99%) was purchased from Sigma-Aldrich and potassium dihydrogen orthophosphate (KH2PO4) (≥99%) from Fisher Scientific.UV-vis spectroscopy
UV-visible spectra were taken on a USB2000+UV-VIS fibre optic spectrometer using a DH-2000-BAL deuterium-halogen light source (Ocean Optics).Dynamic light scattering (DLS) and zeta-potential measurements
A Malvern Zetasizer Nano ZS was operated in backscatter mode (173°), using a 633 nm HeNe laser. The graphene/DOPC dispersions were diluted in ultra-pure water (cG = 3 μg mL−1) and equilibrated by the instrument to 25 °C for each measurement. The zeta potential was calculated by Malvern software using the Smoluchowski model.X-ray photoelectron spectroscopy (XPS)
Spectra were measured using an AXIS Nova (Kratos Analytical, UK) with a monochromatic AlKα source operated at 225 W (15 kV and 15 mA). Survey and high resolution spectra were collected at pass energies of 160 eV (average of 3 sweeps) and 20 eV (average of 10 sweeps) respectively. Analysis was performed using CasaXPS software, utilizing Shirley background subtraction and the appropriate relative sensitivity factors. XPS samples were prepared as drop-cast graphene dispersions on Si/SiO2 substrates (as described for the AFM measurements).Thermogravimetric analysis (TGA)
A 15 mL aliquot of the supernatants was freeze dried in a 50 mL Falcon tube (Fisherbrand, polypropylene) using a Genevac evaporator (EZ-2 series); the resulting solid was transferred to a cellulose filter, repeatedly washed with ultrapure water and allowed to dry in a vacuum oven at 120 °C for 24 h. Reference graphene samples were produced according to a literature procedure9 via exfoliation in NMP, with the suspension twice centrifuged at 6000 rpm for 30 min and then filtered through filter paper, finally drying the dispersed material in a vacuum oven at 120 °C for 24 h. TGA measurements were performed on a Q500 instrument (TA Instruments, USA) under a nitrogen atmosphere and at 10 °C min−1 using a temperature range of 20–1000 °C.Raman spectroscopy
Samples were prepared by drop casting 100 μL of fresh dispersions (cG = 30 μg mL−1) onto 1 cm2 Si/SiO2 substrates (pre-cleaned by sonication in ethanol and acetone for 10 min), followed by drying at 70 °C for approximately 10 min. Spectra were taken on a Renishaw inVia system using 532 nm (2.33 eV) excitation energy (laser power of 1.8 mW) and a 100× objective (numerical aperture of 0.9), giving a spot size of approximately of 1 μm. This system uses a high-resolution grating of 1800 g mm−1, giving a spectral resolution ≤1 cm−1. Spectra represent three accumulations at 5 s of exposure time.Atomic force microscopy (AFM)
Samples were prepared by drop casting 100 μL of fresh dispersion (cG = 3 μg mL−1) onto 1 cm2 clean mica substrates and allowed to dry overnight. AFM images were acquired in tapping mode at a scan rate of 1 Hz using a Molecular Force Probe 3D AFM (MFP-3D, Oxford Instrument Asylum Research, Buckinghamshire, UK). An OTESPA cantilever (Bruker, Camarillo, CA) was used with a nominal spring constant of 42 N m−1 and a tip radius of 7 nm. All images were analysed (height sections and distributions) with Igor-pro (Asylum Research AFM software, version 13, Wavemetrics). In order to obtain the height and CE diameter (the diameter of a circle having the same area of the segmented object) distributions, images were first segmented using the particle analysis routine offered by the Igor-pro software, then generating the height distributions and analysing/plotting it through Origin 8.5 (OriginLab Corporation).Preparation of graphene dispersions
In a typical experiment, phospholipids were dispersed in 10 mL of ultra-pure water (18 MΩ cm, Milli-Q Direct 8, Millipore, USA) at a concentration of 0.2 mg mL−1 in a 20 mL glass vial. Graphite powder was sieved before use through a 500 μm pore-size mesh in order to remove large particles, and subsequently added to the phospholipid/water solution at a concentration of 5 mg mL−1. The mixture was then sonicated in an ultrasound bath for 12 hours (Elmasonic P, Elma, Germany. Frequency = 37 kHz, power equivalent = 40%, true power ≈ 34 W) at a constant bath temperature of 20 °C, after which the resulting dispersion was left standing for 24 hours to allow for the sedimentation of large aggregates. The supernatant was subsequently removed and centrifuged at 6000 rpm for 30 min, with the process being repeated once more to produce a light grey/green-coloured dispersion. The amount of dispersed material was calculated by carefully separating the supernatants from the sediment after centrifugation. The volume of dispersion was measured and the undispersed mass was dried in a vacuum oven (Gallenkamp, UK) at 120 °C.Results and discussion
Using 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) in water, sonication of graphite produced stable, dark suspensions that became clearer and grey/green in colour after removing large aggregates via centrifugation. We have estimated an extinction coefficient lower than for other surfactant-stabilized aqueous graphene dispersions: ε660 = 6.2 mL mg−1 cm−1 (see Fig. S1 in ESI†) vs. 13.90 mL mg−1 cm−1 with sodium dodecylbenzene sulfonate (SDBS).5 Dynamic light scattering (DLS) seemed to exclude the presence of large amounts of the parent DOPC colloids (compare the black curves in Fig. 1A top and bottom), while the objects present in the graphene dispersion showed a considerably broader size distribution (Fig. 1A) peaked at 190 nm; due to the approximations in the technique (e.g. assumption of spherical form), this value should be simply taken as an indication of a size comprised between tens and hundreds of nm. Hereafter, we will refer to these colloids as graphene flakes.18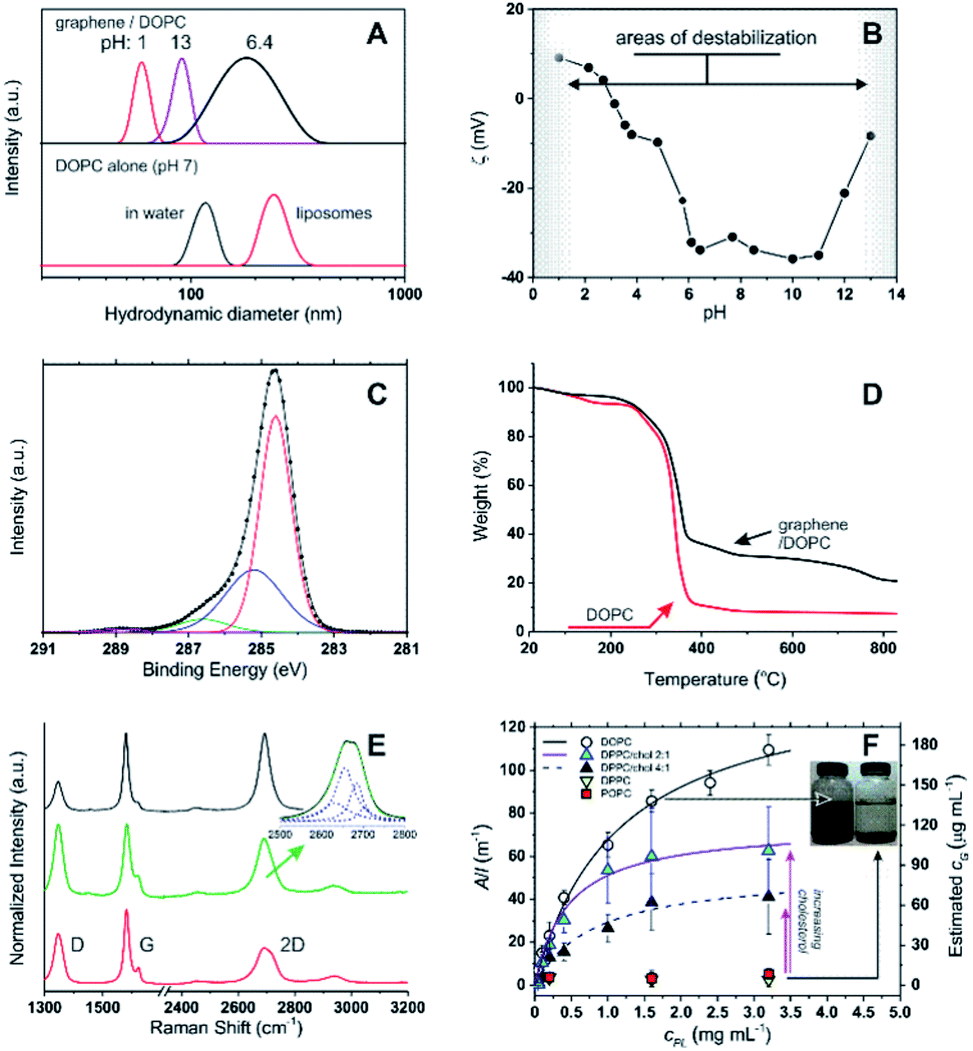 | ||
Fig. 1 (A) Top: size distribution (DLS) for DOPC–graphene dispersions in deionized water (pH ≈ 7) and after adjusting the pH to 1 and 13 via addition of concentrated HCl and NaOH, respectively. Bottom: size distribution of DOPC aggregates in water prior to the addition of graphite (black curve); DOPC liposomes (red curve) are shown for comparison. (B) ζ potential of DOPC/graphene suspensions (cG = 3 μg mL−1) as a function of pH in deionized water; the negative values at neutral pH are comparable to those obtained for DOPC in its initial aggregates in water (−37 mV) or in liposomes (−41 mV). (C) Carbon 1s core-level XPS spectrum of a DOPC/graphene dispersion drop cast onto a Si/SiO2 substrate. In the fitting, the red line corresponds to the C–C sp2 peak, the blue to the C–C sp3 peak, the green to C–O and C–N peaks, the purple to the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak. (D) Thermogravimetric analysis of DOPC/graphene dispersions (black) and of pure DOPC (red). (E) Raman spectra of DOPC/graphene flakes isolated on Si/SiO2 substrates. Top (black): attributed to monolayer. Middle (green): attributed to bilayer, with the inset showing the fitting of the 2D band to a Lorentzian model.8 Bottom (red): attributed to flakes with layer >2 but <6, i.e. few-layer graphene. (F) Dependency of the optical density of dispersed graphene samples on phospholipid concentration. The concentration of dispersed graphene (estimated from A660) can be fitted as a hyperbolic (Langmuir) function of the phospholipid concentration. DPPC and POPC are unable to disperse graphene to any significant extent, unless cholesterol is used; in this case, the asymptotic amount of dispersed graphene increases with cholesterol concentration. The flakes dispersed with DPPC/cholesterol are similar to those obtained with DOPC; e.g. with a 4 O peak. (D) Thermogravimetric analysis of DOPC/graphene dispersions (black) and of pure DOPC (red). (E) Raman spectra of DOPC/graphene flakes isolated on Si/SiO2 substrates. Top (black): attributed to monolayer. Middle (green): attributed to bilayer, with the inset showing the fitting of the 2D band to a Lorentzian model.8 Bottom (red): attributed to flakes with layer >2 but <6, i.e. few-layer graphene. (F) Dependency of the optical density of dispersed graphene samples on phospholipid concentration. The concentration of dispersed graphene (estimated from A660) can be fitted as a hyperbolic (Langmuir) function of the phospholipid concentration. DPPC and POPC are unable to disperse graphene to any significant extent, unless cholesterol is used; in this case, the asymptotic amount of dispersed graphene increases with cholesterol concentration. The flakes dispersed with DPPC/cholesterol are similar to those obtained with DOPC; e.g. with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DOPC/cholesterol molar ratio one obtains a Z-average size of 239 nm and a ζ potential of −22 mV. :1 DOPC/cholesterol molar ratio one obtains a Z-average size of 239 nm and a ζ potential of −22 mV. | ||
The DOPC/graphene dispersions were characterized by a negative ζ potential at neutral pH (−34 mV, Fig. 1B). The rather large value of the ζ potential suggests that the dispersions have long-term stability and indeed their optical density decreased very moderately with time (ca. 11, 25 and 43% reduction in A660 respectively after 22, 41 and 56 days). Further ensuring their stability under biologically relevant conditions, the ζ potential remained constant throughout a broad pH window centred on neutrality (pH = 4–11). We have recorded strong changes only at strongly acidic pH, due to amine protonation and loss of electrostatic stabilization, and at strongly basic pH, most likely due to phospholipid desorption following hydrolysis of the head-groups. Under both conditions graphene precipitated, leaving behind colloids with too small an average size to be considered as being dispersed flakes.
The presence of both DOPC and graphene in the dispersion was qualitatively and quantitatively confirmed through X-ray photoelectron spectroscopy (XPS) and thermogravimetric analysis (TGA). In XPS (Fig. 1C), the C1s spectrum confirmed the presence of DOPC through the peaks at 285.2 (C–C sp3), 286.6 (C–O and C–N) and 288.8 eV (O–CO), which accompany the peak at 284.6 eV (C–C sp2); as it is to be expected for a graphene-based material, the latter is the largest peak in this area of the spectrum (while it is small in the predominantly aliphatic DOPC). The binding energies of C–N and C–O are too similar to allow an effective differentiation into separate peaks, but their combination had a 4:1 ratio with respect to the O–CO peak, as expected from the structure of DOPC. Phosphorous was detected at approximately the same percentage atomic concentration as nitrogen, with the P2p3/2 and P2p1/2 peaks at 133.1 eV and 133.9 eV respectively (constrained at a peak separation of 0.87 eV and 2:1 area ratio) and the N1s peak at 401.9 eV.
Thermogravimetric analysis (TGA) showed that the degradation pattern of freeze-dried dispersions presented features typical of DOPC but also of graphene: the degradation step of DOPC is clearly seen at around 300 °C (compare black and red curves in Fig. 1D). However, the amount of residual material is considerably higher than for pure DOPC and shows a weight loss at temperatures > 500 °C that is typical of graphene thermal degradation19 (see also ESI, Fig. S2†). From the extent of the DOPC-related weight loss we estimate that the graphene corresponded to 24% wt of the dispersed material. It is worth noting that FT-IR analysis did not provide any significant information: DOPC peaks were only marginally affected by the presence of graphene, while graphene does not present any diagnostic band.
We have further confirmed that, for a given exposure to ultrasound, the amount of dispersed graphene depends on the concentration of phospholipid, cPL. For example, a graphene concentration cG = 0.03 or 0.18 mg mL−1 can be obtained respectively with cPL = 0.2 and 3.2 mg mL−1 (Fig. 1F). The dependency, however, is non-linear and could be easily fitted with asymptotic (hyperbolic) models, for example:
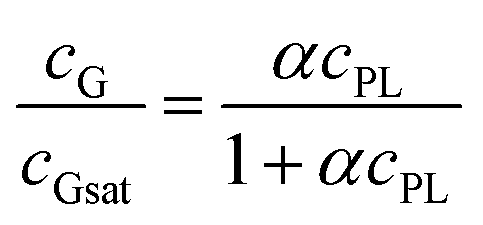
When more rigid phospholipids such as 1,2-dipalmitoyl-sn-glycero-3-phosphocholine and 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (DPPC and POPC, with no or one cis double bond, respectively) were used instead of DOPC, the exfoliation was successful only in the presence of cholesterol and proportionally to the amount of the latter (Fig. 1F). This behaviour is clearly linked to the fluidity of the phospholipid hydrophobic chains: it is indeed known that DPPC forms rigid structures22 and cholesterol increases the fluidity of its bilayers by inducing a gel (Lβ) to liquid-ordered (L0) phase transition.23–25 It is noteworthy that the cholesterol/DPPC mixtures differed from DOPC in the overall amount of dispersible graphene (above all at high phospholipid concentrations), but not significantly in the early ‘linear’ phase; it would seem logical to hypothesize that all mixtures behaved similarly (see ESI, Fig. S3†) from a mechanistic point of view, although characterized by a different dispersing power.
Finally, by using Raman spectroscopy and atomic force microscopy (AFM) we have identified the material as predominantly composed by DOPC-coated few-layer graphene. Raman spectroscopy measurements on dried graphene dispersions showed that the majority of spectra displayed the major graphitic peaks (Fig. 1E, bottom/red) at 1347 (D band), 1582 (G band) and 2695 cm−1 (2D band), with the shape of the 2D bands being quite distinct from that of the parent graphite8 (see ESI, Fig. S4†). The most frequently observed spectra (73% of all the spectra taken; Fig. 1E, bottom/red) were representative of flakes >2, with the lower intensity of the 2D band vs. G (I2D/IG = 0.50) and the broad shape of the 2D band (FWHM = 72 cm−1) suggesting the presence of few-layer structures8,26 (probably varying within the range of 3–5 layers); a distribution in the number of layers is expected with liquid-phase exfoliation of graphite.2,9 Less frequently observed (20% of all spectra taken) were those characteristic of bilayer graphene26 (Fig. 1E, middle/green), with I2D/IG = 0.76 and the 2D band (FWHM = 64 cm−1) fitting to four Lorentzian functions (Fig. 1E inset) peaked at 2669 (FWHM = 46 cm−1), 2688 (FWHM = 33 cm−1), 2706 (FWHM = 30 cm−1) and 2726 cm−1 (FWHM = 32 cm−1). However, in most samples it was possible to isolate individual flakes that displayed 2D peaks at 2694 cm−1 and I2D/IG = 0.99, which suggest the presence of monolayer flakes;26 the 2D peaks in these spectra were best fitted by a single Lorentzian function (FWHM = 42 cm−1; Fig. 1E, top/black spectrum), with the fitting of more than three Lorentzian functions being difficult to accommodate. Due to the small size of the exfoliated flakes (lateral size of a few tens to hundreds of nm), and coupled with the fact that the laser spot size used for the Raman measurements was approximately 1 μm, the D and 2D band intensities will be strongly influenced by both structural defects and the edges of the graphene flakes,27 with one of the expected results being the intensity reduction and broadening of the 2D band.28
Atomic force microscopy (AFM) showed two groups of differently sized structures: flatter 5–10 nm thick flakes (red arrows in Fig. 2A) and larger, ≫ 10 nm-tall aggregates (white arrow in Fig. 2A; white bars and red curve in Fig. 2B). We can exclude the presence of uncoated FLG, which has a different size distribution (black bars and curves in Fig. 2B) even when composed of a similar number of layers.10 On the other hand, we cannot exclude some structures that are entirely composed of DOPC, for example: ‘steps’ of approximately 1 nm are occasionally observed (Fig. 2D and E), which by analogy to the structurally similar dioleoylphosphatidylethanolamine (1.2–1.3 nm monolayers29) can be interpreted as DOPC monolayers. Furthermore, the variability in the thickness of supported DOPC bilayers (recorded between 4 (ref. 30 and 31) and 5 nm (ref. 32 and 33)) is comparable to the reported thickness of ∼0.40 nm/graphene layer,34,35 thus a DOPC bilayer can hardly be distinguished from a DOPC-coated graphene mono- or bilayer. However, the bulk of the 5–10 nm size distribution can only correspond to DOPC-coated FLG. Finally, we noticed that the larger and irregular objects often showed ‘steps’ with a height comparable to the thickness of the flatter flakes (Fig. 2G and H). It therefore seems reasonable to hypothesize that stacking of the latter during deposition onto the mica substrate may give rise to the large aggregates. It is peculiar that these aggregates appear to maintain a comparable aspect ratio independent of their actual dimensions (Fig. 2C), which is possibly a result of a specific balance of interactions between flake borders and flat surfaces during the deposition of the flakes from water dispersions.
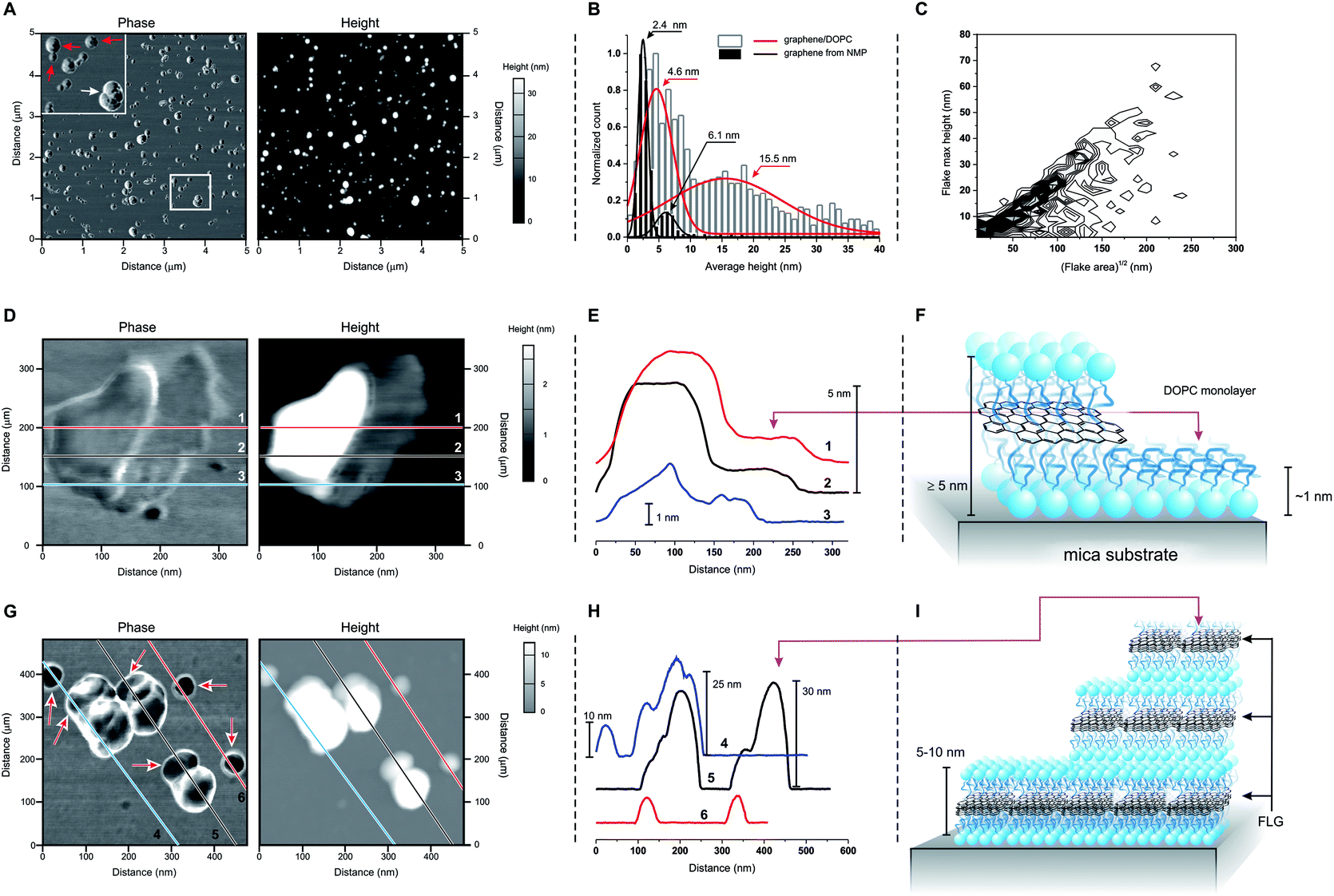 | ||
| Fig. 2 (A) Phase and height images obtained in tapping mode AFM for a sample of DOPC-coated graphene. The top left inset in the phase image shows flatter, smaller and more homogeneous objects (red arrows) co-dispersed with larger and heterogeneous ones (white arrow). (B) Size distributions obtained from AFM height images of DOPC/graphene and of graphene exfoliated in NMP; the latter is FLG10 with a number of layers (4–5) similar to graphene/DOPC (E). The graph also presents Gaussian fittings for an easier view of the distribution. (C) Frequency plot of the maximum height vs. the square root of the area (used as a characteristic lateral dimension) for the materials obtained via deposition of DOPC/graphene on mica from dispersions on deionized water (data for 1240 flakes; the frequency data are calculated with a binning of 2 nm for height and 5 nm for lateral size, and normalized to show the highest frequency in black and lowest frequency in white). (D)–(F) Phase and height images: (D) for a ‘flat’ flake showing the presence of a ∼1 nm-thick layer on the right hand side of the flake; the height profiles corresponding to the lines 1, 2 and 3 are reported in (E) and clearly show the presence of two homogeneous levels, which are ascribed to a DOPC monolayer and of DOPC-coated graphene, as depicted in (F). (G)–(I) Phase and height images: (G) for a field comprising ‘flat’ flakes and agglomerates; the height profiles corresponding to the lines 4, 5 and 6 are reported in (H) and show that the ‘flat’ flakes have a height comprised between 5 and 10 nm, which is similar to that of the lowest steps in the agglomerates; the latter then typically grows in steps with comparable size, as depicted in (I). | ||
Conclusions
In summary, this single-step method produces aqueous dispersions of phospholipid-coated graphene through the exfoliation of graphite in water. The critical step of this procedure appears to be the fluidity of the phospholipid aliphatic chains. We believe that the benign nature of the phospholipid coating can be exploited for applications involving graphene in contact with biological environments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Erwin Hohn and Dr Enrique Lallana for assistance with DLS and zeta potential measurements, Dr Mark Bissett for XPS measurements and Ms Tania Campos Hernández for useful discussions. The authors gratefully acknowledge the Faculty of Medical and Human Sciences of The University of Manchester for funding and the Bio-AFM facility for technical support.Notes and references
- D. Bitounis, H. Ali-Boucetta, B. H. Hong, D.-H. Min and K. Kostarelos, Adv. Mater., 2013, 25, 2258–2268 CrossRef PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef PubMed.
- T. C. Hernandez, A. C. F. Blanco, A. T. Williams, M. Velicky, H. V. Patten, A. Colina and R. A. W. Dryfe, Electroanalysis, 2015, 27, 1026–1034 CrossRef.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef PubMed.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. M. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef PubMed.
- J. S. Y. Chia, M. T. T. Tan, P. S. Khiew, J. K. Chin, H. Lee, D. C. S. Bien and C. W. Siong, Chem. Eng. J., 2014, 249, 270–278 CrossRef.
- J. S. Y. Chia, M. T. T. Tan, P. S. Khiew, J. K. Chin and C. W. Siong, Sci. Adv. Mater., 2016, 8, 1177–1186 CrossRef.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef PubMed.
- A. J. Cooper, N. R. Wilson, I. A. Kinloch and R. A. W. Dryfe, Carbon, 2014, 66, 340–350 CrossRef.
- N. B. Vargaftik, B. N. Volkov and L. D. Voljak, J. Phys. Chem. Ref. Data, 1983, 12, 817–820 CrossRef.
- S. K. Misra, P. Kondaiah, S. Bhattacharya and C. N. R. Rao, Small, 2012, 8, 131–143 CrossRef PubMed.
- A. B. Bourlinos, V. Georgakilas, R. Zboril, T. A. Steriotis, A. K. Stubos and C. Trapalis, Solid State Commun., 2009, 149, 2172–2176 CrossRef.
- M. Fang, K. G. Wang, H. B. Lu, Y. L. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- M. Pykal, K. Safarova, K. M. Siskova, P. Jurecka, A. B. Bourlinos, R. Zboril and M. Otyepka, J. Phys. Chem. C, 2013, 117, 11800–11803 Search PubMed.
- R. Zappacosta, M. Di Giulio, V. Ettorre, D. Bosco, C. Hadad, G. Siani, S. Di Bartolomeo, A. Cataldi, L. Cellini and A. Fontana, J. Mater. Chem. B, 2015, 3, 6520–6527 RSC.
- J. Maatta, S. Vierros and M. Sammalkorpi, J. Phys. Chem. B, 2015, 119, 4020–4032 CrossRef PubMed.
- M. Lotya, A. Rakovich, J. F. Donegan and J. N. Coleman, Nanotechnology, 2013, 24, 265703 CrossRef PubMed.
- V. Chabot, B. Kim, B. Sloper, C. Tzoganakis and A. P. Yu, Sci. Rep., 2013, 3, 1378 CrossRef PubMed.
- M. Alwarawrah, J. A. Dai and J. Y. Huang, J. Phys. Chem. B, 2010, 114, 7516–7523 CrossRef PubMed.
- N. Kucerka, S. Tristram-Nagle and J. F. Nagle, J. Membr. Biol., 2005, 208, 193–202 CrossRef PubMed.
- D. O. Shah and J. H. Schulman, J. Lipid Res., 1967, 8, 215–226 Search PubMed.
- T. T. Mills, J. Y. Huang, G. W. Feigenson and J. F. Nagle, Gen. Physiol. Biophys., 2009, 28, 126–139 CrossRef PubMed.
- J. H. Ipsen, G. Karlstrom, O. G. Mouritsen, H. Wennerstrom and M. J. Zuckermann, Biochim. Biophys. Acta, 1987, 905, 162–172 CrossRef.
- M. R. Vist and J. H. Davis, Biochemistry, 1990, 29, 451–464 CrossRef PubMed.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef.
- S. Gayathri, P. Jayabal, M. Kottaisamy and V. Ramakrishnan, AIP Adv., 2014, 4, 027116 CrossRef.
- L. G. Cancado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- Y. F. Dufrene, W. R. Barger, J. B. D. Green and G. U. Lee, Langmuir, 1997, 13, 4779–4784 CrossRef.
- M. Hirtz, A. Oikonomou, T. Georgiou, H. Fuchs and A. Vijayaraghavan, Nat. Commun., 2013, 4, 2591 CrossRef PubMed.
- S. T. Wang, M. Fukuto and L. Yang, Phys. Rev. E, 2008, 77, 031909 CrossRef PubMed.
- A. Dols-Perez, L. Fumagalli and G. Gomila, Colloids Surf., B, 2014, 116, 295–302 CrossRef PubMed.
- Z. V. Leonenko, E. Finot, H. Ma, T. E. S. Dahms and D. T. Cramb, Biophys. J., 2004, 86, 3783–3793 CrossRef PubMed.
- Y. K. Koh, M.-H. Bae, D. G. Cahill and E. Pop, ACS Nano, 2011, 5, 269–274 CrossRef PubMed.
- P. Nemes-Incze, Z. Osvath, K. Kamaras and L. P. Biro, Carbon, 2008, 46, 1435–1442 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03365j |
| This journal is © The Royal Society of Chemistry 2018 |