
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing carbon dots to moderate the blue emission from zinc vanadium oxide hydroxide hydrate nanoplates
Peng Huang a,
Guodong Yuana,
Tongbo Weia,
Jinmin Li*a and
Michael N. R. Ashfold*b
a,
Guodong Yuana,
Tongbo Weia,
Jinmin Li*a and
Michael N. R. Ashfold*b
aResearch and Development Center for Solid State Lighting, Institute of Semiconductors, Chinese Academy of Sciences, Beijing, China. E-mail: jmli@semi.ac.cn
bSchool of Chemistry, University of Bristol, Bristol, UK BS8 1TS. E-mail: Mike.Ashfold@bristol.ac.uk
First published on 6th June 2018
Abstract
The relative intensity of the blue component of the total emission from light-emitting diodes (LEDs) can be an important factor when assessing their biological safety. Carbon quantum dots (CQDs) are compatible with many materials and present a high density of multiple surface states; the incorporation of such CQDs thus offers a route to modifying the emission from a given LED matrix. Here we report the fabrication of stable CQD/zinc pyrovanadate (Zn3(OH)2V2O7·2H2O) nanoplate composites via a facile hydrothermal route. Structural and morphological analyses confirm that the nanoplates are hexagonal phase and grew normal to the [0001] direction. X-ray photoemission spectroscopy, Raman and infrared spectroscopy demonstrate that the CQDs combine with nanoplates via surface carbon–oxygen bonds. Wavelength resolved photoluminescence measurements show that the relative intensity of the blue (2.93 eV) component of the emission associated with the nanoplates is significantly reduced by incorporating CQDs. We suggest that this reduction arises as a result of preferential trapping of the higher energy photoelectrons by surface defects on the CQDs.
1. Introduction
The biological safety of GaN-based light-emitting diodes (LEDs) is a topic of on-going concern, following animal studies showing that the blue (450–490 nm) light from such sources can damage photoreceptor cells.1,2 Other studies have suggested that chronic blue light exposure may enhance the development of age-related macular degeneration and other pathologies.3,4 Traditional methods for modifying the spectral output of LEDs include: (i) phosphor engineering, using a blue or near ultraviolet chip as the pump source to activate a phosphor. This strategy is heavily dependent on the availability of a suitable host structure and activators for the phosphor, and typically suffers from poor color rendering, poor thermal stability and the cost of rare earth doping; (ii) combining multiple chips, wherein red, green and blue emitting chips are integrated to produce a target spectrum. A challenge with this approach is that each chip contributes a relatively narrow spectral bandwidth to the overall emission, so any differences in their respective aging characteristics results in a color shift; (iii) one-chip LEDs, involving multiple quantum well structures. These are currently hampered by the complicated epitaxy required for their fabrication, a low luminous efficacy and stability issues.5–7 Achieving any of these routes requires sophisticated instrumentation and time-consuming sample preparation. Developing a cost-effective, convenient and broadly applicable method for modifying the relative intensity of the blue component within a polychromatic light source remains a desirable objective.Carbon quantum dots (CQDs) display both size and wavelength dependent photoluminescence (PL), which is inspiring intensive research – particularly in the design and application of multi-functional optical nanocomposites.8–12 CQDs offer the advantages of large surface areas, short effective charge-transfer lengths and available surface defects. The injection and extraction of electrons/holes can be controlled by appropriate surface functionalisation or by combining CQDs with a suitable matrix, thereby offering a route for switching between excitation and recombination processes.13 The present contribution reports on the structure, morphology and optical properties of nanocomposites involving blends of CQDs and zinc vanadium oxide hydroxide hydrate nanoplates (NPs). Zn3(OH)2V2O7·2H2O itself shows fascinating optical properties, owing to its distinctive structure wherein alternate layers of Zn octahedra are connected by pyrovanadate groups.14,15 Previous studies of Zn3(OH)2V2O7·nH2O samples with a range of morphologies have reported strong visible light emission in the range 400–700 nm, and attributed energy levels in the band gap to zinc vacancies.16 Two-dimensional (2-D) ultrathin NP samples in general (of which Zn3(OH)2V2O7·2H2O NPs are an obvious example) are attracting particular interest. These NPs are highly anisotropic and endowed with charge transfer and separation capabilities that are well suited for combining with CQDs in such a way as to modify the PL properties.17,18
Here we report the straight forward hydrothermal synthesis of CQD/Zn3(OH)2V2O7·2H2O NP composites. The structure and morphology of the as-prepared samples were characterized to confirm the incorporation of CQDs and Zn3(OH)2V2O7·2H2O NPs. The PL properties of pure Zn3(OH)2V2O7·2H2O and of the CQD/Zn3(OH)2V2O7·2H2O NP composite samples were investigated, compared and contrasted to demonstrate the effective suppression of blue emission from the latter. Possible mechanisms for the observed modification of the PL properties are discussed.
2. Experimental
Preparation of CQDs
A mixture of 2.5 g sucrose (C12H22O11) dissolved in 50 ml deionized (DI) water was transferred into a 50 ml Teflon-lined stainless steel autoclave and heated at 180 °C for 5 hours. After completion of the reaction, the resulting dark brown solution was centrifuged to yield CQDs.Synthesis of Zn3(OH)2V2O7·2H2O nanoplates
1 mmol vanadium pentoxide (V2O5), 5 mmol hexamethylenetetramine (C6H12N4), 1 mmol zinc acetate dihydrate (CH3COO)2Zn·2H2O and 3 mmol sodium sulfate (Na2SO4) were dissolved in 30 ml DI water. The obtained homogeneous suspension was transferred into a 50 ml Teflon-lined autoclave and heated in an oven at 140 °C for 24 hours. The resulting white product was washed and dried in a vacuum oven (0.08 Mpa) at 60 °C for 12 hours.Fabrication of CQD/Zn3(OH)2V2O7·2H2O NP composites
10 ml of CQD solution and 30 ml of Zn3(OH)2V2O7·2H2O solution with respective concentrations of 1 and 10 mg ml−1 were mixed and then placed in an autoclave. The mixture was heated in an oven at 80 °C for 5 hours. The product was washed with distilled water and dried in a vacuum oven (0.08 MPa) at 50 °C for 12 hours.Characterization
The structure, morphology and composition of the products were characterized by X-ray Diffraction (XRD), Field Emission Scanning Electron Microscopy (FE-SEM), High Resolution Transmission Electron Microscopy and Selected Area Electron Diffraction (HRTEM and SAED), X-ray Photoelectron Spectroscopy (XPS), Raman (532 nm excitation) and Fourier Transform Infrared (FTIR) Spectroscopy. The electronic absorption spectrum of the CQDs in aqueous solution was recorded using a UV-Vis spectrophotometer, and fluorescence emission spectra were measured following steady state/transient illumination at selected wavelengths (λex, at 20 nm intervals in the range 320–500 nm) from the continuum emission of a high pressure xenon lamp. PL spectra of the Zn3(OH)2V2O7·2H2O NP sample and the CQD/Zn3(OH)2V2O7·2H2O NP composites were measured using a Raman spectrometer with a He–Cd (325 nm) excitation laser at room temperature.3. Results and discussion
The microstructure of the as-grown CQD/Zn3(OH)2V2O7·2H2O NP composite samples has been characterized by several complementary techniques. All diffraction peaks in the XRD spectrum match with the documented pattern for hexagonal phase Zn3(OH)2V2O7·2H2O (JCPDs, no. 50-0570). The full-width at half maximum (FWHM) values of the peaks are sensitive to their (hkl) indices, indicating a highly anisotropic structure.19 No diffraction peak attributable to carbon was detected, reflecting the small volume fraction and low crystallinity of the CQDs (Fig. 1(a)). Numerous NPs with smooth surfaces, regular edges and lateral dimensions of the order of several to tens of microns are observable in the SEM image (Fig. 1(b)), which bodes well for applications that might require high levels of surface interactions.20 | ||
| Fig. 1 (a) XRD pattern, (b) SEM, (c) TEM and (d, f and g) HR-TEM images, along with (e) the SAED pattern of the as-prepared CQD/Zn3(OH)2V2O7·2H2O NP composites. | ||
TEM and SAED analysis revealed additional subtleties concerning these CQD/Zn3(OH)2V2O7·2H2O NP composites. The low resolution TEM image (Fig. 1(c)) accords with the SEM results and the associated SAED pattern (Fig. 1(e)) exhibits clear diffraction spots in a hexagonal configuration, showing that the dominant phase is hexagonal (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1) with a high degree of crystallinity, and that the preferred growth direction is normal to [0001]. The HRTEM image (Fig. 1(d)) shows that the CQDs are well dispersed and have small diameters in the range 5–7 nm. Lattice fringes associated with both CQDs (I) and Zn3(OH)2V2O7·2H2O NPs (II) can be seen in the enlarged patterns (Fig. 1(f) and (g), respectively). The 0.27 nm lattice spacing is associated with the CQDs, which is a little larger than the spacing of the (020) plane of graphitic carbon (0.25 nm), while the 0.30 nm spacing corresponds to the (012) plane of the Zn3(OH)2V2O7·2H2O NPs.21
m1) with a high degree of crystallinity, and that the preferred growth direction is normal to [0001]. The HRTEM image (Fig. 1(d)) shows that the CQDs are well dispersed and have small diameters in the range 5–7 nm. Lattice fringes associated with both CQDs (I) and Zn3(OH)2V2O7·2H2O NPs (II) can be seen in the enlarged patterns (Fig. 1(f) and (g), respectively). The 0.27 nm lattice spacing is associated with the CQDs, which is a little larger than the spacing of the (020) plane of graphitic carbon (0.25 nm), while the 0.30 nm spacing corresponds to the (012) plane of the Zn3(OH)2V2O7·2H2O NPs.21
The composition and the surface properties of the CQD/Zn3(OH)2V2O7·2H2O NP composites were also investigated by XPS. The full survey spectrum (Fig. 2(a)) indicates the presence of zinc (Zn 2p), carbon (C 1s), vanadium (V 2p) and oxygen (O 1s). An enlarged view of the C 1s binding energy region is shown in Fig. 2(b), wherein the peaks were fitted as four components corresponding to C–C, C–dots, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.22,23 No peaks attributable to Zn–C or V–C bonds were identified, implying that the CQDs are not present as dopants in the composite material. Fig. 2(c) shows V 2p3/2 and V 2p1/2 peaks at 517.8 and 524.8 eV, respectively, attributable to the V–O bonds in the [V2O7]4− anions. Peaks at binding energies associated with O 1s are identified at 530.2, 531.6 and 533.0 eV (Fig. 2(d)) and ascribed to Zn–O and V–O bonding, to CO bonding and to H2O, respectively.24–26
O bonds, respectively.22,23 No peaks attributable to Zn–C or V–C bonds were identified, implying that the CQDs are not present as dopants in the composite material. Fig. 2(c) shows V 2p3/2 and V 2p1/2 peaks at 517.8 and 524.8 eV, respectively, attributable to the V–O bonds in the [V2O7]4− anions. Peaks at binding energies associated with O 1s are identified at 530.2, 531.6 and 533.0 eV (Fig. 2(d)) and ascribed to Zn–O and V–O bonding, to CO bonding and to H2O, respectively.24–26
 | ||
| Fig. 2 (a) Survey XPS spectrum of the as-prepared CQD/Zn3(OH)2V2O7·2H2O NP composites along with expanded views of the (b) C 1s, (c) V 2p and (d) O 1s binding regions and illustrative deconvolutions as described in the text. | ||
FTIR and Raman spectra of the as-prepared Zn3(OH)2V2O7·2H2O NPs and the CQD/Zn3(OH)2V2O7·2H2O NP composites are compared in Fig. 3. The clear Raman peaks at 250, 329, 434, 478, 796 and 869 cm−1 (Fig. 3(a)) are all characteristic of hexagonal Zn3(OH)2V2O7·2H2O. No extra features attributable to bonding in the CQD/Zn3(OH)2V2O7·2H2O NP composites were observed in this region, indicating the stability of the matrix structure.27,28 FTIR features attributable to intrinsic Zn3(OH)2V2O7·2H2O NPs are found in two wavenumber regions. Peaks in the 600–950 cm−1 region are associated with symmetric and asymmetric V–O–Zn and V–O–V vibrations, while both symmetric stretching and bending vibrations of H2O and the vibrations of any surface adsorbed CO2 molecules typically fall at higher wavenumber (in the range 1600–3600 cm−1).9,29 The additional features at 621 and 1137 cm−1, which do not match with the documented peaks of hexagonal Zn3(OH)2V2O7·2H2O and can be associated with C–O and CO stretching vibrations,30 hint at contributions from the CQDs. Thus it is tempting to propose that the CQD/Zn3(OH)2V2O7·2H2O NP composites are constituted by the formation of C–O and CO bonds between the surfaces of the CQDs and Zn3(OH)2V2O7·2H2O NPs.
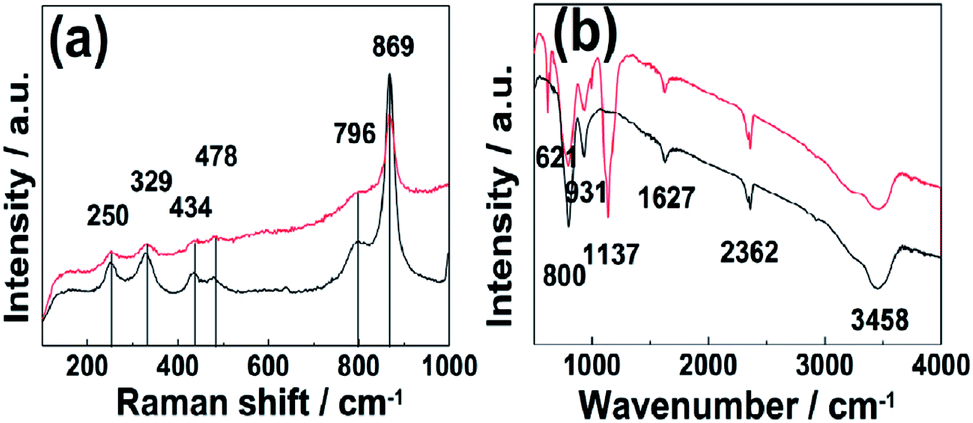 | ||
| Fig. 3 (a) Raman and (b) FTIR spectra of the as-prepared Zn3(OH)2V2O7·2H2O NPs (black) and of the CQD/Zn3(OH)2V2O7·2H2O NP composite (red). | ||
The UV-Vis absorption and fluorescent properties of the CQDs were investigated in aqueous solution: results are shown in Fig. 4. The absorption curve (black line) shows an intense peak at ∼284 nm that can be assigned to CC bond localized π* ← π excitations and a much weaker tail stretching to longer wavelengths consistent with a CO bond localized π* ← n transition.31,32 The PL spectra taken at progressively longer excitation wavelengths (λex), stepping in 20 nm increments between 320 and 500 nm (colored line), reveal a clear λex-dependence to the peak emission wavelength (λem) and its intensity, though we recognize that the latter dependence must be convoluted with the λex-dependent excitation light intensity. As noted elsewhere, such observations point to the existence of a range of emission traps at surface sites on the CQDs.33–35
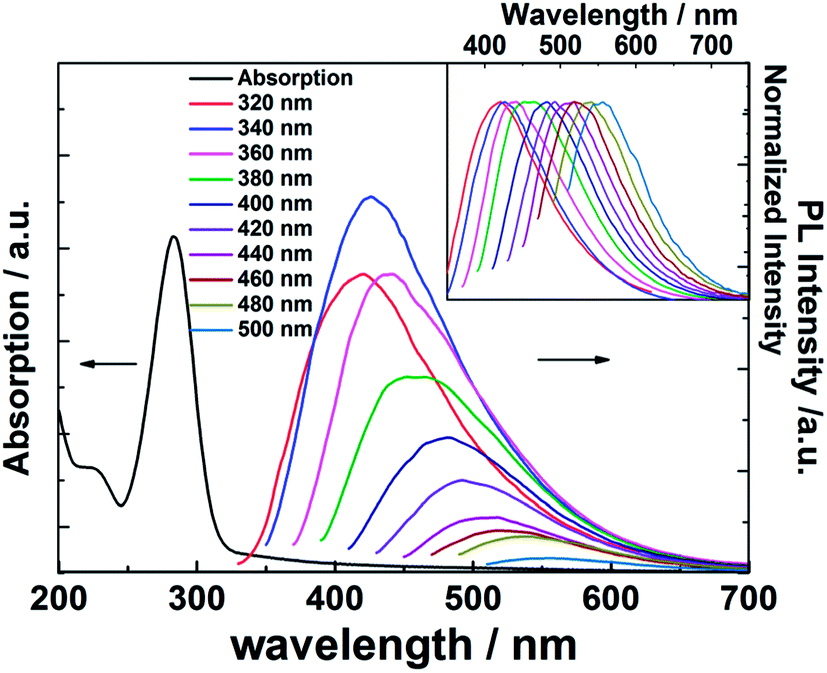 | ||
| Fig. 4 UV-Vis absorption and PL spectra of the as-prepared CQDs following illumination at progressively longer excitation wavelengths (stepped in 20 nm increments across the range 320 ≤ λex ≤ 500 nm). The corresponding normalized emission spectra are shown in the inset. | ||
Fig. 5 shows further analysis of the CQD PL data. In all cases, the energy corresponding to the peak of the emission spectrum (Eem) lies below the excitation energy (Eex), i.e. below the Eem = Eex line, confirming that the Stokes' shift is positive throughout the range 320 ≤ λex ≤ 500 nm. The origin of the phenomenon is not yet entirely understood, but it is generally assumed that the emission and its shift with Eex arise from the radiative recombination of excitons located at surface energy traps.36–38
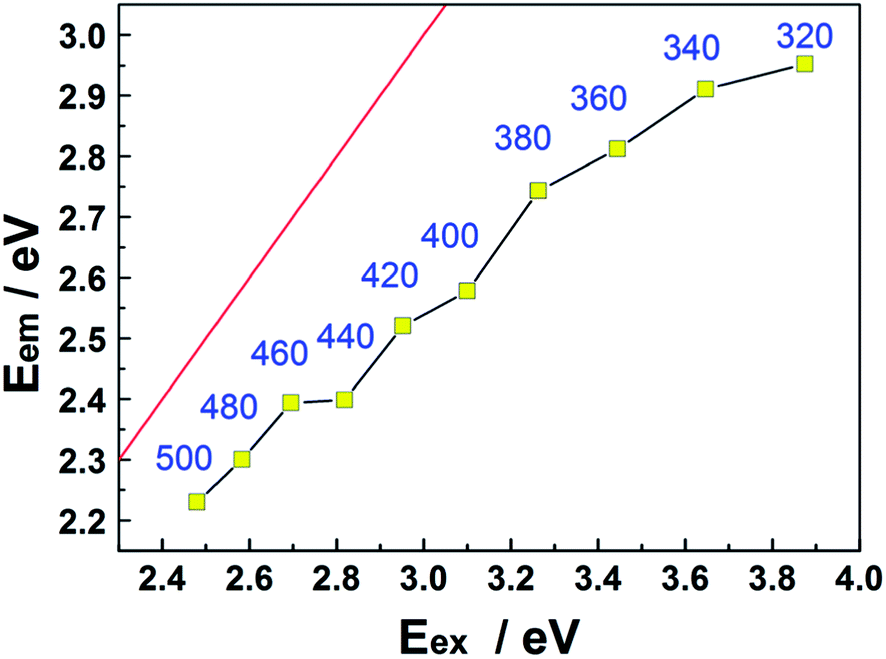 | ||
| Fig. 5 Plot showing the variation of Eem with Eex. Eem = Eex is shown by the red line, and each data point is additionally labelled by the corresponding λex value (in nm). | ||
Data such as those shown in Fig. 4 and 5 have attracted interest, both from a fundamental perspective and in the context of practical applications. These data for the as-prepared CQDs are now compared with the room temperature PL spectra (λex = 325 nm) of a CQD/Zn3(OH)2V2O7·2H2O NP composite sample and of pure Zn3(OH)2V2O7·2H2O shown in Fig. 6 (red and blue curves, respectively). Both spectra show two broad emission bands, centered at energies below the band gap of Zn3(OH)2V2O7·2H2O (∼3.35 eV).14,16,39 As noted elsewhere, these emissions are likely to arise from energy levels in the band gap induced by local defects such as zinc vacancies.16 As Fig. 6 also shows, these PL spectra can both be decomposed as the sum of two Gaussian features centered at 2.33 and 2.93 eV, but the relative peak intensities are substantially modified when CDQs are attached. Specifically, the relative intensity of the higher energy (blue) emission feature is reduced by ∼50% relative to that of the lower energy feature.
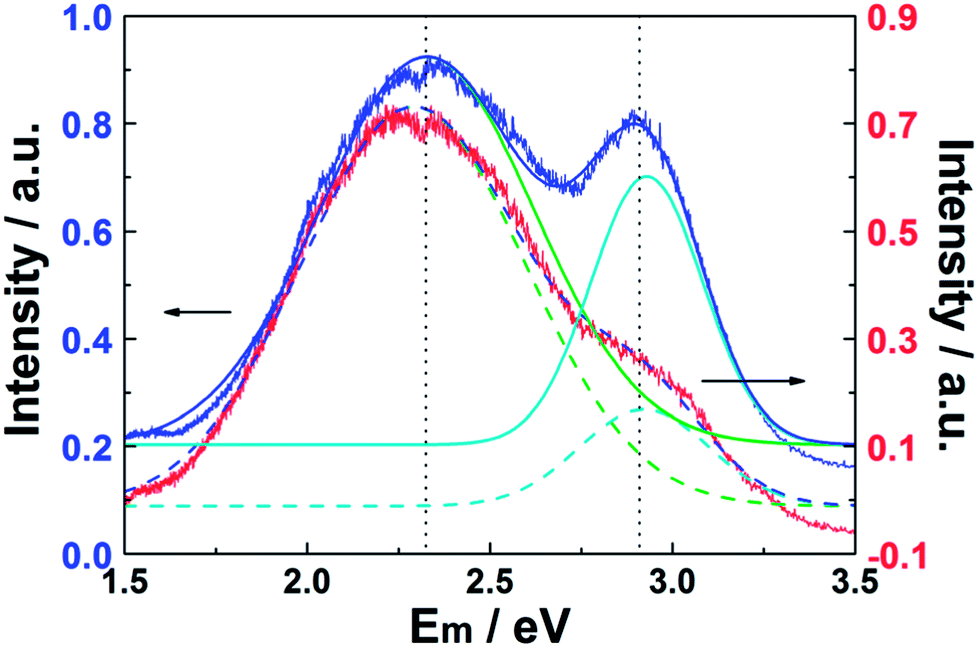 | ||
| Fig. 6 PL spectra (λex = 325 nm) of the as-prepared Zn3(OH)2V2O7·2H2O NPs (blue) and CQD/Zn3(OH)2V2O7·2H2O NP composites (red). The corresponding best-fits in terms of a sum of two Gaussian features centered at 2.33 and 2.93 eV are shown by the solid and dashed lines, respectively. The two PL spectra have been normalized to the same peak intensity, but have been offset vertically by 0.1 a.u. for display purposes. | ||
Fig. 7 suggests, schematically, how the presence of the CQDs could influence the PL spectrum of the CQD/Zn3(OH)2V2O7·2H2O NP composite samples. The CQDs, which are both excellent electron donors and electron acceptors, are well dispersed on the surface of the Zn3(OH)2V2O7·2H2O NPs. Incident 325 nm photons (with energy hν0) are absorbed by the CQD/Zn3(OH)2V2O7·2H2O NP composite, generating photoelectrons. Some of these result in light emission from the Zn3(OH)2V2O7·2H2O NPs themselves at energies hν1 (2.33 eV) and hν2 (2.93 eV), but other photoelectrons transfer to lower energy sites on the surface of the CQDs resulting in (relatively) increased visible and reduced blue emission.11,40–43 Such proposed selective absorption of photoelectrons by the CQDs lacks systematic definition at this time, but we speculate that the CQDs support a high density of energy states (as a result of size quantization, irregularly spaced surface defects and adsorbed functional groups) that offer a wide range of chemical potentials and enable modification of the PL emission. Clearly, there is a pressing need for more detailed investigations of, for example, the excited state dynamics and recombination processes in CQD/Zn3(OH)2V2O7·2H2O NP composite materials.
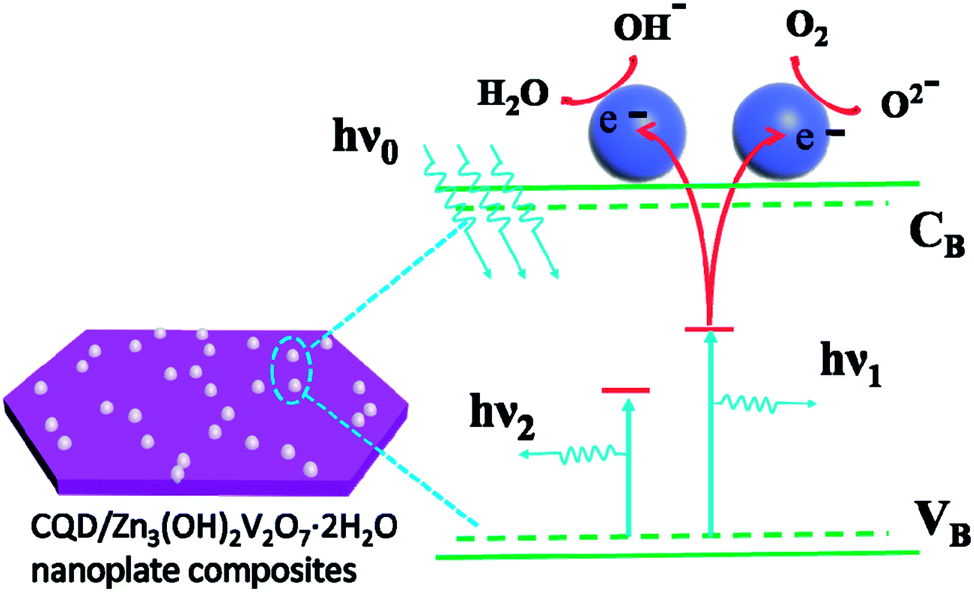 | ||
| Fig. 7 Schematic illustration of the mechanism for the modification of the PL from CQD/Zn3(OH)2V2O7·2H2O NP composite samples. | ||
4. Conclusions
We report the characterization of CQD/Zn3(OH)2V2O7·2H2O NP composites prepared via a simple hydrothermal process. The CQDs appear to be accommodated through C–O and CO bonds to the surfaces of the Zn3(OH)2V2O7·2H2O NPs. Further, PL measurements show that CQDs can be used to modify the PL properties of Zn3(OH)2V2O7·2H2O NPs, specifically reducing the relative intensity of the blue component within the overall emission – thereby ameliorating the possibility of blue-light induced damage to, for example, photoreceptor cells. Such novel composites offer new insights into the design of efficient optoelectrical devices and could serve to inspire future technological applications in this field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. Y612040000). The authors are grateful to Lian C. Wang, Hai Z. Xie, and Jia Q. Pan for their many and varied contributions to the described work.References
- M. Hatori, C. Gronfier, R. N. Van Gelder, P. S. Bernstein, J. Carreras, S. Panda, F. Marks, D. Sliney, C. E. Hunt, T. Hirota, T. Furukawa and K. Tsubota, npj Aging Mech. Dis., 2017, 3, 9 CrossRef PubMed.
- A. Krigel, M. Berdugo, E. Picard, R. Levy-Boukris, I. Jaadane, L. Jonet, M. Dernigoghossian, C. Andrieu-Soler, A. Torriglia and F. Behar-Cohen, Neuroscience, 2016, 339, 296–307 CrossRef PubMed.
- F. Behar-Cohen, C. Martinsons, F. Viénot, G. Zissis, A. Barlier-Salsi, J. P. Cesarini, O. Enouf, M. Garcia, S. Picaud and D. Attia, Prog. Retinal Eye Res., 2011, 30, 239–257 CrossRef PubMed.
- I. Jaadane, P. Boulenguez, S. Chahory, S. Carré, M. Savoldelli, L. Jonet, F. Behar-Cohen, C. Martinsons and A. Torriglia, Free Radical Biol. Med., 2015, 84, 373–384 CrossRef PubMed.
- S. Ye, F. Xiao, Y. X. Pan, Y. Y. Ma and Q. Y. Zhang, Mater. Sci. Eng., R, 2010, 71, 1–34 CrossRef.
- J. Cho, J. H. Park, J. K. Kim and E. F. Schubert, Laser Photonics Rev., 2017, 11, 1600147 CrossRef.
- P. F. Smet and J. J. Joos, Nat. Mater., 2017, 16, 500 CrossRef PubMed.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- J. Pan, Y. Sheng, J. Zhang, J. Wei, P. Huang, X. Zhang and B. Feng, J. Mater. Chem. A, 2014, 2, 18082–18086 Search PubMed.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef.
- B. Cui, X.-T. Feng, F. Zhang, Y.-L. Wang, X.-G. Liu, Y.-Z. Yang and H.-S. Jia, New Carbon Mater., 2017, 32, 385–401 CrossRef.
- P. Namdari, B. Negahdari and A. Eatemadi, Biomed. Pharmacother., 2017, 87, 209–222 CrossRef PubMed.
- X. Wang, L. Cao, F. Lu, M. J. Meziani, H. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y.-P. Sun, Chem. Commun., 2009, 3774–3776, 10.1039/B906252A.
- Q. Wang, L. Zheng, Y. Bai, J. Zhao, F. Wang, R. Zhang, H. Huang and B. Su, Appl. Surf. Sci., 2015, 347, 602–609 CrossRef.
- H. Guo, D. Guo, Z. Zheng, W. Wen and J. Chen, J. Mater. Res., 2014, 29, 2934–2941 CrossRef.
- S. Ni, G. Zhou, X. Wang, X. Sun, F. Yang, Y. Liu and D. He, Mater. Chem. Phys., 2010, 120, 426–430 CrossRef.
- S. Hu and X. Wang, Chem. Soc. Rev., 2013, 42, 5577–5594 RSC.
- S. Han, L. Hu, Z. Liang, S. Wageh, A. A. Al-Ghamdi, Y. Chen and X. Fang, Adv. Funct. Mater., 2014, 24, 5719–5727 CrossRef.
- F. Wang, W. Wu, X. Sun, S. Song, Y. Xing, J. Wang, D. Yu and Z. Su, Mater. Charact., 2013, 86, 139–145 CrossRef.
- R. Mas-Balleste, C. Gomez-Navarro, J. Gomez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
- G. Yang, M. Wu and C. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 23746–23754 Search PubMed.
- H. Yu, Y. Zhao, C. Zhou, L. Shang, Y. Peng, Y. Cao, L.-Z. Wu, C.-H. Tung and T. Zhang, J. Mater. Chem. A, 2014, 2, 3344–3351 Search PubMed.
- Q. Liang, W. Ma, Y. Shi, Z. Li and X. Yang, Carbon, 2013, 60, 421–428 CrossRef.
- H. Guo, J. Chen, W. Weng and S. Li, Appl. Surf. Sci., 2011, 257, 3920–3923 CrossRef.
- R. Gopal and C. Calvo, Can. J. Chem., 1973, 51, 1004–1009 CrossRef.
- S. Ni, G. Zhou, S. Lin, X. Wang, Q. Pan, F. Yang and D. He, Mater. Lett., 2009, 63, 2459–2461 CrossRef.
- E. Hryha, E. Rutqvist and L. Nyborg, Surf. Interface Anal., 2012, 44, 1022–1025 CrossRef.
- L. Yang, Y. Tang, L. Tong, H. Zhou, J. Ding, T. Fan and D. Zhang, Appl. Surf. Sci., 2015, 346, 115–123 CrossRef.
- M. Ren, J. Song, Y. Shi, Y. Xiang and G. Hu, J. Cryst. Growth, 2014, 402, 119–123 CrossRef.
- M. Shim, S. V. Shilov, M. S. Braiman and P. Guyot-Sionnest, J. Phys. Chem. B, 2000, 104, 1494–1496 CrossRef.
- J. Ke, X. Li, Q. Zhao, B. Liu, S. Liu and S. Wang, J. Colloid Interface Sci., 2017, 496, 425–433 CrossRef PubMed.
- X. Jia, J. Li and E. Wang, Nanoscale, 2012, 4, 5572–5575 RSC.
- N. Baker Sheila and A. Baker Gary, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef PubMed.
- S. K. Das, Y. Liu, S. Yeom, D. Y. Kim and C. I. Richards, Nano Lett., 2014, 14, 620–625 CrossRef PubMed.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef PubMed.
- S. Zhuo, M. Shao and S.-T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef PubMed.
- A.-M. Alam, B.-Y. Park, Z. K. Ghouri, M. Park and H.-Y. Kim, Green Chem., 2015, 17, 3791–3797 RSC.
- J. W. Arbogast, C. S. Foote and M. Kao, J. Am. Chem. Soc., 1992, 114, 2277–2279 CrossRef.
- D. L. Wood and J. Tauc, Phys. Rev. B, 1972, 5, 3144–3151 CrossRef.
- K. T. Shimizu, W. K. Woo, B. R. Fisher, H. J. Eisler and M. G. Bawendi, Phys. Rev. Lett., 2002, 89, 117401 CrossRef PubMed.
- A. Henglein, A. Kumar, E. Janata and H. Weller, Chem. Phys. Lett., 1986, 132, 133–136 CrossRef.
- P. V. Kamat, I. Bedja and S. Hotchandani, J. Phys. Chem., 1994, 98, 9137–9142 CrossRef.
- P. V. Kamat, J. Am. Chem. Soc., 1991, 113, 9705–9707 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |