
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDihydroartemisinin induces apoptosis and downregulates glucose metabolism in JF-305 pancreatic cancer cells
Wenhe Zhu†
 a,
Wei Zhang†a,
Na Xu†a,
Yawei Lia,
Junjie Xua,
Hong Zhanga,
Yan Lia,
Shijie Lva,
Wensen Liu*b and
Huiyan Wang*a
a,
Wei Zhang†a,
Na Xu†a,
Yawei Lia,
Junjie Xua,
Hong Zhanga,
Yan Lia,
Shijie Lva,
Wensen Liu*b and
Huiyan Wang*a
aJilin Medical University, Jilin 132013, China. E-mail: jlmpcwhy@163.com; Fax: +86-0432-64560460; Tel: +86-0432-64560460
bInstitute of Military Veterinary Medicine, Academy of Military Medical Sciences, Changchun 130122, China
First published on 6th June 2018
Abstract
Cancer cell promotion of glycolysis provides a promising therapeutic target for cancer treatment. Dihydroartemisinin (DHA) displays cytotoxicity to multiple human tumor cells. However, its effects on pancreatic cancer cells are not well studied. The objective of this study was to investigate the effect of DHA on glucose metabolism and cell viability in JF-305 pancreatic cancer cells. To achieve these goals, cell viability was measured with MTT assay, and the occurrence of apoptosis was detected. Glucose uptake, lactate production, and ATP content were measured. Western blotting was used for the detection of apoptosis-related protein expression. The result showed that DHA caused significant reduction in JF-305 cell viability, arrested the cell phase in G2/M, induced apoptosis, and decreased the mitochondrial membrane potential and accumulated ROS. DHA also inhibited glucose uptake, lactate generation, and ATP production. Western blotting showed that treatment with DHA increased the activity of caspase-9 and caspase-3, downregulated Bcl-2 expression, and upregulated the expression levels of Bax and Cyto C. Meanwhile, DHA downregulated the Akt/mTOR signaling pathway and inhibited glucose transporter 1 expression. Our data suggest that DHA treatment increased the apoptosis of JF-305 pancreatic cancer cells, and the effect of apoptosis may be associated with the inhibition of glycolysis.
1. Introduction
Pancreatic cancer has been the fourth leading cause of cancer death in the last two decades because of various obstacles in its treatment.1,2 The treatment of patients with pancreatic cancer mainly relies on surgery, radiation therapy, chemotherapy, or the combination of these therapeutic methods. However, due to aggressive tumor biology and the lack of specific symptoms during the early stages, pancreatic cancer frequently presents as an incurable disease with approximately two-thirds of patients showing radiographically detectable metastasis at the time of diagnosis.3 Pancreatic ductal adenocarcinomas carry a dismal prognosis, and less than 5% of the patients survives at least 5 years.4 Thus, the discovery of new therapeutic agents and approaches in patients with pancreatic cancer is of paramount importance. Traditional Chinese medicine has been practiced and recorded in medical literature for thousands of years. The rich history of traditional Chinese medicine has prompted a recent surge in clinical research efforts.5–7 Furthermore, its efficacy as an alternative strategy to pharmaceutical-based approaches used in developed countries is evaluated for the prevention and treatment of various diseases, including cancers. Artemisinin is a sesquiterpene lactone isolated from the sweet wormwood Artemisia annua L. and is being used as an antimalarial agent.8 Dihydroartemisinin (DHA) is an important derivative of artemisinin (Fig. 1A).9 Recent studies showed that DHA has profound effect against breast cancer, papillomavirus-expressing cervical cancer, liver cancer, and pancreatic cancer.10 Additionally, DHA exerts anticancer effects in lung carcinomas by the induction of apoptosis without obvious side effects. Aside from its prominent pro-apoptotic effect, DHA affects cancer cell functions, including tumor cell proliferation, angiogenesis, and immune regulation.11,12 However, the exact molecular mechanisms of DHA anticancer effects remain to be fully investigated.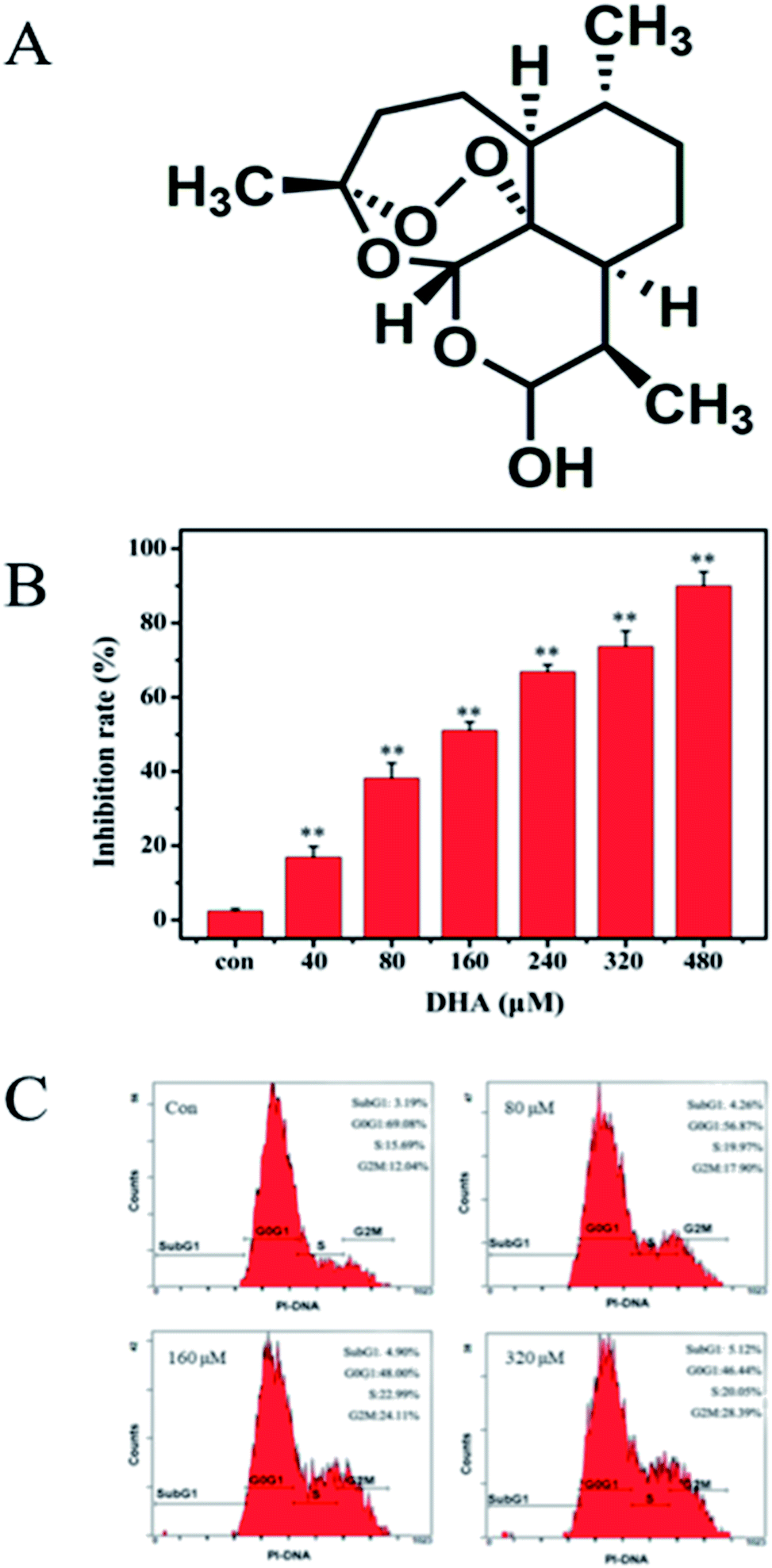 | ||
| Fig. 1 DHA inhibits cell viability and cell cycle in cultured human JF-305 cells. (A) Chemical structures of DHA. (B) DHA induced concentration-dependent reduction of cell viability in JF-305 cells. Cells were incubated with indicated concentrations of DHA for 48 h. Cell growth inhibition activity of DHA was assessed by MTT. (C) JF-305 cells were exposed to various concentrations of DHA for 48 h followed by cell cycle distribution assay. Data are present as mean ± SD, n = 5. **p < 0.01 compared with control. | ||
Metabolic alterations are a hallmark of cancer. Metabolic reprogramming provides nutrient influx for the biomass requirement of rapidly proliferating tumor cells and also facilitates resistance to chemotherapy.13,14 Among the several important metabolic alterations, most cancer cells exhibit enhanced aerobic glycolysis.15,16 To date, accumulating evidence suggests that such alterations in glucose metabolism may contribute to cancer cell proliferation, survival, metastasis, and therapy resistance. As enhanced evidence on glucose glycolysis is a hallmark of cancer cells, research suggests that targeting glucose glycolysis may be an avenue for the development of anticancer drugs.17,18
Resistance to apoptosis is linked to glycolysis and predicts a poor clinical outcome in cancer. However, most studies conducted essentially only focused on using DHA to induce tumor cell apoptosis. As the recent evidence indicates DHA exhibits profound antitumor properties by inducing apoptosis in a wide variety of cancer cells, such as glioblastoma, lung, prostate, hepatocarcinoma cancer.19–22 In particular, Mi et al.23 have reported that DHA can inhibit proliferation, promotes apoptosis, and attenuate the glycolytic metabolism in non-small cell lung carcinoma cells. Thus, the role of DHA on glucose metabolism in the pancreatic cancer cells is rarely investigated and remains largely unknown. Some evidences have suggested that targeting glycolysis will be a good strategy against pancreatic cancer. In this study,24 we examined the capacity of DHA to alter metabolic phenotype in pancreatic cancer JF-305 cells and induce cell growth reduction.
2. Materials and Methods
2.1 Materials
JF-305 Human pancreatic cancer cell line was provided by the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Dulbecco's Modified Eagle's Medium (DMEM) was obtained from Gibco® (USA). Dimethyl sulfoxide (DMSO, analytical grade) was purchased from Beijing Chemical Reagent Factory (Beijing, China). The antibodies against Bcl-2, Bax and cleaved caspase-3, cleaved caspase-9, and Cyto C were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). p-AKT, AKT, p-MTOR, MTOR, Glut1 and β-actin were purchased from Sigma-Aldrich Company (St. Louis, MO, USA).All other chemicals were of analytical grade.
2.2 MTT assay
In vitro anti-tumor activity was examined by an MTT assay, following the procedure in the original report.25 Briefly, JF-305 cells were seeded at a concentration of 5.0 × 104 cells per mL (200 μL total) in a 96-well assay plate. After 24 h incubation at 37 °C under 5% carbon dioxide, the test substance was added. Each concentration was tested five times. The same volume of solution without the test substance was used as a control. The cells were incubated for 48 h, and then 20 μL of MTT solution (5 mg mL−1) was added to each well and incubated for another 4 h. After removal of medium, 150 μL dimethyl sulfoxide (DMSO) was added to each cell, and the optical density (OD) was measured at 490 nm using a Bio-assay reader (Bio-Rad550, USA). The relative cell viability was expressed as the ratio of the absorbance of DHA treated cells to that of the control cells.2.3 Cell cycle
The cells (1 × 106 per ml) were seeded onto 6-well plate and treated with different concentration of DHA. After incubation for 48 h, cells were harvested, washed twice with cold PBS and fixed in ice-cold 70% ethanol overnight at 4 °C. The cells were concentrated by removing ethanol and treated with 0.01% Dnase-free RNase A for 10 min at 37 °C. Cellular DNA was stained with 0.05% propidium iodide (PI) for 20 min at 4 °C in darkness. The cell cycle distribution was detected with flow cytometry (FCM) on a Beckman Coulter Epics XL flow cytometer. The percentage of cells at G0/G1, S, or G2/M phase was documented using Expo32 software (Beckman Coulter, Miami, FL, USA).2.4 Morphological examination for apoptosis
JF-305 cells were seeded on slides at a density of 5 × 104 per mL in 6-well plates. After treatment as mentioned above, cells from all 4 groups were washed twice with PBS, fixed in 4% paraformaldehyde for 10 min, and then stained with Hoechst 33258 for 5 min. Then the cells were observed under a fluorescence microscope. The nuclei of the living cells were a homogeneous blue; those of apoptotic cells were compact, condensed, and whitish-blue.2.5 Transmission electron microscopy (TEM)
JF-305 cells were treated with different concentrations of DHA for 48 h. After DHA treatment, the cells were trypsinized and then fixed in PBS (pH 7.3) containing ice-cold 2.5% glutaraldehyde. After removal of the fixative, each cell mass was washed twice with cold PBS, then dehydrated in graded ethanol, immersed in propylene oxide and embedded in epoxy resin. Ultra thin sections were double-stained with lead citrate/uranyl acetate previously and examined by TEM.2.6 Apoptosis
JF-305 cells treated with different concentration of DHA were harvested by centrifugation at 1000g for 3 min, and washed with ice-cold PBS. The cell suspension (100 μL) was centrifuged at 1000g for 3 min. After that, the supernatant was discarded and the pellet was gently resuspended in 100 μL cell culture medium and incubated with 100 μL Muse Annexin V & Dead Cell reagents in the dark at room temperature for 20 min. Apoptosis was analyzed using a Muse Cell Analyzer (Merck-Millipore).2.7 Detection of change in mitochondria membrane potential
Mitochondrial stability was assessed using a mitochondrial membrane potential assay kit with JC-1. JF-305 cells of 1 × 104 cells per well were cultured in 96-well plates for 24 h and then treated with DHA. After 24 h treatment, the cells were incubated with 62.5 μL JC-1 fluorescent dye for 20 min in the dark at 37 °C. Then, the cells were washed slowly twice with JC-1 dyeing buffer, followed by treating Hoechst (100 μL) for 10 min. The mitochondrial membrane potential was imaged using fluorescence microscopy (Olympus, Tokyo, Japan) at 550 nm excitation and 570 nm emissions for JC-1.2.8 Reactive oxygen species (ROS) detected
Intracellular ROS levels were measured using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) oxidation. For the microplate reader-based assay, a microplate reader (TEcAN M200, TecanInc. Switzerland) was used to measure the absorbance at 525 nm. Untreated or treated cells were stained with 20 μM DCFH-DA for 30 min at 37 °C in the dark and subsequently assayed as described in detail previously.262.9 Caspase-3/7 activation detected
The caspase-3/7 activation was detected by Muse Cell Analyzer. JF-305 cells treated with different concentration of DHA were harvested by centrifugation at 1000g for 3 min, and then the cells were resuspend in 50 μL PBS. Add 5 μL of Muse Caspase-3/7 reagent working solution to each group. Mix thoroughly by pipetting up and incubate samples for 30 minutes in the 37 °C incubator with 5% CO2. After incubation, add 150 μL of Muse™ Caspase 7-AAD working solution to each tube, incubate at room temperature for 5 minutes before detecting by Muse Cell Analyzer.2.10 Measurements of glucose uptake levels and lactate production of cells in the media
JF-305 cells of 1 × 104 cells per well were seeded in 24-well plates, which treated with various concentration of DHA for 48 h incubation. At the end of incubation, cell culture media was collected and analyzed for lactate and glucose concentration. For assessment of lactate production, the media was collected and diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 in lactate assay buffer. The amount of lactate present in the media was then estimated using the Lactate Assay Kit (Sigma, St. Louis, MO, USA) according to the manufacturer's instructions. For assessment of glucose uptake, the media were collected and the content of glucose was immediately measured.
:50 in lactate assay buffer. The amount of lactate present in the media was then estimated using the Lactate Assay Kit (Sigma, St. Louis, MO, USA) according to the manufacturer's instructions. For assessment of glucose uptake, the media were collected and the content of glucose was immediately measured.
2.11 Determination of ATP content
The JF-305 cells were seeded in 6-well plates (1 × 105 cells per mL) and incubated with DHA for 48 h. Subsequently, the cells were washed twice with PBS and lysed by somatic cell ATP releasing reagent. ATP levels were assessed with ATP diagnostic kits (Genmed Scientifics, Wilmington, Delaware, USA) according to step provided by the manufacturer's instruction.2.12 Western blot analysis
After treatment as mentioned above, 5 × 105 cells were harvested and sonicated in RIPS buffer. After centrifugation at 12000 × 1g for 10 min at 4 °C, protein content was estimated according to Bio-Rad protein assay and 50 μg protein/lane were loaded on to 12% polyacrylamide SDS gel. The separated proteins were then transferred electrophoretically to nitrocellulose paper and soaked in transfer buffer (25 mmol L−1 Tris, 192 mmol L−1 glycine) and 20% methanol v/v. Non-specific binding was blocked by incubation of the blots in 5% no-fat dry milk in TBS/0.1% Tween (25 mmol L−1 Tris, 150 mmol L−1 NaCl, 0.1% Tween v/v) for 60 min. After washing, the blots were incubated overnight at 4 °C with the primary antibody. After incubation with the primary antibodies and washing in TBS/0.1% Tween, the appropriate secondary antibody was added and left for 1 h at room temperature. Immunoreactive protein bands were detected by chemiluminescence using enhanced chemiluminescence reagents (ECL). Blots were also stained with anti β-actin antibody as internal control for the amounts of target proteins. The films were then subjected to densitometry analysis using a Gel Doc 2000 system (Bio-Rad).
2.13 Statistical analysis
Results are expressed as mean ± SEM. All data were analyzed by one-way ANOVA by using SPSS version 13 software and expressed in mean ± standard deviation. The value of p < 0.05 was considered to be significant.3. Results
3.1 DHA inhibits JF-305 cell growth and induces cell arrest in G2/M
MTT assay was conducted to evaluate the cytotoxic effects of DHA on JF-305 cells. The JF-305 cells were treated with DHA at concentrations of 0–480 μmol L−1 for 48 h. Fig. 1B shows that proliferation of JF-305 cells was markedly inhibited by DHA treatment in a dose-dependent manner. To determine the effect of DHA on cell cycle progression, we performed flow cytometry (FCM) analysis on cells treated with or without DHA at different concentrations (80, 160, and 320 μmol L−1). The results showed that the cell cycle of DHA-treated cells was arrested in G2/M phase in a dose-dependent manner compared with that in the untreated control cells (Fig. 1C).3.2 DHA induces apoptosis in JF-305 cells
The uniform shape of JF-305 nuclei and well-distributed deep blue fluorescence were revealed by Hoechst 33258 staining. Hoechst 33258 was used as an apoptosis marker, which detected apoptotic nuclei with condensed and/or fragmented DNA. As shown in Fig. 2A, the majority of the nuclei in the control groups had uniform blue chromatin with an organized structure. However, following the treatment with 80, 160, and 320 μmol L−1 DHA for 48 h, the JF-305 cells became scarce and showed reduced nuclear sizes, strong fluorescent spots, and pyknotic nuclei and exhibited extensive blebbing, indicating the presence of condensed chromatin and apoptotic bodies. The apoptotic index increased markedly in the cells treated with DHA. The apoptotic effect of DHA treatment on ultrastructure change of JF-305 cells was observed by transmission electron microscopy. The morphological characteristics of tumor cells were observed, such as clear cellularity, integrated structure of the organelles, uniform distribution of chromatin, and microvilli on the cell membrane in the control group (Fig. 2B). After treatment with different concentrations of DHA for 48 h, the JF-305 cells displayed typical apoptotic morphologies, including decrease in microvilli, chromatin condensation, crescent margination of chromatin against the nuclear envelope, enlargement of the perinuclear space, and apoptotic body formation. Moreover, at increased DHA concentrations, the number of apoptotic cells increased. Similar results were obtained for double fluorescence staining of the Annexin V FITC/PI FCM assay. Fig. 2C shows that the percentage of early apoptotic cells for DHA-treated cell increased significantly (p < 0.01) compared with that in the control group.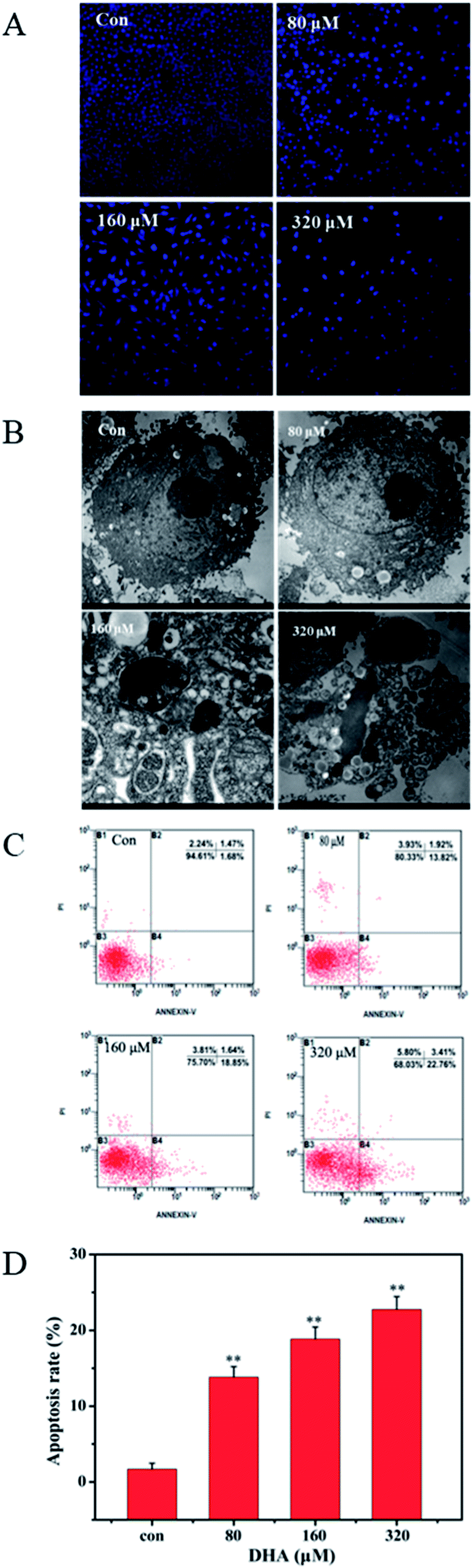 | ||
| Fig. 2 DHA-induced apoptosis in JF-305 cells. (A) JF-305 cells were treated with various concentrations of DHA for 48 h, stained with Hoechst 33258, and viewed by fluorescence microscopy. (B) Transmission electron microscopy detected the morphological change of JF-305 cells treated with different concentration of DHA for 48 h. (C) JF-305 cells were incubated with different concentrations of DHA for 48 h, and then assayed by flow cytometry analysis with Annexin V-FITC staining. (D) Quantification of apoptotic cells. Data are present as mean ± SD, n = 3. **p < 0.01 compared with control. | ||
3.3 Mitochondrial membrane potential and reactive oxygen species (ROS) change
We monitored the cells by using mitochondria-specific probe JC-1 to evaluate changes in mitochondrial membrane potential after 48 h of DHA incubation. As shown in Fig. 3A, the control cells showed mainly an intense red-orange fluorescence coming from JC-1 aggregates. By contrast, DHA-treated cells exhibited green fluorescence coming from JC-1 monomers. The intensity of red-orange fluorescence in the DHA-treated cells was significantly lower than that in the control in a dose-dependent manner, indicating DHA-induced dose-dependent decrease in the mitochondrial membrane potential after 48 h of treatment. To investigate whether ROS are involved in DHA-induced apoptosis, we assessed the levels of intracellular ROS in DHA-treated JF-305 cells by FCM analysis of dichlorodihydrofluorescein (DCF) fluorescence intensity. As shown in Fig. 3B, DHA-induced apoptosis was associated with a concentration-dependent increase in DCF fluorescence intensity. These results suggested that the induction of apoptosis by DHA is positively regulated by intracellular ROS generation.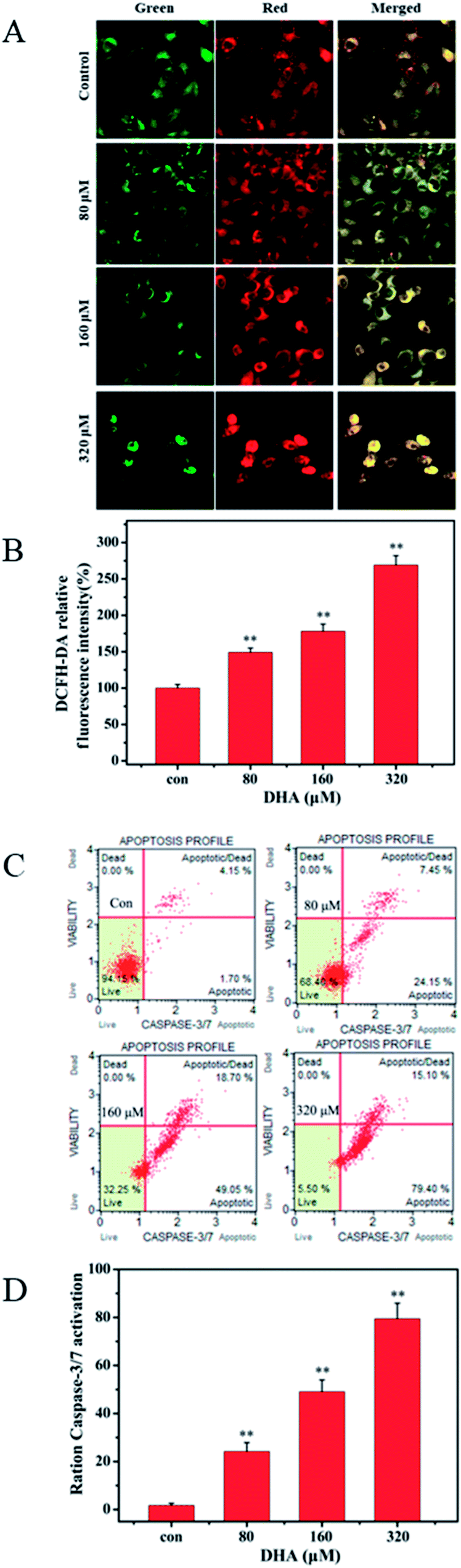 | ||
| Fig. 3 Effect of DHA on change of Mitochondrial Membrane Potential, ROS change and caspase-3/7 activation. (A) JF-305 cells were treated with DHA,mitochondrial membrane potential was using JC-1 detected by laser scanning confocal microscope. (B) The effect of DHA on mitochondrial ROS generation in cells cultured was detected by flow cytometer. (C) After treatment with different concentrations of DHA, the effect of DHA on caspase-3/7 activation was detected by cell analyzer. (D) Quantification of caspase-3/7 activation. Data are present as mean ± SD, n = 3. **p < 0.01 compared with control. | ||
3.4 Caspase-3/7 activation detected result
We selected Muse Caspase-3/7 Kit to determine the effect of DHA on caspase-3/7 activation. Muse cell analyzer was used to quantify stained cells. The details are shown in Fig. 3C. The percentages of cells in the early stages of apoptosis were 1.7%, 24.15%, 49.05%, and 79.40% at DHA concentrations of 0, 80, 160, and 320 μmol L−1, respectively.3.5 Inhibitory effect of DHA on glucose uptake, lactate generation, and adenosine triphosphate (ATP) production
Considering that ATP and lactic acid are the main products during glycolysis, we determined the ATP content in the JF-305 cells and lactic acid content in culture medium to investigate whether DHA induces apoptosis through glycolysis regulation. After 48 h incubation with DHA, the levels of intracellular ATP and extracellular lactic acid both decreased in the JF-305 cells after treatment with different concentrations of DHA compared with that in the control group at the concentrations of 80, 160, and 320 μmol L−1 (Fig. 4A and B). Correspondingly, DHA inhibited glucose uptake in the JF-305 cells in a dose-dependent manner (Fig. 4C).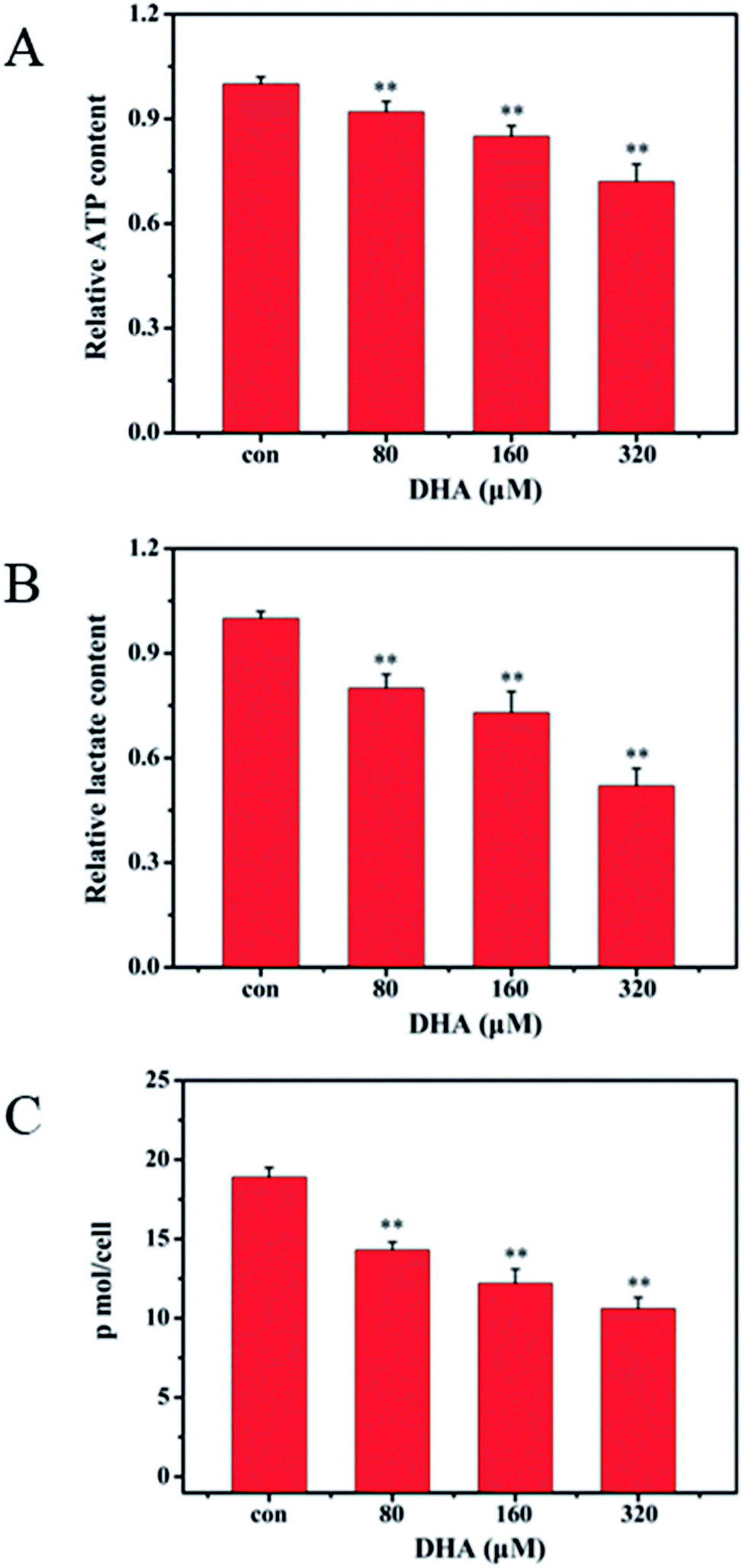 | ||
| Fig. 4 DHA decreases the level of glycolytic metabolism in JF-305 cells. (A) JF-305 cells were treated with indicated concentrations of DHA for 48 h. ATP content was determined using a bioluminescence assay. (B) Relative lactate content was determined as described in Materials and Methods. (C) JF-305 cells were treated with indicated concentrations of DHA for 48 h. The cells were counted and the glucose in the culture media was immediately tested. Data are present as mean ± SD, n = 3. **p < 0.01 compared with control. | ||
3.6 DHA induces apoptosis in JF-305 cells through mitochondrial apoptotic pathway
To investigate the mechanism underlying DHA-induced apoptosis in JF-305 cell, we performed western blot analysis to detect the expression of apoptosis-related proteins in the DHA-treated cells. As shown in Fig. 5, after treatment with different concentrations of DHA for 48 h, the expression of Bax and the activated forms of caspase-3 and caspase-9 remarkably increased. By contrast, DHA markedly decreased Bcl-2 expression, leading to an increase in the Bax/Bcl-2 ratio in a dose-dependent manner. In addition, in DHA-treated JF-305 cells, several Cyto C were released from the mitochondria to cytosol at increased DHA concentration.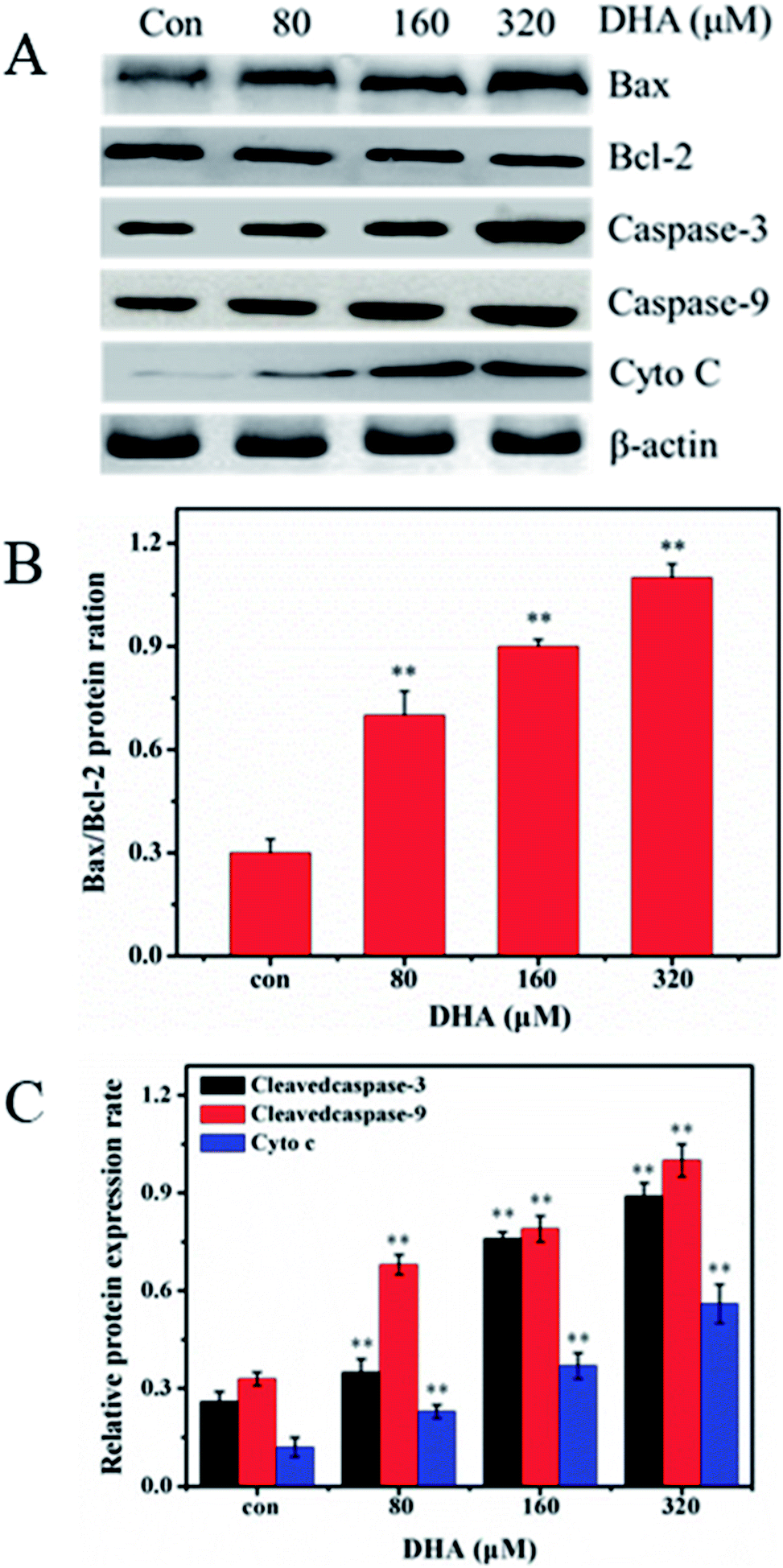 | ||
| Fig. 5 DHA induced cleavage of caspase-3 and caspase-9, increased in Cyto C, Bax, and decrease in Bcl-2 in JF-305 cells. (A) JF-305 cells were incubated with different concentrations of DHA for 48 h and subsequently harvested for western blot analysis. The blots shown are representative of three independent experiments. (B) Quantitation of Bax/Bcl-2. (C) Densitometric values were normalized by β-actin and expressed as mean ± SD, n = 3. **p < 0.01 compared with control. | ||
3.7 DHA downregulates Akt/mTOR signaling pathway and inhibit glycolysis
Akt/mTOR pathway plays a central role in the activation of Warburg effect for cancer cell survival. We further investigated whether DHA repress aerobic glycolysis via the Akt/mTOR signaling pathway in JF-305 cells. After DHA treatment for 48 h, DHA resulted in decreased phosphorylation of Akt and mTOR in a dose-dependent manner (Fig. 6). Furthermore, we detected the expression of glucose transporter 1 (GLUT1), an important glucose transporter. The result showed the DHA treatment can decrease the expression levels of GLUT1 in JF-305 cells.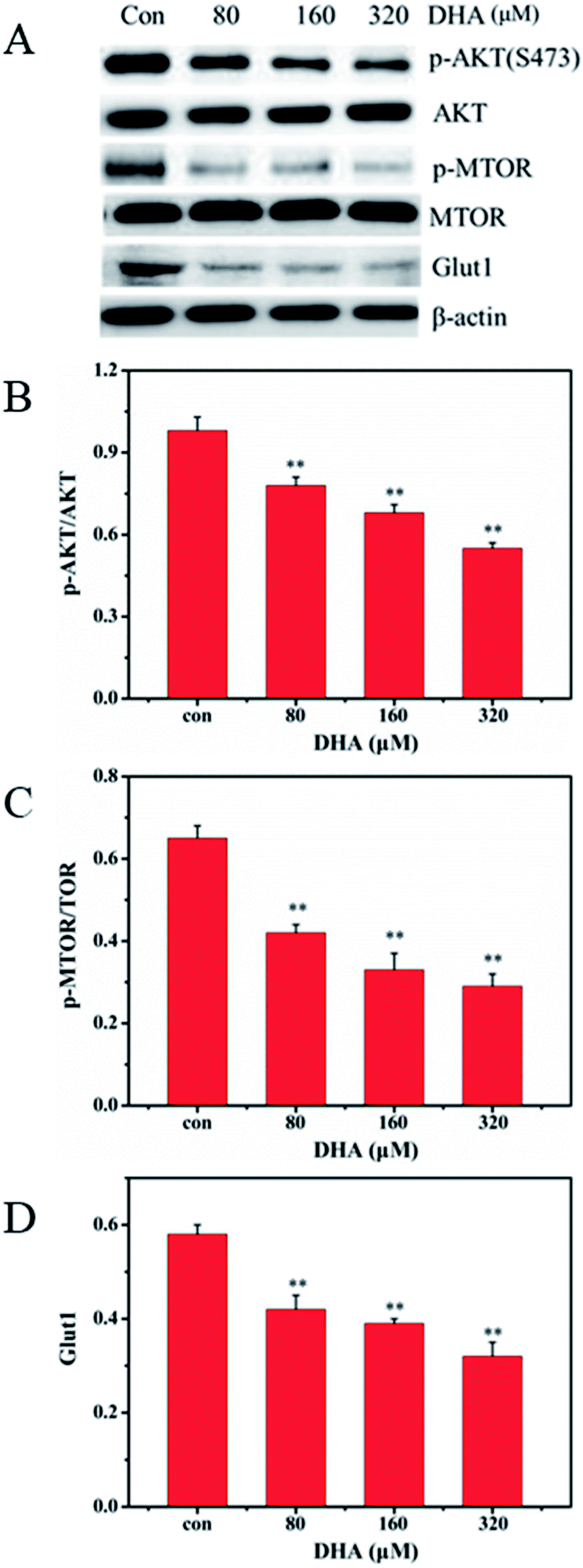 | ||
| Fig. 6 DHA-suppressed glycolytic metabolism is associated with mTOR activation and GLUT1. (A) JF-305 cells were treated with indicated concentrations of DHA for 48 h and subsequently harvested for western blot analysis. (B) Quantitation of p-AKT/AKT. (C) Quantitation of p-MTOR/TOR. (D) Quantitation of GLUT1. Data are the mean ± SD, n = 3; **p < 0.01 compared with control. | ||
4. Discussion
Increasing attention has been focused on traditional Chinese medicine in the search for new anticancer agents with great efficacies against cancer cells and less toxicity to normal cells. DHA, a semisynthetic derivative of artemisinin, displays at least three times more potent antimalarial activity than ART and has therefore been used as the first-line therapeutics against falciparum malaria worldwide.20,21 Similar to other natural products, DHA acts in a multitarget manner against tumors and represents an attractive candidate for cancer therapy.27,28 However, the potential of DHA in treating pancreatic cancer is limited. Li et al.29 reported the microRNA–mRNA regulatory network in DHA treated pancreatic cancer, which improve to understand the possible anti-cancer mechanism. Jia et al.30 research certified the activation of c-Jun NH2-terminal kinase is required for DHA-induced autophagy in pancreatic cancer cells. Thus, in the present study, we will investigate the role of DHA on apoptosis and glucose metabolism in pancreatic cancer cells and explore the possible mechanism.Cell proliferation is considered an important feature of cell growth and development. The present research demonstrated that DHA markedly inhibited the viability of pancreatic cancer cell JF-305 and arrested the cell phase in G2/M in a dose-dependent manner. These effects were not observed in the control group. Programmed cell death (apoptosis) plays an important role in the development, homeostasis, and anticancer protection of multicellular organisms.31,32 Apoptotic cells display characteristic morphological changes, such as cell shrinkage, cytoplasmic vacuolation, chromatin condensation, nuclear fragmentation, and cell blebbing to produce apoptotic bodies. Our research showed that after treatment with DHA, JF-305 cells displayed the typical apoptotic morphology. With increasing concentrations of DHA, the number of apoptotic cells increased.
We further investigated the mechanism involved in DHA-induced apoptosis. As two typical proteins of the Bcl-2 family, Bcl-2 and Bax play key roles in caspase-dependent apoptosis. Bcl-2 had a strong anti-apoptosis effect in inhibiting the Bax-induced caspase-dependent apoptosis.33–35 Thus, a change in the Bax/Bcl-2 ratio indicates the susceptibility of cells to apoptosis.36,37 According to our results, the Bax/Bcl-2 ratio increased in a dose-dependent manner compared with the control. Various signaling pathways for apoptosis exist within an organism, with the mitochondrial pathway as one of the most important. Cyto C is an important mitochondrial protein that induces apoptosis when accumulated in the cytosol in response to diverse stress stimuli.38 Cyto C binds caspase-9 to form a complex that activates other caspase family members, including caspase-3, to induce apoptosis.39
In this study, we demonstrated that treatment with DHA increases caspase-9 activity in the JF-305 cells. This result is consistent with the release of Cyto C into the cytosol from the mitochondria, potentially activating caspase-9. The activated upstream caspase-9 acts on the downstream target of caspase-3 enzymes and subsequently on the activated caspase-3, ultimately inducing apoptosis.
The enhanced aerobic glycolysis in cancer cells is known as the Warburg effect, which is characterized by the dramatic increased rates of glucose uptake and utilization in cancer cells to levels higher than those observed in normal cells. Multiple molecules are involved in the regulation of the Warburg effect. The PI3K/Akt/mTOR signaling pathway is essential in the regulation of glucose metabolism in several types of cells.40 Akt is the best-studied downstream factor of PI3K signaling and an important driver of the glycolytic phenotype tumor. Akt also stimulates ATP generation, ensuring that the bioenergetics capacity required by cells to respond to growth signals. Akt phosphorylates key glycolytic enzymes, such as hexokinase, phosphofructokinase 2, and GLUT1.41,42 Our research showed that after treatment with DHA, the phosphorylation of Akt and mTOR decreased, and the expression of GLUT1 in JF-305 cells was inhibited.
5. Conclusions
In conclusion, our results demonstrated that DHA treatment could inhibit the proliferation and induce apoptosis in JF-305 pancreatic cancer cells. The potential underlying mechanism were associated with several different aspects, including the cell phase arrested in G2/M, decrease the MMP and accumulation of ROS, inhibition glucose uptake, lactate generation, and ATP production, regulation of the proteins associated with apoptosis. Moreover, DHA treatment also downregulated Akt/mTOR signaling pathway and inhibited glucose transporter 1 expression. Our date suggest that DHA treatment increased the apoptosis of JF-305 pancreatic cancer cells, and the effect of apoptosis may be associated with the inhibition of glycolysis. But further studies are still needed to explore more about these mechanisms.Conflicts of interest
The authors declare that there is no conflict of interest associated with this study.Acknowledgements
This work was supported by Excellent Youth Talents Training programs of Jilin City (No. 20156428), the Health and Family Planning Commission of Jilin Province (2015Q041), Department of Education of Jilin Province (JJKH20170419KJ), and China Postdoctoral Science Foundation of China (No. 2017M613412).References
- Q. J. Lin, F. Yang, C. Jin and D. L. Fu, World J. Gastroenterol., 2015, 21, 7988–8003 CrossRef PubMed.
- R. Slotwinski and S. M. Slotwinska, Cent. Eur. J. Immunol., 2016, 41, 392–403 CrossRef PubMed.
- G. Parker and H. Brotchie, J. Nerv. Ment. Dis., 2017, 205, 487–490 CrossRef PubMed.
- M. Ogrendik, Gastrointest. Tumors, 2017, 3, 125–127 CrossRef PubMed.
- G. D. Sun, C. Y. Li, W. P. Cui, Q. Y. Guo, C. Q. Dong, H. B. Zou, S. J. Liu, W. P. Dong and L. N. Miao, J. Diabetes Res., 2016, 2016, 5749857 Search PubMed.
- Y. Jaiswal, Z. Liang and Z. Zhao, J. Ethnopharmacol., 2016, 194, 245–259 CrossRef PubMed.
- R. Gaur, H. S. Cheema, Y. Kumar, S. P. Singh, D. K. Yadav, M. P. Darokar, F. Khan and R. S. Bhakuni, RSC Adv., 2015, 5, 47959–47974 RSC.
- C. J. Paddon, P. J. Westfall, D. J. Pitera, K. Benjamin, K. Fisher, D. McPhee, M. D. Leavell, A. Tai, A. Main, D. Eng, D. R. Polichuk, K. H. Teoh, D. W. Reed, T. Treynor, J. Lenihan, M. Fleck, S. Bajad, G. Dang, D. Dengrove, D. Diola, G. Dorin, K. W. Ellens, S. Fickes, J. Galazzo, S. P. Gaucher, T. Geistlinger, R. Henry, M. Hepp, T. Horning, T. Iqbal, H. Jiang, L. Kizer, B. Lieu, D. Melis, N. Moss, R. Regentin, S. Secrest, H. Tsuruta, R. Vazquez, L. F. Westblade, L. Xu, M. Yu, Y. Zhang, L. Zhao, J. Lievense, P. S. Covello, J. D. Keasling, K. K. Reiling, N. S. Renninger and J. D. Newman, Nature, 2013, 496, 528–532 CrossRef PubMed.
- H. Sun, X. Meng, J. Han, Z. Zhang, B. Wang, X. Bai and X. Zhang, Tumour Biol., 2013, 34, 3791–3800 CrossRef PubMed.
- M. X. Feng, J. X. Hong, Q. Wang, Y. Y. Fan, C. T. Yuan, X. H. Lei, M. Zhu, A. Qin, H. X. Chen and D. Hong, Sci. Rep., 2016, 6, 19074 CrossRef PubMed.
- T. Efferth, Semin. Cancer Biol., 2017, 46, 65–83 CrossRef PubMed.
- S. Slezakova and J. Ruda-Kucerova, Anticancer Res., 2017, 37, 5995–6003 Search PubMed.
- B. Kelly and L. A. O'Neill, Cell Res., 2015, 25, 771–784 CrossRef PubMed.
- G. J. Yoshida, J. Exp. Clin. Cancer Res., 2015, 34, 111 CrossRef PubMed.
- A. A. Fernandez-Ramos, V. Poindessous, C. Marchetti-Laurent, N. Pallet and M. A. Loriot, Biochimie, 2016, 127, 23–36 CrossRef PubMed.
- S. Y. Lunt and M. G. Vander Heiden, Annu. Rev. Cell Dev. Biol., 2011, 27, 441–464 CrossRef PubMed.
- S. Aminzadeh, S. Vidali, W. Sperl, B. Kofler and R. G. Feichtinger, Transl. Pediatr., 2015, 4, 20–32 Search PubMed.
- B. Bhattacharya, S. H. Low, C. Soh, N. Kamal Mustapa, M. Beloueche-Babari, K. X. Koh, J. Loh and R. Soong, Br. J. Pharmacol., 2014, 171, 3255–3267 CrossRef PubMed.
- C. Qu, J. Ma, X. Liu, Y. Xue, J. Zheng, L. Liu, J. Liu, Z. Li, L. Zhang and Y. Liu, Front. Cell. Neurosci., 2017, 11, 310 CrossRef PubMed.
- H. Jin, A. Y. Jiang, H. Wang, Y. Cao, Y. Wu and X. F. Jiang, Mol. Med. Rep., 2017, 16, 3475–3481 CrossRef PubMed.
- S. Du, G. Xu, W. Zou, T. Xiang and Z. Luo, Anti-Cancer Drugs, 2017, 28, 384–391 CrossRef PubMed.
- L. Wu, Y. Pang, G. Qin, G. Xi, S. Wu, X. Wang and T. Chen, PLoS One, 2017, 12, e0171840 Search PubMed.
- Y. J. Mi, G. J. Geng, Z. Z. Zou, J. Gao, X. Y. Luo, Y. Liu, N. Li, C. L. Li, Y. Q. Chen and X. Y. Yu, PLoS One, 2015, 10, e0120426 Search PubMed.
- V. Bhardwaj, N. Rizvi, M. B. Lai, J. C. K. Lai and A. Bhushan, Anticancer Res., 2010, 30, 743–749 Search PubMed.
- P. W. Sylvester, Methods Mol. Biol., 2011, 716, 157 Search PubMed.
- C. Saison, F. Perreault, J. C. Daigle, C. Fortin, J. Claverie, M. Morin and R. Popovic, Aquat. Toxicol., 2010, 96, 109–114 CrossRef PubMed.
- W. Yu, M. Ma, X. Chen, J. Min, L. Li, Y. Zheng, Y. Li, J. Wang and Q. Wang, Am. J. Chin. Med., 2017, 45, 1–12 CrossRef PubMed.
- H. Sebai, M. A. Jabri, A. Souli, K. Hosni, S. Selmi, H. Tounsi, O. Tebourbi, S. Boubaker, J. Elbenna and M. Sakly, RSC Adv., 2017, 4, 281–285 Search PubMed.
- Y. Li, Y. Wang, K. Rui, D. Xue, S. Pan, C. Hua and S. Bei, Oncotarget, 2016, 7, 62460–62473 Search PubMed.
- G. Jia, R. Kong, Z. B. Ma, B. Han, Y. W. Wang, S. H. Pan, Y. H. Li and B. Sun, J. Exp. Clin. Cancer Res., 2014, 33, 8 CrossRef PubMed.
- Z. Zhang, J. Wang, Y. Shen, C. Guo, K. E. Sai, F. Chen, X. Mei, F. U. Han and Z. Chen, Oncol. Lett., 2015, 10, 379 CrossRef PubMed.
- S. Fulda, Methods Enzymol., 2014, 542, 81–90 Search PubMed.
- W. Wu, P. Liu and J. Li, Crit. Rev. Oncol. Hematol., 2012, 82, 249–258 CrossRef PubMed.
- U. Hong-Duck, Oncotarget, 2016, 7, 5193–5203 Search PubMed.
- T. Vervliet, J. B. Parys and G. Bultynck, Oncogene, 2016, 35, 5079 CrossRef PubMed.
- J. H. Zheng, A. V. Follis, R. W. Kriwacki and T. Moldoveanu, FEBS J., 2016, 283, 2690 CrossRef PubMed.
- E. Khodapasand, N. Jafarzadeh, F. Farrokhi, B. Kamalidehghan and M. Houshmand, Iran. Biomed. J., 2015, 19, 69–75 Search PubMed.
- Z. Chang, J. Xing and X. Yu, Tumour Biol., 2014, 35, 753–758 CrossRef PubMed.
- L. Jing, M. T. He, Y. Chang, S. L. Mehta, Q. P. He, J. Z. Zhang and P. A. Li, Int. J. Biol. Sci., 2015, 11, 59 CrossRef PubMed.
- T. Ersahin, N. Tuncbag and R. Cetinatalay, Mol. BioSyst., 2015, 11, 1946 RSC.
- Y. Y. Bao, S. H. Zhou, Z. J. Lu, J. Fan and Y. P. Huang, Oncol. Rep., 2015, 34, 1805 CrossRef PubMed.
- H. Gwak, G. Haegeman, B. K. Tsang and Y. S. Song, Mol. Carcinog., 2015, 54, 1529 CrossRef PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2018 |