
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a palladium acetylide-based tubular microporous polymer monolith via a self-template approach: a potential precursor of supported palladium nanoparticles for heterogeneous catalysis†
Jeongmin Lee and
Ji Young Chang *
*
Department of Materials Science and Engineering, College of Engineering, Seoul National University, Seoul 08826, Korea. E-mail: jichang@snu.ac.kr; Tel: +82 2 880 7190
First published on 16th July 2018
Abstract
A monolithic, palladium acetylide-based conjugated microporous polymer, Pd-CMP, was synthesized from a palladium dichloride and a trialkyne. The polymerization proceeded in two different ways, the dehydrohalogenation reaction between the alkyne and the palladium halide and the homocoupling reaction of the alkyne. Pd-CMP had a rigid hollow tubular structure. The in situ formed crystalline triethylammonium chloride (TEACl) rod played a critical role in the formation of the tubular morphology as a template. Through the attachment of the polymer particles to the surface of the rod and their reactions with soluble alkynes, a core–shell structure with a TEACl core and a polymer shell formed. The TEACl core was removed by washing with methanol to yield a hollow polymer tube. Pd-CMP showed a hierarchical pore structure and reversible compressibility. Supported Pd nanoparticles were prepared by one-step thermolysis of Pd-CMP as a heterogeneous catalyst. The average diameters of NPs in the products thermolyzed at 300 (Pd-CMP300) and 500 °C (Pd-CMP500) were 2.6 and 4.1 nm, respectively. Pd-CMP300 was used in the heterogeneous catalysis of the 4-nitrophenol reduction reaction and Suzuki–Miyaura coupling between iodobenzene and phenylboronic acid. The reaction yields were higher than 95%. The catalyst could be used for a flow reaction and easily recycled without significant activity loss.
Introduction
Microporous materials such as zeolites, activated carbons, metal–organic frameworks and microporous organic polymers (MOPs) have been extensively researched as adsorbents and supports. MOPs have drawn particular interest in recent years because they have flexibility in the architectural design. MOPs can have tuned functionalities to serve specific applications such as selective gas storage and separation,1,2 chemical sensing3 and heterogeneous catalysis.4–6Heterogeneous catalysts are widely used in the chemical industry due to their advantages of easy separation from reaction mixtures, recyclability, tolerance in harsh operating conditions and usability in continuous flow reactors.7,8 Most heterogeneous catalysts are supported catalysts, where active species are immobilized on porous materials.9 MOPs are attractive candidates for catalyst supports. The flexibility in their architectural designs allows the interactions between the active species and the supports to be adjusted. Furthermore, hierarchically porous structures of some MOPs facilitate the mass transfer of reactants, leading to the enhancement of the reaction rates. MOPs with conjugated structures have been used to support metal nanoparticles (NPs) in heterogeneous catalysis. Metal NPs are generally loaded in a MOP support by two step processes, in which soluble metal complexes are first dispersed in the support and then are reduced to metal NPs.10–13 This in situ reduction approach has an advantage in preventing the NP aggregation compared to the direct incorporation of metal NPs, but the even distribution of NPs in the support is still a challenge. When metal precursors are externally added to the microporous support for the reduction, the micropore diffusion resistance can be encountered.
Conjugated polymers having transition metal–carbon σ-bonds in the main chain are an interesting class of materials because of their unique bonding properties.14 Acetylides have strong σ-donating properties towards d-block transition metals with minimal steric hindrance around the donor carbon atom to form stable metal-carbon bonds.15 Many polyyne polymers having group 10 elements such as nickel (Ni), platinum (Pt) and palladium (Pd) have been synthesized by the dehydrohalogenation reaction between diterminal alkynes and metal halides or the oxidative coupling reaction of metal-containing monomers with two terminal alkynyl groups.14–16
In this work, we present the synthesis of a palladium acetylide-based microporous polymer monolith. The polymer monolith showed an interesting morphology, where rigid hollow polymer tubes were piled. We investigated the mechanism of the tubular structure formation and explored the possible use of the polymer as a precursor of a heterogeneous Pd catalyst.
Experimental section
Materials
Copper iodide, 4-nitrophenol (4-NP) and iodobenzene were purchased from Sigma-Aldrich. 1,3,5-Triethynylbenzene, sodium borohydride (NaBH4), triethylamine hydrochloride (TEACl) and phenylboronic acid were purchased from Tokyo Chemical Industry. Trans-PdCl2(PEt3)2 was obtained from Alfa Aesar.Characterization
Fourier-transform infrared (FT-IR) spectra were recorded by a Bruker Vertex 80v spectrometer using a KBr pellet. Solid state 13C NMR and 31P NMR spectra were obtained by a Bruker Avance 400WB spectrometer (100 MHz) equipped with a cross polarization magic angle spinning (CP/MAS) probe. 1H NMR spectra were obtained by a Bruker Avance-300 (300 MHz). Thermogravimetric analysis (TGA) was performed by a TA modulated TGA2050 with a heating rate of 10 °C min−1 under nitrogen and air. Scanning electron microscopy (SEM) images were obtained by a JEOL JSM-6330F microscope. Transmission electron microscope (TEM) images were obtained by a JEOL JEM-2010 microscope at 200 keV. TEM samples were dispersed in methanol by using a sonicator and a drop was placed on a TEM grid. Powder X-ray diffraction (PXRD) data were collected on a Bruker New D8 Advance using a Cu Kα source (λ = 1.5418 Å). N2 adsorption–desorption isotherms were measured at 77 K with a Belsorp-Max (BEL Japan, Inc.) apparatus. The pore size distribution was calculated by nonlocal density functional theory (NL-DFT). Before analysis, the samples were degassed at 120 °C for 12 h under vacuum. Inductively coupled plasma mass spectrometry (ICP-MS) was performed using a Varian 820-MS ICP-MS spectrometer. UV-Vis spectra were recorded with a Sinco S-3150 spectrometer. The compression tests were performed by a KES-FB3 automatic compression tester with a strain rate of 50 mm min−1.Synthesis and thermolysis of a palladium acetylide containing conjugated microporous polymer (Pd-CMP)
1,3,5-Triethynylbenzene (150 mg, 1 mmol) and trans-PdCl2(PEt3)2 (82.3 mg, 0.2 mmol) were dissolved in a mixture of toluene (6 mL) and triethylamine (3 mL) solution. CuI (9 mg, 47.3 μmol) was added to the solution and heated without stirring at 70 °C under aerobic conditions. The solution turned to a brownish gel in about 30 min. The monolithic gel was isolated after the reaction of 24 h and washed with methanol, acetone and THF. After Soxhlet extraction with methanol and THF, the polymer was dried in vacuo to give a light brownish monolith (94% yield).Pd-CMP was heated in a tube furnace under a nitrogen atmosphere for 1 h at 300 and 500 °C to yield the thermolysed products, Pd-CMP300 and Pd-CMP500, respectively.
Reduction of 4-nitrophenol with Pd-CMP300
Pd-CMP300 (60 mg) was fabricated to have a shape that fitted into a 10 mL syringe. The syringe was plugged with Pd-CMP300 and used for the flow reduction reaction of 4-nitrophenol (4-NP). An excess amount of NaBH4 (3 mM) was added to an aqueous solution of 4-NP (0.1 mM). The solution was transferred into the syringe and was passed through Pd-CMP300 at a rate of 2.5 mL min−1 with a syringe pump.Suzuki–Miyaura coupling reaction with Pd-CMP300
Phenylboronic acid (73.2 mg, 0.6 mmol), iodobenzene (102.0 mg, 0.5 mmol), potassium carbonate (138.2 mg, 1 mmol) and Pd-CMP300 (5 mg) were added to an aqueous ethanol solution (4 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). After stirring at 70 °C for 1 h, the reaction mixture was diluted with 10 mL of water and then was extracted with 10 mL of ethyl acetate three times. The organic extract was concentrated under reduced pressure and the product yield was determined by 1H NMR spectroscopy.
:1, v/v). After stirring at 70 °C for 1 h, the reaction mixture was diluted with 10 mL of water and then was extracted with 10 mL of ethyl acetate three times. The organic extract was concentrated under reduced pressure and the product yield was determined by 1H NMR spectroscopy.
For recycling, the catalyst was separated by centrifugation and washed by ethanol and ethyl acetate.
Results and discussion
A palladium acetylide-based conjugated microporous polymer, Pd-CMP, was synthesized by the reaction of 1,3,5-triethynylbenzene with trans-PdCl2(PEt3)2 in the presence of CuI as a catalyst (Fig. 1). The reaction was performed at 70 °C in a co-solvent of toluene and triethylamine without stirring. The reaction mixture turned highly viscous as the reaction proceeded and became a macroscopic gel in 30 min (Fig. S1, ESI†). A self-standing monolith was isolated after 24 h, which maintained its original shape even after solvent washing and drying. The density of the dried Pd-CMP monolith was 26 mg cm−3.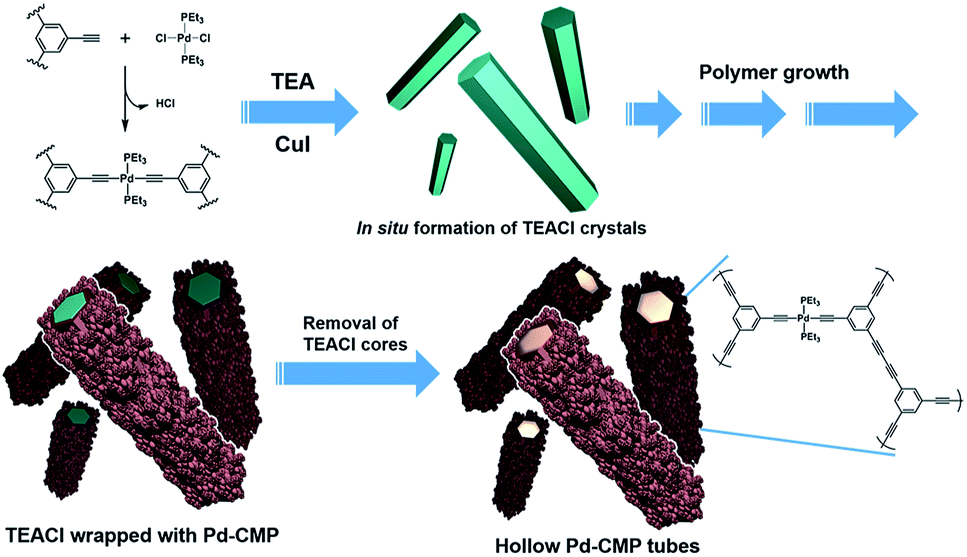 | ||
| Fig. 1 Schematic drawing of the tubular microstructure formation process of Pd-CMP. | ||
A compound having multiple terminal alkyne groups is a versatile building block for the synthesis of microporous polymers. The Sonogashira–Hagihara cross-coupling reaction of terminal alkynes with aryl halides has been used for the synthesis of CMPs.17,18 The oxidative homocoupling reaction of terminal alkynes is also useful for the extension of a carbon skeleton. There have been reported a few examples of CMPs prepared by the homocoupling reaction of terminal alkynes.19,20 Another interesting feature of a terminal alkyne is that it has an acidic hydrogen which can be removed easily to generate an acetylide. A variety of metal acetylides are synthesized by the reaction between metal halides and terminal alkynes. Linear organometallic polymers are obtained when bisalkynes and metal dihalide are used as monomers.21,22
The reaction of 1,3,5-triethynylbenzene with trans-PdCl2(PEt3)2 proceeded in two different ways. Under the basic reaction conditions, copper acetylides likely formed first from copper iodide and terminal alkynes. They could be intermediates of the alkyne–alkyne homocoupling reaction or be used for the Pd–alkyne complexation through the trans-metalation process. The both reactions occurred competitively to produce a highly cross-linked structure.
The Pd-CMP monolith was reversibly compressible. Fig. 2 shows the compression-release test of the monolith at 40% maximum strains and the monolith recovered its own shape after release. We previously reported monolithic and compressible microporous polymers prepared by the Sonogashira–Hagihara cross-coupling reaction of a trialkyne with aromatic dihalide.23,24 It was found that the monolithic and compressible properties of the polymers were originated from their microstructures consisting of hollow polymer fibers. However, the formation mechanism of such intriguing microstructures was not clearly understood.
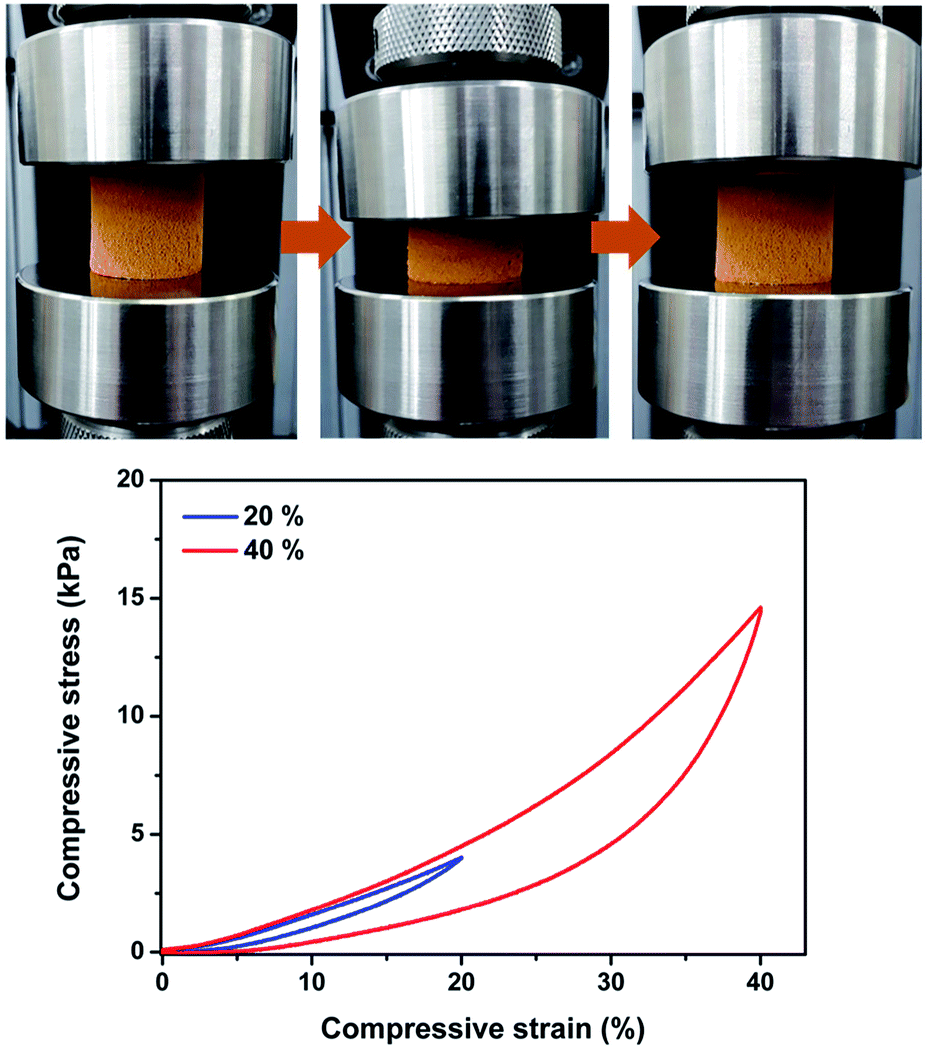 | ||
| Fig. 2 Photo images of the Pd-CMP monolith taken during the compression test and the compressive stress–strain curves (maximum strain = 20 and 40%). | ||
The Pd-CMP monolith also had a tubular microstructure as observed in the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 3). The elemental mapping images of Pd-CMP by TEM (Fig. 4) showed the presence of evenly distributed carbon and palladium elements in the polymer tube. The high angle annular dark field (HAADF) image also exhibited no Pd aggregates in the polymer. In particular, the carbon signal in the tube wall was stronger than in the core region, suggesting the hollow structure of Pd-CMP.25 Although there was a variation in the diameters of the hollow cores from hundreds to thousands nanometers, most tubes had hexagonal ends (Fig. S2, ESI†). The lengths of the polymer tubes were in the range of tens to hundreds micrometers. These long tubes were piled on one another to form a compressible monolith.
 | ||
| Fig. 3 (a–c) The SEM images of Pd-CMP at different magnifications and (d) the TEM image of Pd-CMP. | ||
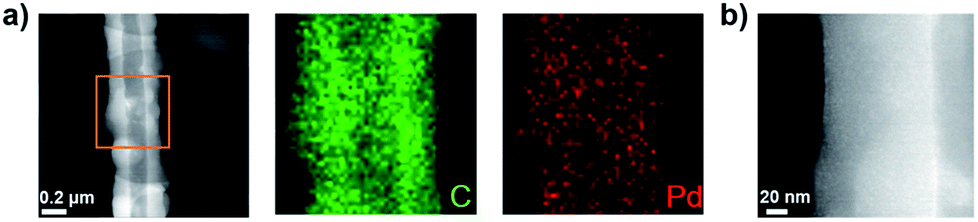 | ||
| Fig. 4 (a) Elemental mapping of the selected area in Pd-CMP and (b) HAADF image of Pd-CMP. | ||
To elucidate the formation mechanism of the tubular structure, the products that formed in the early stages of the reaction were analysed. Yellowish precipitates began to form immediately after the reaction started. They were collected by filtration in 5 min and the morphology was investigated by SEM. The precipitates were hexagonal rods with flat surfaces (Fig. S3a, ESI†). When isolated after 15 min reaction time, the initially flat surface of the hexagonal rods became rough with small particles (Fig. S3b, ESI†). The X-ray diffraction (XRD) analysis showed that the hexagonal rod was crystalline triethylamine hydrochloride (TEACl) (Fig. S3c, ESI†). The FT-IR spectrum of the hexagonal rods was also almost identical with that of the commercial TEACl (Fig. S4, ESI†). TEACl could be formed by the reaction of the solvent, TEA with HCl which was generated as a byproduct in the Pd–alkyne dehydrohalogenation reaction as shown in Fig. 1.
These observations suggested that the TEACl crystals played a critical role in the formation of the hexagonal shaped tubular morphology as a template. The possible mechanism of the tubular structure formation of Pd-CMP is schematically drawn in Fig. 1. Upon the addition of CuI to a solution of PdCl2(PEt3)2 and the trialkyne in TEA/toluene, the dehydrohalogenation reaction occurred rapidly to form a Pd acetylide. TEACl which formed from the evolved HCl and TEA precipitated in the form of a crystalline hexagonal rod. As the reaction proceeded, insoluble cross-linked polymer particles were produced and adhered to the surface of the TEACl rod in the process of minimizing surface energy.26,27 Through the attachment of the polymer particles and their reactions with soluble alkynes on the surface of the rod, a core–shell structure with a TEACl core and a polymer shell eventually formed. The TEACl core was removed by washing with methanol to yield a hollow polymer tube. A tubular structure with many voids was obtained when the polymer was isolated and washed before the completion of the reaction (Fig. S5, ESI†).
The chemical structures produced by the two reactions were confirmed by Fourier transform infrared spectroscopy (FT-IR) and solid-state 13C and 31P CP/MAS NMR spectroscopy (Fig. 5). The 31P NMR spectrum exhibited a singlet peak at δ = 19 ppm, corresponding to the triethylphosphine ligand attached to the Pd atom.28 In the 13C NMR spectrum, the peaks at 16.5 and 7.0 ppm were assigned to the carbons of the triethylphosphine ligand. These results suggested the introduction of palladium elements in the polymer framework. Aromatic carbon peaks appeared at 135, 131 and 123 ppm, and the carbon peaks of acetylene groups showed up at 90 and 80 ppm. The formation of Pd–C bonds was also corroborated by FT-IR spectroscopy. The spectrum showed the characteristic peak of the alkyne group attached to the heavy metal center (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Pd) at 2100 cm−1 in addition to the peak for –CC–C stretching vibration at 2195 cm−1.29 The powder X-ray diffraction (PXRD) pattern of Pd-CMP did not show any discernible signals, indicating that Pd-CMP had an amorphous nature (Fig. S6, ESI†). The mass of Pd in the polymer was determined by TGA in air was 10.5 wt%, which was comparable with the theoretical palladium content (9.9 wt%) calculated from the reactants (Fig. S7, ESI†).
C–Pd) at 2100 cm−1 in addition to the peak for –CC–C stretching vibration at 2195 cm−1.29 The powder X-ray diffraction (PXRD) pattern of Pd-CMP did not show any discernible signals, indicating that Pd-CMP had an amorphous nature (Fig. S6, ESI†). The mass of Pd in the polymer was determined by TGA in air was 10.5 wt%, which was comparable with the theoretical palladium content (9.9 wt%) calculated from the reactants (Fig. S7, ESI†).
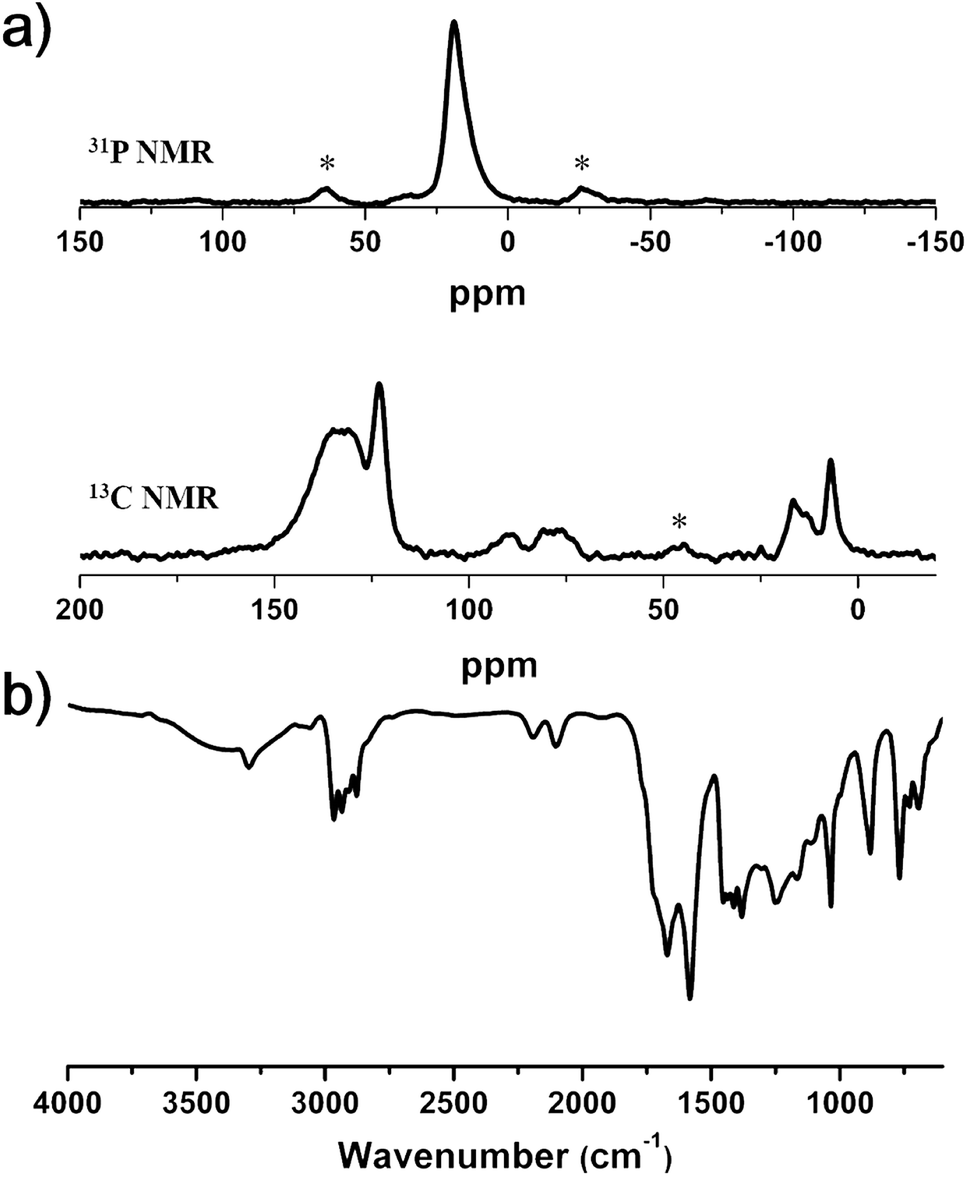 | ||
| Fig. 5 (a) Solid-state 31P (up) and 13C (down) CP/MAS NMR spectra and (b) FT-IR spectrum of Pd-CMP (asterisks denote spinning sidebands). | ||
The porosity of the Pd-CMP was investigated by the nitrogen gas (N2) adsorption–desorption measurement at 77 K (Fig. 6, Table S1, ESI†). The adsorption–desorption curves of Pd-CMP showed type I sorption isotherms with a steep increase of nitrogen uptake at the very low relative pressure region (p/p0 < 0.01), indicating the presence of abundant micropores within the polymer. The calculated Brunauer–Emmett–Teller (BET) surface areas of Pd-CMP was 1236 m2 g−1, and total pore and micropore volumes were 1.04 and 0.44 cm3 g−1, respectively. The pore size distribution of Pd-CMP, determined based on the non-local density functional theory (NL-DFT), showed the presence of micro- and mesopores (Fig. 6, Table S1, ESI†).
 | ||
| Fig. 6 N2 adsorption–desorption isotherms measured at 77 K and NL-DFT pore size distribution of the Pd-CMP (inset). | ||
The controlled thermolysis of microporous Pd-CMP could be an efficient method to prepare supported Pd NPs. Since Pd-CMP contained palladium acetylide units in the polymer backbone as a constituent, its thermolysis was expected to form Pd NPs on the inner pore walls of the microporous polymer. The thermolysis of the Pd-CMP was carried out under a nitrogen atmosphere at two different temperatures, 300 and 500 °C for 1 h. The FT-IR spectrum of the thermolysis product at 300 °C showed no peaks corresponding to Pd–CC bonds at 2100 cm−1, suggesting their complete dissociation (Fig. S8, ESI†). The TEM images of the products showed a uniform distribution of Pd NPs with near-spherical shapes in the polymer matrix (Fig. 7). The average diameters of NPs in the thermolysis products at 300 (Pd-CMP300) and 500 °C (Pd-CMP500) were 2.6 and 4.1 nm, respectively (Fig. S9, ESI†). The increase in an average Pd NP size as the thermolysis temperature increased could be explained by the confinement effect of pores. The particle growth occurred mainly in a porous scaffold at low temperatures, but as the temperature increased, the porous structure would collapse and NPs agglomerated together to form larger particles.
 | ||
| Fig. 7 TEM images of (a and b) Pd-CMP300 and (c and d) Pd-CMP500. | ||
NPs attached on the outer tube surface were not observed by TEM, suggesting that most Pd NPs formed in the inner space of the polymer.30 The powder XRD analysis of Pd NPs (Fig. S10, ESI†) showed the diffraction peaks at 40.2 and 46.8°, which were indexed as the (1 1 1) and (2 0 0) facets of the palladium face-centered cubic (fcc) phase. The broad widths of the peaks with low intensities indicated that Pd NPs were small in size.31 We presume that the reductive elimination occurred to form Pd(0) during the thermolysis, but further study on the elimination mechanism and structural change of the polymer backbone is needed.
Pd-CMP300 having a monolithic shape with less change in the polymer structure was selected for the catalytic application. The BET surface area and total pore volume of Pd-CMP300 were 757 m2 g−1 and 0.35 cm3 g−1 (Fig. S11, ESI†), respectively, which decreased compared with those of Pd-CMP possibly because some pores collapsed partially and some of them were occupied by in situ formed NPs. The Pd content in Pd-CMP300 was 13.5 wt% when measured by TGA in air (Fig. S12, ESI†) and 11.6 wt% when determined by ICP-MS. Pd-CMP300 (60 mg) was fabricated to have a shape that fitted into a 10 mL syringe and used for the flow reduction of 4-nitrophenol (4-NP). An aqueous solution of 4-NP (0.1 mM) and sodium borohydride was transferred in a syringe and pressed to pass through the Pd-CMP300 monolith with a flow rate of 2.5 mL min−1 (Fig. 8). The yellowish solution of 4-nitrophenolate ion turned colorless after passing through the monolith. The UV-Vis spectrum showed that the absorption peak of 4-nitrophenolate at 400 nm disappeared, and a new peak at 300 nm developed from 4-aminophenol, the product of the reduction reaction.
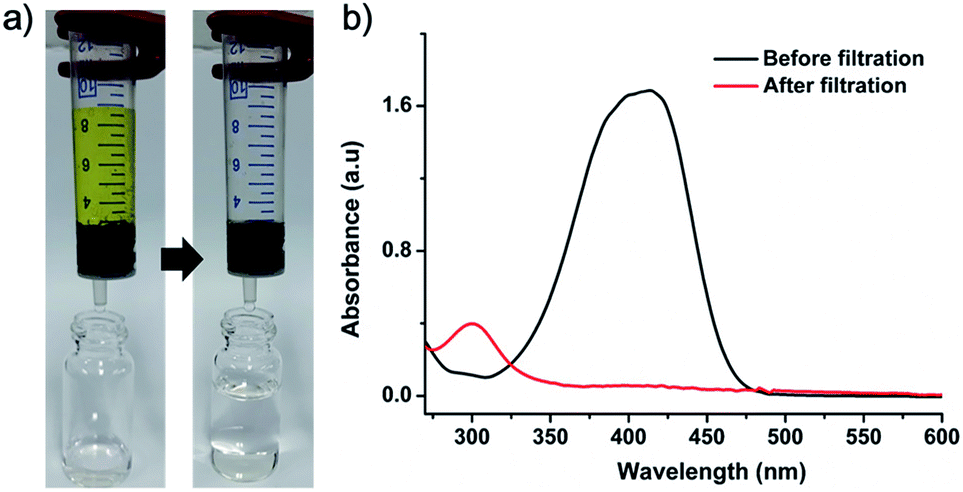 | ||
| Fig. 8 (a) Sequential photographs showing the 4-NP reduction reaction with Pd-CMP300 and (b) UV-Vis spectra of the 4-NP stock solution and the filtered solution. | ||
Pd-CMP300 also successfully catalysed the Suzuki–Miyaura coupling reaction between iodobenzene and phenylboronic acid in an aqueous ethanol solution (Fig. S13, ESI†).32–35 The product yield was higher than 95%. Pd-CMP300 was easily recovered and recycled five times without significant loss of the catalytic activity (Fig. S14, ESI†).
Conclusions
In summary, we demonstrated the use of a palladium acetylide-based microporous polymer as a precursor for the one-step synthesis of a heterogeneous catalyst. Pd-CMP was prepared from a palladium dichloride and a trialkyne. It had a hollow tubular structure constructed by the template-directed process, where an in situ formed TEACl rod acted as a template. The polymer tubes were irregularly piled one upon another to form a monolith. Due to the uniformly distributed palladium acetylide groups in the polymer backbone and the confined effect of the porous structure, well dispersed Pd NPs with a narrow size distribution formed in the polymer support by the thermolysis. Pd-CMP300 showed good catalytic activity in the 4-NP reduction. The catalyst could be used for a flow reaction and easily recycled without significant activity loss. We believe that the successful application of a Pd–acetylide polymer as a heterogeneous catalyst precursor would provide new insights for the catalyst design based on organometallic microporous polymers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2015R1A2A2A01006585).Notes and references
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173 RSC
.
- Y. Zhuang, J. G. Seong, Y. S. Do, W. H. Lee, M. J. Lee, M. D. Guiver and Y. M. Lee, J. Membr. Sci., 2016, 504, 55–65 CrossRef
- X. Wu, H. Li, B. Xu, H. Tong and L. Wang, Polym. Chem., 2014, 5, 4521 RSC
- D. S. Kundu, J. Schmidt, C. Bleschke, A. Thomas and S. Blechert, Angew. Chem., Int. Ed., 2012, 51, 5456–5459 CrossRef PubMed
- M. H. Kim, T. Song, U. R. Seo, J. E. Park, K. Cho, S. M. Lee, H. J. Kim, Y. J. Ko, Y. K. Chung and S. U. Son, J. Mater. Chem. A, 2017, 5, 23612–23619 RSC
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960–1967 CrossRef PubMed
- K. Wu, J. Guo and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 6013–6017 CrossRef PubMed
- J. M. Tobin, J. Liu, H. Hayes, M. Demleitner, D. Ellis, V. Arrighi, Z. Xu and F. Vilela, Polym. Chem., 2016, 7, 6662–6670 RSC
- M. Campanati, G. Fornasari and A. Vaccari, Catal. Today, 2003, 77, 299–314 CrossRef
- H. L. Cao, H. B. Huang, Z. Chen, B. Karadeniz, J. Lü and R. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 5231–5236 CrossRef PubMed
- T. Ishida, Y. Onuma, K. Kinjo, A. Hamasaki, H. Ohashi, T. Honma, T. Akita, T. Yokoyama, M. Tokunaga and M. Haruta, Tetrahedron, 2014, 70, 6150–6155 CrossRef
- B. Li, Z. Guan, W. Wang, X. Yang, J. Hu, B. Tan and T. Li, Adv. Mater., 2012, 24, 3390–3395 CrossRef PubMed
- P. Pachfule, M. K. Panda, S. Kandambeth, S. M. Shivaprasad, D. D. Díaz and R. Banerjee, J. Mater. Chem. A, 2014, 2, 7944–7952 RSC
- K. A. Williams, A. J. Boydston and C. W. Bielawski, Chem. Soc. Rev., 2007, 36, 729–744 RSC
- R. Buschbeck, P. J. Low and H. Lang, Coord. Chem. Rev., 2011, 255, 241–272 CrossRef
- N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2003, 42, 2586–2617 CrossRef PubMed
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef PubMed
- J. H. Park, J. H. Ko, S. Hong, Y. J. Shin, N. Park, S. Kang, S. M. Lee, H. J. Kim and S. U. Son, Chem. Mater., 2015, 27, 5845–5848 CrossRef
- J. J. Chen, T. L. Zhai, Y. F. Chen, S. Geng, C. Yu, J. M. Liu, L. Wang, B. Tan and C. Zhang, Polym. Chem., 2017, 8, 5533–5538 RSC
- R. Du, N. Zhang, H. Xu, N. Mao, W. Duan, J. Wang, Q. Zhao, Z. Liu and J. Zhang, Adv. Mater., 2014, 26, 8053–8056 CrossRef PubMed
- Y. Miyagi, Y. Shibutani, Y. Otaki and F. Sanda, Polym. Chem., 2016, 7, 1070–1078 RSC
- W. Wang and H. B. Yang, Chem. Commun., 2014, 50, 5171–5186 RSC
- Y. Lim, M. C. Cha and J. Y. Chang, Sci. Rep., 2015, 5, 15957 CrossRef PubMed
- J. G. Kim, M. C. Cha, J. Lee, T. Choi and J. Y. Chang, ACS Appl. Mater. Interfaces, 2017, 9, 38081–38088 CrossRef PubMed
- S. Hong, J. Yoo, N. Park, S. M. Lee, J. G. Park, J. H. Park and S. U. Son, Chem. Commun., 2015, 51, 17724–17727 RSC
- Z. Wang, J. Liu, Y. Fu, C. Liu, C. Pan, Z. Liu and G. Yu, Chem. Commun., 2017, 53, 4128–4131 RSC
- L. Zhu, Y. Xu, W. Yuan, J. Xi, X. Huang, X. Tang and S. Zheng, Adv. Mater., 2006, 18, 2997–3000 CrossRef
- V. Wing-Wah Yam and L. Zhang, J. Chem. Soc., Dalton Trans., 2001, 0, 1111–1116 RSC
- J. Lewis, P. R. Raithby and W. Y. Wong, J. Organomet. Chem., 1998, 556, 219–228 CrossRef
- H. Liu, L. Chang, C. Bai, L. Chen, R. Luque and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 5019–5023 CrossRef PubMed
- W. Xu, X. Wang, Q. Zhou, B. Meng, J. Zhao, J. Qiu and Y. Gogotsi, J. Mater. Chem., 2012, 22, 14363 RSC
- S. Paul, M. M. Islam and S. M. Islam, RSC Adv., 2015, 5, 42193–42221 RSC
- I. Maluenda and O. Navarro, Molecules, 2015, 20, 7528–7557 CrossRef PubMed
- D. Roy and Y. Uozumi, Adv. Synth. Catal., 2018, 360, 602–625 CrossRef
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Porosity and characterization data of the polymers and the catalytic reaction results. See DOI: 10.1039/c8ra03275k |
| This journal is © The Royal Society of Chemistry 2018 |